1. Introduction
Biocatalysts (extracellular enzymes or whole-cells) constitute an alternative to traditional catalytic systems, being able to cause transformation of natural or synthetic substrates under mild reaction conditions, with high substance-, stereo-, and regiospecificity, and lack of toxic byproducts [
1,
2]. With the advances in the production of relatively inexpensive free enzymes, the steady increase of their extensive implementation as effective catalysts in many industrial and biomedical applications is beyond any doubt [
3].
The wide use of industrial-scale processes with enzymatic catalysts is restricted by the free enzymes’ inactivation by different denaturant agents and by their difficult or even impossible recovery from the reaction medium after use. These problems can be resolved by immobilization (conjugation) of the enzymes to prefabricated matrices of various nature, geometry, and topography. Immobilized enzymes, apart from their application as reusable heterogeneous biocatalysts, can serve as appropriate platforms for the development of biosensors, biofuel cells, or microscale devices for controlled release of protein drugs [
4].
The most frequently used immobilization techniques can be sorted into five main groups, each with its associated advantages and disadvantages [
5]: (i) non-covalent adsorption or deposition; (ii) immobilization via ionic interactions; (iii) entrapment in a polymeric gel or capsule (encapsulation); (iv) covalent attachment to the support (tethering); and (v) cross-linking of the enzyme. In the first three cases (i–iii), the enzyme immobilization is related to physical phenomena, i.e., entropy effects, electrostatic interactions, or hydrogen bonding. The distinct advantage of these methods is that neither the enzyme, nor the support, needs to be chemically modified or pretreated, which contributes to a cleaner and cost-effective biocatalyst. Moreover, the enzyme retains mobility, thus avoiding unfavorable screening of the active site. However, conjugates based on physical interactions often suffer from enzyme leaching, especially if used in aqueous media. To avoid leakage, immobilization protocols from groups (iv–v) are used. In them, covalent bonds across the enzyme–support interface are produced or cross-linking of the polypeptide moieties of the enzyme is caused by appropriate chemical reagents. These chemical modifications should be carefully directed so as not to cause deactivation of the biomacromolecule. As a rule, the immobilization method is selected on a case-by-case basis, by trial and error, although attempts to optimize this process by computer-aided analysis and molecular modeling have also been communicated [
3,
6].
General requirements for polymer supports are that they should not deactivate the immobilized enzyme, and for many important applications they have to be biocompatible. Therefore, traces of solvents, emulsifiers, unreacted monomers, or other accompanying substances that could be toxic or potentially deactivate the immobilized enzyme must be totally excluded. More specifically, immobilization by enzyme entrapment requires formation of the polymer support in situ, e.g., by low-temperature processes in the presence of the respective enzyme, thus avoiding denaturation. Furthermore, the monomer, the polymer support, and the enzyme should possess suitable functional groups in order to enable covalent or non-covalent interactions before, during, or after the polymerization. Finally, the polymer support should have appropriate physical structure and be formable into controllable shapes and textures at various length scales [
7].
A large number of supports have been employed for enzyme immobilization in the form of beads, porous particles, membranes, and micro- or nanosized fibers made of inorganic materials [
8] or organic polymers such as polyacrylates, cross-linked styrene-based copolymers, smart polymers, and modified polysaccharides [
3]. Smart biocatalysts for industrial and biomedical applications produced by conjugation of enzymes to stimuli-responsive polymer supports have attracted a growing interest [
9]. The activity of the enzyme can be favorably modified by complexation with supramolecular linear-dendritic block copolymers [
10] or amphiphilic block copolymers self-assembling into micelles or physical networks [
11].
Among the big variety of industrially relevant enzymes, the group of laccases (EC 1.10.3.2, benzenediol:oxygen oxidoreductases) have generated significant biotechnological interest since they require molecular oxygen as the final electron acceptor and release only water as a by-product [
12]. Due to their broad specificity laccases are being used in different industrial fields for very diverse purposes, from food additives and beverage processing to biomedical diagnosis, pulp delignification, wood fiber modification, chemical or medicinal synthesis, or in the production of biofuels [
13]. They show great promise as ‘green’ synthesis and polymerization mediators [
14,
15,
16] and catalysts for Bisphenol A removal [
17]. Also, laccases have great potential to biotechnological applications of industrial effluent treatment including textile dye decolorization and degradation [
18,
19]. Enzymes from the laccase group have also been studied intensely in nanobiotechnology for the development of implantable biosensors and biofuel cells, as well as wastewater and soil remediation [
20].
Synthetic polyamides are among the known supports for laccase immobilization since they are cheap and can be easily produced in different forms. Thus, Fatarella et al. [
21] explored the preparation of polyamide 6 (PA6) film or electrospun nanofiber carriers for covalently bonded laccase from
Trametes versicolor, reaching enzyme loadings of ca. 60% and 71%, respectively. As reported, the resultant PA6/laccase conjugates are useful in industrial biocatalyst applications. Furthermore, a nanofibrous membrane for 3,3′-dimethoxybenzidine detoxification was developed by Jasni et al. [
22] on the basis of PA66/laccase/Fe
3+ conjugate produced by covalent bonding of the enzyme to the support by glutaraldehyde crosslinking. The immobilized enzyme displayed enhanced storage stability and a good potential for emerging pollutant detoxification. In a study of Silva et al. [
23], laccase from
Trametes hirsuta was covalently immobilized on woven PA66 supports employing glutaraldehyde and a 1,6-hexanediamine spacer. The PA66-laccase conjugate obtained under optimized conditions displayed higher half-life time than the free enzyme and potential for application in membrane reactors for continuous decolorization of effluents.
To the best of our knowledge, at this point of time, no attempt has been reported on laccase immobilization by polyamide particulate supports. Very recently, Dencheva et al. [
24] synthesized neat and magnetic responsive PA4 [(poly-2-pyrrolidone), PPD] microparticles in good yields, with controlled size, shape, and porosity using activated anionic ring-opening polymerization (AAROP) of 2-pyrrolidone (2PD) carried out at 40 °C. This low-temperature and solventless process produces magnetic PA4 microparticles with controllable size, shape, and structure. There are certain advantageous factors in this technique that can open promising routes towards novel recyclable and efficient biocatalysts: PA4 is biodegradable in various environments [
25,
26], its monomer 2-pyrrolidone can be produced by sustainable biosynthesis [
27], and the polyamide support is synthesized directly in the form of porous microparticles without the need of any additional treatment. Furthermore, the chemical structure of PA4 with its dense amide linkages and final functional groups resembles that of the proteins, thus enhancing the H-bonding between the support and the immobilized enzyme. Evidently, the neat and magnetic responsive PA4 microparticles combine most of the requirements for effective supports of biomolecules. Moreover, they have already been found to be good supports for model proteins [
24] or to be used in molecular imprinting [
28].
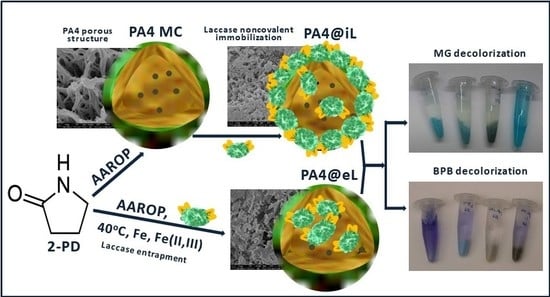
The present work investigates the potential of neat and magnetic-responsive PA4 microparticles obtained by low-temperature AAROP to serve as supports for immobilization of laccase. Two immobilization strategies were employed: (i) physical adsorption of laccase upon preformed PA4 microparticles and (ii) in situ encapsulation (entrapment) of laccase during the AAROP of PA4 synthesis. The morphology and the crystalline structure of the PA4-laccase conjugates obtained by these two methods were analyzed by microscopy, spectral and X-ray scattering techniques, and the respective enzyme activities toward 2,2’-azino-bis (3-ethylbenzothiazoline-6-sulphonic acid (ABTS) were compared. The biocatalytic capability of the synthesized PA4-laccase conjugates, as well as their reusability, were tested in two reactions of environmental interest related to the treatment of effluents from the textile industry. The activity of the immobilized laccase was tested in decolorization reactions of two dyes, widely used as textile coloring agents (malachite green and bromophenol blue).
2. Results and Discussion
Three microparticulate PA4 supports, without and with magnetic susceptibility, designated as PA4, PA4-Fe, and PA4-Fe
3O
4, were synthesized by low-temperature AAROP. They are in the form of fine powders with white, grey or brownish color, respectively. The scheme for the AAROP representing the chemical structure of all substances involved is presented in
Figure S1 of the Supporting Information. When the polymerization is carried out in the presence of magnetic micro/nanoparticles, the PA4 MP become susceptible to external magnetic fields [
24].
The 2PD monomer is a cyclic analog of the amino acids and the chemical composition of PA4 is comparable to that of proteins and enzymes. This will expectedly enable intense H-bond formation between the laccase and the PA4 supports: neat PA4, PA4-Fe, and PA4-Fe3O4. The fact that AAROP was possible at 40 °C allowed the entrapment of active laccase into the shell of the forming PA4 MP during the polymerization, resulting in three PA4@eL samples. Separately, the enzyme was immobilized by physical adsorption on prefabricated PA4 supports, thus preparing the three PA4@iL samples. This work presents a comparative discussion on the morphology, structure, and the catalytic activity of the three conjugates immobilized by adsorption PA4@iL and the three conjugates immobilized by entrapment PA4@eL.
2.1. Synthesis and Morphology of Empty PA4, PA4-Fe, and PA4-Fe3O4 Microparticles
Some basic characteristics of the synthesized neat and magnetic PA4 microparticles (MP) are listed in
Table 1. The yields of polymer were in the 59–63 wt % range meaning that the presence of iron micro- and iron (II, III) oxide nanosized particles in the reaction mixture did not affect the activity of DL/C20 catalytic system and the kinetics of the AAROP reaction. Intrinsic viscosity (η) (see the
Supporting Information) of 0.926 dL/g was found for the neat PA4 MP, which is similar to the values of PA4 microspheres obtained by a different method involving AAROP of 2PD [
29].
The average maximum size of the particles
dmax in all PA4 empty supports based on optical microscopy with image-processing was found to be between 8–20 μm (
Table 1,
Figure S2 of the Supporting Information). The
dmax, of PA4 MP without magnetic load is the smallest, ranging between 8 and 15 µm. Similar values of 10–15 µm are registered for the support with nanosized Fe
3O
4 loads, while in the presence of Fe the upper limit of
dmax grows above 20 µm. The Fe- and Fe
3O
4 containing PA4 microparticles become less spherical, as seen from the roundness parameter
dmax/
dmin (
Table 1,
Figure S2). This means that the PA4-Fe MP would contain in their core up to 4–5 iron microparticles whose proper size is 3–5 µm, while the PA4-Fe
3O
4 core would expectedly contain iron oxide particles in the nanometer length scale [
24,
28]. The
dmax/
dmin in the range of 3–4 found in both magnetic empty supports are attributable to self-assembly of magnetized Fe and Fe
3O
4 to higher aspect ratio aggregates with their subsequent coating with PA4 during the AAROP.
All PA4 supports have porous structure as demonstrated by BET analysis (
Table S1 of the Supporting Information). The neat PA4 microparticles showed the largest specific surface area,
SBET = 2.766 m
2/g with a total pore volume
Vtotal = 0.012 cm
3/g, followed by PA4-Fe
3O
4 (2.542 m
2/g; 0.008 cm
3/g) and PA4-Fe (2.052 m
2/g; 0.004 cm
3/g) samples. The largest average pore size
σave = 85 Å was found for the PA4 MP, the values of PA4-Fe
3O
4 and PA4-Fe being 42 and 32 Å, respectively. These changes in
σave, Vtotal, and
SBET should be attributed to the presence of dense magnetic loads in the microparticles core of the last two samples.
More details on the morphology of the empty supports′ particles can be obtained by SEM (
Figure 1).
