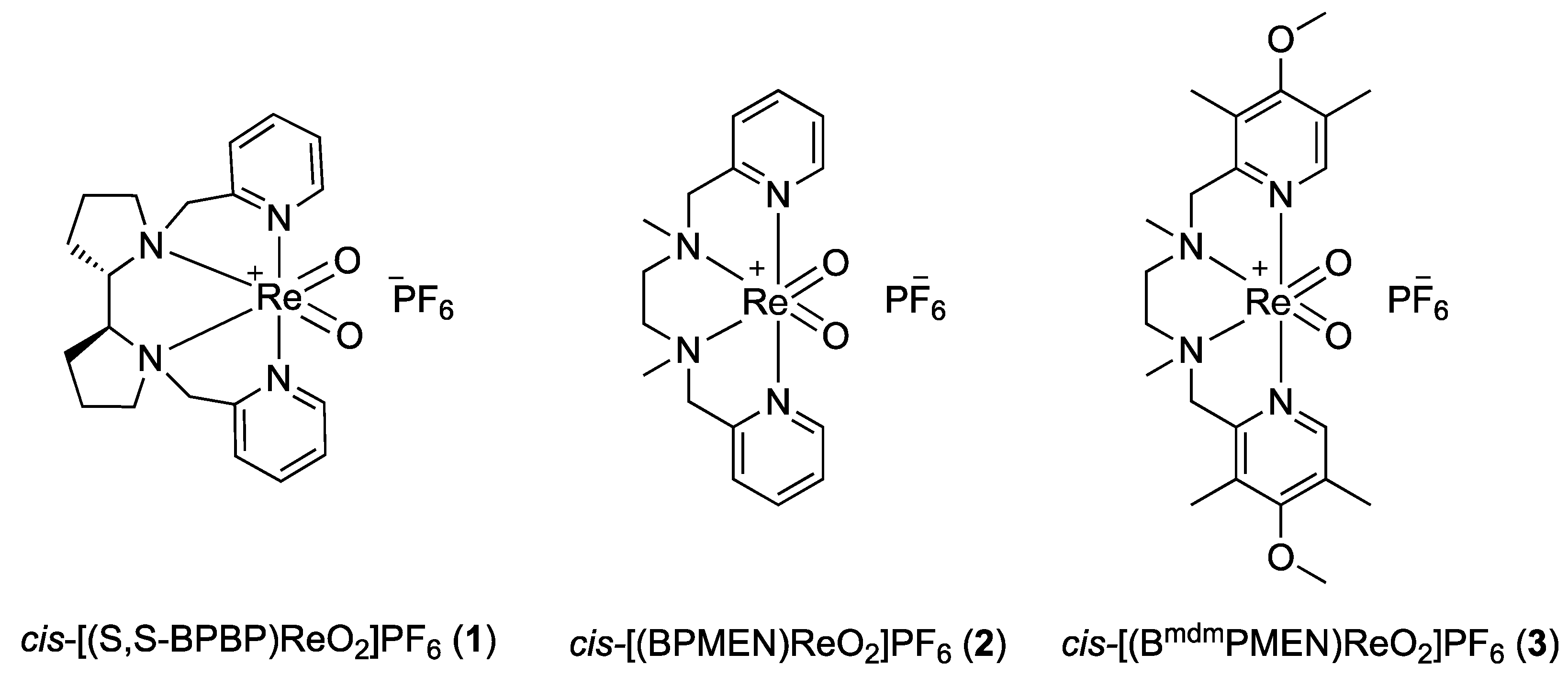
2.1. Synthesis of Rhenium Complexes
For our initial study,
cis-[(
S,
S-BPBP)ReO
2]PF
6 (
1) (
S,
S-BPBP = (2
S,2’
S)-1,1’-bis(pyridin-2-ylmethyl)-2,2’-bipyrrolidine),
cis-[(BPMEN)ReO
2]PF
6 (
2) (BPMEN =
N1,
N2-dimethyl-
N1,
N2-bis(pyridin-2-ylmethyl)ethane-1,2-diamine), and
cis-[(B
mdmPMEN)ReO
2]PF
6 (
3) (B
mdmPMEN =
N1,
N2-bis((4-methoxy-3,5-dimethylpyridin-2-yl)methyl)-
N1,
N2-dimethyl ethane-1,2-diamine) (
Scheme 4) were synthesized according to the protocol developed by Che et al. for the synthesis of
1 [
25]. To this end, reaction of IReO
2(PPh
3)
2 with the corresponding N
2Py
2 ligand in dichloromethane afforded crude
cis-[(N
2Py
2)ReO
2]I, which was then reacted with NH
4PF
6 to afford
2 and
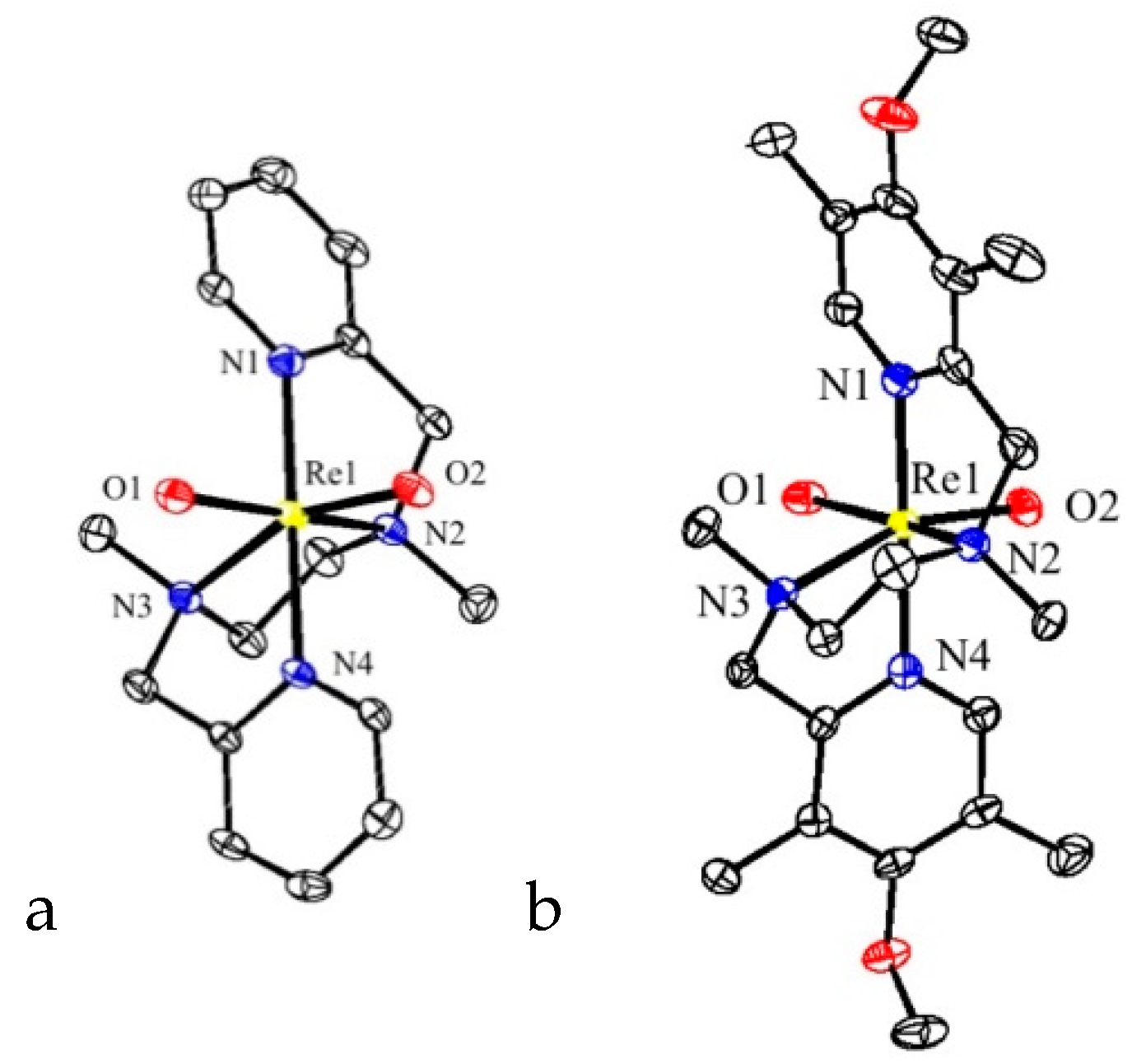
3 in 40% and 31% yields, respectively. The structures of
2 and
3 were established by X-ray crystal structure determination (
Figure 1). Diffraction-quality crystals of
2 were obtained by vapor diffusion of diethyl ether into an acetonitrile solution of the complex, while diffraction-quality crystals of
3 were obtained by vapor diffusion of diethyl ether into a methanol solution of the complex.
These two new
cis-dioxo-rhenium(V) complexes adopt a pseudo-octahedral geometry with a
cis-α configuration of the ligand, similar to the geometry of
1. Also the bond distances and angles are very similar to that of
cis-[(
S,
S-BPBP)ReO
2]
+ reported by Che [
26]. Selected bond distances and angles are given in
Table 1. For all of these three structures, the N(pyridine)–Re distances are shorter than the N(amine)–Re distances and the planes of the pyridine rings are tilted under a small angle with respect to the N(pyridine)–Re bonds (
