Recent Progress with Pincer Transition Metal Catalysts for Sustainability




Abstract
:1. Introduction
2. Dehydrogenation Reactions
2.1. Early Works
2.2. Dehydrogenation Reactions for a Hydrogen Economy
2.2.1. Methanol Dehydrogenation
2.2.2. Formic Acid Dehydrogenation
2.2.3. Other Hydrogen Storage Systems
3. Hydrogenation Reactions
3.1. CO2 Hydrogenation
3.1.1. Early Works
3.1.2. CO2 Hydrogenation to Methanol
3.1.3. CO2 Hydrogenation to Formate Salts
3.2. Nitrogen Fixation
3.3. Valorization of Biomass-Derived Compounds
3.4. Transfer Hydrogenation
Ethanol Upgrading
4. Conclusions
Funding
Conflicts of Interest
References
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice 2000; Oxford University Press: New York, NY, USA, 1998; p. 1546. [Google Scholar]
- Grützmacher, H. Cooperating Ligands in Catalysis. Angew. Chem. Int. Ed. 2008, 47, 1814–1818. [Google Scholar] [CrossRef]
- Askevold, B.; Roesky, H.W.; Schneider, S. Learning from the Neighbors: Improving Homogeneous Catalysts with Functional Ligands Motivated by Heterogeneous and Biocatalysis. ChemCatChem 2012, 4, 307–320. [Google Scholar] [CrossRef]
- Lawrence, M.A.W.; Green, K.A.; Nelson, P.N.; Lorraine, S.C. Review: Pincer ligands—Tunable, versatile and applicable. Polyhedron 2018, 143, 11–27. [Google Scholar] [CrossRef]
- Morales-Morales, D. Pincer Compounds: Chemistry and Applications; Elsevier: Amsterdam, The Netherlands, 2018; ISBN 9780128129326. [Google Scholar]
- Morales-Morales, D.; Jensen, C. The Chemistry of Pincer Compounds; Elsevier: Amsterdam, The Netherlands, 2007; ISBN 9780444531384. [Google Scholar]
- Moulton, B.C.J.; Shaw, B.L. 1020 J.C.S.; Dalton: New York, NY, USA, 1975; pp. 1020–1024. [Google Scholar]
- Liu, C.-C.; Liu, Q.-L.; Wu, Z.-Y.; Chen, Y.-C.; Xie, H.-J.; Lei, Q.-F.; Fang, W.-J. Mechanistic insights into small molecule activation induced by ligand cooperativity in PCcarbeneP nickel pincer complexes: A quantum chemistry study. J. Mol. Model. 2015, 21, 242. [Google Scholar] [CrossRef]
- Zhao, S.; Wu, J.; Chen, W. Organometallic chemistry of bis (N-heterocyclic carbene) ligands containing a heteroarene spacer. J. Organomet. Chem. 2017, 848, 249–280. [Google Scholar] [CrossRef]
- Pugh, D.; Danopoulos, A.A. Metal complexes with ‘pincer’-type ligands incorporating N-heterocyclic carbene functionalities. Coord. Chem. Rev. 2007, 251, 610–641. [Google Scholar] [CrossRef]
- Mejuto, C.; García-Eleno, M.A.; Guisado-Barrios, G.; Spasyuk, D.; Gusev, D.; Peris, E. Ruthenium complexes with an N-heterocyclic carbene NNC-pincer ligand: Preparation and catalytic properties. Org. Chem. Front. 2015, 2, 936–941. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Gendy, C.; Roesler, R. Nickel, Ruthenium, and Rhodium NCN-Pincer Complexes Featuring a Six-Membered N-Heterocyclic Carbene Central Moiety and Pyridyl Pendant Arms. Organometallics 2018, 37, 1123–1132. [Google Scholar] [CrossRef]
- Sung, S.; Wang, Q.; Krämer, T.; Young, R.D. Synthesis and reactivity of a PCcarbeneP cobalt (i) complex: The missing link in the cobalt PXP pincer series (X = B, C, N). Chem. Sci. 2018, 9, 8234–8241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prokopchuk, D.E.; Tsui, B.T.H.; Lough, A.J.; Morris, R.H. Intramolecular C-H/O-h bond cleavage with water and alcohol using a phosphine-free ruthenium carbene NCN Pincer Complex. Chem. A Eur. J. 2014, 20, 16960–16968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimoyama, Y.; Ishizuka, T.; Kotani, H.; Kojima, T. Ruthenium (II) Complexes Having a Pincer-Type Ligand with Two N-Heterocyclic Carbene Moieties. Z. Fur. Anorg. Und. Allg. Chem. 2018, 644, 611–615. [Google Scholar] [CrossRef] [Green Version]
- Charra, V.; de Frémont, P.; Braunstein, P. Multidentate N-heterocyclic carbene complexes of the 3d metals: Synthesis, structure, reactivity and catalysis. Coord. Chem. Rev. 2017, 341, 53–176. [Google Scholar] [CrossRef]
- Andrew, R.E.; González-Sebastián, L.; Chaplin, A.B. NHC-based pincer ligands: Carbenes with a bite. Dalt. Trans. 2016, 45, 1299–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Wang, L.; Wang, P.; Deng, L. High-Oxidation-State 3d Metal (Ti-Cu) Complexes with N-Heterocyclic Carbene Ligation. Chem. Rev. 2018, 118, 9930–9987. [Google Scholar] [CrossRef] [PubMed]
- Liddle, S.T.; Mills, D.P.; Wooles, A.J. Early metal bis (phosphorus-stabilised) carbene chemistry. Chem. Soc. Rev. 2011, 40, 2164–2176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zi, G. N-heterocyclic carbene (NHC) complexes of group 4 transition metals. Chem. Soc. Rev. 2015, 44, 1898–1921. [Google Scholar] [CrossRef] [Green Version]
- Polukeev, A.V.; Wendt, O.F. Cyclohexane-Based Phosphinite Iridium Pincer Complexes: Synthesis, Characterization, Carbene Formation, and Catalytic Activity in Dehydrogenation Reactions. Organometallics 2017, 36, 639–649. [Google Scholar] [CrossRef]
- Gu, S.; Chen, C.; Qiu, H.; Chen, W. Potentially Hemilabile N-Heterocyclic Carbene Palladium Complexes: Synthesis and Catalytic Applications. Curr. Org. Chem. 2011, 15, 3291–3308. [Google Scholar] [CrossRef]
- Peris, E.; Crabtree, R.H. Recent homogeneous catalytic applications of chelate and pincer N-heterocyclic carbenes. Coord. Chem. Rev. 2004, 248, 2239–2246. [Google Scholar] [CrossRef]
- Li, H.; Zheng, B.; Huang, K.W. A new class of PN3-pincer ligands for metal-ligand cooperative catalysis. Coord. Chem. Rev. 2015, 293–294, 116–138. [Google Scholar] [CrossRef] [Green Version]
- Kisala, J.; Ruman, T. Pincer Complexes Based on Phosphinoaminopyridines: Synthesis, Structural Characterization and Catalytic Applications. Curr. Org. Chem. 2011, 15, 3486–3502. [Google Scholar] [CrossRef]
- Benito-Garagorri, D.; Kirchner, K. Modularly designed transition metal PNP and PCP pincer complexes based on aminophosphines: Synthesis and catalytic applications. Acc. Chem. Res. 2008, 41, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Deolka, S.; Tarannam, N.; Fayzullin, R.R.; Kozuch, S.; Khaskin, E. Unusual rearrangement of modified PNP ligand based Ru complexes relevant to alcohol dehydrogenation catalysis. Chem. Commun. 2019, 55, 11350–11353. [Google Scholar] [CrossRef]
- Henrion, M.; Roisnel, T.; Couturier, J.L.; Dubois, J.L.; Sortais, J.B.; Darcel, C.; Carpentier, J.F. Ruthenium complexes bearing amino-bis (phosphinite) or amino-bis (aminophosphine) ligands: Application in catalytic ester hydrogenation. Mol. Catal. 2017, 432, 15–22. [Google Scholar] [CrossRef]
- Yao, C.; Chakraborty, P.; Aresu, E.; Li, H.; Guan, C.; Zhou, C.; Liang, L.C.; Huang, K.W. Monomeric nickel hydroxide stabilized by a sterically demanding phosphorus-nitrogen PN3P-pincer ligand: Synthesis, reactivity and catalysis. Dalt. Trans. 2018, 47, 16057–16065. [Google Scholar] [CrossRef]
- Gradiski, M.V.; Tsui, B.T.H.; Lough, A.J.; Morris, R.H. PNN′ & P 2 NN′ ligands via reductive amination with phosphine aldehydes: Synthesis and base-metal coordination chemistry. Dalt. Trans. 2019, 48, 2150–2159. [Google Scholar]
- Kim, Y.; Lee, J.; Son, Y.-H.; Choi, S.-U.; Alam, M.; Park, S. Novel nickel(II), palladium(II), and platinum(II) complexes having a pyrrolyl-iminophosphine (PNN) pincer: Synthesis, crystal structures, and cytotoxic activity. J. Inorg. Biochem. 2020, 205, 111015. [Google Scholar] [CrossRef]
- Adams, G.M.; Weller, A.S. POP-type ligands: Variable coordination and hemilabile behaviour. Coord. Chem. Rev. 2018, 355, 150–172. [Google Scholar] [CrossRef]
- Leis, W.; Mayer, H.A.; Kaska, W.C. Cycloheptatrienyl, alkyl and aryl PCP-pincer complexes: Ligand backbone effects and metal reactivity. Coord. Chem. Rev. 2008, 252, 1787–1797. [Google Scholar] [CrossRef]
- Morales-Morales, D. Recent Applications of Phosphinite POCOP Pincer Complexes towards Organic Transformations. Mini. Rev. Org. Chem. 2008, 5, 141–152. [Google Scholar] [CrossRef]
- Gelman, D.; Romm, R. PC(sp3)P Transition Metal Pincer Complexes: Properties and Catalytic Applications. In Topics in Organometallic Chemistry; Springer: Berlin, Germany, 2013; pp. 289–317. ISBN 9783642310805. [Google Scholar]
- Leforestier, B.; Gyton, M.R.; Chaplin, A.B. Synthesis and group 9 complexes of macrocyclic PCP and POCOP pincer ligands. Dalt. Trans. 2020, 49, 2087–2101. [Google Scholar] [CrossRef]
- Jensen, C.M. Iridium PCP pincer complexes: Highly active and robust catalysts for novel homogeneous aliphatic dehydrogenations. Chem. Commun. 1999, 3, 2443–2449. [Google Scholar] [CrossRef]
- Barrett, B.J.; Iluc, V.M. Coordination of a Hemilabile Pincer Ligand with an Olefinic Backbone to Mid-to-Late Transition Metals. Inorg. Chem. 2014, 53, 7248–7259. [Google Scholar] [CrossRef]
- Schörgenhumer, J.; Zimmermann, A.; Waser, M. SNS-Ligands for Ru-Catalyzed Homogeneous Hydrogenation and Dehydrogenation Reactions. Org. Process Res. Dev. 2018, 22, 862–870. [Google Scholar] [CrossRef]
- Spasyuk, D.; Smith, S.; Gusev, D.G. Replacing Phosphorus with Sulfur for the Efficient Hydrogenation of Esters. Angew. Chem. Int. Ed. 2013, 52, 2538–2542. [Google Scholar] [CrossRef]
- Valdés, H.; González-Sebastián, L.; Morales-Morales, D. Aromatic para-functionalized NCN pincer compounds. J. Organomet. Chem. 2017, 845, 229–257. [Google Scholar] [CrossRef]
- Fernández-Alvarez, F.J.; Lalrempuia, R.; Oro, L.A. Monoanionic NSiN-type ligands in transition metal coordination chemistry and catalysis. Coord. Chem. Rev. 2017, 350, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Garbe, S.; Krause, M.; Klimpel, A.; Neundorf, I.; Lippmann, P.; Ott, I.; Brünink, D.; Strassert, C.A.; Doltsinis, N.L.; Klein, A. Cyclometalated Pt Complexes of CNC Pincer Ligands: Luminescence and Cytotoxic Evaluation. Organometallics 2020, 39, 746–756. [Google Scholar] [CrossRef]
- Ruan, J.; Wang, D.; Vedernikov, A.N. CH3–X Reductive Elimination Reactivity of PtIVMe Complexes Supported by a Sulfonated CNN Pincer Ligand (X = OH, CF3CO2, PhNMe2+). Organometallics 2020, 39, 142–152. [Google Scholar] [CrossRef]
- Heidebrecht, J.; Gendy, C.; Gelfand, B.S.; Roesler, R. Water-soluble NNN-pincer complexes of cobalt, nickel and palladium: Solid-state structures and catalytic activity. Polyhedron 2018, 143, 138–143. [Google Scholar] [CrossRef]
- Kozlov, V.A.; Aleksanyan, D.V.; Vasilév, A.A.; Odinets, I.L. Thiophosphoryl-, thiophosphoryloxy-, and thiophosphorylamino-benzene derivatives as novel classes of hybrid pincer ligands. Phosphorus Sulfur Silicon Relat. Elem. 2011, 186, 626–637. [Google Scholar] [CrossRef]