The micrographs of the neat PA4 support (
Figure 1a–c) display spheroidal particles with sizes of the individual entity between 5–8 µm, the latter forming also aggregates with average sizes close to 20 µm. At larger magnifications, the PA4 particles display porous, scaffold-like topology with visible pore diameters in the range of 100–150 nm. The use of Fe and Fe
3O
4 particles (
Figure 1d,e,g–i, respectively), seem to decrease the number of surface pores changing their distribution but with no significant change of the pore size. Moreover, as seen from some selected EDX traces in
Figure 1f,i, the FeKα and FeKβ peaks only appear if the electron beam is directed into the pores (location Z1), rather than if it hits a non-porous spot of the particles surface (location Z2). This means that most of the magnetic particulate fillers (especially the much larger Fe particles) are embedded in the core of the respective MP, thus confirming the supposed precipitation–crystallization mechanism of the polyamide MP formation [
24].
2.2. Immobilization of Laccase on PA4 MP Supports
2.2.1. Immobilization by Physical Adsorption
As previously mentioned, owing to the analogous chemical structure of PA4 and the peptide-containing biomolecules, a high capacity toward H-bonding formation is expected between the laccase and the PA4 supports during immobilization by adsorption. Since the laccase isoelectric point is in the range of 3–4, if the adsorption is carried out at pH > 4 the enzyme will be negatively charged. From the Z-potential measurements (see the
Supporting Information, Table S2) it can be seen that under such conditions the three PA4 supports will also be negatively charged with values of –35 eV at pH 7 and between −10 ÷ −12 eV at pH 5. Nevertheless, preliminary adsorption tests confirmed that larger amounts of laccase are adsorbed in DDW at neutral pH than in PB, pH 5. This means that the immobilization by adsorption is not upset by possible electrostatic repulsion between the enzyme and the support, therefore all adsorption experiments were performed at pH 7.
Table 2 shows that, as expected, the particles of the three PA4@iL laccase-adsorbed samples do not change their average size and roundness. Judging from the SEM images in
Figure 2, the topography of all particulate PA4@iL conjugates is noticeably smoother as compared to the starting PA4 support particles, which is better expressed in the case of the PA4-iL sample (
Figure 2c).
The next step in the PA4@iL preparation was to quantify the amount of the adsorbed enzyme. Using the common method for determination of immobilized laccase, after adsorption completion, the residual supernatants were subjected to activity test toward ABTS. All three samples showed no activity, leading to the conclusion that there is no residual laccase in the supernatant, therefore almost 100% of the enzyme was adsorbed upon the PA4 supports (
Table 3). This was a superior immobilization efficiency as compared to previous studies disclosing laccase immobilization by covalent bonding on PA6 [
21] or carbon nanotube supports [
30] with immobilization effectiveness (IE) values of 59–71% or 60–90%, respectively. However, the lack of activity of the residual supernatant means that it is free of active laccase but could contain some amounts of inactive enzyme. Therefore, a second method to verify the laccase content in the supernatant was used.
An alternative method for laccase quantification can be based on its UV absorbance in the 280–290 nm range [
31,
32] due to the presence of aromatic amino acid residues: tryptophan, tyrosine, or phenylalanine. Thus, all supernatants were studied for residual laccase by UV spectroscopy measuring the intensity of the band appearing at 286 nm. As indicated in
Table 3, 19–46% of residual laccase was found in the supernatants, which showed no activity in the previous test with ABTS. A possible reason for this behavior is that during the multiple adsorption/desorption processes in the PA4@iL sample preparation, a structural change occurs leading to lowering of the redox potential of the desorbed laccase, this effect depends on the PA4 support composition. Based on UV spectroscopy data, the maximum amount of laccase (40.3 mg/g or 81% from the enzyme amount) was adsorbed on the PA4-Fe MP support, whereas the PA4-Fe
3O
4 MP displayed the lowest IE of 54%. These IE values were considered more reliable and were therefore used in all further calculations.
Employing the approach of Qui et al. [
33], the data in
Table 3 can provide information about the mechanism of laccase adsorption on the PA4 supports of this study. Thus, since the size of laccase macromolecule is found to be 6.5 × 5.5 × 4.5 nm [
34], its largest footprint on a surface will be ca. 35.8 nm
2. The surface areas of the three PA4@MP samples determined by BET (
Table S1) were in the range of 2.05–2.78 m
2/g. Assuming that laccase molecules arrange in ideal monolayers when adsorbing and that the laccase molecular weight is ca. 85,000 Da, then 1.0 g of PA4 support should theoretically adsorb 8–11 mg enzyme. According to
Table 3, however, the real values vary in the 27–40 mg range, i.e., about 3–4 times larger. Evidently, the laccase adsorption occurs in multilayers, implying lateral intra- and interlayer interactions between adsorption sites. This finding is in good agreement with our previous work on protein adsorption upon similar PA4 microparticles [
28] proving that without special treatment of the PA4 support the adsorption data are consistent with the Freundlich isotherm, thus proving the multilayer adsorption model.
2.2.2. Immobilization by Entrapment
Entrapment is defined as physical retention of enzymes in a porous solid matrix [
35]. In our case, the laccase entrapment occurs during AAROP. The enzyme is first suspended in the monomer solution, and a subsequent polymerization process keeps the biomolecule trapped, preventing direct contact with the environment. To obtain laccase-entrapped PA4 MP with or without magnetic particles, the same low temperature AAROP was used as for the preparation of the empty PA4 supports, however carried out in the presence of the enzyme. The final 2PD conversion to PA4 was in the 45–58 wt % range (
Table 4), i.e., in average 5–10% lower than of the empty PA4 supports (
Table 1). This is explained with the more rigorous washing/purification procedure to eliminate not entrapped enzyme and oligomers with lower conversion to PA4. It can therefore be concluded that the presence of laccase in the AAROP reaction mixture did not significantly upset the catalytic system permitting to use the same protocol for synthesis and purification as for the empty PA4 particulate supports. However,
Table 4 indicates larger average particle sizes of 15–25 µm and lesser roundness values of 1.2–1.3 in the PA4@eL samples than in the respective empty and laccase-adsorbed supports (
Table 2).
The possibility to carry out AAROP to PA4 in the presence of laccase at 40 °C had two major advantages. First, no denaturation or other disruption of the enzyme′s secondary or tertiary structure caused by temperature will occur during the polymerization process. Second, in AAROP of all lactams, the chain propagation is accompanied by PA4 crystallization that affixes the topology of the enzyme-loaded microparticles and results in their precipitation from the reaction medium, thus facilitating the entrapment.
As seen from
Table 5 showing the laccase quantification in the PA4@eL series, the total amount of entrapped laccase was 45–58 mg, corresponding to 13–18 mg enzyme per gram of MP. These concentrations are up to 2.5 times lower than in the case of immobilization by adsorption (
Table 3). As to the entrapment factor EF, it is dependent on the 2PD conversion to PA4. Thus, the highest degree of 2PD conversion of ca. 70% was achieved with the PA4-Fe
3O
4@eL sample, which accounted for the minimum amount of entrapped enzyme per gram support in this case.
Table 4 indicates larger average particle sizes of 15–25 µm and lesser roundness values of 1.2–1.3 in the PA4@eL samples than in the respective empty and laccase-adsorbed supports.
Figure 3 displays selected SEM images at different magnifications of the three PA4 supports carrying entrapped enzyme.
Unlike the three empty PA4 supports or the magnetic-loaded laccase conjugates PA4@iL, the particles′ shapes in PA4@eL samples are far from being spherical or elliptical. Most probably, this is due to the fact that upon its dispersion in the 2PD monomer the lyophilized enzyme produced aggregates that were subsequently covered by PA4. The growing PA4 molecules wind up upon these aggregates and, after reaching a critical molecular weight, crystallize upon them interacting by H-bonds with the enzyme. As a result, the specific topography of the PA4@eL conjugates is produced with visible average pore diameters up to 300–350 nm, being almost twice as large as in the other samples and with quite distinct shape.
2.3. Structure Characterization of PA4 Empty Supports and Laccase Conjugates
The PA4 porous microparticles obtained by AAROP are used for the first time as enzyme supports. The laccase-entrapped PA4@eL samples are also synthesized for the first time. Therefore, some initial structural characterization of all samples of this study was necessary in order to be able to explain their catalytic activity and decide about their potential as enzyme supports.
2.3.1. FTIR Spectroscopy
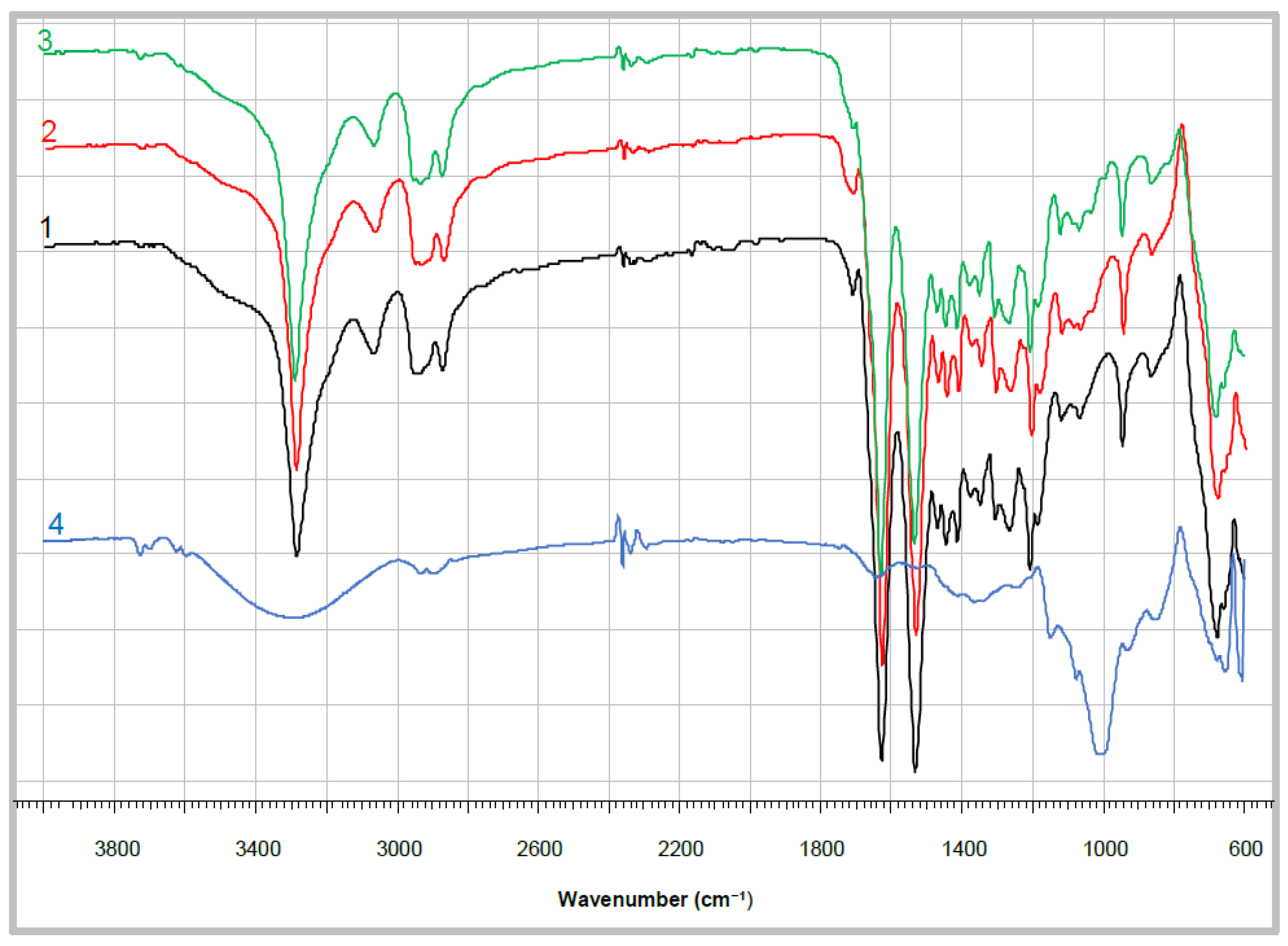
A FT-IR spectra comparison between the empty PA4 microparticles and the laccase-adsorbed and entrapped samples are presented in
Figure 4. In all samples, the bands at 3300 cm
−1 were assigned to the valence stretching vibrations of hydrogen atoms in secondary NH groups. The shoulder with a maximum at 3450 cm
−1 was attributed to stretching vibrations in primary amines corresponding to terminal amine groups. Also, the spectra show well defined peaks for Amide I at 1631.5 cm
−1 and Amide II at 1535.0 cm
−1 with almost identical intensities. This is a clear indication for fixation of the trans-conformation of the NH–CO group, being typical for high molecular weight polyamides and proteins.
