Stereoselective Synthesis of Terpenoids through Lipase-Mediated Resolution Approaches



{kind=link}
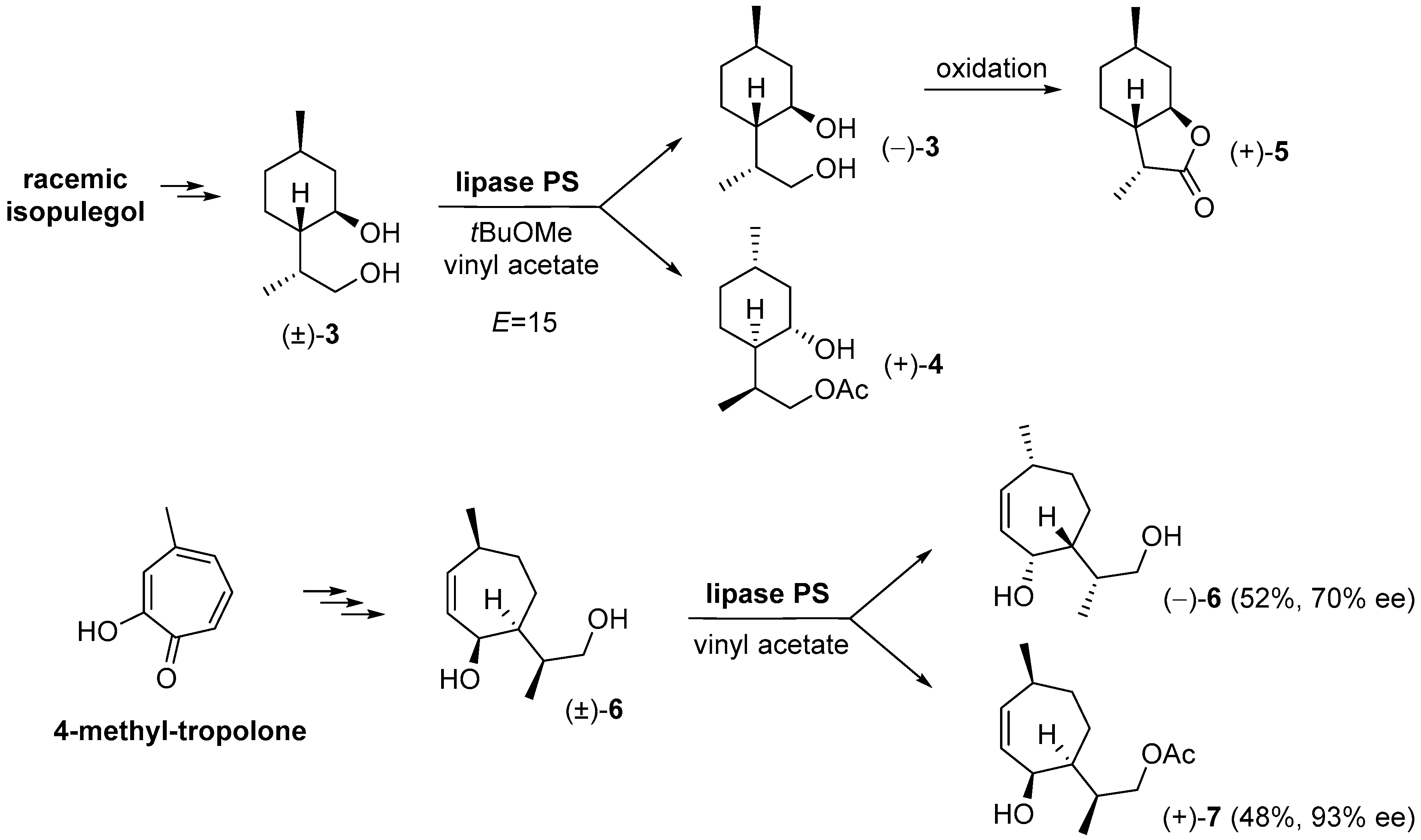{kind=link}
{kind=link}
{kind=link}
{kind=link}
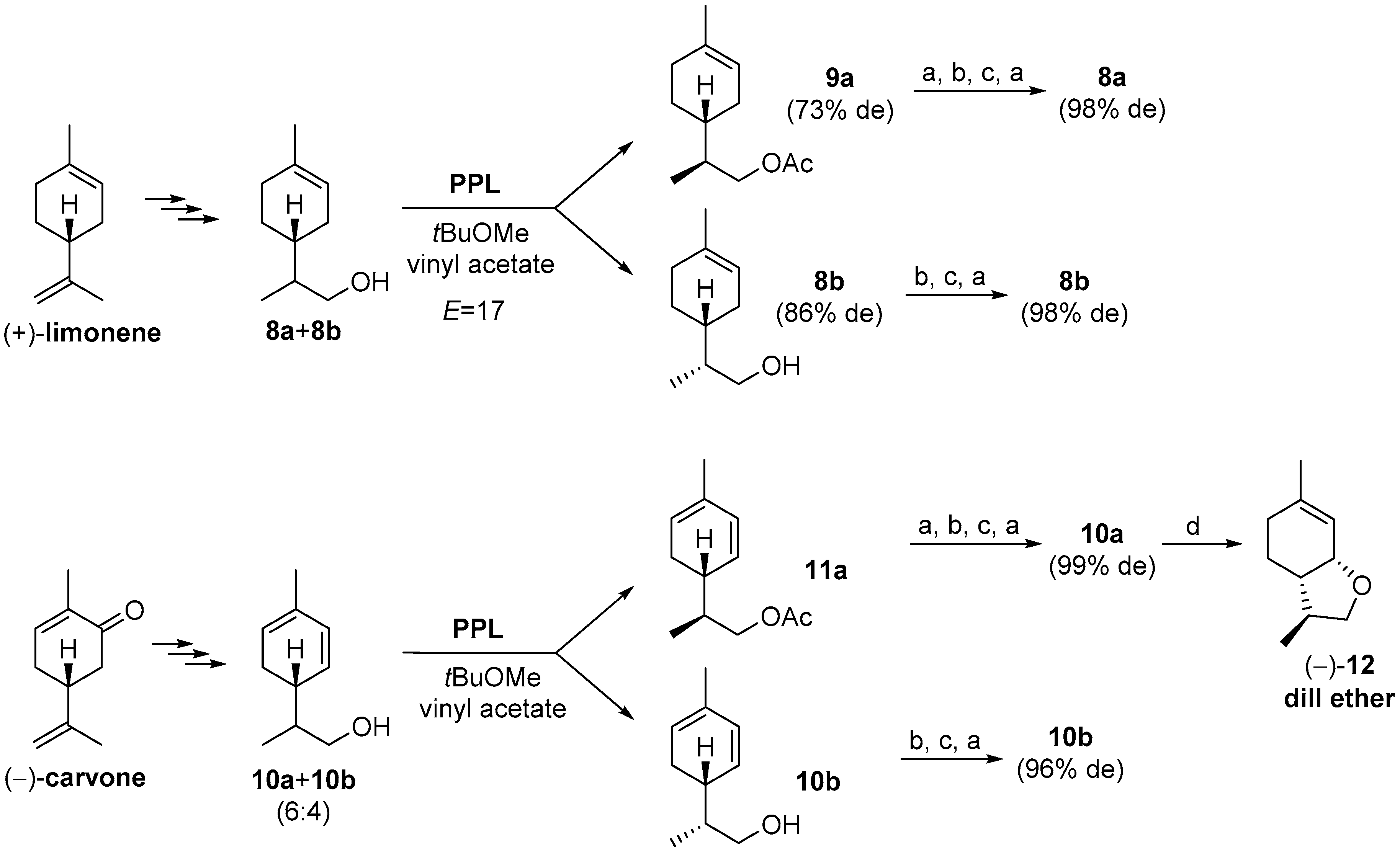
{kind=link}
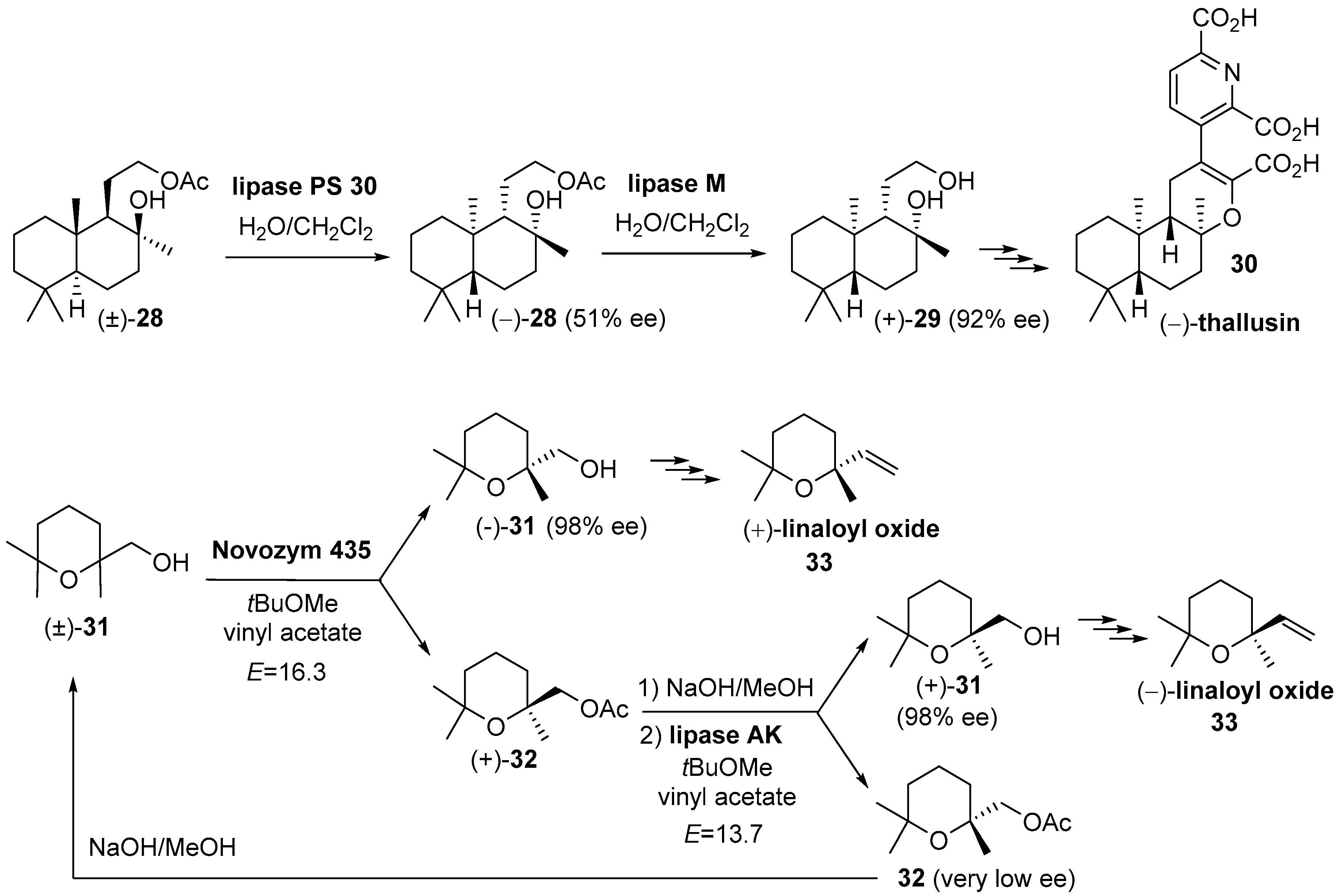
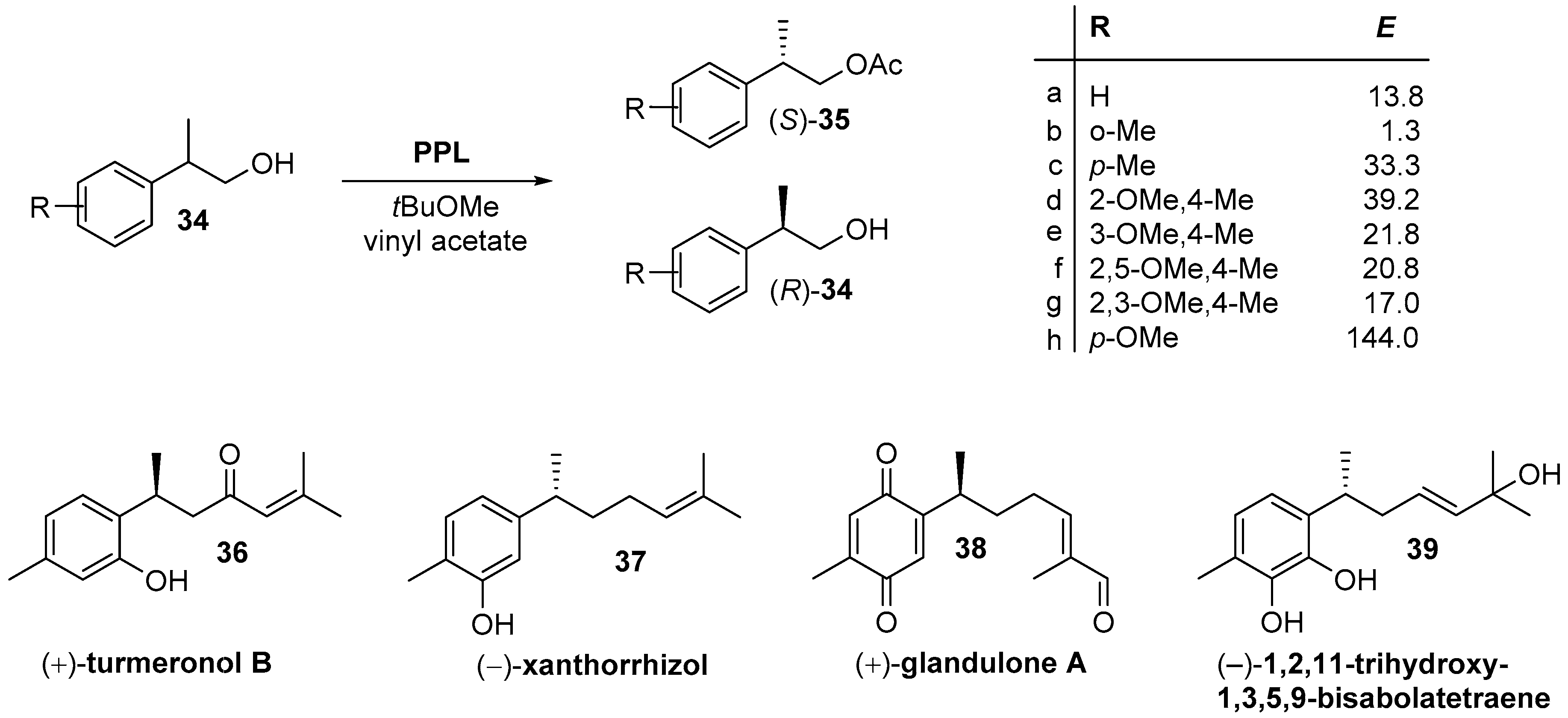{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Lipase-Mediated Resolution of Racemic Alcohols or Their Esters
2.1. Primary Alcohols
2.2. Secondary Alcohols
2.3. Tertiary Alcohols
3. Lipase-Mediated Resolution of Racemic Acid Derivatives
4. Lipase-Mediated Desymmetrization of Meso Compounds
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PPL | porcine pancreas lipase; |
| Novozyme 435 | immobilized lipase B from Candida antartica; |
| lipase PS | lipase from Burkholderia cepacia; |
| CAL-B | Candida antartica lipase B; |
| lipase PL-266 | lipase from Alcaligenes sp.; |
| lipase QL | lipase from Alcaligenes sp.; |
| lipase P | lipase from Pseudomonas cepacia; |
| lipase PS-30 | lipase from Pseudomonas cepacia; |
| lipase M | lipase from Mucor javanicus; |
| lipase AK | lipase from Pseudomonas fluorescens; |
| lipase OF 360 | lipase from Candida cylindracea; |
| CRL | Candida rugosa lipase; |
| CCL | Candida cylindracea lipase; |
| CRL LIP1 | Candida rugosa lipase; |
| Chirazyme L-2 | immobilized lipase from Candida Antarctica; |
| lipase PS-C | lipase from Burkholderia cepacia immobilized on ceramic; |
| lipase PS-IM | lipase from Burkholderia cepacia immobilized on diatomaceous heart. |
References
- Schmid, R.D.; Verger, R. Lipases: Interfacial enzymes with attractive applications. Angew. Chem. Int. Edit. 1998, 37, 1608–1633. [Google Scholar] [CrossRef]
- Carrea, G.; Riva, S. Properties and synthetic applications of enzymes in organic solvents. Angew. Chem. Int. Edit. 2000, 39, 2226–2254. [Google Scholar] [CrossRef]
- Filho, D.G.; Silva, A.G.; Guidini, C.Z. Lipases: Sources, immobilization methods, and industrial applications. Appl. Microbiol. Biotechnol. 2019, 103, 7399–7423. [Google Scholar] [CrossRef]
- Theil, F. Lipase-Supported Synthesis of Biologically Active Compounds. Chem. Rev. 1995, 95, 2203–2227. [Google Scholar] [CrossRef]
- Patel, R.N. Biocatalysis: Synthesis of Key Intermediates for Development of Pharmaceuticals. ACS Catal. 2011, 1, 1056–1074. [Google Scholar] [CrossRef]
- Norjannah, B.; Ong, H.C.; Masjuki, H.H.; Juan, J.C.; Chong, W.T. Enzymatic transesterification for biodiesel production: A comprehensive review. RSC Adv. 2016, 6, 60034–60055. [Google Scholar] [CrossRef]
- Abate, A.; Brenna, E.; Fuganti, C.; Gatti, F.G.; Serra, S. Lipase-catalysed preparation of enantiomerically enriched odorants. J. Mol. Catal. B Enzym. 2004, 32, 33–51. [Google Scholar] [CrossRef]
- Serra, S.; Fuganti, C.; Brenna, E. Biocatalytic preparation of natural flavours and fragrances. Trends Biotechnol. 2005, 23, 193–198. [Google Scholar] [CrossRef]
- Chênevert, R.; Pelchat, N.; Jacques, F. Stereoselective Enzymatic Acylations (Transesterifications). Curr. Org. Chem. 2006, 10, 1067–1094. [Google Scholar] [CrossRef]
- Monti, H.; Audran, G. Lipases-Promoted Enantioselective Syntheses of Monocyclic Natural Products. Mini Rev. Org. Chem. 2005, 2, 265–281. [Google Scholar] [CrossRef]