- Al-Noaimi, M.; Awwadi, F.F.; Talib, W.H.; Atia, S.; Hammud, H.H. Cis and trans- palladium (II) complexes derived from SNN amidrazone pincer ligand: Synthesis, crystal structures and biological evaluation. J. Mol. Struct. 2019, 1197, 282–291. [Google Scholar] [CrossRef]
- Okamoto, K.; Kuwabara, J.; Kanbara, T. Secondary Thioamides as Multidentate Ligands for Functional Metal Complexes. Chem. Lett. 2015, 44, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.; Breher, F. Multidentate silyl ligands in transition metal chemistry. Dalt. Trans. 2017, 46, 7976–7997. [Google Scholar] [CrossRef]
- Zhou, Y.P.; Mo, Z.; Luecke, M.P.; Driess, M. Stereoselective Transfer Semi-Hydrogenation of Alkynes to E-Olefins with N-Heterocyclic Silylene–Manganese Catalysts. Chem. A Eur. J. 2018, 24, 4780–4784. [Google Scholar] [CrossRef]
- Kumar, A.; Rao, G.K.; Saleem, F.; Singh, A.K. Organoselenium ligands in catalysis. Dalt. Trans. 2012, 41, 11949–11977. [Google Scholar] [CrossRef]
- Sharma, K.N.; Satrawala, N.; Srivastava, A.K.; Ali, M.; Joshi, R.K. Palladium (ii) ligated with a selenated (Se, CNHC, N-) -type pincer ligand: An efficient catalyst for Mizoroki-Heck and Suzuki-Miyaura coupling in water. Org. Biomol. Chem. 2019, 17, 8969–8976. [Google Scholar] [CrossRef]
- Kameo, H.; Nakazawa, H. Recent developments in the coordination chemistry of multidentate ligands featuring a boron moiety. Chem. Asian J. 2013, 8, 1720–1734. [Google Scholar] [CrossRef]
- Yamashita, M. The Organometallic Chemistry of Boron-Containing Pincer Ligands based on Diazaboroles and Carboranes. Bull. Chem. Soc. Jpn. 2016, 89, 269–281. [Google Scholar] [CrossRef] [Green Version]
- Balakrishna, M.S. Unusual and rare pincer ligands: Synthesis, metallation, reactivity and catalytic studies. Polyhedron 2018, 143, 2–10. [Google Scholar] [CrossRef]
- Van Der Boom, M.E.; Milstein, D. Cyclometalated phosphine-based pincer complexes: Mechanistic insight in catalysis, coordination, and bond activation. Chem. Rev. 2003, 103, 1759–1792. [Google Scholar] [CrossRef] [PubMed]
- Peris, E.; Crabtree, R.H. Key factors in pincer ligand design. Chem. Soc. Rev. 2018, 47, 1959–1968. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Suzuki, S.; Kuwata, S. Metallo-supramolecular assembly of protic pincer-type complexes: Encapsulation of dinitrogen and carbon disulfide into a multiproton-responsive diruthenium cage. Chem. Commun. 2019, 55, 1028–1031. [Google Scholar] [CrossRef]
- Nelson, D.J.; Nolan, S.P. Hydroxide complexes of the late transition metals: Organometallic chemistry and catalysis. Coord. Chem. Rev. 2017, 353, 278–294. [Google Scholar] [CrossRef] [Green Version]
- Maser, L.; Vondung, L.; Langer, R. The ABC in pincer chemistry—From amine- to borylene- and carbon-based pincer-ligands. Polyhedron 2018, 143, 28–42. [Google Scholar] [CrossRef]
- Gusev, D.G.; Madott, M.; Dolgushin, F.M.; Lyssenko, K.A.; Antipin, M.Y. Agostic bonding in pincer complexes of ruthenium. Organometallics 2000, 19, 1734–1739. [Google Scholar] [CrossRef]
- Annibale, V.T.; Song, D. Multidentate actor ligands as versatile platforms for small molecule activation and catalysis. RSC Adv. 2013, 3, 11432–11449. [Google Scholar] [CrossRef]
- Ananthnag, G.S.; Shetti, V.S. Synthesis, structure and catalysis of organometallic porphyrin-pincer hybrids: A review. Dalt. Trans. 2017, 46, 14062–14082. [Google Scholar] [CrossRef]
- Gunanathan, C.; Milstein, D. Bond Activation and Catalysis by Ruthenium Pincer Complexes. Chem. Rev. 2014, 114, 12024–12087. [Google Scholar] [CrossRef]
- Younus, H.A.; Ahmad, N.; Su, W.; Verpoort, F. Ruthenium pincer complexes: Ligand design and complex synthesis. Coord. Chem. Rev. 2014, 276, 112–152. [Google Scholar] [CrossRef]
- Younus, H.A.; Su, W.; Ahmad, N.; Chen, S.; Verpoort, F. Ruthenium pincer complexes: Synthesis and catalytic applications. Adv. Synth. Catal. 2015, 357, 283–330. [Google Scholar] [CrossRef]
- Werkmeister, S.; Junge, K.; Beller, M. Catalytic hydrogenation of carboxylic acid esters, amides, and nitriles with homogeneous catalysts. Org. Process Res. Dev. 2014, 18, 289–302. [Google Scholar] [CrossRef]
- Gusev, D.G.; Lough, A.J. Experimental and computational study of pincer complexes of ruthenium with Py, CO, and N2 ligands. Organometallics 2002, 21, 5091–5099. [Google Scholar] [CrossRef]
- Gusev, D.G.; Dolgushin, F.M.; Antipin, M.Y. Hydride, borohydride, and dinitrogen pincer complexes of ruthenium. Organometallics 2000, 19, 3429–3434. [Google Scholar] [CrossRef]
- Abbel, R.; Abdur-Rashid, K.; Faatz, M.; Hadzovic, A.; Lough, A.J.; Morris, R.H. A succession of isomers of ruthenium dihydride complexes. Which one is the ketone hydrogenation catalyst? J. Am. Chem. Soc. 2005, 127, 1870–1882. [Google Scholar] [CrossRef]
- Bruneau, C.; Dixneuf, P.H. Ruthenium in Catalysis; Springer: Berlin, Germany, 2014; ISBN 9783319084817. [Google Scholar]
- Dub, P.A.; Ikariya, T. Quantum chemical calculations with the inclusion of nonspecific and specific solvation: Asymmetric transfer hydrogenation with bifunctional ruthenium catalysts. J. Am. Chem. Soc. 2013, 135, 2604–2619. [Google Scholar] [CrossRef]
- Bertoli, M.; Choualeb, A.; Lough, A.J.; Moore, B.; Spasyuk, D.; Gusev, D.G. Osmium and ruthenium catalysts for dehydrogenation of alcohols. Organometallics 2011, 30, 3479–3482. [Google Scholar] [CrossRef]
- Chelucci, G.; Baldino, S.; Baratta, W. Recent advances in osmium-catalyzed hydrogenation and dehydrogenation reactions. Acc. Chem. Res. 2015, 48, 363–379. [Google Scholar] [CrossRef]
- Bertoli, M.; Choualeb, A.; Gusev, D.G.; Lough, A.J.; Major, Q.; Moore, B. PNP pincer osmium polyhydrides for catalytic dehydrogenation of primary alcohols. Dalt. Trans. 2011, 40, 8941. [Google Scholar] [CrossRef]
- Gusev, D.G.; Lough, A.J.; Double, C.-H. activation on osmium and ruthenium centers: Carbene vs olefin products. Organometallics 2002, 21, 2601–2603. [Google Scholar] [CrossRef]
- Choi, J.; MacArthur, A.H.R.; Brookhart, M.; Goldman, A.S. Dehydrogenation and Related Reactions Catalyzed by Iridium Pincer Complexes. Chem. Rev. 2011, 111, 1761–1779. [Google Scholar] [CrossRef]
- Choualeb, A.; Lough, A.J.; Gusev, D.G. Hemilabile pincer-type hydride complexes of iridium. Organometallics 2007, 26, 5224–5229. [Google Scholar] [CrossRef]
- Clarke, Z.E.; Maragh, P.T.; Dasgupta, T.P.; Gusev, D.G.; Lough, A.J.; Abdur-Rashid, K. A family of active iridium catalysts for transfer hydrogenation of ketones. Organometallics 2006, 25, 4113–4117. [Google Scholar] [CrossRef]
- Meiners, J.; Scheibel, M.G.; Lemée-Cailleau, M.H.; Mason, S.A.; Boeddinghaus, M.B.; Fässler, T.F.; Herdtweck, E.; Khusniyarov, M.M.; Schneider, S. Square-planar iridium (II) and iridium (III) amido complexes stabilized by a PNP pincer ligand. Angew. Chem. Int. Ed. 2011, 50, 8184–8187. [Google Scholar] [CrossRef]
- Goldberg, J.M.; Wong, G.W.; Brastow, K.E.; Kaminsky, W.; Goldberg, K.I.; Heinekey, D.M. The Importance of Steric Factors in Iridium Pincer Complexes. Organometallics 2015, 34, 753–762. [Google Scholar] [CrossRef]
- Scheibel, M.G.; Wu, Y.; Stückl, A.C.; Krause, L.; Carl, E.; Stalke, D.; De Bruin, B.; Schneider, S. Synthesis and reactivity of a transient, terminal nitrido complex of rhodium. J. Am. Chem. Soc. 2013, 135, 17719–17722. [Google Scholar] [CrossRef]
- Sundermann, A.; Uzan, O.; Milstein, D.; Martin, J.M.L. Selective C-C vs C-H bond activation by rhodium (I) PCP pincer complexes. A computational study. J. Am. Chem. Soc. 2000, 122, 7095–7104. [Google Scholar] [CrossRef]
- Urgoitia, G.; Galdón, G.; Churruca, F.; SanMartin, R.; Herrero, M.T.; Domínguez, E. Aerobic oxidation of secondary benzyl alcohols catalyzed by phosphinite-based palladium pincer complexes. Environ. Chem. Lett. 2018, 16, 1101–1108. [Google Scholar] [CrossRef]
- Albrecht, M.; Van Koten, G. Platinum group organometallics based on “pincer” complexes: Sensors, switches, and catalysts. Angew. Chem. Int. Ed. 2001, 40, 3750–3781. [Google Scholar] [CrossRef]
- Selander, N.; Szabó, K.J. Catalysis by palladium pincer complexes. Chem. Rev. 2011, 111, 2048–2076. [Google Scholar] [CrossRef]
- González-Sebastián, L.; Morales-Morales, D. Cross-coupling reactions catalysed by palladium pincer complexes. A review of recent advances. J. Organomet. Chem. 2019, 893, 39–51. [Google Scholar] [CrossRef]
- Esteruelas, M.A.; López, A.M.; Oliván, M. Polyhydrides of Platinum Group Metals: Nonclassical Interactions and σ-Bond Activation Reactions. Chem. Rev. 2016, 116, 8770–8847. [Google Scholar] [CrossRef] [Green Version]
- Therrien, J.A.; Wolf, M.O.; Patrick, B.O. Synthesis and comparison of nickel, palladium, and platinum bis (N-heterocyclic carbene) pincer complexes for electrocatalytic CO2 reduction. Dalt. Trans. 2018, 47, 1827–1840. [Google Scholar] [CrossRef]
- Bauer, G.; Hu, X. Recent developments of iron pincer complexes for catalytic applications. Inorg. Chem. Front. 2016, 3, 741–765. [Google Scholar] [CrossRef] [Green Version]
- Dai, H.; Guan, H. Iron Dihydride Complexes: Synthesis, Reactivity, and Catalytic Applications. Isr. J. Chem. 2017, 57, 1170–1203. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Guan, H. Synthesis and catalytic applications of iron pincer complexes. Comments Inorg. Chem. 2011, 32, 88–112. [Google Scholar] [CrossRef]
- Balaraman, E.; Nandakumar, A.; Jaiswal, G.; Sahoo, M.K. Iron-catalyzed dehydrogenation reactions and their applications in sustainable energy and catalysis. Catal. Sci. Technol. 2017, 7, 3177–3195. [Google Scholar] [CrossRef]
- Benito-Garagorri, D.; Puchberger, M.; Mereiter, K.; Kirchner, K. Stereospecific and reversible CO binding at iron pincer complexes. Angew. Chem. Int. Ed. 2008, 47, 9142–9145. [Google Scholar] [CrossRef]
- Junge, K.; Schröder, K.; Beller, M. Homogeneous catalysis using iron complexes: Recent developments in selective reductions. Chem. Commun. 2011, 47, 4849. [Google Scholar] [CrossRef]
- Bernskoetter, W.H.; Hazari, N. Hydrogenation and dehydrogenation reactions catalyzed by iron pincer compounds. In Pincer Compounds; Elsevier Inc.: Amsterdam, The Netherland, 2018; ISBN 9780128129326. [Google Scholar]
- Rohit, K.R.; Radhika, S.; Saranya, S.; Anilkumar, G. Manganese-Catalysed Dehydrogenative Coupling—An Overview. Adv. Synth. Catal. 2020, 362, 1602–1650. [Google Scholar] [CrossRef]