The IR spectra in
Figure 4 indicate undoubtedly that even at polymerization temperatures as low as 40 °C AAROP of 2DP led to high molecular weight polyamide in all samples studied. This was important to confirm for the PA4@eL and PA4@iL samples, in which the determination of (η) by viscosimetry is technically impossible. All curves display also a weak peak at 1710–1712 cm
−1 attributable to terminal carboxyl groups. In the neat PA4 sample (curve 1) this narrow and well-resolved band belongs to the –COOH terminus of the polyamide macromolecule. In the enzyme-adsorbed and entrapped samples (curves 2 and 3) a broader composite peak appears due to superposition of the –COOH signal of PA4 with such belonging to laccase carboxyl groups. Curve 4 indicates that a weak and broad band centered at 1641.0 cm
−1 really exists in the free laccase spectrum. However, FT-IR cannot provide more detailed quantitative or qualitative information about the entrapped or adsorbed laccase.
2.3.2. DSC and TGA
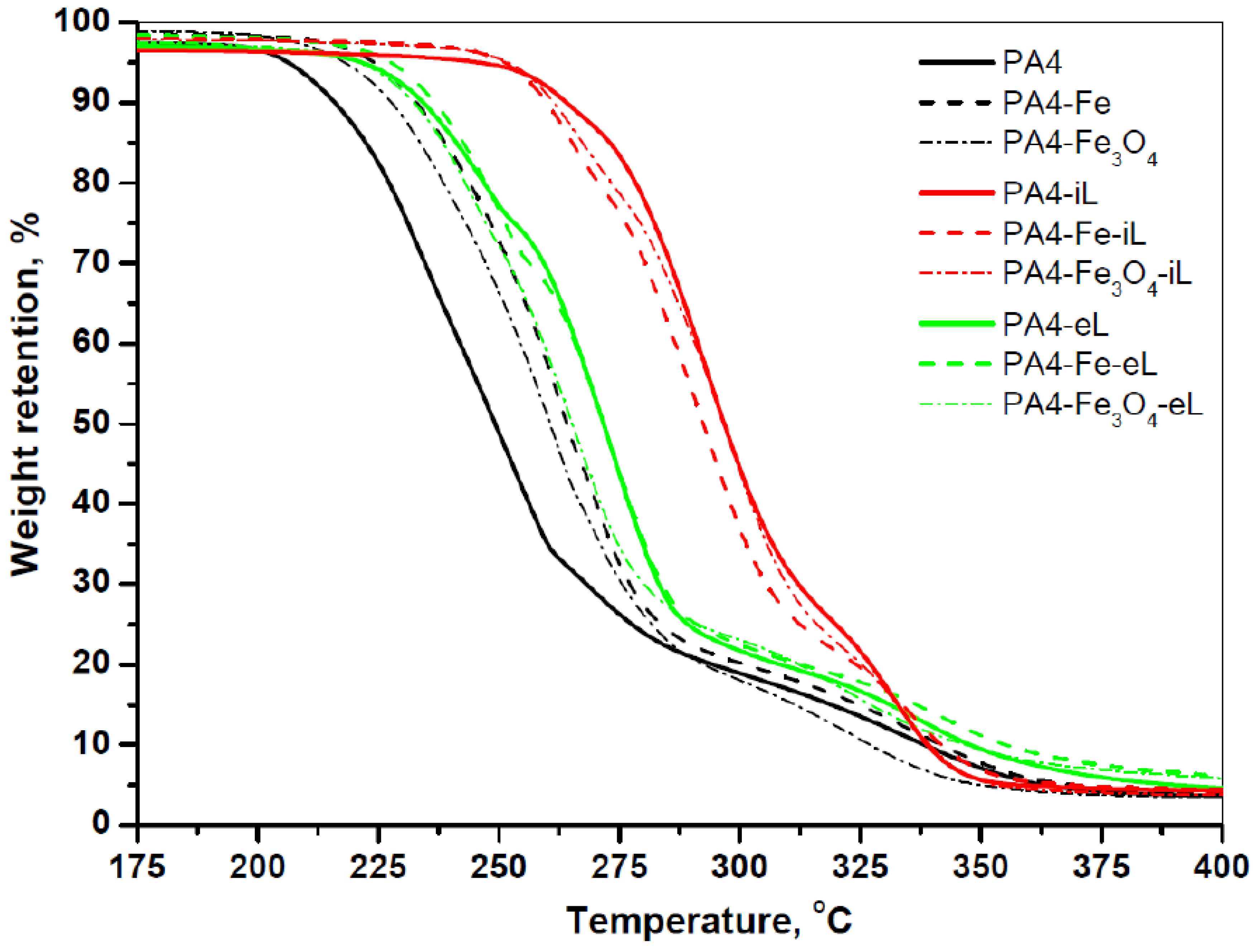
A typical shortcoming of PA4 is the intense degradation that precedes or accompanies the melting occurring normally in the broad range of 230–260 °C [
24,
36]. As shown previously, the presence of a model protein adsorbed onto PA4 microparticles can significantly increase the thermal resistance of the latter [
28]. The TGA traces of all samples in this work displayed in
Figure 5 confirm that the presence of laccase in the PA4@iL series can also considerably improve the thermal stability of the supports. Thus, the temperature of initial degradation
of PA4-iL is 255 °C, which is 38 °C higher than of the respective PA4 empty support particles. For the PA4-Fe-iL/PA4-Fe and PA4-Fe
3O
4-iL/PA4-Fe
3O
4 pairs
20 and 26 °C, respectively. Similar behavior was observed in PA4 obtained by AAROP of 2PD with subsequent conversion of the terminal –COOH into –NH
2 groups [
37], which in our case may have occurred during the heating scan in the TGA.
It should be noted that the presence of magnetic particles alone in PA4 MP increases the
with 15–20 °C in comparison with the neat PA4 MP, most probably due to heat dissipation phenomena. At the same time, the
for the entrapped PA4-eL/PA4 pair is only 12 °C. It can be hypothesized that in the case of the laccase-entrapped PA4@eL samples the said conversion of PA4 terminal groups is difficult or impossible. The TGA study permitted also to determine the carbonized residue at 600 °C of each sample of this study and to calculate on this basis the real content of Fe or Fe
3O
4 by means of Equation (2) (see Materials and Methods—
Section 3.2).
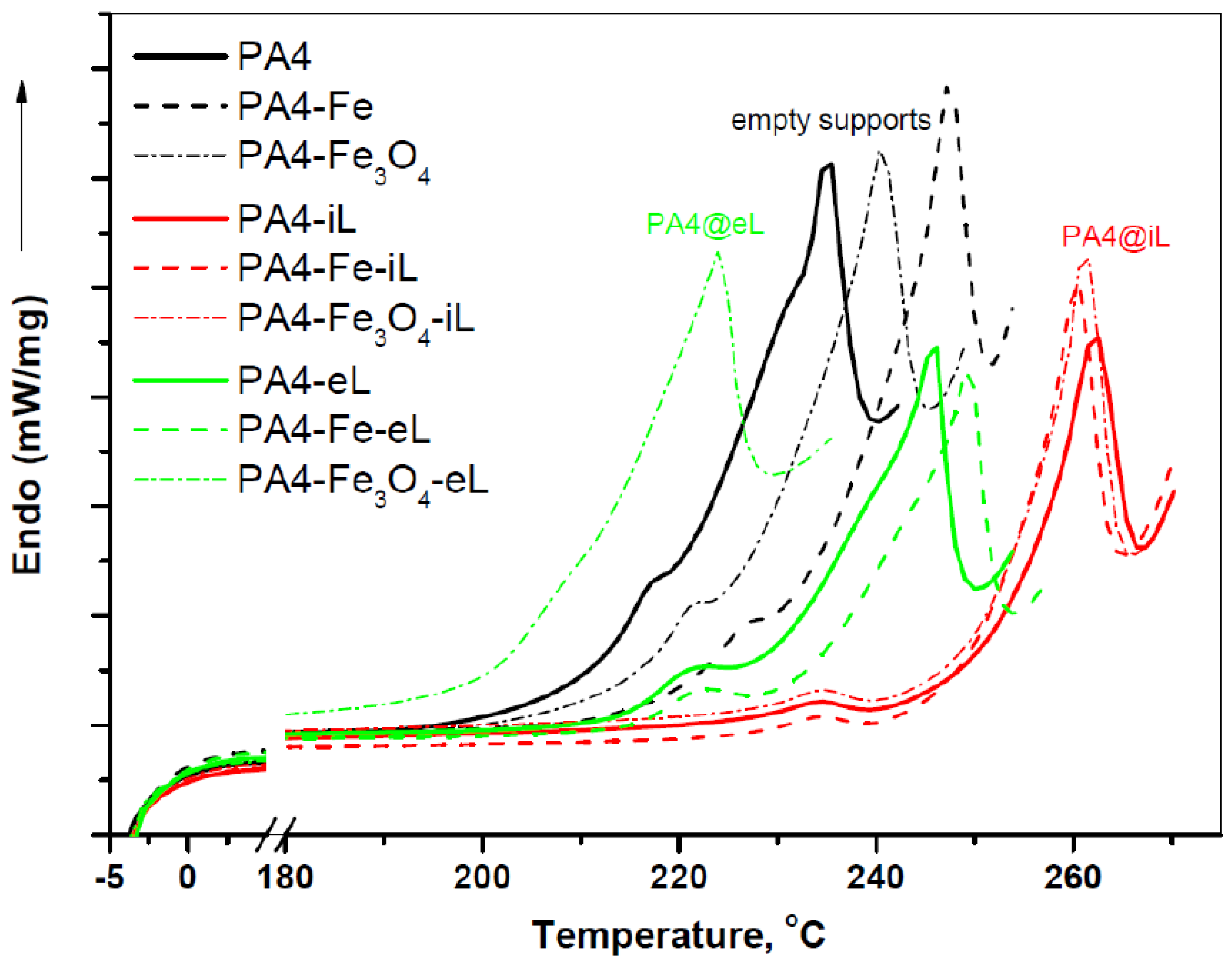
The DSC curves of all samples presented in
Figure 6 and the consolidated data extracted from them (
Table S3 in the Supporting Information) display a trend of increasing the melting temperature
Tm of PA4 in the presence of enzyme. After immobilization by adsorption in the PA4@iL series, the
Tm of the three resulting conjugates goes over 260 °C irrespective of the support type. This is definitely higher than the
Tm of the three empty supports, being in the range of 235–247 °C. As for the laccase-entrapped samples, the two of them i.e., PA4-eL and PA4-Fe-eL melt at slightly higher or similar
Tm, whereas the PA4-Fe
3O
4-eL sample is the only one with a
Tm with 11 °C lower than the respective empty support. The fact that both TGA and DSC display similar degradation behavior and melting temperatures for the empty supports and the laccase-entrapped samples means that the molecular weight of PA4 in these two sets should be similar and relatively high.
2.3.3. Synchrotron WAXS
The activity of laccase physically adsorbed onto or entrapped into PA4 MP and the efficiency of the immobilization process in each case will directly depend on the nature and strength of the interactions at the enzyme/PA4 support interface. The postulated intense H-bond formation between the laccase and its structural analogue PA4 should have some influence on the enzyme configuration, or the crystalline structure of the polymeric support that can be probed by X-ray scattering techniques. It is important to know also whether or not the interior pores and channels of the PA4 microparticles that are impossible to access for direct SEM observation are filled with enzyme. In an attempt to evaluate these factors in the empty PA4 supports and in the respective adsorbed or entrapped PA4-laccase conjugates, synchrotron WAXS and SAXS were employed. Moreover, no structure studies about the PA4 crystalline structure by synchrotron X-ray were reported up to now.