Table 2). Complexes
1,
2, and
3 are chiral at the amine nitrogens. Complexes
1 and
2 are enantiopure in the crystal, while complex
3 is racemic in the crystal. The tetradentate N
2Py
2 ligands of these complexes are well-known ligands for non-heme iron and manganese oxidation catalysts. Interestingly, for both N
2Py
2 supported high-valent rhenium complexes and N
2Py
2 supported low-valent iron/manganese complexes, the same pseudo-octahedral geometry is observed. Further characterization of complexes
2 and
3 included
1H NMR,
13C NMR, ESI-MS, and elemental analysis (see
Supplementary Material).
2.2. Initial Catalytic Activity Investigation of Complexes 1–3
Next, we investigated the use of
1,
2, and
3 as catalysts in the DODH of vicinal diols by using 1,2-octanediol as a benchmark substrate and triphenylphosphine (PPh
3) as reductant. Only trace amounts of 1-octene formed when the reaction was performed at 135 °C using 2 mol%
1 as catalyst and 1.1 equiv. of PPh
3 as reductant. The reaction temperature was then gradually increased from 135 °C to 180 °C. The yield of 1-octene jumped from 11% to quantitative when the temperature was increased from 165 °C to 180 °C (
Table 3, entries 1–4). Similarly, when
2 was used as catalyst, an elevated reaction temperature was also necessary; the yield of 1-octene dramatically increased when the reaction temperature was increased from 150 °C to 180 °C (
Table 3, entries 5, 6). For all three complexes, full conversion and quantitative 1-octene yield were achieved when the reaction was performed at 180 °C for 15 h under N
2 (
Table 3, entries 4, 5, and 7).