- Waiba, S.; Maji, B. Manganese Catalyzed Acceptorless Dehydrogenative Coupling Reactions. ChemCatChem 2019, 12, 1891–1902. [Google Scholar] [CrossRef]
- Maji, B.; Barman, M.K. Recent Developments of Manganese Complexes for Catalytic Hydrogenation and Dehydrogenation Reactions. Synthesis 2017, 49, 3377–3393. [Google Scholar] [CrossRef]
- Garbe, M.; Junge, K.; Beller, M. Homogeneous Catalysis by Manganese-Based Pincer Complexes. Eur. J. Org. Chem. 2017, 2017, 4344–4362. [Google Scholar] [CrossRef]
- Eberhardt, N.A.; Guan, H. Nickel Hydride Complexes. Chem. Rev. 2016, 116, 8373–8426. [Google Scholar] [CrossRef]
- Gafurov, Z.N.; Kagilev, A.A.; Kantyukov, A.O.; Balabaev, A.A.; Sinyashin, O.G.; Yakhvarov, D.G. Classification and synthesis of nickel pincer complexes. Russ. Chem. Bull. 2018, 67, 385–394. [Google Scholar] [CrossRef]
- Klein, A.; Sandleben, A.; Vogt, N. Synthesis, Structure and Reactivity of Cyclometalated Nickel (II) Complexes: A Review and Perspective. Proc. Natl. Acad. Sci. India Sect. A Phys. Sci. 2016, 86, 533–549. [Google Scholar] [CrossRef]
- Gutsulyak, D.V.; Piers, W.E.; Borau-Garcia, J.; Parvez, M. Activation of water, ammonia, and other small molecules by PC carbeneP nickel pincer complexes. J. Am. Chem. Soc. 2013, 135, 11776–11779. [Google Scholar] [CrossRef]
- LaPierre, E.A.; Clapson, M.L.; Piers, W.E.; Maron, L.; Spasyuk, D.M.; Gendy, C. Oxygen Atom Transfer to Cationic PCPNi(II) Complexes Using Amine-N-Oxides. Inorg. Chem. 2018, 57, 495–506. [Google Scholar] [CrossRef]
- Chapman, G.; Nicholas, K.M. Vanadium-catalyzed deoxydehydration of glycols. Chem. Commun. 2013, 49, 8199. [Google Scholar] [CrossRef]
- Gopaladasu, T.V.; Nicholas, K.M. Carbon Monoxide (CO)- and Hydrogen-Driven, Vanadium-Catalyzed Deoxydehydration of Glycols. ACS Catal. 2016, 6, 1901–1904. [Google Scholar] [CrossRef]
- Hanson, S.K.; Baker, R.T.; Gordon, J.C.; Scott, B.L.; Thorn, D.L. Aerobic Oxidation of Lignin Models Using a Base Metal Vanadium Catalyst. Inorg. Chem. 2010, 49, 5611–5618. [Google Scholar] [CrossRef] [PubMed]
- Junge, K.; Papa, V.; Beller, M. Cobalt–Pincer Complexes in Catalysis. Chem. A Eur. J. 2019, 25, 122–143. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Sahoo, B.; Junge, K.; Beller, M. Cobalt Complexes as an Emerging Class of Catalysts for Homogeneous Hydrogenations. Acc. Chem. Res. 2018, 51, 1858–1869. [Google Scholar] [CrossRef] [PubMed]
- Ai, W.; Zhong, R.; Liu, X.; Liu, Q. Hydride Transfer Reactions Catalyzed by Cobalt Complexes. Chem. Rev. 2019, 119, 2876–2953. [Google Scholar] [CrossRef] [PubMed]
- Lagaditis, P.O.; Schluschaß, B.; Demeshko, S.; Würtele, C.; Schneider, S. Square-Planar Cobalt (III) Pincer Complex. Inorg. Chem. 2016, 55, 4529–4536. [Google Scholar] [CrossRef]
- Midya, S.P.; Pitchaimani, J.; Landge, V.G.; Madhu, V.; Balaraman, E. Direct access to: N -alkylated amines and imines via acceptorless dehydrogenative coupling catalyzed by a cobalt (ii)-NNN pincer complex. Catal. Sci. Technol. 2018, 8, 3469–3473. [Google Scholar] [CrossRef]
- Ge, H.; Jing, Y.; Yang, X. Computational Design of Cobalt Catalysts for Hydrogenation of Carbon Dioxide and Dehydrogenation of Formic Acid. Inorg. Chem. 2016, 55, 12179–12184. [Google Scholar] [CrossRef]
- Gorgas, N.; Kirchner, K. Isoelectronic Manganese and Iron Hydrogenation/Dehydrogenation Catalysts: Similarities and Divergences. Acc. Chem. Res. 2018, 51, 1558–1569. [Google Scholar] [CrossRef]
- Reed-Berendt, B.G.; Polidano, K.; Morrill, L.C. Recent advances in homogeneous borrowing hydrogen catalysis using earth-abundant first row transition metals. Org. Biomol. Chem. 2019, 17, 1595–1607. [Google Scholar] [CrossRef] [Green Version]
- Anastas, P.T.; Zimmerman, J.B. The periodic table of the elements of green and sustainable chemistry. Green Chem. 2019, 6545–6566. [Google Scholar] [CrossRef]
- Kallmeier, F.; Kempe, R. Manganese Complexes for (De) Hydrogenation Catalysis: A Comparison to Cobalt and Iron Catalysts. Angew. Chem. Int. Ed. 2018, 57, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Morin, Y.; Zhang, L.; Trivelli, X.; Capet, F.; Paul, S.; Desset, S.; Dumeignil, F.; Gauvin, R.M. Oxidative Transformations of Biosourced Alcohols Catalyzed by Earth-Abundant Transition Metals. ChemCatChem 2017, 9, 2652–2660. [Google Scholar] [CrossRef]
- Mukherjee, A.; Milstein, D. Homogeneous Catalysis by Cobalt and Manganese Pincer Complexes. ACS Catal. 2018, 8, 11435–11469. [Google Scholar] [CrossRef]
- Zell, T.; Langer, R. From Ruthenium to Iron and Manganese—A Mechanistic View on Challenges and Design Principles of Base-Metal Hydrogenation Catalysts. ChemCatChem 2018, 10, 1930–1940. [Google Scholar] [CrossRef]
- Filonenko, G.A.; Van Putten, R.; Hensen, E.J.M.; Pidko, E.A. Catalytic (de) hydrogenation promoted by non-precious metals-Co, Fe and Mn: Recent advances in an emerging field. Chem. Soc. Rev. 2018, 47, 1459–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alig, L.; Fritz, M.; Schneider, S. First-Row Transition Metal (De) Hydrogenation Catalysis Based on Functional Pincer Ligands. Chem. Rev. 2019, 119, 2681–2751. [Google Scholar] [CrossRef]
- Zhang, Z.; Butt, N.A.; Zhou, M.; Liu, D.; Zhang, W. Asymmetric Transfer and Pressure Hydrogenation with Earth-Abundant Transition Metal Catalysts. Chin. J. Chem. 2018, 36, 443–454. [Google Scholar] [CrossRef]
- Riisager, A.; Fehrmann, R.; Haumann, M.; Wasserscheid, P. Supported Ionic Liquid Phase (SILP) catalysis: An innovative concept for homogeneous catalysis in continuous fixed-bed reactors. Eur. J. Inorg. Chem. 2006, 695–706. [Google Scholar] [CrossRef]
- Selvam, T.; MacHoke, A.; Schwieger, W. Supported ionic liquids on non-porous and porous inorganic materials-A topical review. Appl. Catal. A Gen. 2012, 445–446, 92–101. [Google Scholar] [CrossRef]
- Brünig, J.; Csendes, Z.; Weber, S.; Gorgas, N.; Bittner, R.W.; Limbeck, A.; Bica, K.; Hoffmann, H.; Kirchner, K. Chemoselective Supported Ionic-Liquid-Phase (SILP) Aldehyde Hydrogenation Catalyzed by an Fe(II) PNP Pincer Complex. ACS Catal. 2018, 8, 1048–1051. [Google Scholar] [CrossRef]
- Castro-Amoedo, R.; Csendes, Z.; Brünig, J.; Sauer, M.; Foelske-Schmitz, A.; Yigit, N.; Rupprechter, G.; Gupta, T.; Martins, A.M.; Bica, K.; et al. Carbon-based SILP catalysis for the selective hydrogenation of aldehydes using a well-defined Fe(ii) PNP complex. Catal. Sci. Technol. 2018, 8, 4812–4820. [Google Scholar] [CrossRef] [Green Version]
- Sheludko, B.; Cunningham, M.T.; Goldman, A.S.; Celik, F.E. Continuous-Flow Alkane Dehydrogenation by Supported Pincer-Ligated Iridium Catalysts at Elevated Temperatures. ACS Catal. 2018, 8, 7828–7841. [Google Scholar] [CrossRef]
- Barman, M.K.; Waiba, S.; Maji, B. Manganese-Catalyzed Direct Olefination via an Acceptorless Dehydrogenative Coupling of Methyl Heteroarenes with Primary Alcohols. Synlett 2019, 30, 12–20. [Google Scholar]
- Thiyagarajan, S.; Gunanathan, C. Ruthenium-Catalyzed α-Olefination of Nitriles Using Secondary Alcohols. ACS Catal. 2018, 8, 2473–2478. [Google Scholar] [CrossRef]
- Zhang, G.; Irrgang, T.; Dietel, T.; Kallmeier, F.; Kempe, R. Manganese-Catalyzed Dehydrogenative Alkylation or α-Olefination of Alkyl-Substituted N-Heteroarenes with Alcohols. Angew. Chem. Int. Ed. 2018, 57, 9131–9135. [Google Scholar] [CrossRef] [PubMed]
- Bauer, E.B.; Andavan, G.T.S.; Hollis, T.K.; Rubio, R.J.; Cho, J.; Kuchenbeiser, G.R.; Helgert, T.R.; Letko, C.S.; Tham, F.S. Air-and water-stable catalysts for hydroamination/cyclization. Synthesis and application of CCC-NHC pincer complexes of RH and Ir. Org. Lett. 2008, 10, 1175–1178. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Hollis, T.K.; Valente, E.J.; Trate, J.M. CCC-N-heterocyclic carbene pincer complexes: Synthesis, characterization and hydroamination activity of a hafnium complex. J. Organomet. Chem. 2011, 696, 373–377. [Google Scholar] [CrossRef]
- Castonguay, A.; Spasyuk, D.M.; Madern, N.; Beauchamp, A.L.; Zargarian, D. Regioselective hydroamination of acrylonitrile catalyzed by cationic pincer complexes of nickel (II). Organometallics 2009, 28, 2134–2141. [Google Scholar] [CrossRef]
- Takaya, J.; Iwasawa, N. Hydrocarboxylation of allenes with CO2 catalyzed by silyl pincer-type palladium complex. J. Am. Chem. Soc. 2008, 130, 15254–15255. [Google Scholar] [CrossRef]
- Serra, D.; Cao, P.; Cabrera, J.; Padilla, R.; Rominger, F.; Limbach, M. Development of platinum (II) and -(IV) CNC pincer complexes and their application in a hydrovinylation reaction. Organometallics 2011, 30, 1885–1895. [Google Scholar] [CrossRef]
- Mastalir, M.; Pittenauer, E.; Allmaier, G.; Kirchner, K. Manganese-Catalyzed Aminomethylation of Aromatic Compounds with Methanol as a Sustainable C1 Building Block. J. Am. Chem. Soc. 2017, 139, 8812–8815. [Google Scholar] [CrossRef]
- Zhang, Y.; Fang, H.; Yao, W.; Leng, X.; Huang, Z. Synthesis of Pincer Hydrido Ruthenium Olefin Complexes for Catalytic Alkane Dehydrogenation. Organometallics 2016, 35, 181–188. [Google Scholar] [CrossRef]
- Kumar, A.; Bhatti, T.M.; Goldman, A.S. Dehydrogenation of Alkanes and Aliphatic Groups by Pincer-Ligated Metal Complexes. Chem. Rev. 2017, 117, 12357–12384. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Kumar, A. Alkane Dehydrogenation Reactions Catalyzed by Pincer-Metal Complexes, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2019; ISBN 9780128171172. [Google Scholar]
- Renkema, K.B.; Kissin, Y.V.; Goldman, A.S. Mechanism of alkane transfer-dehydrogenation catalyzed by a pincer-ligated iridium complex. J. Am. Chem. Soc. 2003, 125, 7770–7771. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Achord, P.D.; Zhang, X.; Krogh-Jespersen, K.; Goldman, A.S. Highly effective pincer-ligated iridium catalysts for alkane dehydrogenation. DFT calculations of relevant thermodynamic, kinetic, and spectroscopic properties. J. Am. Chem. Soc. 2004, 126, 13044–13053. [Google Scholar] [CrossRef] [PubMed]
- Göttker-Schnetmann, I.; White, P.; Brookhart, M. Iridium Bis (phosphinite) p -XPCP Pincer Complexes: Highly Active Catalysts for the Transfer Dehydrogenation of Alkanes. J. Am. Chem. Soc. 2004, 126, 1804–1811. [Google Scholar] [CrossRef] [PubMed]