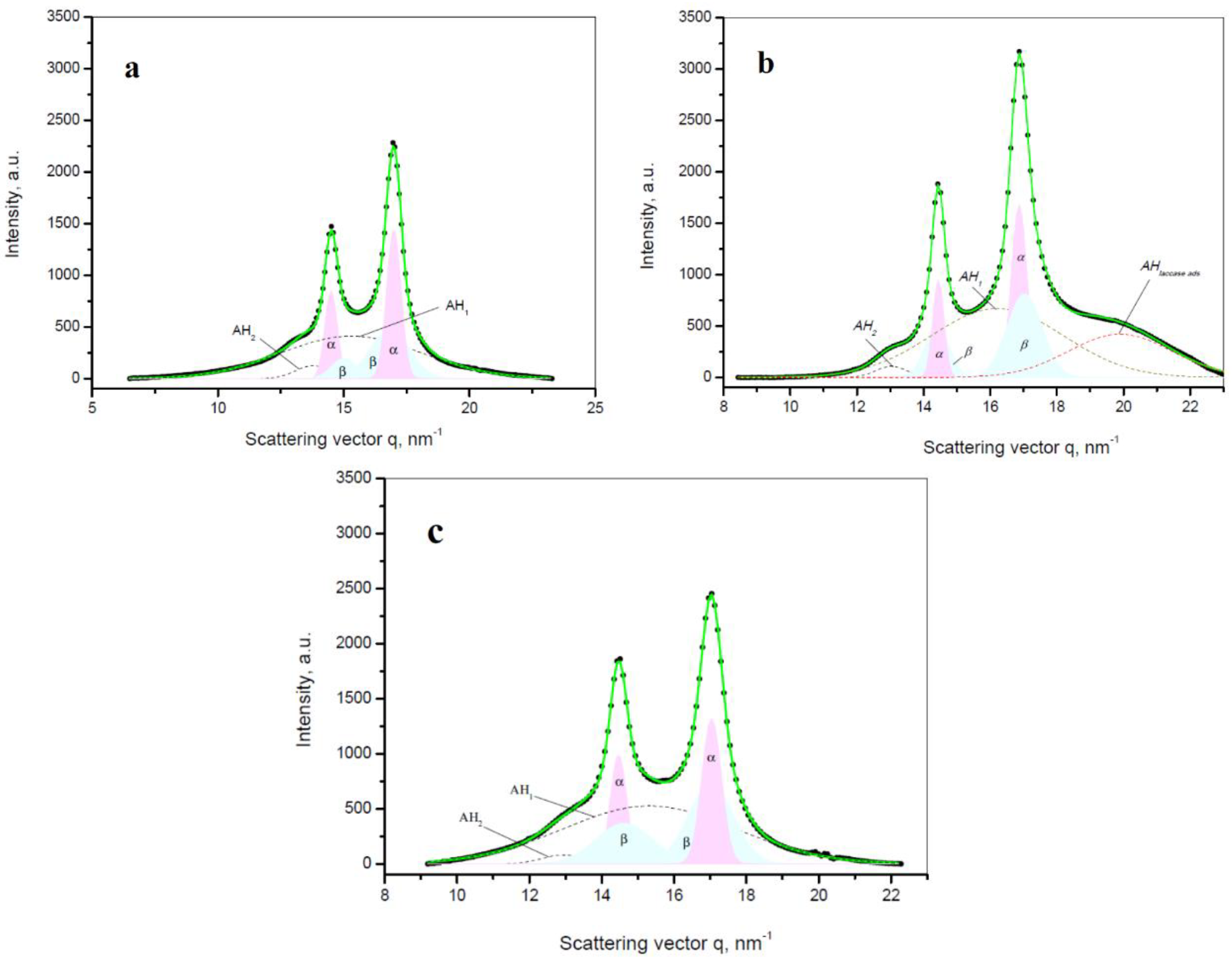
Figure 7 shows a comparison between the linear WAXS patterns of samples representing the three empty supports, as well as the three PA4@iL immobilized and the three PA4@eL entrapped samples. All WAXS patterns display two strong reflections at
q ≈ 14.5 nm
−1 and 17.0 nm
−1 that, according to Bellinger et al. [
38], should be ascribed to the monoclinic unit cell of the α-PA4 with d
α[200] = 4.33 Å and d
α[020] = 3.69 Å. Moreover, in accordance with the same study, it should be postulated that the PA4 chains are parallel to the lamellar normal and that an amide group is incorporated in the fold. Such incorporation does not take place in PA6 or PA66, however it is very typical for the β-bends in proteins [
39]. Consequently, PA4 is really a closer structural analogue to all protein-containing biomolecules than other polyamides.
The visual inspection of the WAXS patterns of the three PA4 supports in
Figure 7a suggest that the presence of Fe or Fe
3O
4 fillers does not seem to change the PA4 crystalline structure leaving the angular position and the intensities of the two α-PA4 reflections unaffected.
Figure 7a displays also the pattern of a free laccase producing a wide diffuse scattering peak (halo) typical of amorphous materials and centered at
, i.e., the free laccase is amorphous at the length scale of various angstroms that is probed by WAXS.
The patterns of PA4@iL samples (
Figure 7b) represent a superposition of the laccase and PA4 support scatterings. The two α-PA4 reflections apparently maintain their form and position, but the amorphous laccase halo appears centered at
. It is attributable to the adsorbed bulk laccase deposited on the surface and within the pores of the PA4 particulate support. The shift Δ
q ≈ 7 nm
−1 is significant and can be explained as follows. Since the amorphous halo in WAXS is related to intermolecular interactions [
40], its position must be dependent on the degree of packing of the molecules (i.e., the density) of the respective amorphous phase. As pointed out by Alexander [
41], the dependence of the amorphous halo angular position
qa on the intermolecular distance
ra is given as
which is the reciprocal dependence typical of all diffraction phenomena. Thus, the larger the scattering vector
qa, value is, the smaller the intermolecular distance and consequently the higher the density of this phase will be. This means that the adsorbed enzyme in the PA4@iL samples has a denser packing as compared to the free one, confirming the supposition for intensive interaction between the laccase and PA4 via multiple H-bonds.
The patterns of the entrapped PA4@eL samples (
Figure 7c) do not show any difference in comparison to the empty PA4 supports in
Figure 7a. This observation leads to two conclusions: (i) the enzyme arrested in the PA4 particles during AAROP cannot form a separate amorphous reflection and (ii) the entrapped enzyme does not upset the crystallization of the α-PA4 polymorph. It can be therefore hypothesized that, in the PA4@eL series, the enzyme macromolecules are distributed within the amorphous phase of the semi-crystalline PA4 support quite homogeneously with no significant interaction between one another.
Further information about the crystalline structure of the samples can be extracted after deconvolution of the WAXS patterns in
Figure 8 by peak fitting. This procedure and the subsequent quantification of the α- and β-PA4 crystalline phases is made according to earlier publications, resolving the crystalline parameters and polymorph structure of PA4 [
38] and PA6 [
42]. All structural information from the fitted WAXS patterns is presented in
Table 6.
All example deconvolutions in
Figure 8 show that excellent fits with regression coefficients R
2 ≥ 0.99 were only possible if along with the two peaks of α−PA4 and the two amorphous halos AH
1 and AH
2 two more crystalline peaks were considered that should be assigned to an additional monoclinic phase designated by β-PA4. It was first described in the early work of Frederiks et al. [
43] and later on shown to co-exist with α-PA4 [
24].
Table 6 shows that the values of the long spacings of these two PA4 phases almost coincide. Thus, the reflections related to d
β(002) and d
β(200) are quite away from each other, which is typical of a monoclinic unit cell, contrary to the two γ-PA6 peaks that are often described as belonging to a pseudo-hexagonal unit cell [
42,
44].
Table 6 displays the structural data of all studied samples.
The α/β ratio in the empty PA4 supports and in the laccase-entrapped samples PA4@eL (
Table 6) seems to be relatively constant varying between 0.70–0.85. The α/β ratio rises to ca. 0.90 upon physical adsorption of laccase in the PA4@iL samples, meaning that the PA4-enzyme interaction via hydrogen bonds may cause some slight β−α transition.
According to
Table 6, the WAXS crystallinity indices
of the empty PA4 supports vary in the narrow range of 52–55%, i.e., values relatively high for polyamides. Entrapping laccase by in situ AAROP drops the crystallinity with about 5%, being within the margin of the experimental error of the deconvolution method. More fluctuations in
appear in the laccase-adsorbed samples. We are inclined to explain this with an increased error in the deconvolution due to interactions between the laccase halo
AHlaccase and AH
2 diffuse peak related to the PA4 own amorphous fraction, rather than to alteration of the crystallinity index.
As regards the Δ
qa values displayed in
Table 6 for all samples with laccase adsorption, it should be noted that this difference is larger with the PA4-Fe-iL, followed by the PA4-iL and the PA4-Fe
3O
4-iL samples. In accordance with Equation (1), in this sequence the density of the adsorbed bulk laccase is expected to decrease as a function of the polymer support composition.
2.3.4. Synchrotron SAXS
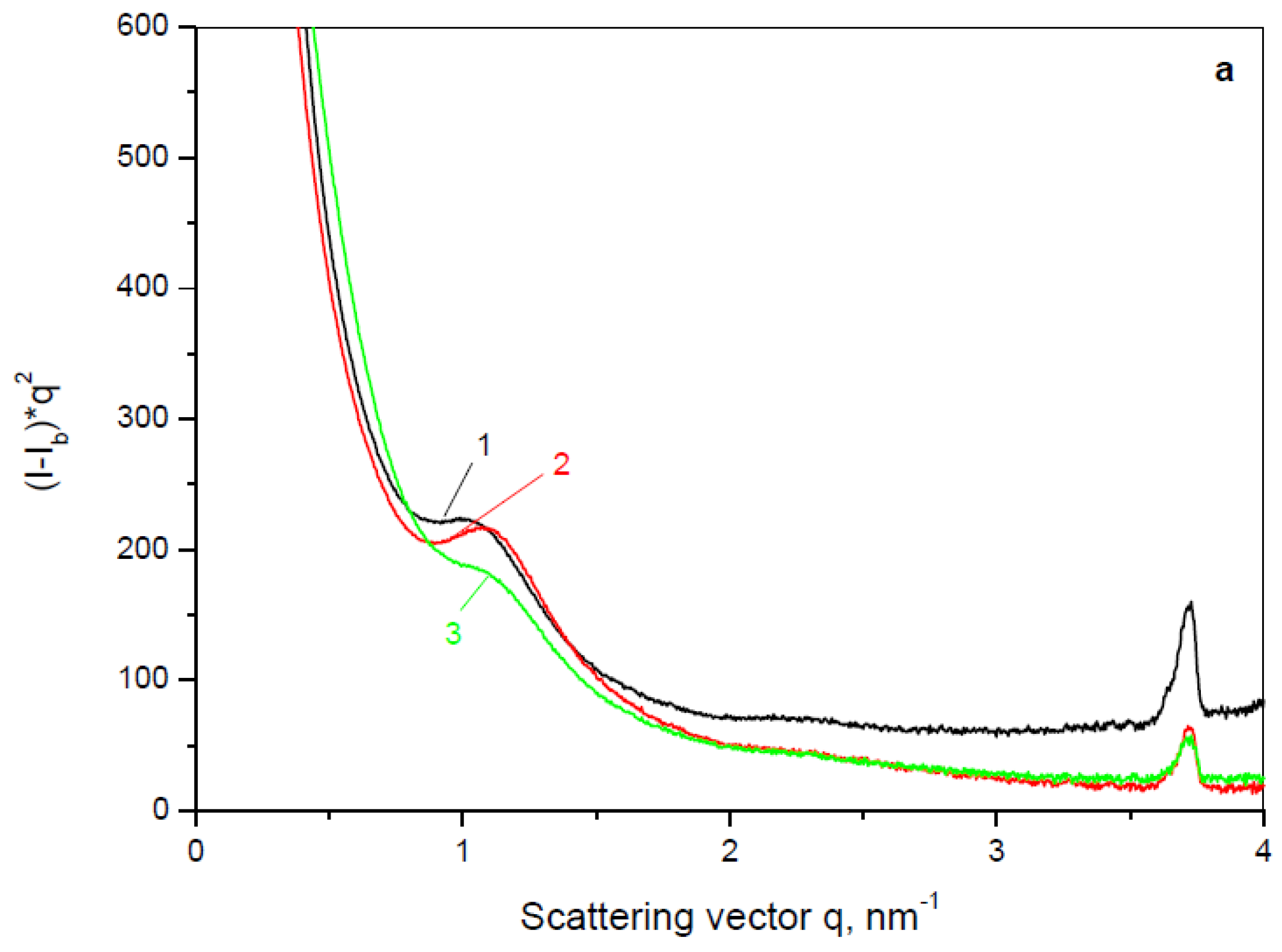
The use of synchrotron SAXS allows further clarification of the structure of the PA4 MP before and after laccase immobilization or entrapment. This method probes density periodicities with dimensions in the 20–250 angstroms range, which includes the sizes of the crystalline lamellae typically found in semi-crystalline polymers.