- Serra, S. Enzyme-mediated synthesis of sesquiterpenes. Nat. Prod. Commun. 2015, 10, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.; Rathod, V.K. Enzyme catalyzed synthesis of cosmetic esters and its intensification: A review. Process. Biochem. 2015, 50, 1793–1806. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Woodley, J.M. Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 2017, 118, 801–838. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A. Trends in lipase-catalyzed asymmetric access to enantiomerically pure/enriched compounds. Tetrahedron 2007, 63, 1721–1754. [Google Scholar] [CrossRef]
- García-Urdiales, E.; Alfonso, I.; Gotor, V. Enantioselective Enzymatic Desymmetrizations in Organic Synthesis. Chem. Rev. 2005, 105, 313–354. [Google Scholar] [CrossRef] [PubMed]
- Sakauchi, H.; Kiyota, H.; Oritani, T.; Kuwahara, S.; Takigawa, S.-Y. Enzymatic Resolution and Odor Description of Both Enantiomers of Lavandulol, a Fragrance of Lavender Oil. Chem. Biodivers. 2005, 2, 1183–1186. [Google Scholar] [CrossRef]
- Cross, H.; Marriott, R.; Grogan, G. Enzymatic esterification of lavandulol—A partial kinetic resolution of (S)-lavandulol and preparation of optically enriched (R)-lavandulyl acetate. Biotechnol. Lett. 2004, 26, 457–460. [Google Scholar] [CrossRef]
- Olsen, T.; Kerton, F.; Marriott, R.; Grogan, G. Biocatalytic esterification of lavandulol in supercritical carbon dioxide using acetic acid as the acyl donor. Enzym. Microb. Technol. 2006, 39, 621–625. [Google Scholar] [CrossRef]
- Levi−Zada, A.; Harel, M. Enzymatic transesterification of racemic lavandulol: Preparation of the two enantiomeric alcohols and of the two enantiomers of lavandulyl senecioate. Tetrahedron Asymmetry 2004, 15, 2339–2343. [Google Scholar] [CrossRef]
- Levi−Zada, A.; Dunkelblum, E. A convenient resolution of racemic lavandulol through lipase-catalyzed acylation with succinic anhydride: Simple preparation of enantiomerically pure (R)-lavandulol. Tetrahedron Asymmetry 2006, 17, 230–233. [Google Scholar] [CrossRef]
- Serra, S.; Fuganti, C. Enzyme-Mediated Preparation of Enantiomerically Pure p-Menthan- 3,9-diols and Their Use for the Synthesis of Natural p-Menthane Lactones and Ethers. Helv. Chim. Acta 2002, 85, 2489–2502. [Google Scholar] [CrossRef]
- Shimoma, F.; Kusaka, H.; Wada, K.; Azami, H.; Yasunami, M.; Suzuki, T.; Hagiwara, H.; Ando, M. A Novel Synthetic Method of the (±)-(3aα,8aα)-Ethyl 8β-Hydroxy-6β-methyl-2-oxooctahydro-2H-cyclohepta[b]furan-3α-carboxylate and Its Chemical Transformation to (±)-(3aα,8aα)-3α,6β-Dimethyl-3,3a,4,5,6,8a-hexahydro-2H-cyclohepta[b]furan-2-one, (+)- and (−)-7β-(2-Acetoxy-1α-methylethyl)-4β-methyl-2-cyclohepten-1β-ol, and (+)- and (−)-7β-(2-Acetoxy-1α-methylethyl)-4β-methyl-2-cyclohepten-1-one. Possible Common Synthetic Intermediates for Pseudoguaianolides, 4,5-Secopseudoguaianolides, Guaianolides, 4,5-Secoguaianolides and Octalactins. J. Org. Chem. 1998, 63, 920–929. [Google Scholar] [CrossRef]
- Serra, S.; Fuganti, C.; Gatti, F.G. A Chemoenzymatic, Preparative Synthesis of the Isomeric Forms of p-Menth-1-en-9-ol: Application to the Synthesis of the Isomeric Forms of the Cooling Agent 1-Hydroxy-2,9-cineole. Eur. J. Org. Chem. 2008, 2008, 1031–1037. [Google Scholar] [CrossRef]
- Serra, S.; Nobile, I. Chemoenzymatic preparation of the p-menth-1,5-dien-9-ol stereoisomers and their use in the enantiospecific synthesis of natural p-menthane monoterpenes. Tetrahedron Asymmetry 2011, 22, 1455–1463. [Google Scholar] [CrossRef]