- Nawara-Hultzsch, A.J.; Hackenberg, J.D.; Punji, B.; Supplee, C.; Emge, T.J.; Bailey, B.C.; Schrock, R.R.; Brookhart, M.; Goldman, A.S. Rational design of highly active “hybrid” phosphine-phosphinite pincer iridium catalysts for alkane metathesis. ACS Catal. 2013, 3, 2505–2514. [Google Scholar] [CrossRef]
- Chakraborty, S.; Gellrich, U.; Diskin-Posner, Y.; Leitus, G.; Avram, L.; Milstein, D. Manganese-Catalyzed N-Formylation of Amines by Methanol Liberating H2: A Catalytic and Mechanistic Study. Angew. Chem. 2017, 129, 4293–4297. [Google Scholar] [CrossRef]
- Daw, P.; Chakraborty, S.; Leitus, G.; Diskin-Posner, Y.; Ben-David, Y.; Milstein, D. Selective N-Formylation of Amines with H2 and CO2 Catalyzed by Cobalt Pincer Complexes. ACS Catal. 2017, 7, 2500–2504. [Google Scholar] [CrossRef]
- El-Sepelgy, O.; Matador, E.; Brzozowska, A.; Rueping, M. C-Alkylation of Secondary Alcohols by Primary Alcohols through Manganese-Catalyzed Double Hydrogen Autotransfer. ChemSusChem 2019, 12, 3099–3102. [Google Scholar] [CrossRef]
- Freitag, F.; Irrgang, T.; Kempe, R. Cobalt-Catalyzed Alkylation of Secondary Alcohols with Primary Alcohols via Borrowing Hydrogen/Hydrogen Autotransfer. Chem. A Eur. J. 2017, 23, 12110–12113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barman, M.K.; Jana, A.; Maji, B. Phosphine-Free NNN-Manganese Complex Catalyzed α-Alkylation of Ketones with Primary Alcohols and Friedländer Quinoline Synthesis. Adv. Synth. Catal. 2018, 360, 3233–3238. [Google Scholar] [CrossRef]
- Peña-López, M.; Piehl, P.; Elangovan, S.; Neumann, H.; Beller, M. Manganese-Catalyzed Hydrogen-Autotransfer C–C Bond Formation: α-Alkylation of Ketones with Primary Alcohols. Angew. Chem. Int. Ed. 2016, 55, 14967–14971. [Google Scholar] [CrossRef] [PubMed]
- Elangovan, S.; Neumann, J.; Sortais, J.B.; Junge, K.; Darcel, C.; Beller, M. Efficient and selective N-alkylation of amines with alcohols catalysed by manganese pincer complexes. Nat. Commun. 2016, 7, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastalir, M.; Tomsu, G.; Pittenauer, E.; Allmaier, G.; Kirchner, K. Co(II) PCP Pincer Complexes as Catalysts for the Alkylation of Aromatic Amines with Primary Alcohols. Org. Lett. 2016, 18, 3462–3465. [Google Scholar] [CrossRef] [PubMed]
- Rösler, S.; Ertl, M.; Irrgang, T.; Kempe, R. Cobalt-Catalyzed Alkylation of Aromatic Amines by Alcohols. Angew. Chem. Int. Ed. 2015, 54, 15046–15050. [Google Scholar] [CrossRef]
- Mastalir, M.; Stöger, B.; Pittenauer, E.; Puchberger, M.; Allmaier, G.; Kirchner, K. Air Stable Iron(II) PNP Pincer Complexes as Efficient Catalysts for the Selective Alkylation of Amines with Alcohols. Adv. Synth. Catal. 2016, 358, 3824–3831. [Google Scholar] [CrossRef]
- Homberg, L.; Roller, A.; Hultzsch, K.C. A Highly Active PN 3 Manganese Pincer Complex Performing N-Alkylation of Amines under Mild Conditions. Org. Lett. 2019, 21, 3142–3147. [Google Scholar] [CrossRef]
- Landge, V.G.; Mondal, A.; Kumar, V.; Nandakumar, A.; Balaraman, E. Manganese catalyzed N-alkylation of anilines with alcohols: Ligand enabled selectivity. Org. Biomol. Chem. 2018, 16, 8175–8180. [Google Scholar] [CrossRef]
- Li, J.; Lutz, M.; Klein Gebbink, R.J.M. N,N,O-Coordinated tricarbonylrhenium precatalysts for the aerobic deoxydehydration of diols and polyols. Catal. Sci. Technol. 2020, 10, 3782–3788. [Google Scholar] [CrossRef]
- Siu, T.C.; Silva, I.; Lunn, M.J.; John, A. Influence of the pendant arm in deoxydehydration catalyzed by dioxomolybdenum complexes supported by amine bisphenolate ligands. New J. Chem. 2020, 44, 9933–9941. [Google Scholar] [CrossRef]
- Petersen, A.R.; Fristrup, P. New Motifs in Deoxydehydration: Beyond the Realms of Rhenium. Chem. A Eur. J. 2017, 23, 10235–10243. [Google Scholar] [CrossRef] [PubMed]
- Tshibalonza, N.N.; Monbaliu, J.-C.M. The deoxydehydration (DODH) reaction: A versatile technology for accessing olefins from bio-based polyols. Green Chem. 2020. [Google Scholar] [CrossRef]
- Chen, F.; Wang, N.; Lei, H.; Guo, D.; Liu, H.; Zhang, Z.; Zhang, W.; Lai, W.; Cao, R. Electrocatalytic Water Oxidation by a Water-Soluble Copper(II) Complex with a Copper-Bound Carbonate Group Acting as a Potential Proton Shuttle. Inorg. Chem. 2017, 56, 13368–13375. [Google Scholar] [CrossRef] [PubMed]
- Lant, H.M.C.; Michaelos, T.K.; Sharninghausen, L.S.; Mercado, B.Q.; Crabtree, R.H.; Brudvig, G.W. N,N,O Pincer Ligand with a Deprotonatable Site That Promotes Redox-Leveling, High Mn Oxidation States, and a Mn2O2 Dimer Competent for Catalytic Oxygen Evolution. Eur. J. Inorg. Chem. 2019, 2019, 2115–2123. [Google Scholar] [CrossRef]
- Kohl, S.W.; Weiner, L.; Schwartsburd, L.; Konstantinovski, L.; Shimon, L.J.W.; Ben-David, Y.; Iron, M.A.; Milstein, D. Consecutive Thermal H 2 and Light-Induced O 2 Evolution from Water Promoted by a Metal Complex. Science 2009, 324, 74–77. [Google Scholar] [CrossRef]
- Sandhya, K.S.; Suresh, C.H. Water splitting promoted by a ruthenium(II) PNN complex: An alternate pathway through a dihydrogen complex for hydrogen production. Organometallics 2011, 30, 3888–3891. [Google Scholar] [CrossRef]
- Sandhya, K.S.; Suresh, C.H. DFT study on the mechanism of water-assisted dihydrogen elimination in group 6 octahedral metal hydride complexes. Dalt. Trans. 2012, 41, 11018–11025. [Google Scholar] [CrossRef]
- Sandhya, K.S.; Remya, G.S.; Suresh, C.H. Pincer Ligand Modifications to Tune the Activation Barrier for H2 Elimination in Water Splitting Milstein Catalyst. Inorg. Chem. 2015, 54, 11150–11156. [Google Scholar] [CrossRef]
- Ma, C.; Piccinin, S.; Fabris, S. Reaction mechanisms of water splitting and H2 evolution by a Ru(II)-pincer complex identified with Ab initio metadynamics simulations. ACS Catal. 2012, 2, 1500–1506. [Google Scholar] [CrossRef]
- Yang, X.; Hall, M.B. Mechanism of water splitting and oxygen-oxygen bond formation by a mononuclear ruthenium complex. J. Am. Chem. Soc. 2010, 132, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M. Catalyst Kinetics and Stability in Homogeneous Alcohol Acceptorless Dehydrogenation. In Advanced Chemical Kinetics; InTech: London, UK, 2018; Chapter 6; ISBN 978-953-51-3816-7. [Google Scholar]
- Nielsen, M. Hydrogen production by homogeneous catalysis: Alcohol acceptorless dehydrogenation. In Hydrogen Production and Remediation of Carbon and Pollutants; Springer: Basel, Switzerland, 2015; Chapter 1; ISBN 9783319193755. [Google Scholar]
- Valdés, H.; García-Eleno, M.A.; Canseco-Gonzalez, D.; Morales-Morales, D. Recent Advances in Catalysis with Transition-Metal Pincer Compounds. ChemCatChem 2018, 10, 3136–3172. [Google Scholar] [CrossRef]
- Shende, V.S.; Saptal, V.B.; Bhanage, B.M. Recent Advances Utilized in the Recycling of Homogeneous Catalysis. Chem. Rec. 2019, 19, 2022–2043. [Google Scholar] [CrossRef] [PubMed]
- Dixneuf, P.H. Organometallics for Green Catalysis; Springer: Basel, Switzerland, 2019; ISBN 9783030109547. [Google Scholar]
- Werkmeister, S.; Neumann, J.; Junge, K.; Beller, M. Pincer-Type Complexes for Catalytic (De) Hydrogenation and Transfer (De) Hydrogenation Reactions: Recent Progress. Chem. A Eur. J. 2015, 21, 12226–12250. [Google Scholar] [CrossRef]
- Crabtree, R.H. Homogeneous Transition Metal Catalysis of Acceptorless Dehydrogenative Alcohol Oxidation: Applications in Hydrogen Storage and to Heterocycle Synthesis. Chem. Rev. 2017, 117, 9228–9246. [Google Scholar] [CrossRef]
- Gunanathan, C.; Milstein, D. Applications of Acceptorless Dehydrogenation and Related Transformations in Chemical Synthesis. Science 2013, 341, 1229712. [Google Scholar] [CrossRef]
- Spasyuk, D.; Smith, S.; Gusev, D.G. From esters to alcohols and back with ruthenium and osmium catalysts. Angew. Chem. Int. Ed. 2012, 51, 2772–2775. [Google Scholar] [CrossRef]
- Spasyuk, D.; Gusev, D.G. Acceptorless dehydrogenative coupling of ethanol and hydrogenation of esters and imines. Organometallics 2012, 31, 5239–5242. [Google Scholar] [CrossRef]
- Nielsen, M.; Junge, H.; Kammer, A.; Beller, M. Towards a green process for bulk-scale synthesis of ethyl acetate: Efficient acceptorless dehydrogenation of ethanol. Angew. Chem. Int. Ed. 2012, 51, 5711–5713. [Google Scholar] [CrossRef]
- Sponholz, P.; Mellmann, D.; Cordes, C.; Alsabeh, P.G.; Li, B.; Li, Y.; Nielsen, M.; Junge, H.; Dixneuf, P.; Beller, M. Efficient and Selective Hydrogen Generation from Bioethanol using Ruthenium Pincer-type Complexes. ChemSusChem 2014, 7, 2419–2422. [Google Scholar] [CrossRef]
- Li, Y.; Sponholz, P.; Nielsen, M.; Junge, H.; Beller, M. Iridium-catalyzed hydrogen production from monosaccharides, disaccharide, cellulose, and lignocellulose. ChemSusChem 2015, 8, 804–808. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Trivelli, X.; Capet, F.; Swesi, Y.; Favre-Réguillon, A.; Vanoye, L.; Dumeignil, F.; Gauvin, R.M. Deeper Mechanistic Insight into Ru Pincer-Mediated Acceptorless Dehydrogenative Coupling of Alcohols: Exchanges, Intermediates, and Deactivation Species. ACS Catal. 2018, 8, 4719–4734. [Google Scholar] [CrossRef]
- Pandey, P.; Dutta, I.; Bera, J.K. Acceptorless Alcohol Dehydrogenation: A Mechanistic Perspective. Proc. Natl. Acad. Sci. India Sect. A Phys. Sci. 2016, 86, 561–579. [Google Scholar] [CrossRef]
- Anaby, A.; Schelwies, M.; Schwaben, J.; Rominger, F.; Hashmi, A.S.K.; Schaub, T. Study of Precatalyst Degradation Leading to the Discovery of a New Ru0 Precatalyst for Hydrogenation and Dehydrogenation. Organometallics 2018, 37, 2193–2201. [Google Scholar] [CrossRef]
- Dub, P.A.; Gordon, J.C. The mechanism of enantioselective ketone reduction with Noyori and Noyori-Ikariya bifunctional catalysts. Dalt. Trans. 2016, 45, 6756–6781. [Google Scholar] [CrossRef] [PubMed]
- Remya, G.S.; Suresh, C.H. Hydrogen elimination reactivity of ruthenium pincer hydride complexes: A DFT study. New J. Chem. 2019, 43, 14634–14642. [Google Scholar] [CrossRef]
- Handgraaf, J.-W.; Meijer, E.J. Realistic Modeling of Ruthenium-Catalyzed Transfer Hydrogenation. J. Am. Chem. Soc. 2007, 129, 3099–3103. [Google Scholar] [CrossRef]
- Hou, C.; Zhang, Z.; Zhao, C.; Ke, Z. DFT Study of Acceptorless Alcohol Dehydrogenation Mediated by Ruthenium Pincer Complexes: Ligand Tautomerization Governing Metal Ligand Cooperation. Inorg. Chem. 2016, 55, 6539–6551. [Google Scholar] [CrossRef]
- Awasthi, M.K.; Singh, S.K. Ruthenium Catalyzed Dehydrogenation of Alcohols and Mechanistic Study. Inorg. Chem. 2019, 58, 14912–14923. [Google Scholar] [CrossRef]