Figure 9 presents the background-subtracted and Lorentz-corrected SAXS linear profiles of the three empty PA4 supports (neat PA4 MP and such containing Fe or Fe
3O
4 fillers) and the respective laccase adsorbed (PA4@iL) and entrapped (PA4@eL) samples. To enable comparison,
Figure 9b also contains the SAXS curve of the free laccase.
It can be seen that the three empty PA4 supports (
Figure 9a) show relatively well resolved Bragg peaks, indicating lamellar stack morphology. The neat PA4 displays a long spacing value
LB = 62 Å, i.e., close to the 63–66 Å established previously in PA4 bulk samples [
38]. The presence of Fe or Fe
3O
4 slightly reduces
LB to 57 Å. Having in mind that the total WAXS crystallinity index of the three empty supports is above 50% (
Table 6), it should be concluded that it is the crystalline phase which is predominant in the lamellae. All the three PA4 empty supports display well-expressed and narrow SAXS peaks at
qmax ≈ 3.75 nm
−1 corresponding to
LB = 1.5–1.7 nm that should be related to nanoporosity in the PA4 microparticles not observable directly by SEM but undoubtedly established by BET (
Table S1). It is important to note that, as expected, this nanoporosity-related SAXS peak disappears if the empty support particles undergo melting and recrystallization (
Figure S3 of the Supporting Information, the curves at 270 °C and at 30 °C after heating up to 270 °C).
As seen from
Figure 9b, the free laccase does not show any periodicity in the SAXS q-range, which agrees with the WAXS results. Nevertheless, the laccase-adsorbed PA4@iL samples display very well resolved SAXS peaks with
LB = 67–70 Å, suggesting a much better phase contrast between the densities of the amorphous and crystalline regions, as compared to the respective neat PA4 supports (
Figure 9a).
It should be noted that no significant changes in the crystalline structure of the PA4 supports could possibly occur during the physical adsorption of laccase. Therefore, the SAXS curves in
Figure 9b allow the conclusion that after laccase immobilization most of the pores and channels of the empty supports get filled with enzyme whose density should be comparable to that of the amorphous PA4. This creates a clearer density gradient between the amorphous and crystalline fractions of the lamellar periodicity resulting in better resolved SAXS peaks of the PA4@iL samples. Notably, the narrow SAXS peaks at high
q-values of the empty supports disappear completely after laccase adsorption, confirming its relation to the PA4 nanoporosity.
Considering the SAXS patterns in
Figure 9c with their clear peaks with
LB = 65–67Å leads to the conclusion that a lamellar stack system similar to that of the empty PA4 supports is built after AAROP of 2PD in the presence of laccase that produces the PA4@eL samples. Let us note that the SAXS peaks with
qma ≈ 3.75 nm
−1 remain present in all samples with enzyme entrapment, being the most intense in the PA4-Fe-eL sample. This finding can be logically explained if the enzyme is arrested within the amorphous phase of the PA4 support, leaving its nanoporosity unobstructed. At the same time, in the laccase adsorbed samples, all enzyme seems to be located within the cavities of the particulate support, completely obstructing its nanoporosity. This is an important structural conclusion that will be used in the explanation of the enzyme activities of the two types PA4-laccase conjugates.
2.4. Specific Activity of Laccase Immobilized by Adsorption and Entrapment
The catalytic activity of all PA4-laccase conjugates was studied using ABTS as a substrate and calculating its rate of oxidation in each case. As shown in preliminary studies, the free laccase displays highest activity at pH 5, so this condition was assumed for all activity tests (0.0125 M PB, 25 °C).
Table 7 contains data on the total, specific, and relative activities of each PA4@iL and PA4@eL samples, taking the free laccase activity as 100%. As expected, the laccases immobilized by either adsorption or entrapment, was less active compared to the free laccase, the reduction of specific activity being different for the two immobilization strategies. The laccase-adsorbed samples (PA4@iL) were about 1.7–2.6 times less active, whereas the entrapped laccases displayed 2.4–6.2 times lower specific activity, as compared to the free laccase. Notably, the surface-immobilized laccase samples were from 1.4 to 3 times more active than the corresponding entrapped counterparts.
These results were expected having in mind the SEM and SAXS studies of the PA4@iL samples. After the adsorption, there is a large amount of ‘exposed’ laccase covering the surface of the PA4@iL and entering into their pores/cavities located near the surface. As seen directly from SEM and indirectly from the SAXS data, the topography of the entrapped samples (PA4@eL) is completely different. During the AAROP the laccase macromolecules are covered by a PA4 shell so there should be much less (or no) ‘exposed’ laccase in these samples. The porous shell makes more difficult the access of the ABTS substrate molecules to the active site of the entrapped enzyme. Moreover, the resulting oxidized cation-radical should return to the aqueous medium where it is quantified by UV–VIS, which is also hampered by the PA4 MP shell.
As seen from
Table 7, the conjugate with the highest laccase activity is the PA4-Fe-iL sample, in which the laccase immobilization effectiveness is the highest (ca. 81%,
Table 4). Logically, the enzyme densification measured by WAXS data (
Table 6, Δ
q values) is the largest in this sample. Among the entrapped samples, it is also the Fe-containing PA4-Fe-eL that displays the highest activity. These data suggest some synergism between the laccase activity and the presence of Fe
0 in the PA4 support.
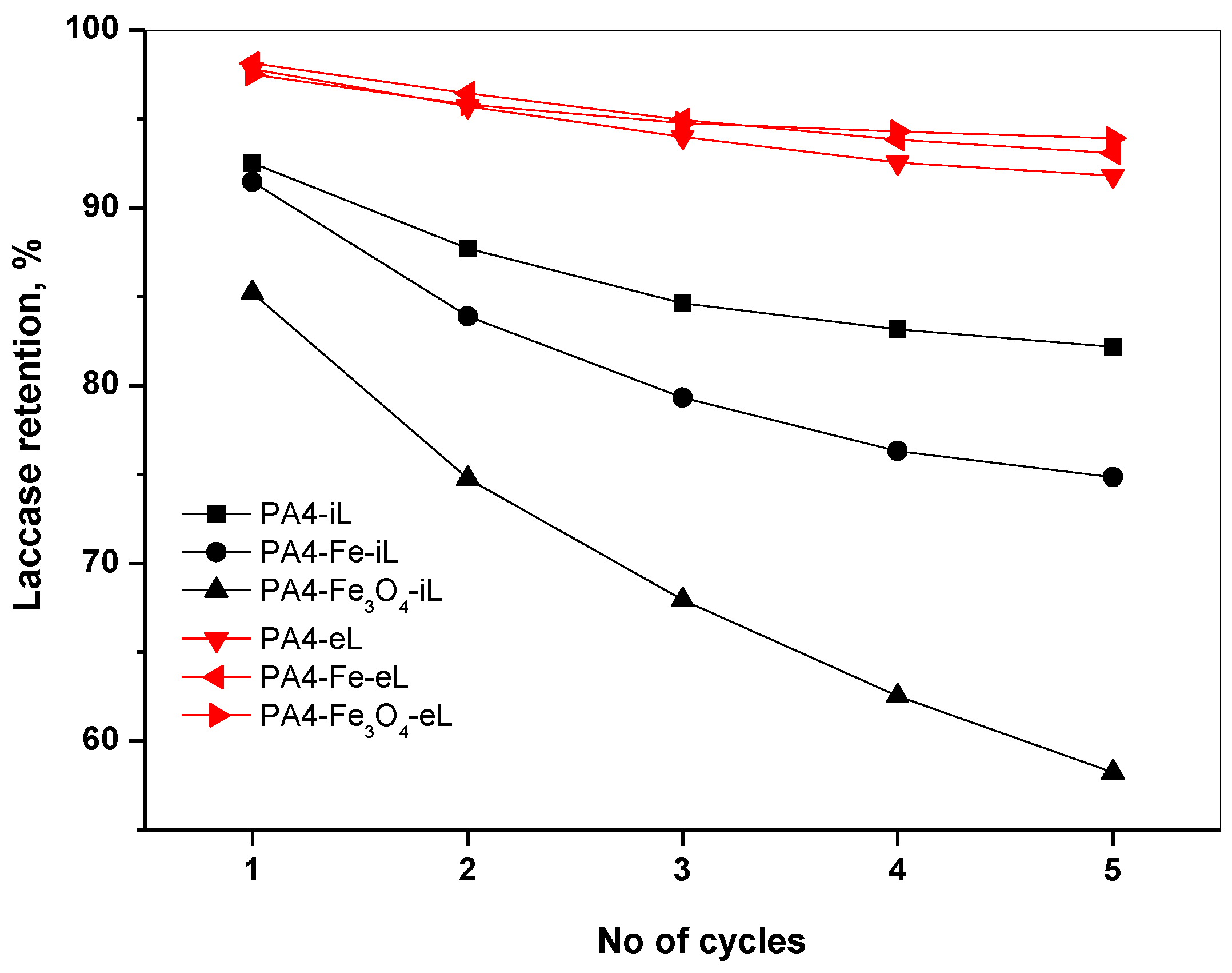
2.5. Laccase Retention Studies
Since both of our immobilization strategies count on H-bond formation between the PA4 support and laccase, leaching of enzyme will be an inevitable feature of PA4@iL and PA4@eL samples, as it is in all conjugates with no covalent bonding [
33].
Figure 10 displays the laccase retention in each conjugate type as a function of the support composition, in five consecutive application cycles.
The amount of laccase leached was below the detection limit of the spectroscopy method measuring the absorbance at λ = 286 nm. Therefore, the supernatants after every application cycle were subjected to activity testing, in which the higher the rate of laccase-catalyzed oxidation of ABTS substrate, the higher the percentage of leached laccase (
Section 2.3.4). The enzyme content in the starting conjugates (
Table 4 and
Table 5) was considered 100%.
Figure 10 shows that the leaching from the PA4@eL samples is quite low and even after the fifth cycle the enzyme retention is close to 95%, not depending on the PA4 support. This can be explained with the steric hindrance of the enzyme entrapped deep in the PA4 shell. The laccase release from the PA4@iL samples is notably higher, whereby the strongest drop is observed during cycles 1–3. Apparently, the laccase that is closer to the surface leaches first at higher rates, and after that a stabilization is attained. Moreover, the leaching rate in this series clearly depends on the PA4 support composition. The leaching is the strongest in the PA4-Fe
3O
4-iL sample ranging from 15% (cycle 1) to a total 42% (after cycle 5). The other two samples of this series display leaching percentages of 15–22% after the last cycle. In summary, the entrapment strategy affords better enzyme retention than immobilization by absorption.