- Luparia, M.; Boschetti, P.; Piccinini, F.; Porta, A.; Zanoni, G.; Vidari, G. Enantioselective Synthesis and Olfactory Evaluation of 13-Alkyl-Substituted α-Ionones. Chem. Biodivers. 2008, 5, 1045–1057. [Google Scholar] [CrossRef]
- Fujii, M.; Morimoto, Y.; Ono, M.; Akita, H. Preparation of (S)-γ-cyclogeraniol by lipase-catalyzed transesterification and synthesis of (+)-trixagol and (+)-luffarin-P. J. Mol. Catal. B Enzym. 2016, 123, 160–166. [Google Scholar] [CrossRef]
- Serebryakov, E.P.; Gamalevich, G.D.; Kulcitki, V.N.; Ungur, N.D.; Vlad, P.F. Superacidic cyclisation–lipase-mediated kinetic resolution as a short route from achiral linear isoprenoid alcohols to scalemic cyclic isomers. Mendeleev Commun. 2002, 12, 59–60. [Google Scholar] [CrossRef]
- Akita, H.; Nozawa, M.; Mitsuda, A.; Ohsawa, H. A convenient synthesis of (+)-albicanol based on enzymatic function: Total syntheses of (+)-albicanyl acetate, (−)-albicanyl 3,4-dihydroxycinnamate, (−)-drimenol, (−)-drimenin and (−)-ambrox. Tetrahedron Asymmetry 2000, 11, 1375–1388. [Google Scholar] [CrossRef]
- Fujii, M.; Ishii, S.; Saito, R.; Akita, H. Enzymatic resolution of albicanol and its application to the synthesis of (−)-copalic acid. J. Mol. Catal. B Enzym. 2009, 59, 254–260. [Google Scholar] [CrossRef]
- Ishii, S.; Fujii, M.; Akita, H. First Syntheses of (−)-Tauranin and Antibiotic (−)-BE-40644 Based on Lipase-Catalyzed Optical Resolution of Albicanol. Chem. Pharm. Bull. 2009, 57, 1103–1106. [Google Scholar] [CrossRef] [Green Version]
- Serra, S. Recent Advances in the Synthesis of Carotenoid-Derived Flavours and Fragrances. Molecules 2015, 20, 12817–12840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, S.; Piccioni, O. A new chemo-enzymatic approach to the stereoselective synthesis of the flavors tetrahydroactinidiolide and dihydroactinidiolide. Tetrahedron Asymmetry 2015, 26, 584–592. [Google Scholar] [CrossRef]
- Okazaki, R.; Kiyota, H.; Oritani, T. Enzymatic Resolution of (±)-Epoxy-β-cyclogeraniol, a Synthetic Precursor for Abscisic Acid Analogs. Biosci. Biotechnol. Biochem. 2000, 64, 1444–1447. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Takagi, Y.; Oshiro, T.; Mitsuyama, T.; Sasaki, I.; Yamasaki, N.; Yamada, A.; Kenmoku, H.; Matsuo, Y.; Kasai, Y.; et al. Total Synthesis of (−)-Thallusin: Utilization of Enzymatic Hydrolysis Resolution. J. Org. Chem. 2014, 79, 8850–8855. [Google Scholar] [CrossRef]
- Serra, S.; De Simeis, D. Two Complementary Synthetic Approaches to the Enantiomeric Forms of the Chiral Building Block (2,6,6-Trimethyltetrahydro-2H-pyran-2-yl)methanol: Application to the Stereospecific Preparation of the Natural Flavor Linaloyl Oxide. Catalysts 2018, 8, 362. [Google Scholar] [CrossRef] [Green Version]
- Serra, S. Bisabolane Sesquiterpenes: Synthesis of (R)-(+)-Sydowic Acid and (R)-(+)-Curcumene Ether. Synlett 2000, 2000, 890–892. [Google Scholar] [CrossRef]
- Serra, S. Lipase-mediated resolution of substituted 2-aryl-propanols: Application to the enantioselective synthesis of phenolic sesquiterpenes. Tetrahedron Asymmetry 2011, 22, 619–628. [Google Scholar] [CrossRef]
- Serra, S. Preparation and use of enantioenriched 2-aryl-propylsulfonylbenzene derivatives as valuable building blocks for the enantioselective synthesis of bisabolane sesquiterpenes. Tetrahedron Asymmetry 2014, 25, 1561–1572. [Google Scholar] [CrossRef]