- Chen, X.; Yang, X. Mechanistic Insights and Computational Design of Transition-Metal Catalysts for Hydrogenation and Dehydrogenation Reactions. Chem. Rec. 2016, 16, 2364–2378. [Google Scholar] [CrossRef]
- Tao, J.; Wen, L.; Lv, X.; Qi, Y.; Yin, H. Ruthenium(II)-PNN pincer complex catalyzed dehydrogenation of benzyl alcohol to ester: A DFT study. J. Mol. Struct. 2016, 1110, 24–31. [Google Scholar] [CrossRef]
- Ji, M.; Dong, C.; Yang, X. Density functional theory prediction of cobalt pincer complexes for catalytic dehydrogenation of ethanol. J. Coord. Chem. 2016, 69, 1380–1387. [Google Scholar] [CrossRef]
- Dub, P.A.; Gordon, J.C. Metal-ligand bifunctional catalysis: The “Accepted” mechanism, the issue of concertedness, and the function of the ligand in catalytic cycles involving hydrogen atoms. ACS Catal. 2017, 7, 6635–6655. [Google Scholar] [CrossRef]
- Vicent, C.; Gusev, D.G. ESI-MS Insights into Acceptorless Dehydrogenative Coupling of Alcohols. ACS Catal. 2016, 6, 3301–3309. [Google Scholar] [CrossRef] [Green Version]
- Dub, P.A.; Gordon, J.C. The role of the metal-bound N–H functionality in Noyori-type molecular catalysts. Nat. Rev. Chem. 2018, 2, 396–408. [Google Scholar] [CrossRef]
- Schneider, S.; Meiners, J.; Askevold, B. Cooperative aliphatic PNP amido pincer ligands-versatile building blocks for coordination chemistry and catalysis. Eur. J. Inorg. Chem. 2012, 412–429. [Google Scholar] [CrossRef]
- Charman, H.B. Hydride transfer reactions catalysed by metal complexes. J. Chem. Soc. B Phys. Org. 1967, 36, 629–632. [Google Scholar] [CrossRef]
- Morton, D.; Cole-Hamilton, D.J. ChemInform Abstract: Molecular Hydrogen Complexes in Catalysis: Highly Efficient Hydrogen Production from Alcoholic Substrates Catalyzed by Ruthenium Complexes. ChemInform 2016, 20, 1154–1156. [Google Scholar] [CrossRef]
- Morton, D.; Cole-Hamilton, D.J.; Schofield, J.A.; Pryce, R.J. Rapid thermal hydrogen production from 2,3-butanediol catalyzed by homogeneous rhodium catalysis. Polyhedron 1987, 6, 2187–2189. [Google Scholar] [CrossRef]
- Dobscn, A.; Robinson, S.D. Catalytic dehydrogenation of primary and secondary alcohols by Ru(OCOCF3)2(CO)(PPh3)2. J. Organomet. Chem. 1975, 87, 52–53. [Google Scholar] [CrossRef]
- Dobson, A.; Robinson, S.D. Complexes of the Platinum Metals. 7. Homogeneous Ruthenium and Osmium Catalysts for the Dehydrogenation of Primary and Secondary Alcohols. Inorg. Chem. 1977, 16, 137–142. [Google Scholar] [CrossRef]
- Zhang, J.; Gandelman, M.; Shimon, L.J.W.; Rozenberg, H.; Milstein, D. Electron-rich, bulky ruthenium PNP-type complexes. Acceptorless catalytic alcohol dehydrogenation. Organometallics 2004, 23, 4026–4033. [Google Scholar] [CrossRef]
- Zhang, J.; Leitus, G.; Ben-David, Y.; Milstein, D. Facile conversion of alcohols into esters and dihydrogen catalyzed by new ruthenium complexes. J. Am. Chem. Soc. 2005, 127, 10840–10841. [Google Scholar] [CrossRef] [PubMed]
- Milstein, D. Discovery of environmentally benign catalytic reactions of alcohols catalyzed by pyridine-based pincer Ru complexes, based on metal-ligand cooperation. Top. Catal. 2010, 53, 915–923. [Google Scholar] [CrossRef]
- Gunanathan, C.; Milstein, D. Metal-ligand cooperation by aromatization-dearomatization: A new paradigm in bond activation and “green” catalysis. Acc. Chem. Res. 2011, 44, 588–602. [Google Scholar] [CrossRef]
- Milstein, D. Metal-ligand cooperation by aromatization-dearomatization as a tool in single bond activation. Phil. Trans. R. Soc. A 2015, 373, 20140189. [Google Scholar] [CrossRef] [Green Version]
- Khusnutdinova, J.R.; Milstein, D. Metal-Ligand Cooperation. Angew. Chem. Int. Ed. 2015, 54, 12236–12273. [Google Scholar] [CrossRef]
- Zell, T.; Milstein, D. Hydrogenation and Dehydrogenation Iron Pincer Catalysts Capable of Metal–Ligand Cooperation by Aromatization/Dearomatization. Acc. Chem. Res. 2015, 48, 1979–1994. [Google Scholar] [CrossRef]
- Hale, L.V.A.; Szymczak, N.K. Hydrogen Transfer Catalysis beyond the Primary Coordination Sphere. ACS Catal. 2018, 8, 6446–6461. [Google Scholar] [CrossRef]
- Gelman, D.; Musa, S. Coordination Versatility of sp 3-Hybridized Pincer Ligands toward Ligand–Metal Cooperative Catalysis. ACS Catal. 2012, 2, 2456–2466. [Google Scholar] [CrossRef]
- Dub, P.A.; Ikariya, T. Catalytic Reductive Transformations of Carboxylic and Carbonic Acid Derivatives Using Molecular Hydrogen. ACS Catal. 2012, 2, 1718–1741. [Google Scholar] [CrossRef]
- Eisenstein, O.; Crabtree, R.H. Outer sphere hydrogenation catalysis. New J. Chem. 2013, 37, 21–27. [Google Scholar] [CrossRef]
- Yang, X. Hydrogenation of carbon dioxide catalyzed by PNP pincer iridium, iron, and cobalt complexes: A computational design of base metal catalysts. ACS Catal. 2011, 1, 849–854. [Google Scholar] [CrossRef]
- Hasanayn, F.; Baroudi, A.; Bengali, A.A.; Goldman, A.S. Hydrogenation of dimethyl carbonate to methanol by trans-[Ru(H)2(PNN)(CO)] catalysts: DFT evidence for ion-pair-mediated metathesis paths for C-OMe bond cleavage. Organometallics 2013, 32, 6969–6985. [Google Scholar] [CrossRef]
- Li, H.; Hall, M.B. Computational mechanistic studies on reactions of transition metal complexes with noninnocent pincer ligands: Aromatization-dearomatization or not. ACS Catal. 2015, 5, 1895–1913. [Google Scholar] [CrossRef]
- Gusev, D.G. Revised Mechanisms of the Catalytic Alcohol Dehydrogenation and Ester Reduction with the Milstein PNN Complex of Ruthenium. Organometallics 2020, 39, 258–270. [Google Scholar] [CrossRef]
- Junge, H.; Beller, M. Ruthenium-catalyzed generation of hydrogen from iso-propanol. Tetrahedron Lett. 2005, 46, 1031–1034. [Google Scholar] [CrossRef]
- Nielsen, M.; Kammer, A.; Cozzula, D.; Junge, H.; Gladiali, S.; Beller, M. Efficient hydrogen production from alcohols under mild reaction conditions. Angew. Chem. Int. Ed. 2011, 50, 9593–9597. [Google Scholar] [CrossRef]
- Kuriyama, W.; Matsumoto, T.; Ogata, O.; Ino, Y.; Aoki, K.; Tanaka, S.; Ishida, K.; Kobayashi, T.; Sayo, N.; Saito, T. Catalytic hydrogenation of esters. Development of an efficient catalyst and processes for synthesising (R)-1,2-propanediol and 2-(l-Menthoxy)ethanol. Org. Process Res. Dev. 2012, 16, 166–171. [Google Scholar] [CrossRef]
- Gunanathan, C.; Ben-David, Y.; Milstein, D. Direct Synthesis of Amides from Alcohols and Amines with Liberation of H2. Science 2007, 317, 790–792. [Google Scholar] [CrossRef]
- Kumar, A.; Espinosa-Jalapa, N.A.; Leitus, G.; Diskin-Posner, Y.; Avram, L.; Milstein, D. Direct Synthesis of Amides by Dehydrogenative Coupling of Amines with either Alcohols or Esters: Manganese Pincer Complex as Catalyst. Angew. Chem. Int. Ed. 2017, 56, 14992–14996. [Google Scholar] [CrossRef] [PubMed]
- Gusev, D.G. Rethinking the dehydrogenative amide synthesis. ACS Catal. 2017, 7, 6656–6662. [Google Scholar] [CrossRef]
- Schley, N.D.; Dobereiner, G.E.; Crabtree, R.H. Oxidative synthesis of amides and pyrroles via dehydrogenative alcohol oxidation by ruthenium diphosphine diamine complexes. Organometallics 2011, 30, 4174–4179. [Google Scholar] [CrossRef]
- Lane, E.M.; Uttley, K.B.; Hazari, N.; Bernskoetter, W. Iron-Catalyzed Amide Formation from the Dehydrogenative Coupling of Alcohols and Secondary Amines. Organometallics 2017, 36, 2020–2025. [Google Scholar] [CrossRef]
- Esteruelas, M.A.; Honczek, N.; Oliván, M.; Oñate, E.; Valencia, M. Direct access to pop-type osmium(II) and osmium(IV) complexes: Osmium a promising alternative to ruthenium for the synthesis of imines from alcohols and amines. Organometallics 2011, 30, 2468–2471. [Google Scholar] [CrossRef]
- Fertig, R.; Irrgang, T.; Freitag, F.; Zander, J.; Kempe, R. Manganese-Catalyzed and Base-Switchable Synthesis of Amines or Imines via Borrowing Hydrogen or Dehydrogenative Condensation. ACS Catal. 2018, 8, 8525–8530. [Google Scholar] [CrossRef]
- Mastalir, M.; Glatz, M.; Gorgas, N.; Stöger, B.; Pittenauer, E.; Allmaier, G.; Veiros, L.F.; Kirchner, K. Divergent Coupling of Alcohols and Amines Catalyzed by Isoelectronic Hydride MnIand FeIIPNP Pincer Complexes. Chem. A Eur. J. 2016, 22, 12316–12320. [Google Scholar] [CrossRef]
- Borghs, J.C.; Azofra, L.M.; Biberger, T.; Linnenberg, O.; Cavallo, L.; Rueping, M.; El-Sepelgy, O. Manganese-Catalyzed Multicomponent Synthesis of Pyrroles through Acceptorless Dehydrogenation Hydrogen Autotransfer Catalysis: Experiment and Computation. ChemSusChem 2019, 12, 3083–3088. [Google Scholar] [CrossRef]
- Gnanaprakasam, B.; Balaraman, E.; Gunanathan, C.; Milstein, D. Synthesis of polyamides from diols and diamines with liberation of H2. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 1755–1765. [Google Scholar] [CrossRef]
- Michlik, S.; Kempe, R. A sustainable catalytic pyrrole synthesis. Nat. Chem. 2013, 5, 140–144. [Google Scholar] [CrossRef]
- Srimani, D.; Ben-David, Y.; Milstein, D. Direct Synthesis of Pyrroles by Dehydrogenative Coupling of β-Aminoalcohols with Secondary Alcohols Catalyzed by Ruthenium Pincer Complexes. Angew. Chem. 2013, 125, 4104–4107. [Google Scholar] [CrossRef]
- Daw, P.; Chakraborty, S.; Garg, J.A.; Ben-David, Y.; Milstein, D. Direct Synthesis of Pyrroles by Dehydrogenative Coupling of Diols and Amines Catalyzed by Cobalt Pincer Complexes. Angew. Chem. Int. Ed. 2016, 55, 14373–14377. [Google Scholar] [CrossRef] [PubMed]
- Midya, S.P.; Landge, V.G.; Sahoo, M.K.; Rana, J.; Balaraman, E. Cobalt-catalyzed acceptorless dehydrogenative coupling of aminoalcohols with alcohols: Direct access to pyrrole, pyridine and pyrazine derivatives. Chem. Commun. 2017, 54, 90–93. [Google Scholar] [CrossRef]
- Borghs, J.C.; Lebedev, Y.; Rueping, M.; El-Sepelgy, O. Sustainable Manganese-Catalyzed Solvent-Free Synthesis of Pyrroles from 1,4-Diols and Primary Amines. Org. Lett. 2019, 21, 70–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michlik, S.; Kempe, R. Regioselectively functionalized pyridines from sustainable resources. Angew. Chem. Int. Ed. 2013, 52, 6326–6329. [Google Scholar] [CrossRef] [PubMed]
- Gnanaprakasam, B.; Balaraman, E.; Ben-David, Y.; Milstein, D. Synthesis of peptides and pyrazines from β-amino alcohols through extrusion of h 2 catalyzed by ruthenium pincer complexes: Ligand-controlled selectivity. Angew. Chem. Int. Ed. 2011, 50, 12240–12244. [Google Scholar] [CrossRef]