2.6. Decolorization of Dyestuff Employing PA4-Laccase Conjugates
Malachite green (MG) is a cationic (i.e., positively charged) dyestuff widely used in the pigment industry and in agriculture. Bromophenol blue (BPB) is negatively charged with major applications as industrial or laboratory dyestuff and color marker. Both dyes contain polyaromatic hydrocarbon moieties (
Figure S4 of Supporting Information) that have carcinogenic and mutagenic health effects. MG and BPB are frequently found in effluents from the textile industry and agriculture, so their neutralization is an important problem. As was proven in a previous report [
45], MG and BPB can be successfully degraded by laccase oxidation. Therefore, the two dyes were selected in this study as substrates for decolorization with both PA4@iL and PA4@eL samples, produced by two different immobilization strategies. In both cases, the oxidizing activity of the conjugates was evaluated without any mediator.
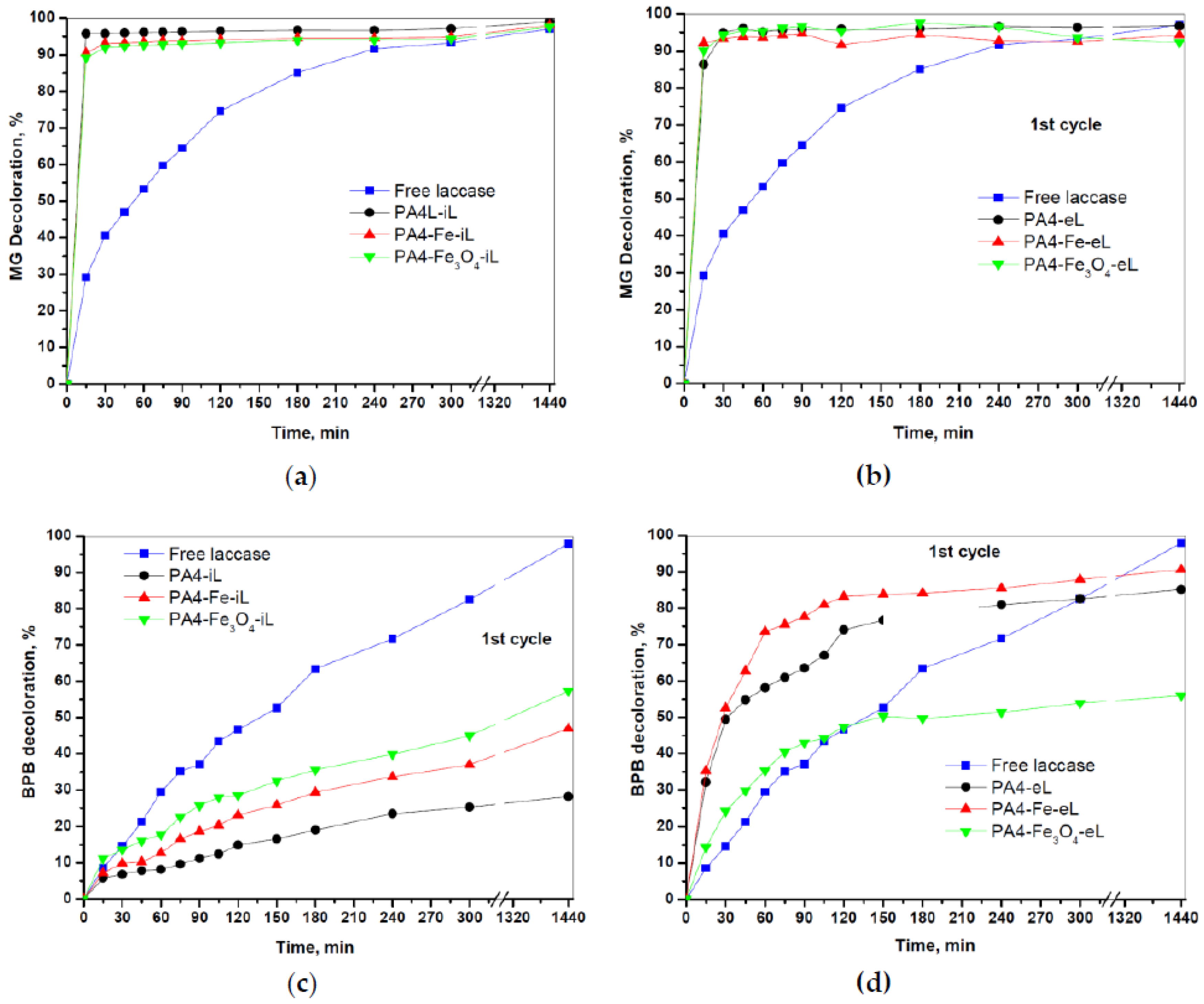
Figure 11 illustrates the decolorization kinetics of MG and BPB by the PA4@iL and PA4@eL samples. For comparison, the action of the free laccase was also studied at the same conditions. All PA4-laccase conjugates showed 90–95% effectiveness of the MG decolorization after 15 min only, irrespective of the immobilization strategy, whereas about 300 min were necessary for the free laccase to reach similar values (
Figure 11a,b). These yields are worth comparing to the data of Bagewadi et al. [
46] who, in a similar experiment, reported 95–100% MG decolorization, but after 960 min using laccase from
Trichoderma harzianum immobilized in a sol–gel matrix and 1-hydroxybenzotriazole (HBT) mediator.
The decolorization of BPB occurred quite differently with a clear dependence on the way of enzyme incorporation into the PA4 support microparticles. The adsorption-immobilized PA4@iL samples were less active than the free laccase, showing a lower decolorization effectiveness within the whole 24 h period that peaked only around 25–50% (
Figure 11c,d). At the same time, the two laccase-entrapped samples PA4-eL and PA4-Fe-eL displayed values of 70–80% in 120 min, the free laccase value being 45% at this time. A final decolorization of 80–90% was achieved after 24 h against 100% of the free enzyme. The PA4-Fe
3O
4-eL sample maintains an effectiveness of ca. 50% after 150 min that almost coincides with that of the free laccase. From this point on, the decolorization rate of the free enzyme continues to increase linearly, whereas that of the Fe
3O
4-containing conjugate saturates. Our results on the BPB decolorization can be favorably compared to the data of Forootanfar et al. [
47] who performed decolorization studies of BPB with free laccase from
Trametes versicolor and reached effectiveness values of 20% after 180 min without mediator and 32% with HBT mediator
Comparing the rates of decolorization of the two dyes by free laccase (
Figure 11) suggests that the MG substrate is more susceptible to oxidative degradation than BPB. This experimental fact can be related to their different chemical structure. Thus, BPB contains bulky and heavy groups (four Br atoms and one SO
3 group) and its molecular weight is almost twice as high as that of MG, making BPB more difficult to degrade [
46].
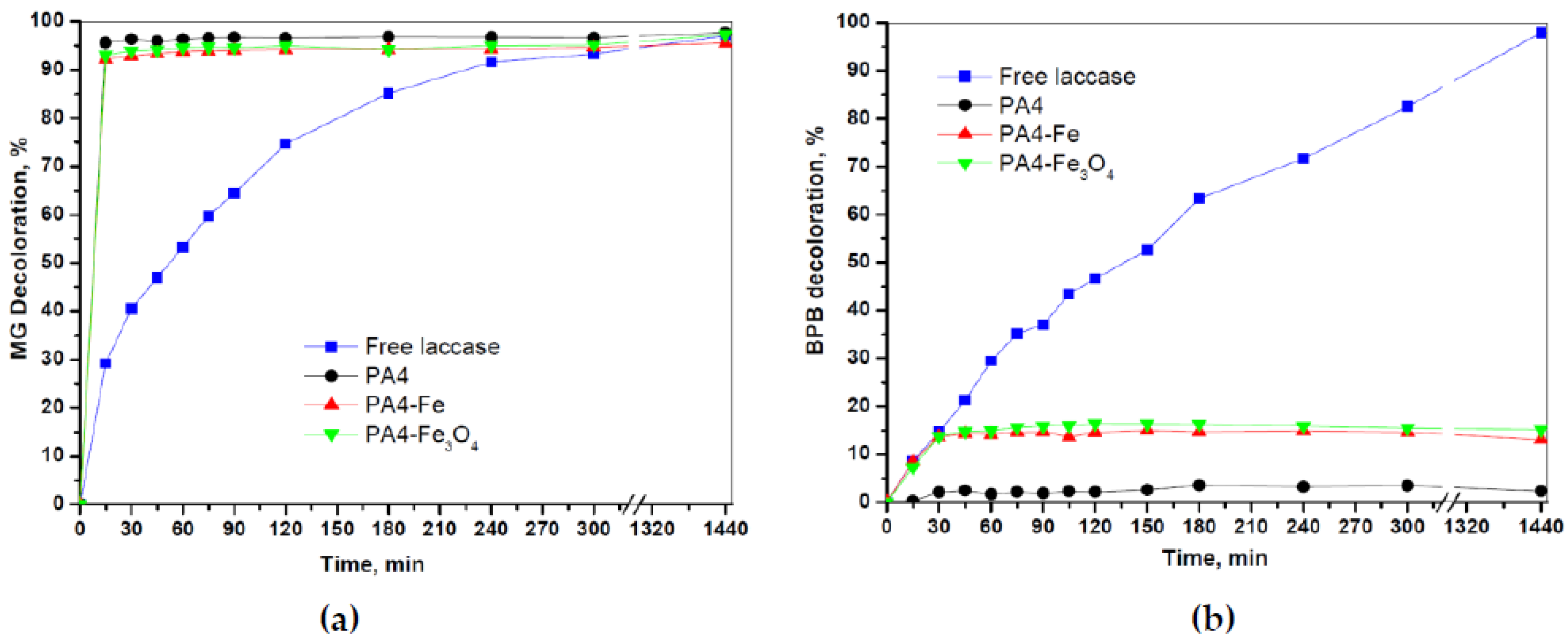
When immobilized enzymes are used, theoretically, the disappearance of the color of the initial dye solution can be caused both by enzymatic action and by adsorption in the support, whereby the contribution of the latter could be quite significant and must be taken into account [
48]. To assess the contribution of the physical adsorption in the above decolorization experiments, the empty PA4 supports were incubated in DDW MG and BPB solutions. Further treatment was exactly the same as indicated in
Section 2.4. The results for MG and BPB are presented in
Figure 12.
In the case of MG (
Figure 12a), a ramp to 95% decolorization effectiveness was achieved within 15 min time with all three empty supports. This effect can only be due to the extremely high adsorption capacity of the PA4 microparticles (Z-potential of −36 eV at pH7) toward the MG cation. Clearly, this vigorous adsorption process is much faster than the enzymatic oxidative degradation of MG by free laccase. In the case of BPB, however (
Figure 12b), the PA4 particles of the support adsorb only 2–3% dye (PA4 MP) or ca. 15% (Fe and Fe
3O
4-containing PA4 MP), these amounts being constant after 30 min exposure. The lower adsorption of BPB should be attributed to the electrostatic repulsion between the negatively charged dye molecules and PA4 microparticles. Evidently, the presence of Fe
0 or Fe
2+,3+ in the PA4 support slightly enhances the BPB removal by physical adsorption.