- Serra, S.; Fronza, G. Unambiguous Characterization of the Sesquiterpene (7R,9E)-1,2,11-Trihydroxy-1,3,5,9-bisabolatetraene through Its Enantioselective Synthesis. Eur. J. Org. Chem. 2012, 2012, 4838–4843. [Google Scholar] [CrossRef]
- Serra, S. Enantioselective synthesis of natural trinorsesquiterpene tetralones by chemo-enzymatic approaches. Nat. Prod. Commun. 2013, 8, 863–868. [Google Scholar] [CrossRef] [Green Version]
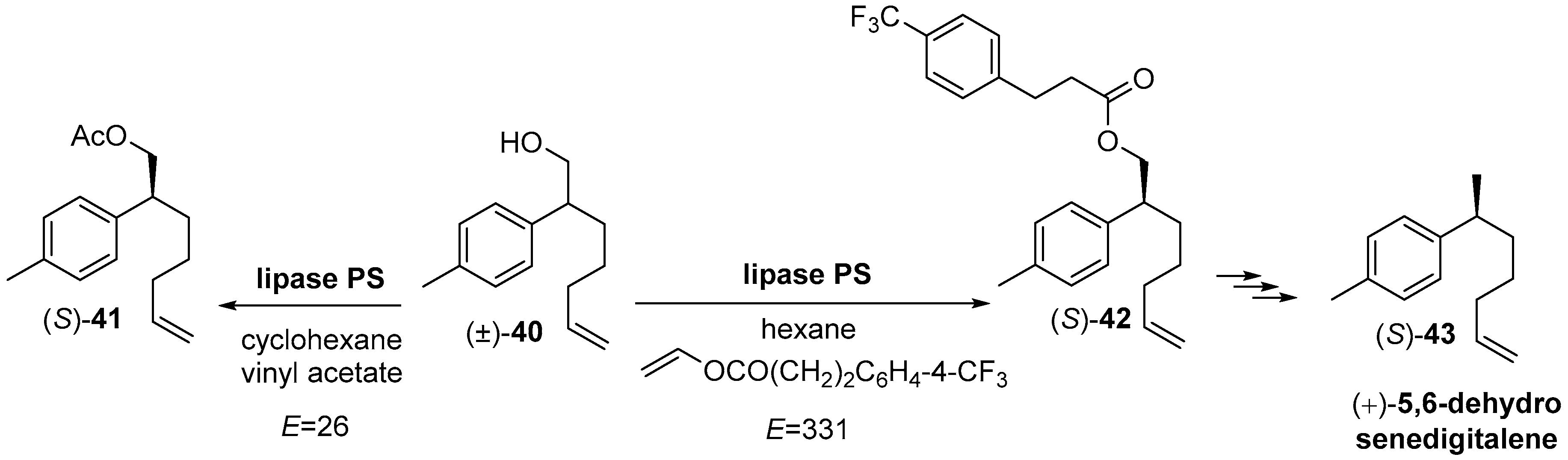
- Kawasaki, M.; Hayashi, Y.; Kakuda, H.; Toyooka, N.; Tanaka, A.; Goto, M.; Kawabata, S.; Kometani, T. Asymmetric synthesis of 5,6-dehydrosenedigitalene using lipase-catalyzed highly enantioselective transesterification of a primary alcohol with vinyl 3-(4-trifluoromethylphenyl)propanoate. Tetrahedron Asymmetry 2005, 16, 4065–4072. [Google Scholar] [CrossRef]
- Tanikawa, S.; Ono, M.; Akita, H. Enzymatic Resolution of (±)-5-Acetoxy-4-aryl-(2E)-pentenoate Derivatives. Chem. Pharm. Bull. 2005, 53, 565–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
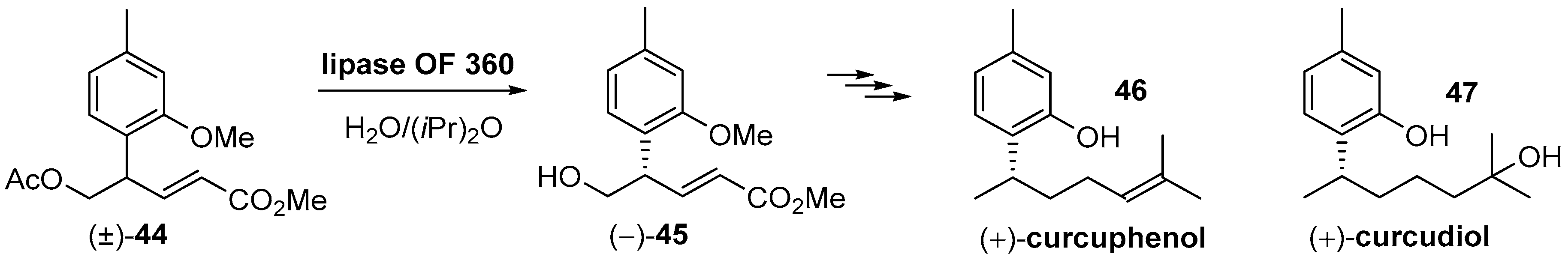
- Ono, M.; Ogura, Y.; Hatogai, K.; Akita, H. Total synthesis of (S)-(+)-curcudiol, and (S)-(+)- and (R)-(-)-curcuphenol. Chem. Pharm. Bull. 2001, 49, 1581–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
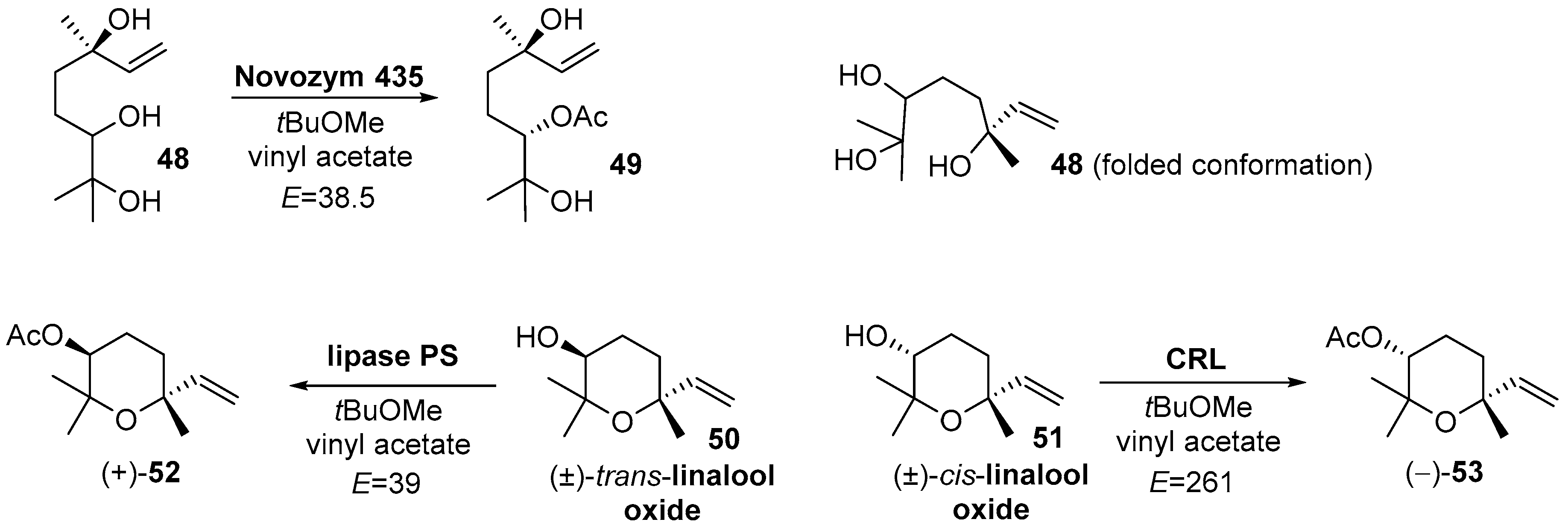
- Serra, S.; De Simeis, D. A Study on the Lipase-catalysed Acylation of 6,7-Dihydroxy-linalool. Nat. Prod. Commun. 2016, 11, 1217–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, S.; De Simeis, D.; Brenna, E. Lipase mediated resolution of cis- and trans-linalool oxide (pyranoid). J. Mol. Catal. B Enzym. 2016, 133, S420–S425. [Google Scholar] [CrossRef]
- Brenna, E.; Negri, C.D.; Fuganti, C.; Gatti, F.G.; Serra, S. Enantioselective synthesis of cis-7-methoxy-calamenene via Claisen rearrangement of an enzymatically resolved allyl alcohol. Tetrahedron Asymmetry 2004, 15, 335–340. [Google Scholar] [CrossRef]
- Brenna, E.; Fuganti, C.; Grasselli, P.; Serra, S. Enzyme-mediated synthesis of (S)- and (R)-verapamil. Eur. J. Org. Chem. 2001, 2001, 1349–1357. [Google Scholar] [CrossRef]
- Brenna, E.; Fuganti, C.; Serra, S. Applications of biocatalysis in fragrance chemistry: The enantiomers of α-, β-, and γ-irones. Chem. Soc. Rev. 2008, 37, 2443. [Google Scholar] [CrossRef]
- Brenna, E.; Fuganti, C.; Grasselli, P.; Redaelli, M.; Serra, S. Enzyme-mediated synthesis of (R)- and (S )-α-ionone. J. Chem. Soc. Perkin Trans. 1 1998, 30, 4129–4134. [Google Scholar] [CrossRef]
- Serra, S.; Fuganti, C. Synthesis of the enantiomeric forms of α- and γ-damascone starting from commercial racemic α-ionone. Tetrahedron Asymmetry 2006, 17, 1573–1580. [Google Scholar] [CrossRef]
- Fuganti, C.; Serra, S.; Zenoni, A. Synthesis and olfactory evaluation of (+)- and (−)-gamma-ionone. Helv. Chim. Acta 2000, 83, 2761–2768. [Google Scholar] [CrossRef]
- Brenna, E.; Fuganti, C.; Serra, S. Synthesis and olfactory evaluation of the enantiomerically enriched forms of 7,11-epoxymegastigma-5(6)-en-9-one and 7,11-epoxymegastigma-5(6)-en-9-ols isomers, identified in Passiflora edulis. Tetrahedron Asymmetry 2005, 16, 1699–1704. [Google Scholar] [CrossRef]
- Vorlová, S.; Bornscheuer, U.T.; Gatfield, I.; Hilmer, J.-M.; Bertram, H.-J.; Schmid, R.D. Enantioselective hydrolysis of D,L-menthyl benzoate to L-(–)-menthol by recombinant Candida rugosa lipase LIP1. Adv. Synth. Catal. 2002, 344, 1152–1155. [Google Scholar] [CrossRef]