In order to compare the reactivity of
1,
2, and
3, a shorter reaction time was chosen. After 3 h of reaction at 180 °C, significantly lower olefin yields (27–41%) were found than for a reaction time of 15 h at this reaction temperature, albeit at 100% 1-octene selectivity. These findings show that the nature of N
2Py
2 ligand has an effect on the proficiency of the dioxo-rhenium complexes in catalysis and that these complexes do not show any isomerization of the olefin product under the current conditions. Remarkably, in DODH reactions catalyzed by Cp-based trioxo-rhenium complexes, olefin isomers are formed when the reactions are performed at higher temperatures (180 °C) [
27].
The precursor of these dioxo complexes, i.e., iododioxobis(triphenylphosphine)rhenium (IReO
2(PPh
3)
2), was also investigated for its DODH activity and gave a 1-octene yield of only 14% with 2% of octene isomers being formed.
trans-[(Py
4)ReO
2]PF
6 was also tested for this DODH reaction, aiming at providing information on the effect of the configuration of the oxo ligands (
cis or
trans) on catalysis. Surprisingly, this complex showed the best activity among all dioxo-rhenium complexes tested in our study (
Table 3, compare entries 8–11). After the reaction mixture was heated at 180 °C for 3 h, a full substrate conversion and quantitative 1-octene product yield was achieved with
trans-[(Py
4)ReO
2]PF
6 (entry 11), while the reactions using the three N
2Py
2-ligated
cis-dioxo-rhenium complexes reached much lower conversions (entries 8–10). As mentioned in the introduction section, Nicholas et al. have carried out a stochiometric reaction between 1,2-decanediol (1.6 equiv.) and
trans-[(Py
4)ReO
2]Cl (1.0 equiv.) to form 1.0 equiv. of 1-decene and no diol oxidation products. From this observation, they concluded that the starting Re(V) complex is converted to a [(Py)
nRe(VII)O
3]
+ species, which would also involve pyridine ligand(s) dissociation, and that DODH catalysis using this Re(V) dioxo complex would proceed through a Re(V)↔Re(VII) cycle [
25]. On the basis of these considerations, we assume that [(N
2Py
2)ReO
2]
+-catalyzed DODH reactions can also proceed through a Re(V)↔Re(VII) cycle. In this case though, the formal oxidation to a Re(VII)-trioxo species (resulting from a single DODH reaction) would be more difficult compared to
trans-[(Py
4)ReO
2]PF
6, since the tetradentate N
2Py
2 ligand is more strongly coordinated to the Re center than the monodentate pyridine donors in
trans-[(Py
4)ReO
2]PF
6. This could then explain why
trans-[(Py
4)ReO
2]PF
6 has a higher catalytic activity compared to complexes
1–
3.
As a last aspect of these initial studies, we investigated the sensitivity of DODH catalysis by complexes
1–
3. Accordingly, the reaction mixtures of entries 7–9 were then heated for another 4 h under air in the closed reaction vessel. Interestingly, the presence of O
2 does not seem to hamper the reactivity and might even slightly promote the reactions (
Table 3, entries 13–15).
2.3. Initial Mechanistic Studies
After the initial study of the catalytic DODH activity of complexes 1–3, the time-course profiles of the DODH of 1,2-octanediol by these N2Py2-ligated dioxo-rhenium complexes were investigated. In situ 1H NMR experiments were carried out using 1,2-octanediol (0.05 mmol), PPh3 (0.055 mmol, 1.1 equiv.), Re-catalyst (0.005 mmol, 10 mol%), and mesitylene (0.05 mmol, 1.0 equiv., internal standard) in toluene-D8 (0.5 mL) at 180 °C under an inert N2 atmosphere.
Except
cis-[(BPMEN)ReO
2]PF
6 (
2),
cis-[(BP
mdmMEN)ReO
2]PF
6 (
3), and
trans-[(Py
4)ReO
2]PF
6, the dioxo-rhenium complex