The results shown in
Figure 12 help assess the contribution of the adsorption by the supports in the experiments in
Figure 11. The instant decolorization of the MG substrate by PA4@iL or PA4@eL conjugates in
Figure 11a,b should be explained by strong adsorption in the support particles, which influences the MG removal more than the oxidative degradation by laccase. In the case of BPB, the results in
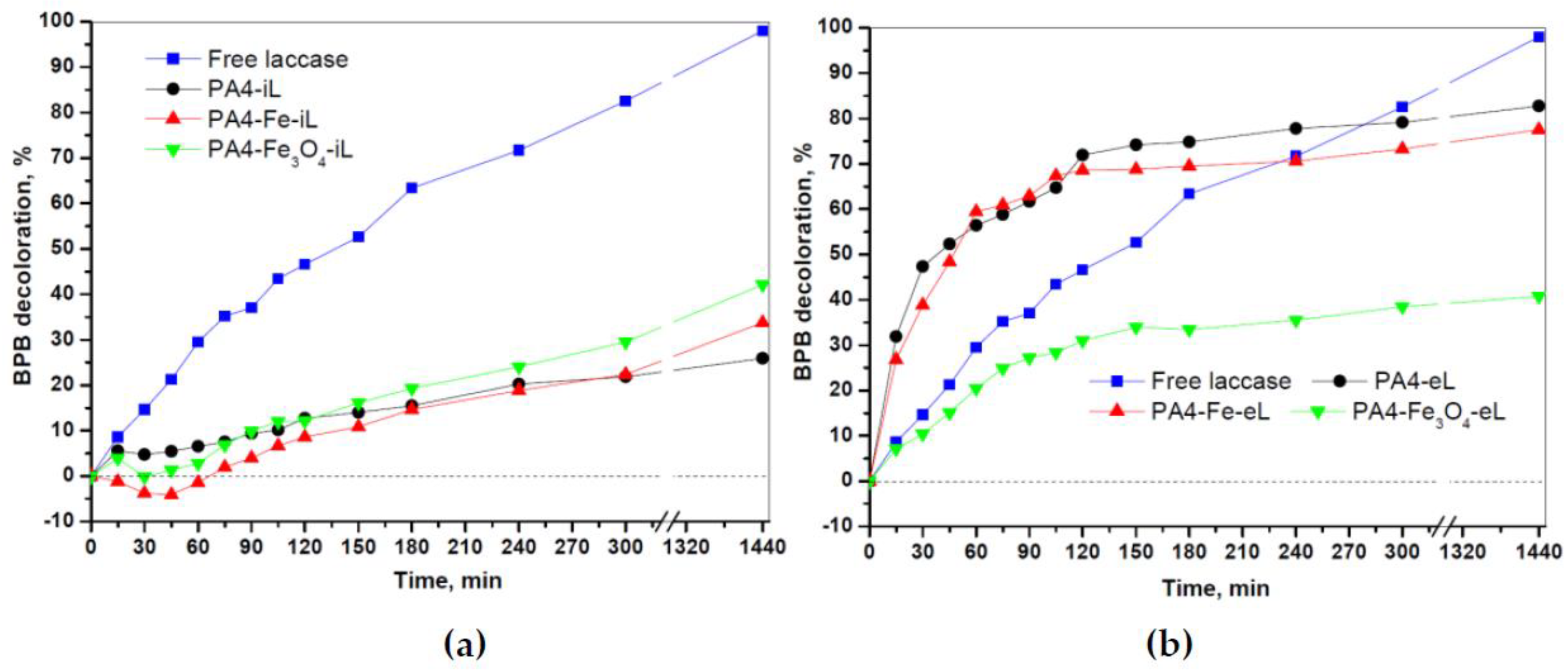
Figure 11c,d could be corrected by subtraction of the adsorption-caused dye removal thus allowing the evaluation of the sole enzymatic decolorization. The corrected data is presented in
Figure 13.
The PA4@iL samples display slower decolorization kinetics than the free laccase in the whole 24 h interval studied. The Fe0 and Fe2+,3+ containing samples of this series display negative values of ΔA in the beginning of the experiment due to predominant adsorption-caused dye removal. As to the laccase entrapped samples, the PA4-eL and the PA4-Fe-eL samples show excellent decolorization kinetics, being faster than that of the free laccase during the first 3 h of the experiment. The maximum decolorization effectiveness of BPB due only to the enzyme action was between 65–75%. The Fe2+,3+-containing support displayed a clear negative deviation from this behavior. Since the specific and relative laccase activities in the PA4-Fe3O4-eL sample are even slightly higher than those of the PA4-eL sample, the significantly lower decolorization kinetics in the former case cannot be attributed to lesser amount or lesser starting activity of the enzyme. Most probably, some laccase inactivation occurs by BPB functional groups during the decolorization process.
On the other hand, as seen from
Table 7, the samples with entrapped laccase displayed lower activity toward the ABTS substrate, as compared to both free and PA4-immobilized laccases. The results in
Figure 12 can be explained with some deactivation of the free laccase and that in the iL samples, while the catalytic activity of the eL samples remained constant. It should be noted that all samples were stored at exactly the same conditions and for the same time duration.
The potential of practical application of the PA4@iL and PA4@eL conjugates will be directly related to the possibility to remove them rapidly and completely from the reaction mixture and use them in several consecutive cycles.
Figure S5 in the Supporting Information visualizes the removal of the PA4-Fe-eL sample from its suspension in DDW by means of a constant magnet.
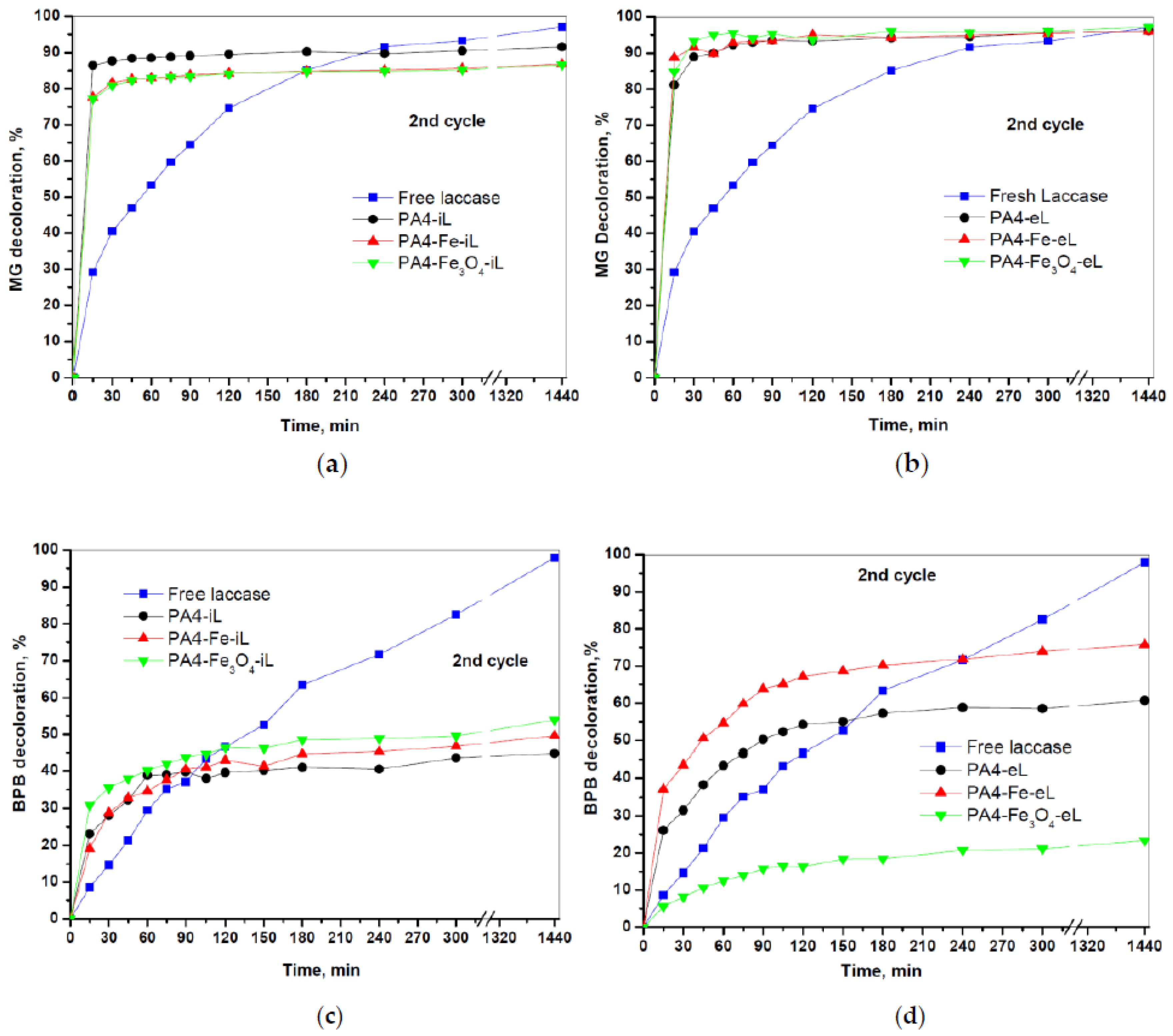
Figure 14 displays the second decolorization cycle of MG and BPB dyes by PA4@iL and PA4@eL conjugates.
The comparison of
Figure 11 and
Figure 14 show that in the case of MG both the PA4@iL and PA4@eL conjugates lose less than 5% of their decolorization capacity during the second cycle, which means they can be reused in many additional decolorizing cycles. However, in this case, the contribution of the enzymatic reaction cannot be evaluated due to the very rapid adsorption process. In regard to the BPB decolorization, where the contribution of the adsorption is negligible, the laccase-entrapped samples lose ~10% of their decolorization effectiveness. Supposing that this rate of loss is maintained in subsequent cycles, at least 3–5 more uses will be possible for PA4@iL and PA4@eL series, respectively, without adding free laccase.
3. Materials and Methods
3.1. Materials
The 2PD functional monomer for the PA4 synthesis and all solvents used in this work are of analytical grade and were supplied by Merck Life Science, Algés, Portugal. The laccase from Trametes versicolor (≥0.5 U/mg) was also purchased from the same supplied and used without further purification. As activator of the anionic polymerization of 2PD the commercial product Brüggolen C20 from Brüggemann Chemical GmbH, Heilbronn, Germany was employed containing, according to the manufacturer, 80 wt % of aliphatic diisocyanate blocked in ε-caprolactam. The polymerization initiator sodium dicaprolactamato-bis-(2-methoxyethoxo)-aluminate (Dilactamate, DL), which is also a commercial product was supplied by Katchem, Prague, Czech Republic and used without further treatment. Soft, non-insulated iron particles (Fe content >99.8%), with average diameters of 3–5 µm were kindly donated by the manufacturer BASF, Ludwigshafen, Germany. The Fe3O4 magnetic particles are a product of Merck, Algés, Portugal with >99% purity and grain sizes of 50–80 nm. Diammonium 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonate) (ABTS) with a purity of ≥98% (HPLC), as well as malachite green and bromophenol blue dyes, were purchased from the same supplier. All buffer solutions in this work were prepared with double-distilled water (DDW).
3.2. Instrumentation and Methods
Fourier-transform infra-red spectroscopy with attenuated total reflection (FTIR-ATR): The FTIR-ATR spectra were collected in a Perkin-Elmer Spectrum 100 apparatus (Waltham, MA, USA) using a horizontal ATR attachment with ZnSe crystal. Spectra were acquired between 4000 and 600 cm−1 accumulating up to 16 spectra with a resolution of 2 cm−1. The PA4 samples were studied in the form of fine powders.
Ultraviolet–visible spectroscopy (UV–VIS): The UV–VIS analysis was performed on a Shimadzu UV-2501 PC spectrometer (Kyoto, Japan). The absorbance at λ = 286 nm of aqueous solutions placed in quartz cuvettes was measured to determine the protein content and calculate the immobilization efficiency (IE). The assessment of the laccase activity was performed using ABTS as substrate, measuring the absorbance at λ = 414 nm as previously indicated by Claus et al. [
19]. One unit of laccase activity expressed in µkatals corresponds to the amount of enzyme transforming 1 μmol ABTS per second at pH 5.0 and 25 °C. The absorbance in decolorization experiments was measured at the respective wavelength of each dye that was λ = 616 nm for malachite green and λ = 591 nm for bromophenol blue.
Scanning electron microscopy (SEM): The SEM studies were performed in a NanoSEM-200 apparatus of FEI Nova (Hillsboro, OR, USA) using mixed secondary electron/back-scattered electron in-lens detection. The pulverulent samples were observed after sputter-coating with Au/Pd alloy in a 208 HR equipment of Cressington Scientific Instruments (Watford, UK) with high-resolution thickness control.