- Chaplin, J.A.; Gardiner, N.S.; Mitra, R.K.; Parkinson, C.J.; Portwig, M.; Mboniswa, B.A.; Evans-Dickson, M.D.; Brady, D.; Marais, S.F.; Reddy, S. Process for Preparing (−)-Menthol and Similar Compounds. U.S. Patent 7026114 B2, 11 April 2006. [Google Scholar]
- Serra, S.; Brenna, E.; Fuganti, C.; Maggioni, F. Lipase-catalyzed resolution of p-menthan-3-ols monoterpenes: Preparation of the enantiomer-enriched forms of menthol, isopulegol, trans- and cis-piperitol, and cis-isopiperitenol. Tetrahedron Asymmetry 2003, 14, 3313–3319. [Google Scholar] [CrossRef]
- Lu, Z.; Sun, W.; Han, Y.; Wang, Y.; Liu, J. Enzymatic esterification of DL-menthol with propionic acid by lipase from Candida cylindracea. J. Chem. Technol. Biotechnol. 2005, 80, 1365–1370. [Google Scholar] [CrossRef]
- Bai, S.; Guo, Z.; Liu, W.; Sun, Y. Resolution of (±)-menthol by immobilized Candida rugosa on superparamagnetic nanoparticles. Food Chem. 2006, 96, 1–7. [Google Scholar] [CrossRef]
- Othman, S.S.; Basri, M.; Hussein, M.Z.; Rahman, M.B.A.; Rahman, R.N.Z.R.A.; Salleh, A.B.; Jasmani, H. Production of highly enantioselective (−)-menthyl butyrate using Candida rugosa lipase immobilized on epoxy-activated supports. Food Chem. 2008, 106, 437–443. [Google Scholar] [CrossRef]
- Zhang, D.-H.; Bai, S.; Ren, M.-Y.; Sun, Y. Optimization of lipase-catalyzed enantioselective esterification of (±)-menthol in ionic liquid. Food Chem. 2008, 109, 72–80. [Google Scholar] [CrossRef]
- Brady, D.; Reddy, S.; Mboniswa, B.; Steenkamp, L.; Rousseau, A.L.; Parkinson, C.; Chaplin, J.; Mitra, R.; Moutlana, T.; Marais, S.; et al. Biocatalytic enantiomeric resolution of l-menthol from an eight isomeric menthol mixture through transesterification. J. Mol. Catal. B Enzym. 2012, 75, 1–10. [Google Scholar] [CrossRef]
- Chen, H.; Wu, J.-P.; Yang, L.; Xu, G. Improving Pseudomonas alcaligenes lipase’s diastereopreference in hydrolysis of diastereomeric mixture of menthyl propionate by site-directed mutagenesis. Biotechnol. Bioprocess Eng. 2014, 19, 592–604. [Google Scholar] [CrossRef]
- Belafriekh, A.; Secundo, F.; Serra, S.; Djeghaba, Z. Enantioselective enzymatic resolution of racemic alcohols by lipases in green organic solvents. Tetrahedron Asymmetry 2017, 28, 473–478. [Google Scholar] [CrossRef]
- De Yan, H.; Wang, Z.; Li, Q. Efficient kinetic resolution of (±)-menthol by a lipase from Thermomyces lanuginosus. Biotechnol. Appl. Biochem. 2015, 64, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Craveiro, R.; Meneses, L.; Durazzo, L.; Rocha, Â.; Silva, J.M.; Reis, R.L.; Barreiros, S.; Duarte, A.R.C.; Paiva, A. Deep Eutectic Solvents for Enzymatic Esterification of Racemic Menthol. ACS Sustain. Chem. Eng. 2019, 7, 19943–19950. [Google Scholar] [CrossRef]
- Brenna, E.; Fuganti, C.; Gatti, F.G.; Perego, M.; Serra, S. Enzyme-mediated preparation of enantioenriched forms of trans- and cis-p-menthan-1,8-dien-5-ol. Tetrahedron Asymmetry 2006, 17, 792–796. [Google Scholar] [CrossRef]
- Gatti, F.G.; Serra, S. New Stereoselective Synthesis of Paeonilactone B. Synthesis 2009, 2009, 1287–1290. [Google Scholar] [CrossRef]