cis-[(BPMEN)ReO
2]BPh
4 (
6) was also included in these studies in order to investigate a possible effect of the counter anion on catalyst activity. For complexes
2,
3, and
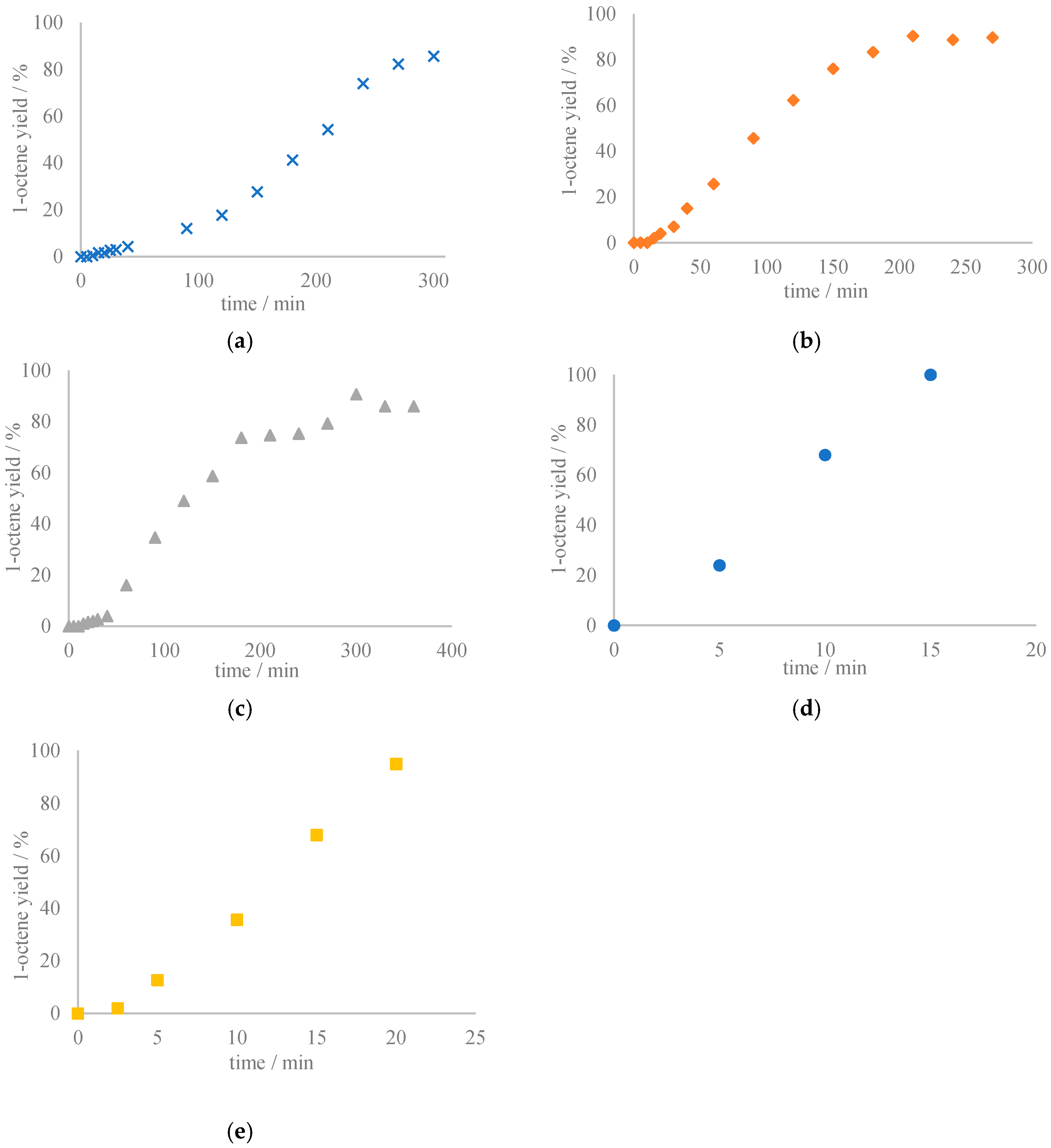
6, an induction period was observed. For the reaction catalyzed by
2, a typical sigmoidal trend for 1-octene formation was found. No 1-octene formation was observed during the initial 5 min., followed by a gradual increase in 1-octene formation until 120 min. After this induction period, formation of 1-octene proceeded at a much higher rate, with the highest yield (86%) reached after 300 min (
Scheme 5a). Modification of the N
2Py
2 ligand framework lead to changes in this reaction profile. When
cis-[(BP
mdmMEN)ReO
2]PF
6 (
3) was used as catalyst, the sigmoidal reaction curve showed a much shorter induction period. In this case the highest product yield (90%) was reached after 210 min (
Scheme 5b). Furthermore, changing the counter anion also resulted in a different reaction profile, i.e., the induction period was significantly shorter for the BPh
4– salt
6 than for PF
6– salt
2 (
Scheme 5a,c). However, the three complexes
2,
3, and
6 give a very similar final 1-octene yield and reached this yield within approximately the same time.
In the reaction profile for the DODH reaction catalyzed by trans-[(Py4)ReO2]PF6 no induction period was observed and 24% of 1-octene had formed after 5 min, while full conversion was achieved after 15 min. The reaction rate in this case was much higher than that for the other three complexes. In all of these four reactions, no formation of olefin products resulting from 1-octene isomerization was observed.
After the investigation of the time-course profiles of these “ReO
2”-catalyzed deoxydehydrations, the reaction mixtures were analyzed by means of ESI-MS measurements. For the DODH reaction of 1,2-octanediol catalyzed by
1, both protonated ligand (ES+
m/z 323.2225; calc. 323.2230) and ReO
4– (ES–
m/z 250.8320; calc. 250.9360) signals were detected by ESI-MS measurements on the crude reaction mixture after the reaction (
Figure 2). The signal for the protonated ligand signal was also detected when subjecting pristine complex
1 to ESI-MS analysis, in addition to the signal of the [(
S,
S-BPBP)ReO
2] cation
(m/z 541.1534; calcd. 541.1608). The signal for the intact [(
S,
S-BPBP)ReO
2] cation was, however not observed in the analysis of the crude DODH reaction mixture. The absence of
1, as well as the appearance of protonated ligand and ReO
4–, indicates that the starting dioxo-rhenium complex decomposes under the conditions of the DODH reaction and that perrhenate is formed during the reaction. In case of the DODH of 1,2-octanediol catalyzed by
1, besides the signal for the protonated ligand, signals at
m/z 480.2455, 576.2290, 669.2780, 857.2693, were detected in positive ion mode. While the signals at
m/z 480.2455, 576.2290, 857.2693 did not match the isotope distribution of either mono- or multi-rhenium species, the signal at
m/z 669.2780 can be assigned to the [(
S,
S-BPBP)ReO(1,2-octanediolate)]
+ ion (calcd. 669.2809), which represents the product formed upon the condensation of [(
S,
S-BPBP)ReO
2]
+ and 1,2-octanediol. This diolate could either form 1-octene by olefin extrusion and generate [(
S,
S-BPBP)ReO
3]
+, or be reduced by PPh
3 to form [(
S,
S-BPBP)Re(1,2-octanediolate)]
+, followed by olefin extrusion to regenerate [(
S,
S-BPBP)ReO
2]
+. In the former case, a 20e
– species [(
S,
S-BPBP)ReO
3]
+ would be formed, which could set the stage for N
2Py
2 ligand dissociation and formal decomposition of the starting dioxo-complex.