Thermo-gravimetric analysis (TGA): The real iron load,
RL, and the thermal stability of all neat PA4 micro-particulate supports and laccase@PA4 MP samples were established by means of thermo-gravimetric analysis (TGA) in a Q500 gravimetric balance (TA Instruments, New Castle, DE, USA), heating the samples in the 40–600 °C range at 20 °C/min in a nitrogen atmosphere. The
RL was calculated according to Equation (2)
where
RPA4 is the carbonized residue at 600 °C of the neat PA4 particles and
Ri represents the carbonized residue of the respective Fe- or Fe
3O
4—containing PA4 MP or laccase@PA4 MP samples.
Differential scanning calorimetry (DSC): The DSC measurements were carried out in a 200 F3 equipment of Netzsch (Selb, Germany) at a heating/cooling rate of 10 °C/min under nitrogen purge. The samples were heated to 290 °C, cooled down to 0 °C, and then heated back to 290 °C. The typical sample weights were in the 10–15 mg range.
Synchrotron X-ray studies: Synchrotron wide- (WAXS) and small-angle X-ray scattering (SAXS) measurements were performed in the NCD-SWEET beamline of the ALBA synchrotron facility in Barcelona, Spain [
49]. Two-dimensional detectors were used, namely LH255-HS (Rayonix, Evanston, IL, USA) and Pilatus 1M (Dectris, Baden Daettwil, Switzerland) for registering the WAXS and SAXS patterns, respectively. The sample-to-detector distance was set to 111.7 mm for WAXS and 2700 mm for SAXS measurements, the λ of the incident beam being 0.1 nm and the beam size 0.35 × 0.38 mm (h × v). The 2D data were reduced to 1D data using pyFAI software [
50]. For processing of the WAXS and SAXS patterns, the commercial package Peakfit 4.12 (2016) by SYSTAT (San Jose, CA, USA) was implemented.
3.3. Sample Preparation and Activity Testing
3.3.1. Synthesis of Empty or Magnetic PA4 MP
The low-temperature AAROP of 2PD to empty PA4 MP or to PA4 MP with magnetic response was described in detail in a previous publication [
24]. Generally, 0.2 mol of 2PD were stirred with 1 wt % of Fe micro- or Fe
3O
4 nanoparticles at 25 °C for 30 min. Subsequently, the C20 activator and the DL initiator were added under stirring, in an inert atmosphere. Then, the temperature of the reaction mixture was set to 40 °C and the pressure to 50 mbar for the next 6 h. After AAROP completion, the resulting reaction mixture was dispersed in acetone and filtered, followed by a two-fold wash with methanol, thus removing most of the unreacted monomer. In order to eliminate the PA4 oligomers, the resulting fine powders were extracted with methanol in a Soxhlet for 4 h, dried in vacuum, and stored in a desiccator. For the synthesis of the empty PA4 MP, the procedure was the same but without addition of any magnetic nanoparticles. Altogether, three types of PA4 MP supports were synthesized: PA4, PA4-Fe, and PA4-Fe
3O
4. Some basic characteristics of these samples are presented in
Table 1.
3.3.2. Immobilization of Laccase by Physical Adsorption
A typical immobilization was carried out by first preparing the laccase solution in DDW with concentration of 2 mg/mL. 200 mg of each PA4 MP sample were introduced into 5 mL of this laccase solution and the three sample tubes were incubated at room temperature for 24 h using a laboratory orbital shaker. Thereafter, the samples were centrifuged, the supernatant was decanted, and the laccase-immobilized PA4 MP samples were washed two times with distilled water to remove the non-adsorbed laccase. UV analysis was performed to determine the residual laccase and calculate the immobilization efficiency, IE, expressed as
where
C0 is the starting laccase concentration and
Cs is the laccase content in the resultant supernatant after completion of the immobilization process. Two methods were applied for the determination of
Cs. In the first one, the absorbance at λ = 286 nm characteristic for the proteic part of the enzyme was measured and the laccase concentration was determined using a standard calibration plot. In the second method, the laccase activity toward ABTS was tested and
Cs was assessed using an activity standard calibration plot. Altogether, three samples of laccase immobilized by adsorption on PA4 MP were prepared and designated as PA4-iL, PA4-Fe-iL, and PA4-Fe
3O
4-iL. They were stored in semi-dried conditions at 4 ºC with basic characteristics listed in
Table 2.
3.3.3. Immobilization of Laccase by Entrapment
The entrapment of laccase into the PA4 MP was carried out in situ during their synthesis by AAROP. Thus, 1.0 wt % of the enzyme in respect to the monomer was used, stirring their mixture for 30 min in an inert atmosphere at room temperature. Thereafter, the respective magnetic micro/nano particles (if necessary) and the DL/C20 catalytic system were introduced and the AAROP was conducted under the conditions described in
Section 2.3.1. After 6 h of polymerization, the raw product was dispersed in acetone and filtered. The resulting laccase-entrapped PA4 MP samples were washed three times with DDW and stored at 4 °C. The water from each washing was stored separately and further subjected to UV analysis to determine the efficiency of the laccase entrapment (EE) using the equation
where
C0 is the starting laccase concentration in the reaction mixture and
Ci (
i = 1–3) is the laccase content in each DDW washing. To assess the content of the non-entrapped laccase, the two methods already described in
Section 2.3.2. were applied. Thus, three PA4 MP samples with entrapped laccase were synthesized, namely: PA4-eL, PA4-Fe-eL, and PA4-Fe3O4-eL, with basic characteristics given in
Table 3.
3.3.4. Laccase Activity Assay
Native laccase and all conjugates of laccase-immobilized PA4@-iL and entrapped PA4@-eL were assayed for their activities using ABTS as a color-generating substrate. The rate of the formation of ABTS-cation radical (ABTS+·) due to the catalytic action of laccase was proportional to the enzyme activity. In a typical assay of free laccase, 0.9 mL of the enzyme solution (1.0 mg/mL in 0.0125 M phosphate buffer, PB, pH 5) were introduced into the spectrometer cell, followed by 0.1 mL of freshly prepared 5 mM solution of ABTS in DDW. During the next 2 min the absorbance at 414 nm (ε414 = 36,000 M−1·cm−1) was measured every 5 s. To determine the activity of the PA4@iL and PA4@eL conjugates, 20 mg of each sample were added to 1.0 mL of 0.0125 M PB, pH 5. The mixtures were stirred for 5 min followed by addition of 0.1 mL of ABTS solution. Then, the samples were centrifuged and 1 mL from the decanted supernatant was subjected to UV analysis to measure the absorbance at 414 nm for 10 min. In order to assay the amount and the activity of the laccase that possibly leached from both adsorption-immobilized and entrapped laccase samples, they were subjected to the following procedure: 50 mg of the respective PA4@iL or PA4@eL sample were dispersed in 5 mL PB, pH 0.0125M, pH 5, and shaken at 37 °C for 15 min. Thereafter, the system was centrifuged and the supernatant decanted and subjected to UV–VIS to determine the laccase concentration based on activity test calibration curve. Then, new portion of fresh PB was added and the above procedure was repeated up to five consecutive cycles.
In all tests the enzyme activities were expressed in microkatals. One microkatal of enzyme activity (μkat) was defined as the amount of enzyme that converted 1.0 μmol of ABTS to ABTS+· per second, at pH 5 and 25 °C. The enzyme activity assay was always performed in duplicate, and the standard deviations in measurements were consistently below 3%. Throughout this study, enzyme activities are expressed in μkat per milliliter (μkat·mL−1).
3.3.5. Application of Free, Adsorbed, and Entrapped Laccase
The enzyme-catalyzed decolorization of two structurally different dyes -malachite green (
λmax = 616 nm) and bromophenol blue (
λmax = 591 nm) were studied in the presence of free, immobilized and entrapped laccases without any mediator. The process was followed using UV–VIS spectroscopy by measuring the decrease in the
λmax absorbance of each dye. The reaction mixture for the decolorization contained a final concentration of 0.0153 mg/mL of individual dye in 0.0125 M PB, pH 5, and 1.0 mg free laccase (0.5 U) or 10–30 mg of adsorption immobilized or entrapped PA4@laccase conjugates (~0.5 U) in a total volume of 1.3 mL. All reaction mixtures were incubated at 30 °C for 24 h under shaking. Aliquots of 1 mL were withdrawn from each sample at certain intervals to measure the residual dye concentration. After the UV–VIS measurement the analyte was returned to the reaction mixture. After 24 h the immobilized or entrapped samples were reused for a second decolorization cycle. For that purpose, the liquid phase was drained, the immobilized or entrapped laccase conjugates were washed consecutively with 2 × 1 mL ethanol, DDW and 0.0125M PB, pH 5 to remove the adsorbed dyes, and then introduced into a fresh dye solution. All UV–VIS measurements were performed twice to determine the standard deviation. The percentage of decolorization ΔA was calculated as
where,
Ai is the initial absorbance of the dye at the respective wavelength, and
At is the absorbance of the test sample.
4. Conclusions
This is the first study reporting on PA4 porous microparticles as effective supports for enzyme immobilization and describes PA4-laccase conjugates with all aspects of their preparation and characterization. The main objective was to investigate three new micron-sized particulate porous supports based on PA4 obtained by low-temperature AAROP, with and without magnetic susceptibility, establishing whether or not they are suitable for laccase immobilization. The structure and morphology were investigated by microscopic, spectral, thermal, and synchrotron WAXS/SAXS technics.
Two immobilization strategies—i.e., by adsorption or entrapment—were applied. Comparing the adsorption-immobilized (PA4@iL) and entrapped (PA4@eL) samples, an important difference in the synthetic procedure of the PA4@eL should be noted. There, the polymerization was carried out in the presence of the enzyme, whereby the PA4 is formed around the laccase molecules leading to their distribution within the shell of the porous microparticles. In the case of PA4@iL, the enzyme is adsorbed onto the surface and fills the pores of prefabricated PA4 microparticles. The SEM studies confirm these morphological differences. On the other hand, the WAXS/SAXS studies provide evidence for the different way of immobilization of the enzyme in PA4@eL and PA4@iL.
The adsorption strategy resulted in high content of immobilized laccase upon the PA4 supports (27–40 mg/g), the immobilization efficiency being in the range of 54–81%. The entrapment strategy produced 60–70% immobilization efficiency with enzyme content of 14–18 mg/g support. The laccase-adsorbed samples PA4@iL showed up to three times higher specific activity as compared to their entrapped laccase counterparts PA4@eL, whereby the highest specific activity of 0.09 µkat·mL−1 was registered with PA4-Fe-iL sample. The relative laccase activity of the PA4-based conjugates determined in relation to the free enzyme was between 16–41% for the PA4@eL samples and 38–60% for the PA4@iL series. The PA4@eL samples displayed >93% enzyme retention after five cycles of incubation, whereas for the PA4@iL series this value was ~60%. The potential application of the adsorption- and entrapment-immobilized PA4-laccase conjugates was tested in the enzymatic decolorization of two synthetic dyes. All laccase conjugates displayed excellent decolorization of malachite green dyestuff reaching ~100% in 15 min, which was mostly due to dye adsorption upon the PA4 support. The decolorization after 24 h of the bromophenol reached 55% by PA4@iL and 85% by PA4@eL samples. The reuse of the laccase-PA4 conjugates in a second consecutive decolorization test resulted in only up to 10% decrease in their effectiveness. Supposing that this will be the decrease after each cycle of utilization, it seems that the laccase-PA4 conjugates will be sufficiently active to use for 3–5 more consecutive cycles.
The present study justifies further investigations on PA4 microparticles as effective enzyme supports for biotechnological applications. Compared to the common polyamides as PA6, PA66, and PA12, PA4 has the advantage of biodegradability and possesses a crystalline structure, which is a closer structural analogue of proteic biomolecules. The possibility to apply different immobilization strategies and to use magnetic field as external stimulus could open the way to the use of PA4 microparticulate supports of this study in smart green catalytic systems.


 ,
, 

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}