- Serra, S.; Fuganti, C. Natural p-Menthene Monoterpenes: Synthesis of the Enantiomeric Forms of Wine Lactone, Epi-wine Lactone, Dill Ether, and Epi-dill Ether Starting from a Common Intermediate. Helv. Chim. Acta 2004, 87, 2100–2109. [Google Scholar] [CrossRef]
- More, G.P.; Bhat, S.V. Facile lipase catalysed syntheses of (S)-(+)-4-hydroxy-β-ionone and (S)-(+)-4-hydroxy-β-damascone: Chiral flavorants and synthons. Tetrahedron Lett. 2013, 54, 4148–4149. [Google Scholar] [CrossRef]
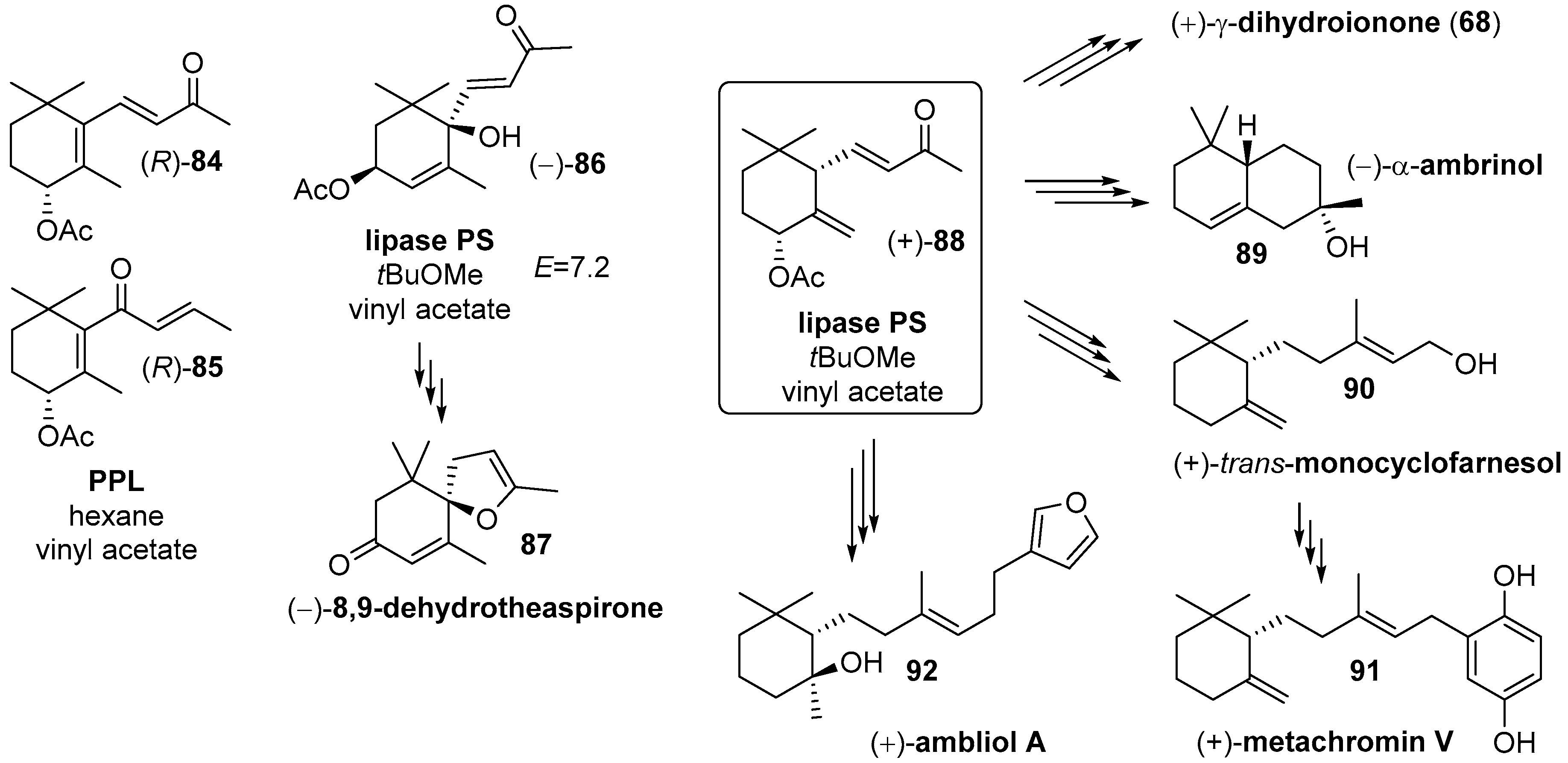
- Serra, S.; Barakat, A.; Fuganti, C. Chemoenzymatic resolution of cis- and trans-3,6-dihydroxy-α-ionone. Synthesis of the enantiomeric forms of dehydrovomifoliol and 8,9-dehydrotheaspirone. Tetrahedron Asymmetry 2007, 18, 2573–2580. [Google Scholar] [CrossRef]
- Serra, S.; Fuganti, C.; Brenna, E. Synthesis, Olfactory Evaluation, and Determination of the Absolute Configuration of the 3,4-Didehydroionone Stereoisomers. Helv. Chim. Acta 2006, 89, 1110–1122. [Google Scholar] [CrossRef]
- Serra, S. An expedient preparation of enantio-enriched ambergris odorants starting from commercial ionone alpha. Flavour Fragr. J. 2012, 28, 46–52. [Google Scholar] [CrossRef]
- Serra, S. A Practical, Enantiospecific Synthesis of (S)-trans-γ-Monocyclofarnesol. Nat. Prod. Commun. 2012, 7, 1569–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, S.; Cominetti, A.A.; Lissoni, V. A general synthetic approach to hydroquinone meroterpenoids: Stereoselective synthesis of (+)-(S)-metachromin V and alliodorol. Nat. Prod. Commun. 2014, 9, 303–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, S.; Cominetti, A.A.; Lissoni, V. Use of (S)-trans-γ-Monocyclofarnesol as a Useful Chiral Building Block for the Stereoselective Synthesis of Diterpenic Natural Products. Nat. Prod. Commun. 2014, 9, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Serra, S.; Lissoni, V. First Enantioselective Synthesis of Marine Diterpene Ambliol-A. Eur. J. Org. Chem. 2015, 2015, 2226–2234. [Google Scholar] [CrossRef]
- Fujiwara, N.; Kinoshita, M.; Akita, H. Chemoenzymatic synthesis of (S)- and (R)-γ-cyclogeraniols. J. Mol. Catal. B Enzym. 2006, 40, 64–72. [Google Scholar] [CrossRef]
- Serra, S.; Gatti, F.G.; Fuganti, C. Lipase-mediated resolution of the hydroxy-cyclogeraniol isomers: Application to the synthesis of the enantiomers of karahana lactone, karahana ether, crocusatin C and γ-cyclogeraniol. Tetrahedron Asymmetry 2009, 20, 1319–1329. [Google Scholar] [CrossRef]
- Serra, S. A General Strategy for the Stereoselective Synthesis of the Furanosesquiterpenes Structurally Related to Pallescensins 1–2. Mar. Drugs 2019, 17, 245. [Google Scholar] [CrossRef] [Green Version]
- Galano, J.-M.; Audran, G.; Monti, H. Straightforward enantioselective synthesis of both enantiomers of karahana lactone using a domino ring-closure sequence. Tetrahedron 2000, 56, 7477–7481. [Google Scholar] [CrossRef]
- Takikawa, H.; Ueda, K.; Sasaki, M. The first synthesis and absolute configuration of glaucescenolide. Tetrahedron Lett. 2004, 45, 5569–5571. [Google Scholar] [CrossRef]
- Ito, S.; Tosaka, A.; Hanada, K.; Shibuya, M.; Ogasawara, K.; Iwabuchi, Y. Chemoenzymatic preparation of functionalized bicycle [3.2.1] octenone and practical utilization. Tetrahedron Asymmetry 2008, 19, 176–185. [Google Scholar] [CrossRef]
- Michalak, K.; Wicha, J. A convenient preparation of (S)-(−)-4-hydroxy-2-methylcyclopent-2-en-1-one and its application as a chiral synthetic equivalent of 2-methylcyclopent-2-en-1-one in the terpenoid synthesis. Tetrahedron 2014, 70, 5073–5081. [Google Scholar] [CrossRef]