- Daw, P.; Kumar, A.; Espinosa-Jalapa, N.A.; Diskin-Posner, Y.; Ben-David, Y.; Milstein, D. Synthesis of Pyrazines and Quinoxalines via Acceptorless Dehydrogenative Coupling Routes Catalyzed by Manganese Pincer Complexes. ACS Catal. 2018, 8, 7734–7741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, K.; Mondal, A.; Srimani, D. Phosphine free Mn-complex catalysed dehydrogenative C-C and C-heteroatom bond formation: A sustainable approach to synthesize quinoxaline, pyrazine, benzothiazole and quinoline derivatives. Chem. Commun. 2018, 54, 10582–10585. [Google Scholar] [CrossRef] [Green Version]
- Deibl, N.; Kempe, R. Manganese-Catalyzed Multicomponent Synthesis of Pyrimidines from Alcohols and Amidines. Angew. Chem. Int. Ed. 2017, 56, 1663–1666. [Google Scholar] [CrossRef]
- Das, U.K.; Ben-David, Y.; Diskin-Posner, Y.; Milstein, D. N-Substituted Hydrazones by Manganese-Catalyzed Coupling of Alcohols with Hydrazine: Borrowing Hydrogen and Acceptorless Dehydrogenation in One System. Angew. Chem. Int. Ed. 2018, 57, 2179–2182. [Google Scholar] [CrossRef]
- Mastalir, M.; Glatz, M.; Pittenauer, E.; Allmaier, G.; Kirchner, K. Sustainable Synthesis of Quinolines and Pyrimidines Catalyzed by Manganese PNP Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 15543–15546. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Leitus, G.; Milstein, D. Iron-Catalyzed Mild and Selective Hydrogenative Cross-Coupling of Nitriles and Amines to Form Secondary Aldimines. Angew. Chem. 2017, 129, 2106–2110. [Google Scholar] [CrossRef]
- Mukherjee, A.; Nerush, A.; Leitus, G.; Shimon, L.J.W.; Ben David, Y.; Espinosa Jalapa, N.A.; Milstein, D. Manganese-Catalyzed Environmentally Benign Dehydrogenative Coupling of Alcohols and Amines to Form Aldimines and H2: A Catalytic and Mechanistic Study. J. Am. Chem. Soc. 2016, 138, 4298–4301. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Luo, Q.; Meng, X.; Li, R.; Zhang, J.; Peng, T. Ru(II) complexes bearing 2,6-bis(benzimidazole-2-yl)pyridine ligands: A new class of catalysts for efficient dehydrogenation of primary alcohols to carboxylic acids and H2 in the alcohol/CsOH system. J. Organomet. Chem. 2017, 830, 11–18. [Google Scholar] [CrossRef]
- Zhang, L.; Nguyen, D.H.; Raffa, G.; Trivelli, X.; Capet, F.; Desset, S.; Paul, S.; Dumeignil, F.; Gauvin, R.M. Catalytic Conversion of Alcohols into Carboxylic Acid Salts in Water: Scope, Recycling, and Mechanistic Insights. ChemSusChem 2016, 9, 1413–1423. [Google Scholar] [CrossRef]
- Luo, Q.; Dai, Z.; Luo, Q.; Jiang, H.; Li, H.; Zhang, J.; Peng, T. Ni(ii)-N′NN′ pincer complexes catalyzed dehydrogenation of primary alcohols to carboxylic acids and H2 accompanied by alcohol etherification. Catal. Sci. Technol. 2017, 7, 2506–2511. [Google Scholar]
- Shao, Z.; Wang, Y.; Liu, Y.; Wang, Q.; Fu, X.; Liu, Q. A general and efficient Mn-catalyzed acceptorless dehydrogenative coupling of alcohols with hydroxides into carboxylates. Org. Chem. Front. 2018, 5, 1248–1256. [Google Scholar] [CrossRef]
- Bhatia, A.; Muthaiah, S. Well-Defined Ruthenium Complex for Acceptorless Alcohol Dehydrogenation in Aqueous Medium. ChemistrySelect 2018, 3, 3737–3741. [Google Scholar] [CrossRef]
- Huang, F.; Liu, Z.; Yu, Z. C-alkylation of ketones and related compounds by alcohols: Transition-metal-catalyzed dehydrogenation. Angew. Chem. Int. Ed. 2016. [Google Scholar] [CrossRef]
- Wang, Z.; Pan, B.; Liu, Q.; Yue, E.; Solan, G.A.; Ma, Y.; Sun, W.H. Efficient acceptorless dehydrogenation of secondary alcohols to ketones mediated by a PNN-Ru(II) catalyst. Catal. Sci. Technol. 2017, 7, 1654–1661. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Raffa, G.; Nguyen, D.H.; Swesi, Y.; Corbel-Demailly, L.; Capet, F.; Trivelli, X.; Desset, S.; Paul, S.; Paul, J.F.; et al. Acceptorless dehydrogenative coupling of alcohols catalysed by ruthenium PNP complexes: Influence of catalyst structure and of hydrogen mass transfer. J. Catal. 2016, 340, 331–343. [Google Scholar] [CrossRef]
- Srimani, D.; Balaraman, E.; Gnanaprakasam, B.; Ben-David, Y.; Milstein, D. Ruthenium pincer-catalyzed cross-dehydrogenative coupling of primary alcohols with secondary alcohols under neutral conditions. Adv. Synth. Catal. 2012, 354, 2403–2406. [Google Scholar] [CrossRef]
- De Boer, S.Y.; Korstanje, T.J.; La Rooij, S.R.; Kox, R.; Reek, J.N.H.; Van Der Vlugt, J.I. Ruthenium PNN(O) Complexes: Cooperative Reactivity and Application as Catalysts for Acceptorless Dehydrogenative Coupling Reactions. Organometallics 2017, 36, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Trivelli, X.; Capet, F.; Paul, J.F.; Dumeignil, F.; Gauvin, R.M. Manganese Pincer Complexes for the Base-Free, Acceptorless Dehydrogenative Coupling of Alcohols to Esters: Development, Scope, and Understanding. ACS Catal. 2017, 7, 2022–2032. [Google Scholar] [CrossRef]
- Das, U.K.; Ben-David, Y.; Leitus, G.; Diskin-Posner, Y.; Milstein, D. Dehydrogenative Cross-Coupling of Primary Alcohols to Form Cross-Esters Catalyzed by a Manganese Pincer Complex. ACS Catal. 2019, 9, 479–484. [Google Scholar] [CrossRef]
- Gunanathan, C.; Shimon, L.J.W.; Milstein, D. Direct conversion of alcohols to acetals and H2 catalyzed by an acridine-based ruthenium pincer complex. J. Am. Chem. Soc. 2009, 131, 3146–3147. [Google Scholar] [CrossRef]
- Das, U.K.; Chakraborty, S.; Diskin-Posner, Y.; Milstein, D. Direct Conversion of Alcohols into Alkenes by Dehydrogenative Coupling with Hydrazine/Hydrazone Catalyzed by Manganese. Angew. Chem. 2018, 130, 13632–13636. [Google Scholar] [CrossRef]
- Musa, S.; Shaposhnikov, I.; Cohen, S.; Gelman, D. Ligand-metal cooperation in pcppincer complexes: Rational design and catalytic activity in acceptorless dehydrogenation of alcohols. Angew. Chem. Int. Ed. 2011, 50, 3533–3537. [Google Scholar] [CrossRef]
- Peña-López, M.; Neumann, H.; Beller, M. Iron(II) Pincer-Catalyzed Synthesis of Lactones and Lactams through a Versatile Dehydrogenative Domino Sequence. ChemCatChem 2015, 7, 865–871. [Google Scholar] [CrossRef]
- Zhao, J.; Hartwig, J.F. Acceptorless, Neat, Ruthenium-Catalyzed Dehydrogenative Cyclization of Diols to Lactones. Organometallics 2005, 24, 2441–2446. [Google Scholar] [CrossRef]
- Chase, P.A.; Gossage, R.A.; Van Koten, G. Modern organometallic multidentate ligand design strategies: The birth of the privileged “pincer” ligand platform. In The Privileged Pincer-Metal Platform: Coordination Chemistry & Applications; Springer: Basel, Switzerland, 2015; ISBN 9783319229270. [Google Scholar]
- Van Koten, G.; Gossage, R.A. The Privileged Pincer-Metal Platform: Coordination Chemistry & Applications; Springer: Basel, Switzerland, 2015; ISBN 9783319229270. [Google Scholar]
- Van Koten, G. Organometallic Pincer Chemistry; van Koten, G., Milstein, D., Eds.; Topics in Organometallic Chemistry; Springer: Berlin/Heidelberg, Germany, 2013; ISBN 978-3-642-31080-5. [Google Scholar]
- Szabó, K.J.; Wendt, O.F. Pincer and Pincer-Type Complexes: Applications in Organic Synthesis and Catalysis; Wiley-VCH: Weinheim, Germany, 2014; ISBN 9783527681303. [Google Scholar]
- van Koten, G. Pincer ligands as powerful tools for catalysis in organic synthesis. J. Organomet. Chem. 2013, 730, 156–164. [Google Scholar] [CrossRef]
- Niermann, M.; Beckendorff, A.; Kaltschmitt, M.; Bonhoff, K. Liquid Organic Hydrogen Carrier (LOHC)—Assessment based on chemical and economic properties. Int. J. Hydrog. Energy 2019, 44, 6631–6654. [Google Scholar] [CrossRef]
- Zhu, Q.-L.; Xu, Q. Liquid organic and inorganic chemical hydrides for high-capacity hydrogen storage. Energy Environ. Sci. 2015, 8, 478–512. [Google Scholar] [CrossRef]
- Biniwale, R.B.; Rayalu, S.; Devotta, S.; Ichikawa, M. Chemical hydrides: A solution to high capacity hydrogen storage and supply. Int. J. Hydrog. Energy 2008, 33, 360–365. [Google Scholar] [CrossRef]
- Di Profio, P.; Arca, S.; Rossi, F.; Filipponi, M. Comparison of hydrogen hydrates with existing hydrogen storage technologies: Energetic and economic evaluations. Int. J. Hydrog. Energy 2009, 34, 9173–9180. [Google Scholar] [CrossRef]
- Sotoodeh, F.; Huber, B.J.M.; Smith, K.J. Dehydrogenation kinetics and catalysis of organic heteroaromatics for hydrogen storage. Int. J. Hydrog. Energy 2012, 37, 2715–2722. [Google Scholar] [CrossRef]
- Züttel, A. Materials for hydrogen storage. Mater. Today 2003, 6, 24–33. [Google Scholar] [CrossRef]
- He, T.; Pei, Q.; Chen, P. Liquid organic hydrogen carriers. J. Energy Chem. 2015, 24, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Preuster, P.; Papp, C.; Wasserscheid, P. Liquid organic hydrogen carriers (LOHCs): Toward a hydrogen-free hydrogen economy. Acc. Chem. Res. 2017, 50, 74–85. [Google Scholar] [CrossRef]
- Stark, K.; Emelyanenko, V.N.; Zhabina, A.A.; Varfolomeev, M.A.; Verevkin, S.P.; Müller, K.; Arlt, W. Liquid Organic Hydrogen Carriers: Thermophysical and Thermochemical Studies of Carbazole Partly and Fully Hydrogenated Derivatives. Ind. Eng. Chem. Res. 2015, 54, 7953–7966. [Google Scholar] [CrossRef]
- Müller, K.; Stark, K.; Emelyanenko, V.N.; Varfolomeev, M.A.; Zaitsau, D.H.; Shoifet, E.; Schick, C.; Verevkin, S.P.; Arlt, W. Liquid Organic Hydrogen Carriers: Thermophysical and Thermochemical Studies of Benzyl- and Dibenzyl-toluene Derivatives. Ind. Eng. Chem. Res. 2015, 54, 7967–7976. [Google Scholar] [CrossRef]
- Sotoodeh, F.; Smith, K.J. An overview of the kinetics and catalysis of hydrogen storage on organic liquids. Can. J. Chem. Eng. 2013, 91, 1477–1490. [Google Scholar] [CrossRef]
- Sotoodeh, F.; Smith, K.J. Kinetics of hydrogen uptake and release from heteroaromatic compounds for hydrogen storage. Ind. Eng. Chem. Res. 2010, 49, 1018–1026. [Google Scholar] [CrossRef]
- Müller, K.; Aslam, R.; Fischer, A.; Stark, K.; Wasserscheid, P.; Arlt, W. Experimental assessment of the degree of hydrogen loading for the dibenzyl toluene based LOHC system. Int. J. Hydrog. Energy 2016, 41, 22097–22103. [Google Scholar] [CrossRef]
- He, T.; Pachfule, P.; Wu, H.; Xu, Q.; Chen, P. Hydrogen carriers. Nat. Rev. Mater. 2016, 1, 16059. [Google Scholar] [CrossRef]
- Müller, K.; Völkl, J.; Arlt, W. Thermodynamic Evaluation of Potential Organic Hydrogen Carriers. Energy Technol. 2013, 1, 20–24. [Google Scholar] [CrossRef]
- Crabtree, R.H. Hydrogen storage in liquid organic heterocycles. Energy Environ. Sci. 2008, 1, 134. [Google Scholar] [CrossRef]