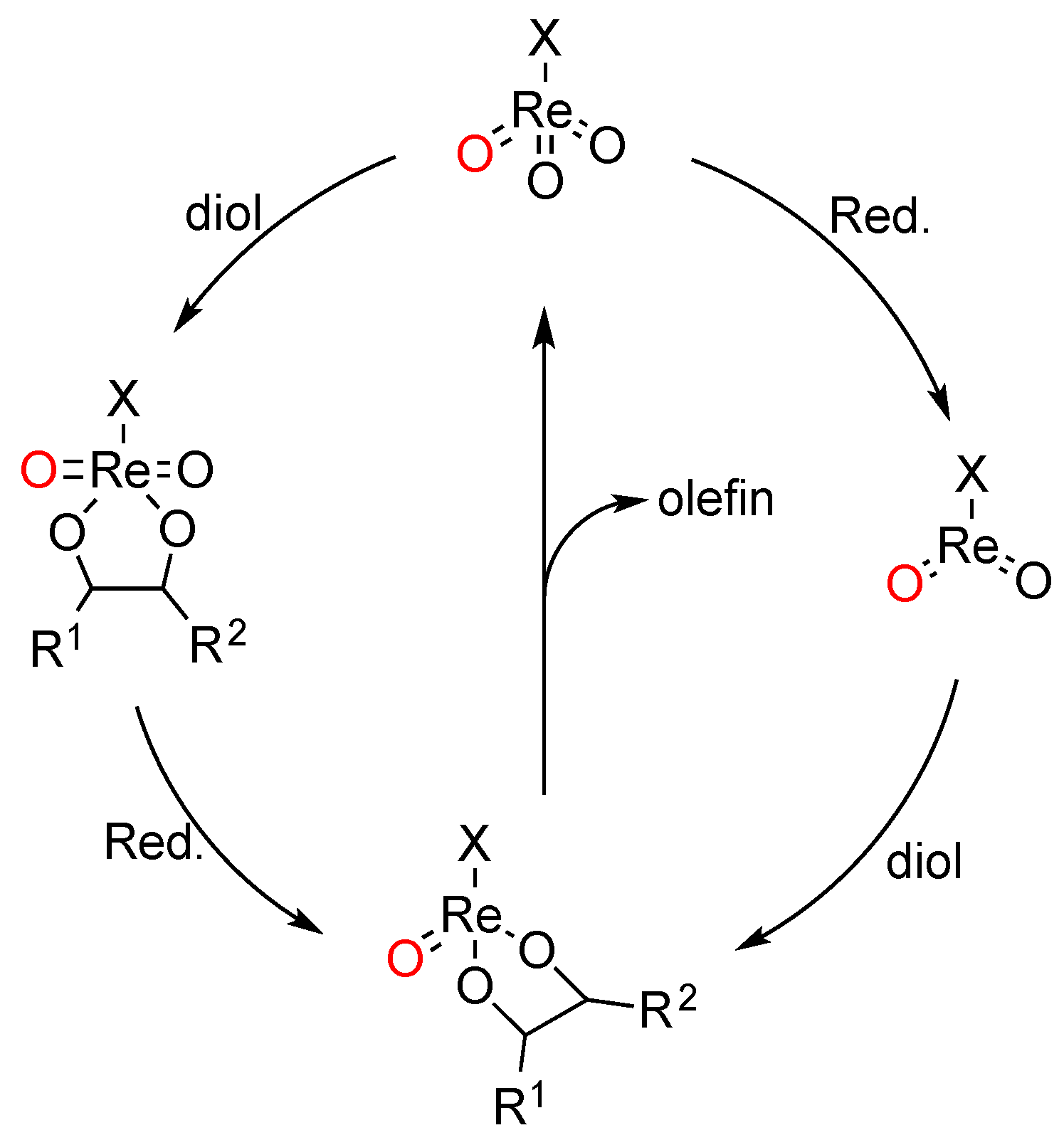
These combined MS and reaction profile observations indicate that complexes 2, 3, and 6 act as pre-catalysts in DODH reactions. Our initial assumption was that the N2Py2-supported complexes show slower kinetics due the more difficult formation of a putative Re(VII) trioxo species as a result of stronger ligand chelation (vide supra). The MS data now show the presence of protonated ligand and the perrhenate anion ReO4– in the crude DODH reaction mixtures of these complexes, suggesting that full ligand dissociation and rhenium oxidation may take place as part of precatalyst activation.
Based on these observations, we decided to investigate the DODH activity of the pyridinium perrhenate salt [PyH][ReO
4]. Pyridinium perrhenate was earlier reported as catalyst for the DODH of diols by Love and coworkers [
28]. In their work, aromatic vicinal diols were converted to the corresponding olefins with moderate to excellent yields (22% to quantitative) at a relatively low temperature (90 °C) in chloroform using 5 mol% lutidinium perrhenate as catalyst and 1.09 equiv. PPh
3 as reductant. For aliphatic vicinal diol substrates, a higher reaction temperature of 140 °C was needed for the DODH reaction to proceed, and only moderate olefin yields (21–51%) were obtained after 16 h. When carrying out the DODH reaction of 1,2-octanediol with [PyH][ReO
4] as catalyst under our reaction conditions, we found quantitative 1-octene formation within approx. 20 min without the formation of octane isomers. The time-course profile of this reaction showed a very short induction period and indicated that the reaction rate was not as high as for
trans-[(Py
4)ReO
2]PF
6 (
Scheme 5d,e). This comparison between time-course profiles seems to suggest that perrhenate is the active species that is formed when
trans-[(Py
4)ReO
2]PF
6 is used in DODH catalysis. On the other hand, the difference in reaction rates does not rule out the possible involvement of yet another active species.