- Acherar, S.; Audran, G.; Fotiadu, F.; Monti, H. Lipase-Promoted Access to Phenolic Herbertane-Type Sesquiterpenes: (+)-1,14-Herbertenediol, (-)-α-Herbertenol, (-)-Herbertenediol and Their Enantiomers. Eur. J. Org. Chem. 2004, 2004, 5092–5099. [Google Scholar] [CrossRef]
- Acherar, S.; Audran, G.; Vanthuyne, N.; Monti, H. Use of lipase-catalyzed kinetic resolution for the enantioselective approach toward sesquiterpenes containing quaternary centers: The cuparane family. Tetrahedron Asymmetry 2003, 14, 2413–2418. [Google Scholar] [CrossRef]
- Acherar, S.; Audran, G.; Cecchin, F.; Monti, H. Enantioselective synthesis of natural (−)-tochuinyl acetate, (−)-dihydrotochuinyl acetate and (+)-β-cuparenone using both enantiomers of the same building block. Tetrahedron 2004, 60, 5907–5912. [Google Scholar] [CrossRef]
- Galano, J.-M.; Audran, G.; Mikolajezyk, L.; Monti, H. Enzyme-Assisted Enantioselective Synthesis of Natural (−)-β-Necrodol and Its Enantiomer. J. Org. Chem. 2001, 66, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Srinivas, K. Enantioselective total syntheses of the novel tricyclic sesquiterpene hydrocarbons (+)- and (−)-kelsoene. Absolute configuration of the natural product. Tetrahedron Lett. 2001, 42, 2855–2857. [Google Scholar] [CrossRef] [Green Version]
- Zanoni, G.; Agnelli, F.; Meriggi, A.; Vidari, G. Enantioselective syntheses of isoprostane and iridoid lactones intermediates by enzymatic transesterification. Tetrahedron Asymmetry 2001, 12, 1779–1784. [Google Scholar] [CrossRef]
- Santangelo, E.M.; Rotticci, D.; Liblikas, I.; Norin, T.; Unelius, C.R. Resolution of an Iridoid Synthon, Gastrolactol, by Means of Dynamic Acetylation and Lipase-Catalyzed Alcoholysis. J. Org. Chem. 2001, 66, 5384–5387. [Google Scholar] [CrossRef]
- Roy, A. Chemoenzymatic synthesis of homochiral (R)- and (S)-karahanaenol from (R)-limonene. J. Agric. Food Chem. 1999, 47, 5209–5210. [Google Scholar] [CrossRef]
- Kitayama, T.; Nagao, R.; Masuda, T.; Hill, R.K.; Morita, M.; Takatani, M.; Sawada, S.; Okamoto, T. The chemistry of Zerumbone IV. J. Mol. Catal. B Enzym. 2002, 17, 75–79. [Google Scholar] [CrossRef]
- Kourist, R.; De Maria, P.D.; Bornscheuer, U. Enzymatic Synthesis of Optically Active Tertiary Alcohols: Expanding the Biocatalysis Toolbox. ChemBioChem 2008, 9, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Kourist, R.; Bornscheuer, U. Biocatalytic synthesis of optically active tertiary alcohols. Appl. Microbiol. Biotechnol. 2011, 91, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Özdemirhan, D. Optically Active Tertiary Alcohols by Biocatalysis. Synth. Commun. 2016, 47, 629–645. [Google Scholar] [CrossRef]
- Deng, D.; Zhang, Y.; Sun, A.; Sai, K.; Hu, Y. Functional Characterization of a New Antarctic Microbial Esterase EST112-2 and Its Use in the Preparation of Chiral Tertiary Alcohol (S)-Linalool. Chin. J. Org. Chem. 2018, 38, 1185–1192. [Google Scholar] [CrossRef]
- Oshikubo, S.; Nozaki, M. Production of Optical Active Linalool. JPH09278 (A) 1997. Available online: https://worldwide.espacenet.com/ (accessed on 28 April 2020).
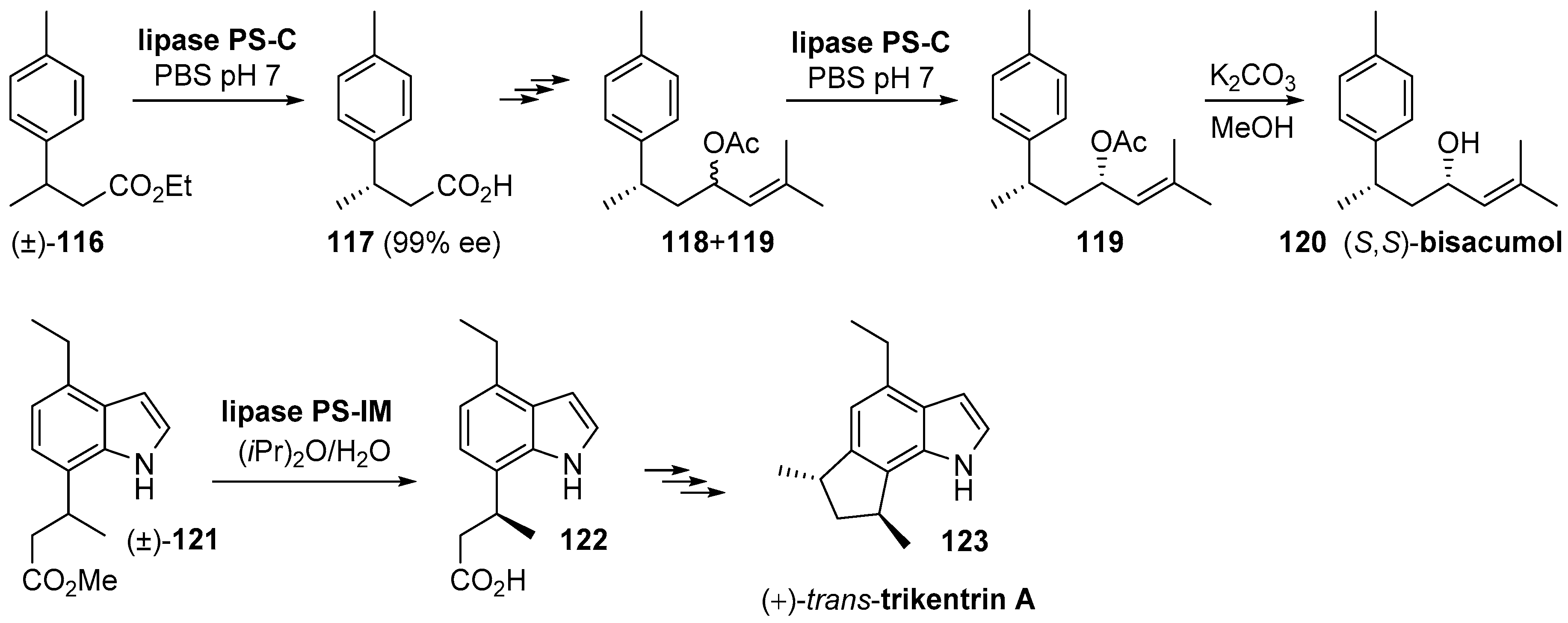