- Markiewicz, M.; Zhang, Y.Q.; Bösmann, A.; Brückner, N.; Thöming, J.; Wasserscheid, P.; Stolte, S. Environmental and health impact assessment of Liquid Organic Hydrogen Carrier (LOHC) systems—Challenges and preliminary results. Energy Environ. Sci. 2015, 8, 1035–1045. [Google Scholar] [CrossRef] [Green Version]
- Teichmann, D.; Arlt, W.; Wasserscheid, P.; Freymann, R. A future energy supply based on Liquid Organic Hydrogen Carriers (LOHC). Energy Environ. Sci. 2011, 4, 2767. [Google Scholar] [CrossRef]
- Teichmann, D.; Stark, K.; Müller, K.; Zöttl, G.; Wasserscheid, P.; Arlt, W. Energy storage in residential and commercial buildings via Liquid Organic Hydrogen Carriers (LOHC). Energy Environ. Sci. 2012, 5, 9044. [Google Scholar] [CrossRef] [Green Version]
- Teichmann, D.; Arlt, W.; Wasserscheid, P. Liquid Organic Hydrogen Carriers as an efficient vector for the transport and storage of renewable energy. Int. J. Hydrog. Energy 2012, 37, 18118–18132. [Google Scholar] [CrossRef]
- Ahmed, A.; Al-Amin, A.Q.; Ambrose, A.F.; Saidur, R. Hydrogen fuel and transport system: A sustainable and environmental future. Int. J. Hydrog. Energy 2016, 41, 1369–1380. [Google Scholar] [CrossRef]
- Eypasch, M.; Schimpe, M.; Kanwar, A.; Hartmann, T.; Herzog, S.; Frank, T.; Hamacher, T. Model-based techno-economic evaluation of an electricity storage system based on Liquid Organic Hydrogen Carriers. Appl. Energy 2017, 185, 320–330. [Google Scholar] [CrossRef]
- Johnson, T.C.; Morris, D.J.; Wills, M. Hydrogen generation from formic acid and alcohols using homogeneous catalysts. Chem. Soc. Rev. 2010, 39, 81–88. [Google Scholar] [CrossRef]
- Friedrich, A.; Schneider, S. Acceptorless dehydrogenation of alcohols: Perspectives for synthesis and H2 storage. ChemCatChem 2009, 1, 72–73. [Google Scholar] [CrossRef]
- Trincado, M.; Banerjee, D.; Grützmacher, H. Molecular catalysts for hydrogen production from alcohols. Energy Environ. Sci. 2014, 7, 2464–2503. [Google Scholar] [CrossRef]
- Campos, J. Dehydrogenation of alcohols and polyols from a hydrogen production perspective. Phys. Sci. Rev. 2018, 3, 1–25. [Google Scholar]
- Shimbayashi, T.; Fujita, K. ichi Metal-catalyzed hydrogenation and dehydrogenation reactions for efficient hydrogen storage. Tetrahedron 2020, 76, 130946. [Google Scholar] [CrossRef]
- Onishi, N.; Laurenczy, G.; Beller, M.; Himeda, Y. Recent progress for reversible homogeneous catalytic hydrogen storage in formic acid and in methanol. Coord. Chem. Rev. 2018, 373, 317–332. [Google Scholar] [CrossRef]
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols. Chem. Rev. 2018, 118, 372–433. [Google Scholar] [CrossRef]
- Trincado, M.; Grützmacher, H.; Prechtl, M.H.G. CO2-based hydrogen storage—Hydrogen generation from formaldehyde/water. Phys. Sci. Rev. 2018, 3, 1–19. [Google Scholar]
- Langer, R.; Thomas, Z.; Schaub, T. CO2-based hydrogen storage: CO2 hydrogenation to formic acid, formaldehyde and methanol. Phys. Sci. Rev. 2018, 3, 1–14. [Google Scholar]
- Boddien, A.; Gärtner, F.; Nielsen, M.; Losse, S.; Junge, H. Hydrogen Generation from Formic Acid and Alcohols; Elsevier: Amsterdam, The Netherland, 2013; Volume 6, pp. 587–603. ISBN 9780080965291. [Google Scholar]
- Enthaler, S.; von Langermann, J.; Schmidt, T. Carbon dioxide and formic acid—The couple for environmental-friendly hydrogen storage? Energy Environ. Sci. 2010, 3, 1207. [Google Scholar] [CrossRef]
- Eppinger, J.; Huang, K.W. Formic Acid as a Hydrogen Energy Carrier. ACS Energy Lett. 2017, 2, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Olah, G.A.; Goeppert, A.; Prakash, G.K.S. The “Methanol Economy”: General Aspects. In Beyond Oil and Gas; Wiley: Weinheim, Germany, 2018. [Google Scholar]
- Olah, G.A. After oil and gas: Methanol economy. Catal. Lett. 2004, 93, 1–2. [Google Scholar] [CrossRef]
- Stephan, D.W. A step closer to a methanol economy. Nature 2013, 495, 54–55. [Google Scholar] [CrossRef]
- Salvi, B.L.; Subramanian, K.A.; Panwar, N.L. Alternative fuels for transportation vehicles: A technical review. Renew. Sustain. Energy Rev. 2013, 25, 404–419. [Google Scholar] [CrossRef]
- Piola, L.; Fernández-Salas, J.A.; Nahra, F.; Poater, A.; Cavallo, L.; Nolan, S.P. Ruthenium-catalysed decomposition of formic acid: Fuel cell and catalytic applications. Mol. Catal. 2017, 440, 184–189. [Google Scholar] [CrossRef]
- Muradov, N.; Veziroglu, T. “Green” path from fossil-based to hydrogen economy: An overview of carbon-neutral technologies. Int. J. Hydrog. Energy 2008, 33, 6804–6839. [Google Scholar] [CrossRef]
- Olah, G.A.; Prakash, G.K.S.; Goeppert, A. Anthropogenic Chemical Carbon Cycle for a Sustainable Future. J. Am. Chem. Soc. 2011, 133, 12881–12898. [Google Scholar] [CrossRef]
- Fu, H.C.; You, F.; Li, H.R.; He, L.N. CO2 Capture and in situ Catalytic Transformation. Front. Chem. 2019, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef] [PubMed]
- Klankermayer, J.; Wesselbaum, S.; Beydoun, K.; Leitner, W. Selective Catalytic Synthesis Using the Combination of Carbon Dioxide and Hydrogen: Catalytic Chess at the Interface of Energy and Chemistry. Angew. Chem. Int. Ed. 2016, 55, 7296–7343. [Google Scholar] [CrossRef] [PubMed]
- Langer, R.; Thomas, Z.; Suárez, A. Hydrogenation of carbonyl compounds of relevance to hydrogen storage in alcohols. Phys. Sci. Rev. 2018, 3, 1–31. [Google Scholar]
- Song, C. Global challenges and strategies for control, conversion and utilization of CO2 for sustainable development involving energy, catalysis, adsorption and chemical processing. Catal. Today 2006, 115, 2–32. [Google Scholar] [CrossRef]
- Rumayor, M.; Dominguez-Ramos, A.; Irabien, A. Formic Acid manufacture: Carbon dioxide utilization alternatives. Appl. Sci. 2018, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Obama, B. The irreversible momentum of clean energy. Science 2017, 355, 126–129. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Nielsen, M.; Li, B.; Dixneuf, P.H.; Junge, H.; Beller, M. Ruthenium-catalyzed hydrogen generation from glycerol and selective synthesis of lactic acid. Green Chem. 2015, 17, 193–198. [Google Scholar] [CrossRef]
- Sharninghausen, L.S.; Mercado, B.Q.; Crabtree, R.H.; Hazari, N. Selective conversion of glycerol to lactic acid with iron pincer precatalysts. Chem. Commun. 2015, 51, 16201–16204. [Google Scholar] [CrossRef]
- Chakraborty, S.; Lagaditis, P.O.; Förster, M.; Bielinski, E.A.; Hazari, N.; Holthausen, M.C.; Jones, W.D.; Schneider, S. Well-Defined Iron Catalysts for the Acceptorless Reversible Dehydrogenation-Hydrogenation of Alcohols and Ketones. ACS Catal. 2014, 4, 3994–4003. [Google Scholar] [CrossRef]
- Lane, E.M.; Hazari, N.; Bernskoetter, W.H. Iron-catalyzed urea synthesis: Dehydrogenative coupling of methanol and amines. Chem. Sci. 2018, 9, 4003–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Hu, P.; Ben-David, Y.; Milstein, D. A Reversible Liquid Organic Hydrogen Carrier System Based on Methanol-Ethylenediamine and Ethylene Urea. Angew. Chem. Int. Ed. 2019, 58, 5105–5109. [Google Scholar] [CrossRef] [PubMed]
- Alberico, E.; Nielsen, M. Towards a methanol economy based on homogeneous catalysis: Methanol to H2 and CO2 to methanol. Chem. Commun. 2015, 51, 6714–6725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kothandaraman, J.; Kar, S.; Goeppert, A.; Sen, R.; Prakash, G.K.S. Advances in Homogeneous Catalysis for Low Temperature Methanol Reforming in the Context of the Methanol Economy. Top. Catal. 2018, 61, 542–559. [Google Scholar] [CrossRef]
- Simon Araya, S.; Liso, V.; Cui, X.; Li, N.; Zhu, J.; Sahlin, S.L.; Jensen, S.H.; Nielsen, M.P.; Kær, S.K. A Review of the Methanol Economy: The Fuel Cell Route. Energies 2020, 13, 596. [Google Scholar] [CrossRef] [Green Version]
- Cortright, R.D.; Davda, R.R.; Dumesic, J.A. Hydrogen from catalytic reforming of biomass-derived hydrocarbons in liquid water. In Materials for Sustainable Energy; Macmillan Publishers Ltd.: London, UK, 2010; pp. 289–292. [Google Scholar]
- Shabaker, J. Aqueous-phase reforming of methanol and ethylene glycol over alumina-supported platinum catalysts. J. Catal. 2003, 215, 344–352. [Google Scholar] [CrossRef]
- Navarro, R.M.; Peña, M.A.; Fierro, J.L.G. Hydrogen Production Reactions from Carbon Feedstocks: Fossil Fuels and Biomass. Chem. Rev. 2007, 107, 3952–3991. [Google Scholar] [CrossRef]
- Palo, D.R.; Dagle, R.A.; Holladay, J.D. Methanol Steam Reforming for Hydrogen Production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef] [PubMed]
- Sá, S.; Silva, H.; Brandão, L.; Sousa, J.M.; Mendes, A. Catalysts for methanol steam reforming—A review. Appl. Catal. B Environ. 2010, 99, 43–57. [Google Scholar] [CrossRef]
- Iulianelli, A.; Ribeirinha, P.; Mendes, A.; Basile, A. Methanol steam reforming for hydrogen generation via conventional and membrane reactors: A review. Renew. Sustain. Energy Rev. 2014, 29, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Alberico, E.; Baumann, W.; Drexler, H.J.; Junge, H.; Gladiali, S.; Beller, M. Low-temperature aqueous-phase methanol dehydrogenation to hydrogen and carbon dioxide. Nature 2013, 495, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Lugo, R.E.; Trincado, M.; Vogt, M.; Tewes, F.; Santiso-Quinones, G.; Grützmacher, H. A homogeneous transition metal complex for clean hydrogen production from methanol—Water mixtures. Nat. Chem. 2013, 5, 342–347. [Google Scholar] [CrossRef]
- Fujita, K.; Kawahara, R.; Aikawa, T.; Yamaguchi, R. Hydrogen Production from a Methanol-Water Solution Catalyzed by an Anionic Iridium Complex Bearing a Functional Bipyridonate Ligand under Weakly Basic Conditions. Angew. Chem. Int. Ed. 2015, 54, 9057–9060. [Google Scholar] [CrossRef]
- Alberico, E.; Sponholz, P.; Cordes, C.; Nielsen, M.; Drexler, H.J.; Baumann, W.; Junge, H.; Beller, M. Selective hydrogen production from methanol with a defined Iron pincer catalyst under mild conditions. Angew. Chem. Int. Ed. 2013, 52, 14162–14166. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Diskin-Posner, Y.; Ben-David, Y.; Milstein, D. Reusable homogeneous catalytic system for hydrogen production from methanol and water. ACS Catal. 2014, 4, 2649–2652. [Google Scholar] [CrossRef]