2.4. Investigation of the Active Species
In order to further probe the nature of the active species in DODH reactions catalyzed by dioxo-rhenium complexes, we decided to explore the DODH reaction of erythritol using these complexes. Erythritol is an interesting substrate in DODH chemistry, since it can lead to the formation of 1,3-butadiene as the reaction product starting from a bio-based source. In addition, this tetraol substrate can engage in a number of side-reactions, including dehydrative cyclisation and internal deoxydehydration, and its product distribution profile has been used to investigate the involvement of different active species during DODH catalysis. For the Cp
*ReO
3-catalyzed DODH of erythritol using PPh
3 as reductant and PhCl as solvent, instead of 2,5-dihydrofuran,
cis-2-butene-1,4-diol and 3-butene-1,2-diol were detected as byproduct. Also, for the Cp
tttReO
3-catalyzed DODH of erythritol using PPh
3 as reductant and PhCl as solvent, both
cis-2-butene-1,4-diol and
trans-2-butene-1,4-diol were detected as byproduct while 2,5-dihydrofuran was not detected [
29]. However, the formation of 2,5-dihydrofuran is highly dependent on the solvent and reductant; when 3-octanol was used as solvent and reductant, 2,5-dihydrofuran was the only byproduct detected with either Cp
tttReO
3, Cp
ttReO
3, or MTO as catalyst. [
21,
22,
30]
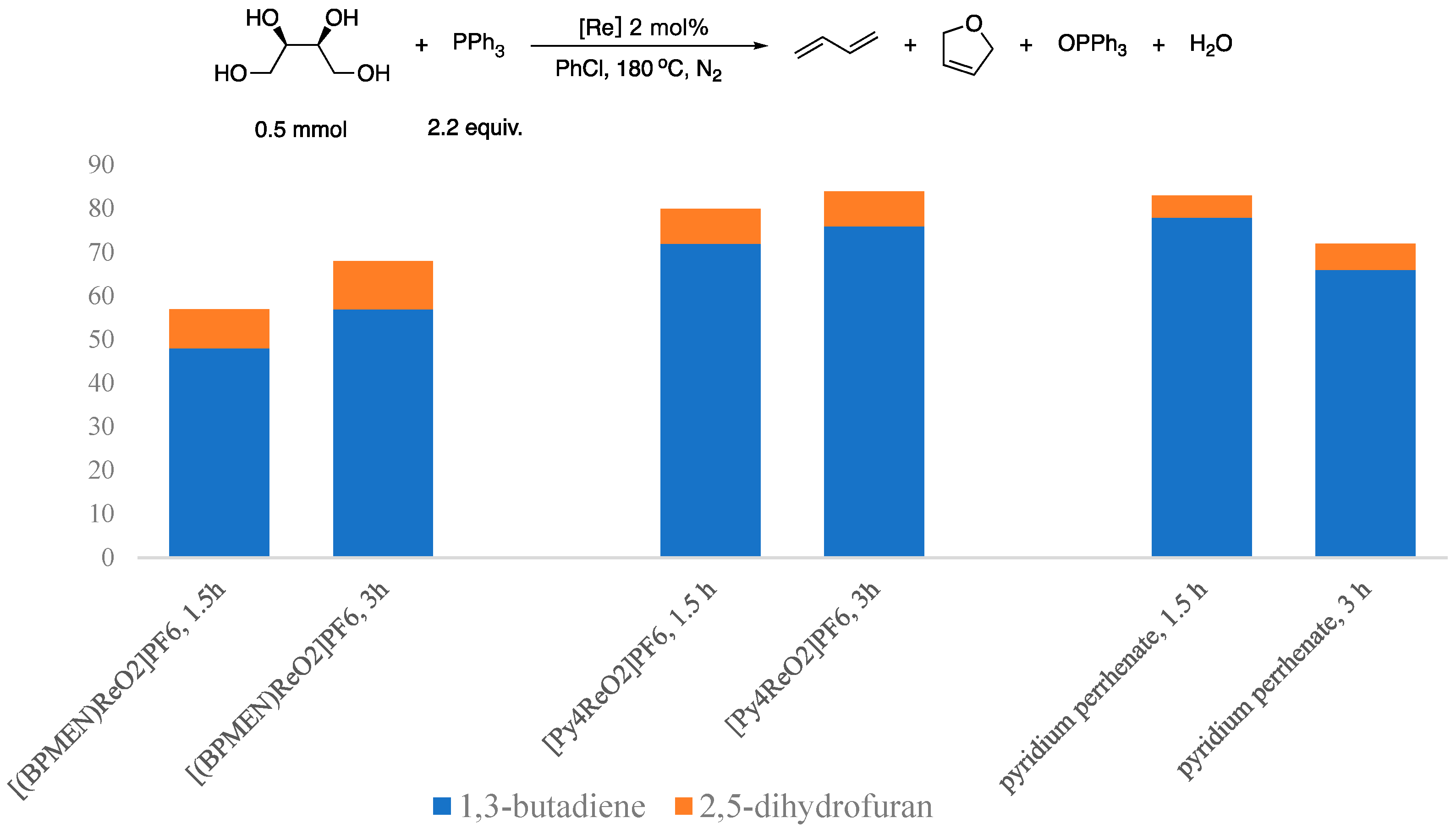
cis-[(BPMEN)ReO
2]PF
6 (
2),
trans-[(Py
4)ReO
2]PF
6 (
4), and pyridinium perrhenate were then tested for the DODH of erythritol using PPh
3 as reductant and PhCl as solvent. The reactions were performed at 180 °C for 1.5 h under N
2. The total yield of 1,3-butadiene plus 2,5-dihydrofuran was 57%, 80%, and 83%, respectively. In addition, different product distributions were observed; the ratio of 1,3-butadiene and 2,5-dihydrofuran products was 5.3:1, 9.0:1, and 15.6:1, respectively (
Scheme 6). Upon prolonging the reaction from 1.5 h to 3 h, the total yield of 1,3-butadiene plus 2,5-dihydrofuran increased to 68% and 84% for
2 and
trans-[(Py
4)ReO
2]PF
6, while a lower 72% was observed for the perrhenate catalyzed reaction. The latter decrease could be the result of secondary reactions of the rather reactive butadiene product under the reactions conditions in the presence of [PyH][ReO
4]. The ratio of 1,3-butadiene and 2,5-dihydrofuran products slightly changed to 5.2:1, 9.5:1, and 11:1 for the prolonged reactions.