- Kamal, A.; Malik, M.S.; Azeeza, S.; Bajee, S.; Shaik, A.A. Total synthesis of (R)- and (S)-turmerone and (7S,9R)-bisacumol by an efficient chemoenzymatic approach. Tetrahedron Asymmetry 2009, 20, 1267–1271. [Google Scholar] [CrossRef]
- Tebeka, I.R.M.; Longato, G.B.; De Carvalho, J.E.; Ruiz, A.L.T.G.; Silva, L.F.; Craveiro, M.V. Total Synthesis of (+)-trans-Trikentrin A. Chem. A Eur. J. 2012, 18, 16890–16901. [Google Scholar] [CrossRef]
- Shishido, K.; Bando, T. Lipase-mediated asymmetric acetylation of prochiral diols directed towards total syntheses of biologically active molecules. J. Mol. Catal. B Enzym. 1998, 5, 183–186. [Google Scholar] [CrossRef]
- Kishuku, H.; Shindo, M.; Shishido, K. Enantioselective total synthesis of (-)-heliannuol A. Chem. Commun. 2003, 350–351. [Google Scholar] [CrossRef]
- Takabatake, K.; Nishi, I.; Shindo, M.; Shishido, K. Enantioselective total synthesis of heliannuols D and A. J. Chem. Soc. Perkin Trans. 1 2000, 1, 1807–1808. [Google Scholar] [CrossRef]
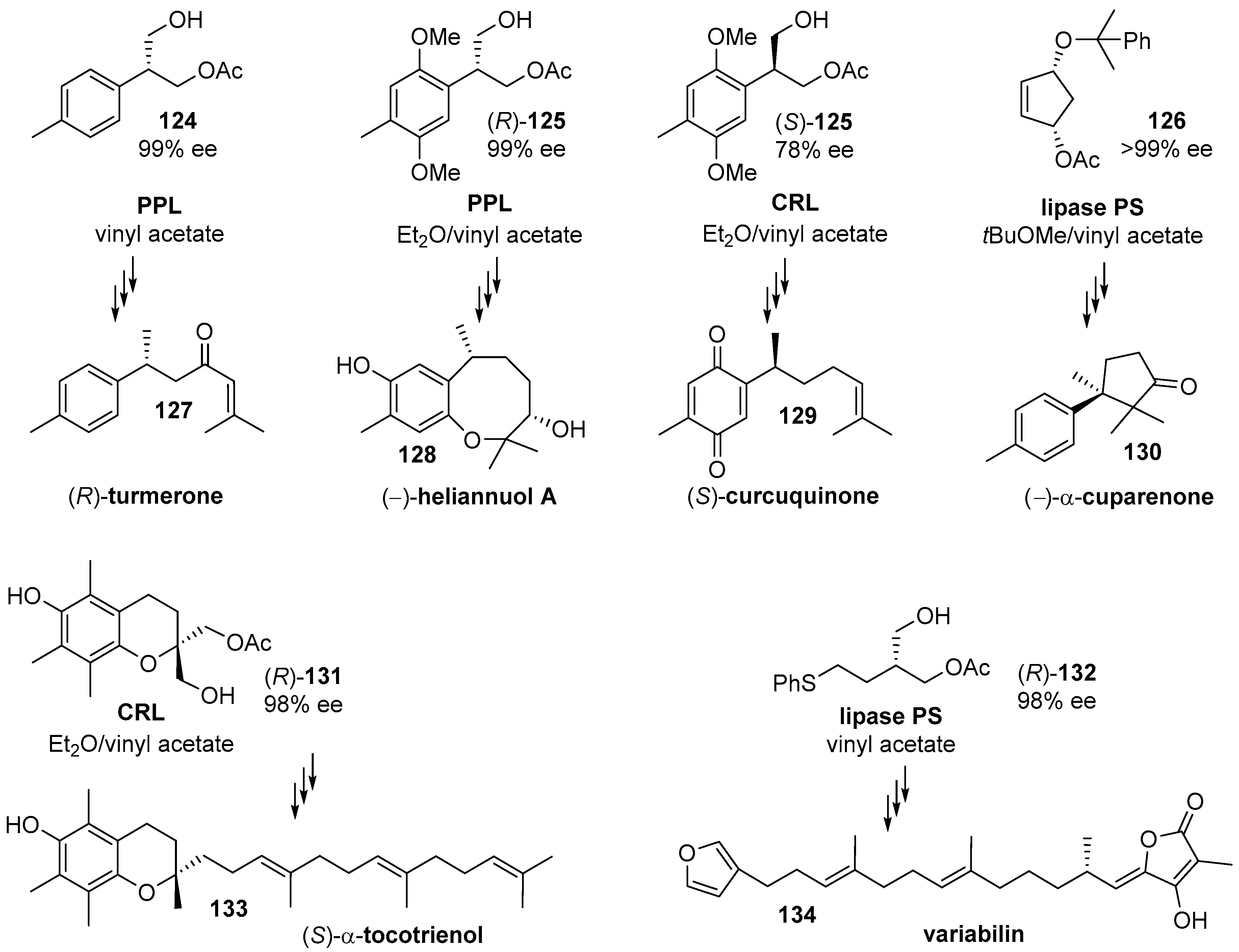
- Yoshimura, T. Enantiocontrolled synthesis of (+)-curcuquinone and (-)-curcuhydroquinone. Arkivoc 2003, 2003, 247. [Google Scholar] [CrossRef] [Green Version]
- Kamei, T.; Shindo, M.; Shishido, K. First enantioselective total synthesis of (−)-heliannuol C. Tetrahedron Lett. 2003, 44, 8505–8507. [Google Scholar] [CrossRef]
- Morimoto, S.; Shindo, M.; Yoshida, M.; Shishido, K. Syntheses of heliannuols G and H; structure revision of the natural products. Tetrahedron Lett. 2006, 47, 7353–7356. [Google Scholar] [CrossRef]
- Shishido, K.; Morimoto, S.; Shindo, M. Enantioselective Syntheses of Heliannuols G and H. Heterocycles 2005, 66, 69. [Google Scholar] [CrossRef]
- Miyawaki, A.; Osaka, M.; Kanematsu, M.; Yoshida, M.; Shishido, K. Enantioselective syntheses of the assigned structures of the helibisabonols A and B. Tetrahedron 2011, 67, 6753–6761. [Google Scholar] [CrossRef]
- Nakashima, H.; Sato, M.; Taniguchi, T.; Ogasawara, K. An enantiodivergent route to α-cuparenone utilizing chiral cyclopentenol having a latent meso structure. Tetrahedron Lett. 2000, 41, 2639–2642. [Google Scholar] [CrossRef]
- Chênevert, R.; Courchesne, G. Synthesis of (S)-α-tocotrienol via an enzymatic desymmetrization of an achiral chroman derivative. Tetrahedron Lett. 2002, 43, 7971–7973. [Google Scholar] [CrossRef]
- Takabe, K.; Hashimoto, H.; Sugimoto, H.; Nomoto, M.; Yoda, H. First asymmetric synthesis of the marine furanosesterterpene natural product, (18S)-variabilin, employing enzymatic desymmetrization of propanediol derivatives. Tetrahedron Asymmetry 2004, 15, 909–912. [Google Scholar] [CrossRef]


















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serra, S.; De Simeis, D. Stereoselective Synthesis of Terpenoids through Lipase-Mediated Resolution Approaches. Catalysts 2020, 10, 504. https://doi.org/10.3390/catal10050504
Serra S, De Simeis D. Stereoselective Synthesis of Terpenoids through Lipase-Mediated Resolution Approaches. Catalysts. 2020; 10(5):504. https://doi.org/10.3390/catal10050504
Chicago/Turabian StyleSerra, Stefano, and Davide De Simeis. 2020. "Stereoselective Synthesis of Terpenoids through Lipase-Mediated Resolution Approaches" Catalysts 10, no. 5: 504. https://doi.org/10.3390/catal10050504
APA StyleSerra, S., & De Simeis, D. (2020). Stereoselective Synthesis of Terpenoids through Lipase-Mediated Resolution Approaches. Catalysts, 10(5), 504. https://doi.org/10.3390/catal10050504