Overall, the product distribution was quite consistent for these three catalysts. The differences observed in product formation and distribution in these experiments do not provide solid proof that perrhenate is the (only) active species in “ReO2” catalyzed DODH reactions. In addition, the absence of cis-2-butene-1,4-diol, trans-2-butene-1,4-diol, and 3-butene-1,2-diol byproducts indicated that these three reactions do not proceed through the same active species and/or do not form the same Re-based side products as Cp*ReO3 and CptttReO3 in the PPh3/PhCl system. On the other hand, as mentioned above, in all cases of using a secondary alcohol as both solvent and reductant, 2,5-dihydrofuran was the only detected byproduct, just like for our experiments using the PPh3/PhCl conditions; indicating that for the dioxo-rhenium and perrhenate systems the same or similar active species could actually be involved as for the Re-trioxo systems.
Since neither the time-course profile nor the product distribution studies could provide full evidence that perrhenate is the active species in dioxo-rhenium-catalyzed DODH reaction, we have sought for other means of probing the nature of the active species in these reactions. On basis of the time-course profile the DODH reaction catalyzed by
2 (
Scheme 5a), the activation/formation of the active species could proceed in a two-step manner. After a small amount of olefin product had formed, a second phase in product formation was observed before the rate of product formation really took on. Accordingly, we considered the possibility that the active species might be generated through the involvement of the initial rhenium complexes and one of the products formed in the DODH reaction, i.e., next to olefin also water and triphenylphosphine oxide (OPPh
3).
We first turned our attention to the possible role of water on the speciation of rhenium in the DODH reactions. It is known that in the present of water methyltrioxorhenium (MTO) will form a gold-colored rhenium oligomer {H
0.5[(CH
3)
0.92ReO
3]} in high yield (ca. 70%) at 70 °C, in addition to the formation of O
2, HReO
4, and methane (ca. 30% in total) [
31,
32]. Thus, we were curious about the species formed from the dioxo-rhenium complexes and water. Accordingly,
cis-[(BPMEN)ReO
2]BPh
4 (
6) was heated in CD
3CN at 180 °C for 2 h in the presence of 50 equiv. of water.
1H NMR analysis of the reaction after this period did not indicate the presence of non-coordinated BPMEN ligand. Next, this “water-treated” complex, i.e., the resulting reaction mixture, was used as catalyst in the DODH of 1,2-octanediol under our standard reaction conditions. In this case, 86% of 1-octene formed after 2 h at 180 °C and again without the formation of olefin isomers. The time-course profile of this reaction shows that only 3% of 1-octene had formed after 10 min, but that the overall induction period was significantly shorter than using pristine
cis-[(BPMEN)ReO
2]BPh
4 as catalyst (
Figure 3). After the induction period, the two reaction profiles are rather similar, which indicates that the same active species might be involved and the formation of this species might involve a reaction with water at high temperature. Comparing the time-course profile of DODH reactions using complex
6 and “water-treated”
6 as catalyst, the latter one is much faster. In both cases though, no olefin isomers were formed during the reaction and the final yield of 1-octene was the same (95%).
Next, we compared the IR spectra of
6 before and after treatment with water to that of pyridinium perrhenate (
Figure 4). The IR spectrum of pristine
cis-[(BPMEN)ReO
2]BPh
4 shows a sharp and intense signal at 813 cm
−1 assigned to Re = O stretching, while the IR of pyridinium perrhenate shows relatively broad and intense signals at 863, 741, and 672 cm
−1 assigned to Re = O stretching of the perrhenate anion. In the IR spectrum of “water-treated”
cis-[(BPMEN)ReO
2]BPh
4 the most intense signal in the 500–1000 cm
−1 range was found at 907 cm
−1, which again was assigned to Re = O stretching, albeit at significantly higher wave numbers compared to pristine
6. Two less intense bands were also observed in this range, at 761 and 706 cm
−1. Comparing these three spectra, it is obvious that no
cis-[(BPMEN)ReO
2]BPh
4 is left after water treatment. The similarities in the 500–1000 cm
−1 range for pyridinium perrhenate and “water-treated”
cis-[(BPMEN)ReO
2]BPh
4 suggest that ReO
4− is formed upon water treatment of
6. In the range of 1000–2000 cm
−1, very different signals were observed, likely due to the presence of different organic ligands.
Recently, Marrone and d’Alessandro et al. have reported a study on the active species in rhenium-catalyzed DODH reactions using secondary alcohols as reductant [
33]. An induction period was observed for the DODH of glycerol using methyltrioxorhenium (MTO), trioxo-rhenium (ReO
3), rhenium pentachloride (ReCl
5), hepta-oxo-dirhenium (Re
2O
7), rhenium triiodide (ReI
3), and IReO
2(PPh
3)
2 as catalyst and 2,4-dimethyl-3-pentanol (DMP) as reductant. After removal of the volatile reaction products and the DMP reductant, the residues of these reactions were used as the catalyst in subsequent DODH experiments and no induction periods were observed. IR spectra of these active residues were collected and compared. A few intense and broad signals were detected in the 1800–3750 cm
−1 range, and these signals could be assigned to C–H and O–H stretching vibrations. In our IR spectra of “water-treated”
cis-[(BPMEN)ReO
2]BPh
4, these kind of intense and broad signals (3346, 3053, 2852 cm
−1) were also observed in this range. In the range of 1000–1750 cm
−1, multiple intense signals were detected by Marrone and d’Alessandro while in our case, much less intense signals were observed. Since this range is where C–O stretching and C–C–H, C–O–H bending vibrations are located, these differences indicate that related species may have formed but these are not identical. In both studies, the most intense signals appeared in the 600–1000 cm
−1 range. In the study of Marrone and d’Alessandro, the most intense signal appears at 920 cm
−1 for the Re = O stretching vibration in all cases studied, independent from the rhenium sources. In our case this vibration is found at 907 cm
−1. A less intense band around 700 cm
−1 was also observed in all cases by Marrone and d’Alessandro, and assigned to either the out-of-plane bending of O–H bonds or the stretching of the Re–O–Re oxo-bridges. Overall, rather similar spectra were obtained in these two studies. By further investigating the MTO-catalyzed DODH of glycerol, Marrone and d’Alessandro proposed a Re(VII) alkoxide oligomer as the actual active species could form alongside methane from the reaction of MTO and the secondary alcohol reductant. A very important observation from their study is the formation of a catalytically active black solid in all cases. In our present study though, we did not observe the formation of black precipitates.
As described in Marrone and d’Alessandro’s work, an active DODH species is formed from the reaction of the Re source and a secondary alcohol. Although in our system, we do not use a secondary alcohol reductant, an active species seems to form from a reaction with water, and obviously a vicinal diol is present as the substrate. A possibility would be that in our case a rhenium-oxide/hydroxide oligomer is formed instead of a Re(VII) alkoxide oligomer as proposed by Marrone and d’Alessandro. The differences seen in the 1000–1750 cm
−1 range in IR analysis would support this assumption. As mentioned before, IR analysis of the {H
0.5[(CH
3)
0.92ReO
3]} oligomer, the reaction product of MTO and water, shows signals at 912 (vs), 881 (sh), 851 (sh), and 758 cm
−1 (m). [
32] These signals could be assigned to Re = O stretching vibrations. Although not identical, we observed quite similar IR vibrations in the 600–1000 cm
−1 region (
Table 4). For the {H
0.5[(CH
3)
0.92ReO
3]} oligomer, a methyl group is bound to the rhenium center, although not in a 1:1 ratio. In our case, no coordinated ligand was observed after the water treatment at 180 °C. This comparison lends further credit to the proposed formation of a rhenium-oxide/hydroxide oligomer as the active species in DODH catalysis.
2.5. Substrate Scope
Previously,
trans-[(Py
4)ReO
2]PF
6 (10 mol%) was used by Nicholas et al. as catalyst in the DODH of 1,2-decanediol using Zn as reductant and benzene as solvent to give 67% of 1-decene with full-substrate conversion at 150 °C in 24 h. In our system, 1,2-octanediol was quantitively converted to 1-octene at 180 °C in 3 h using only 2 mol% of
trans-[(Py
4)ReO
2]PF
6. Remarkably, upon increase of the catalyst loading under our conditions to 10 mol%, the reaction was done in 15 min at 180 °C. For the Cp-based trioxorhenium-catalyzed DODH systems, much longer reaction times are needed to realize the full conversion of 1,2-octanediol. As mentioned before, no isomers were observed under our reaction conditions, while small amount of isomers were observed under the same conditions for Cp-based trioxorhenium catalysts [
27]. Furthermore, for the deoxydehydration of erythritol (
vide supra),
trans-[(Py
4)ReO
2]PF
6 was quite a competitive catalyst (
Scheme 6). This initial catalytic capacity test of
trans-[(Py
4)ReO
2]PF
6 encouraged us to apply this precatalyst to other substrates (
Table 5). When the aromatic vicinal diol 1-phenylethane-1,2-diol was used as substrate, a quantitative styrene yield was obtained (
Table 5, entry 1). For the DODH of 1,4-anhydroerythritol, 97% of 2,5-dihydrofuran was formed (
Table 5, entry 2); this 2,5-dihydrofuran yield is competitive to the one obtained from the MTO/3-pentanol system, which provides 95% of 2,5-dihydrofuran [
30]. Glycerol, a biomass-derived triol, was also investigated in our reaction system and gave a quantitative amount of allyl alcohol (entry 3). Although quantitative allyl alcohol formation is also obtained by the Cp
ttReO
3/3-octanol system [
23], the air-stable property of
trans-[(Py
4)ReO
2]PF
6 makes this catalyst system more attractive.
trans-[(Py
4)ReO
2]PF
6 also catalyzes the DODH of biomass-derived L-(+)-tartaric acid and mucic acid. When using L-(+)-tartaric acid as substrate, there was no product detected using PPh
3 as reductant and PhCl as solvent. This is most likely due to the poor substrate solubility in PhCl. Using 3-pentanol as both solvent and reductant gave 84% of the corresponding olefins as a mixture of fumaric acid and fumarate esters. Along a similar vein, a total yield of muconic acid and muconates of 65% was obtained using mucic acid as substrate (
Table 5, entry 5). In the case of Cp
ttReO
3-catalyzed DODH of mucic acid, a slightly higher product yield of 71% was found, albeit at 5 mol% catalyst loading. Overall,
trans-[(Py
4)ReO
2]PF
6 is a good pre-catalyst for the DODH of diols and biomass-derived polyols.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