- Monney, A.; Barsch, E.; Sponholz, P.; Junge, H.; Ludwig, R.; Beller, M. Base-free hydrogen generation from methanol using a bi-catalytic system. Chem. Commun. 2014, 50, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Agapova, A.; Junge, H.; Beller, M. Developing Bicatalytic Cascade Reactions: Ruthenium-catalyzed Hydrogen Generation from Methanol. Chem. A Eur. J. 2019, 25, 9345–9349. [Google Scholar] [CrossRef] [PubMed]
- Bielinski, E.A.; Förster, M.; Zhang, Y.; Bernskoetter, W.H.; Hazari, N.; Holthausen, M.C. Base-Free Methanol Dehydrogenation Using a Pincer-Supported Iron Compound and Lewis Acid Co-catalyst. ACS Catal. 2015, 5, 2404–2415. [Google Scholar] [CrossRef]
- Alberico, E.; Lennox, A.J.J.; Vogt, L.K.; Jiao, H.; Baumann, W.; Drexler, H.J.; Nielsen, M.; Spannenberg, A.; Checinski, M.P.; Junge, H.; et al. Unravelling the Mechanism of Basic Aqueous Methanol Dehydrogenation Catalyzed by Ru-PNP Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 14890–14904. [Google Scholar] [CrossRef] [Green Version]
- Pingen, D.; Choi, J.-H.; Allen, H.; Murray, G.; Ganji, P.; van Leeuwen, P.W.N.M.; Prechtl, M.H.G.; Vogt, D. Amide versus amine ligand paradigm in the direct amination of alcohols with Ru-PNP complexes. Catal. Sci. Technol. 2018, 8, 3969–3976. [Google Scholar] [CrossRef]
- Dub, P.A.; Scott, B.L.; Gordon, J.C. Why does alkylation of the N-H functionality within M/NH bifunctional Noyori-type catalysts lead to turnover? J. Am. Chem. Soc. 2017, 139, 1245–1260. [Google Scholar] [CrossRef]
- Choi, J.H.; Heim, L.E.; Ahrens, M.; Prechtl, M.H.G. Selective conversion of alcohols in water to carboxylic acids by in situ generated ruthenium trans dihydrido carbonyl PNP complexes. Dalt. Trans. 2014, 43, 17248–17254. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; De Aguirre, A.; Junge, K.; Beller, M.; Jiao, H. Exploring the mechanisms of aqueous methanol dehydrogenation catalyzed by defined PNP Mn and Re pincer complexes under base-free as well as strong base conditions. Catal. Sci. Technol. 2018, 8, 3649–3665. [Google Scholar] [CrossRef]
- Jiang, Y.Y.; Xu, Z.Y.; Yu, H.Z.; Fu, Y. A self-catalytic role of methanol in PNP-Ru pincer complex catalysed dehydrogenation. Sci. China Chem. 2016, 59, 724–729. [Google Scholar] [CrossRef]
- Lei, M.; Pan, Y.; Ma, X. The nature of hydrogen production from aqueous-phase methanol dehydrogenation with ruthenium pincer complexes under mild conditions. Eur. J. Inorg. Chem. 2015, 2015, 794–803. [Google Scholar] [CrossRef]
- Yang, X. Mechanistic insights into ruthenium-catalyzed production of H2 and CO2 from methanol and water: A DFT study. ACS Catal. 2014, 4, 1129–1133. [Google Scholar] [CrossRef]
- Strobel, V.; Schuster, J.J.; Braeuer, A.S.; Vogt, L.K.; Junge, H.; Haumann, M. Shining light on low-temperature methanol aqueous-phase reforming using homogeneous Ru-pincer complexes-operando Raman-GC studies. React. Chem. Eng. 2017, 2, 390–396. [Google Scholar] [CrossRef]
- Andérez-Fernández, M.; Vogt, L.K.; Fischer, S.; Zhou, W.; Jiao, H.; Garbe, M.; Elangovan, S.; Junge, K.; Junge, H.; Ludwig, R.; et al. A Stable Manganese Pincer Catalyst for the Selective Dehydrogenation of Methanol. Angew. Chem. Int. Ed. 2017, 56, 559–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prichatz, C.; Alberico, E.; Baumann, W.; Junge, H.; Beller, M. Iridium–PNP Pincer Complexes for Methanol Dehydrogenation at Low Base Concentration. ChemCatChem 2017, 9, 1891–1896. [Google Scholar] [CrossRef]
- Agapova, A.; Alberico, E.; Kammer, A.; Junge, H.; Beller, M. Catalytic Dehydrogenation of Formic Acid with Ruthenium-PNP-Pincer Complexes: Comparing N-Methylated and NH-Ligands. ChemCatChem 2019, 11, 1910–1914. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, C.H.; Agapova, A.; Junge, H.; Haumann, M. Immobilization of a selective Ru-pincer complex for low temperature methanol reforming–Material and process improvements. Catal. Today 2020, 342, 178–186. [Google Scholar] [CrossRef]
- Laurenczy, G.; Dyson, P.J. Homogeneous Catalytic Dehydrogenation of Formic Acid: Progress towards a Hydrogen-Based Economy. J. Braz. Chem. Soc. 2014, 25, 2157–2163. [Google Scholar] [CrossRef]
- Treigerman, Z.; Sasson, Y. Further Observations on the Mechanism of Formic Acid Decomposition by Homogeneous Ruthenium Catalyst. ChemistrySelect 2017, 2, 5816–5823. [Google Scholar] [CrossRef]
- Müller, K.; Brooks, K.; Autrey, T. Hydrogen Storage in Formic Acid: A Comparison of Process Options. Energy Fuels 2017, 31, 12603–12611. [Google Scholar] [CrossRef]
- Mellmann, D.; Sponholz, P.; Junge, H.; Beller, M. Formic acid as a hydrogen storage material-development of homogeneous catalysts for selective hydrogen release. Chem. Soc. Rev. 2016, 45, 3954–3988. [Google Scholar] [CrossRef] [PubMed]
- Guan, C.; Pan, Y.; Zhang, T.; Ajitha, M.J.; Huang, K.W. An Update on Formic Acid Dehydrogenation by Homogeneous Catalysis. Chem. Asian J. 2020, 15, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Enthaler, S.; Loges, B. The Rise of the Iron Age in Hydrogen Evolution? ChemCatChem 2012, 4, 323–325. [Google Scholar] [CrossRef]
- Grasemann, M.; Laurenczy, G. Formic acid as a hydrogen source—Recent developments and future trends. Energy Environ. Sci. 2012, 5, 8171. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Yamada, Y.; Suenobu, T.; Ohkubo, K.; Kotani, H. Catalytic mechanisms of hydrogen evolution with homogeneous and heterogeneous catalysts. Energy Environ. Sci. 2011, 4, 2754. [Google Scholar] [CrossRef]
- Loges, B.; Boddien, A.; Gärtner, F.; Junge, H.; Beller, M. Catalytic Generation of Hydrogen from Formic acid and its Derivatives: Useful Hydrogen Storage Materials. Top. Catal. 2010, 53, 902–914. [Google Scholar] [CrossRef]
- Fukuzumi, S. Bioinspired Energy Conversion Systems for Hydrogen Production and Storage. Eur. J. Inorg. Chem. 2008, 2008, 1351–1362. [Google Scholar] [CrossRef]
- Scotti, N.; Psaro, R.; Ravasio, N.; Zaccheria, F. A new Cu-based system for formic acid dehydrogenation. RSC Adv. 2014, 4, 61514–61517. [Google Scholar] [CrossRef]
- Enthaler, S.; Brück, A.; Kammer, A.; Junge, H.; Irran, E.; Gülak, S. Exploring the reactivity of nickel pincer complexes in the decomposition of formic acid to CO2/H2 and the hydrogenation of NaHCO3 to HCOONa. ChemCatChem 2015, 7, 65–69. [Google Scholar] [CrossRef]
- Jiang, K.; Xu, K.; Zou, S.; Cai, W.-B. B-Doped Pd Catalyst: Boosting Room-Temperature Hydrogen Production from Formic Acid–Formate Solutions. J. Am. Chem. Soc. 2014, 136, 4861–4864. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-H.; Ertem, M.Z.; Xu, S.; Onishi, N.; Manaka, Y.; Suna, Y.; Kambayashi, H.; Muckerman, J.T.; Fujita, E.; Himeda, Y. Highly Robust Hydrogen Generation by Bioinspired Ir Complexes for Dehydrogenation of Formic Acid in Water: Experimental and Theoretical Mechanistic Investigations at Different pH. ACS Catal. 2015, 5, 5496–5504. [Google Scholar] [CrossRef]
- Guerriero, A.; Bricout, H.; Sordakis, K.; Peruzzini, M.; Monflier, E.; Hapiot, F.; Laurenczy, G.; Gonsalvi, L. Hydrogen Production by Selective Dehydrogenation of HCOOH Catalyzed by Ru-Biaryl Sulfonated Phosphines in Aqueous Solution. ACS Catal. 2014, 4, 3002–3012. [Google Scholar] [CrossRef]
- Oldenhof, S.; De Bruin, B.; Lutz, M.; Siegler, M.A.; Patureau, F.W.; Van Der Vlugt, J.I.; Reek, J.N.H. Base-free production of H2 by dehydrogenation of formic acid using an iridium-bisMETAMORPhos complex. Chem. A Eur. J. 2013, 19, 11507–11511. [Google Scholar] [CrossRef] [PubMed]
- Tedsree, K.; Li, T.; Jones, S.; Chan, C.W.A.; Yu, K.M.K.; Bagot, P.A.J.; Marquis, E.A.; Smith, G.D.W.; Tsang, S.C.E. Hydrogen production from formic acid decomposition at room temperature using a Ag-Pd core-shell nanocatalyst. Nat. Nanotechnol. 2011, 6, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Boddien, A.; Loges, B.; Junge, H.; Gärtner, F.; Noyes, J.R.; Beller, M. Continuous hydrogen generation from formic acid: Highly active and stable ruthenium catalysts. Adv. Synth. Catal. 2009, 351, 2517–2520. [Google Scholar] [CrossRef]
- Fellay, C.; Dyson, P.J.; Laurenczy, G. A viable hydrogen-storage system based on selective formic acid decomposition with a ruthenium catalyst. Angew. Chem. Int. Ed. 2008, 47, 3966–3968. [Google Scholar] [CrossRef] [PubMed]
- Loges, B.; Boddien, A.; Junge, H.; Beller, M. Controlled generation of hydrogen from formic acid amine adducts at room temperature and application in H2/O2 fuel cells. Angew. Chem. Int. Ed. 2008, 47, 3962–3965. [Google Scholar] [CrossRef] [PubMed]
- Czaun, M.; Goeppert, A.; May, R.; Haiges, R.; Prakash, G.K.S.; Olah, G.A. Hydrogen Generation from Formic Acid Decomposition by Ruthenium Carbonyl Complexes. Tetraruthenium Dodecacarbonyl Tetrahydride as an Active Intermediate. ChemSusChem 2011, 4, 1241–1248. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Kobayashi, T.; Suenobu, T. Efficient Catalytic Decomposition of Formic Acid for the Selective Generation of H2 and H/D Exchange with a Water-Soluble Rhodium Complex in Aqueous Solution. ChemSusChem 2008, 1, 827–834. [Google Scholar] [CrossRef]
- Onishi, M. Decomposition of formic acid catalyzed by hydrido (phosphonite) cobalt (I) under photoirradiation. J. Mol. Catal. 1993, 80, 145–149. [Google Scholar] [CrossRef]
- Zhou, X.; Huang, Y.; Xing, W.; Liu, C.; Liao, J.; Lu, T. High-quality hydrogen from the catalyzed decomposition of formic acid by Pd–Au/C and Pd–Ag/C. Chem. Commun. 2008, 3540. [Google Scholar] [CrossRef] [PubMed]
- Sponholz, P.; Mellmann, D.; Junge, H.; Beller, M. Towards a Practical Setup for Hydrogen Production from Formic Acid. ChemSusChem 2013, 6, 1172–1176. [Google Scholar] [CrossRef]
- Loges, B.; Boddien, A.; Junge, H.; Noyes, J.R.; Baumann, W.; Beller, M. Hydrogen generation: Catalytic acceleration and control by light. Chem. Commun. 2009, 28, 4185. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Kobayashi, T.; Suenobu, T. Unusually Large Tunneling Effect on Highly Efficient Generation of Hydrogen and Hydrogen Isotopes in pH-Selective Decomposition of Formic Acid Catalyzed by a Heterodinuclear Iridium–Ruthenium Complex in Water. J. Am. Chem. Soc. 2010, 132, 1496–1497. [Google Scholar] [CrossRef] [PubMed]
- Himeda, Y. Highly efficient hydrogen evolution by decomposition of formic acid using an iridium catalyst with 4,4′-dihydroxy-2,2′-bipyridine. Green Chem. 2009, 11, 2018. [Google Scholar] [CrossRef]
- Tanaka, R.; Yamashita, M.; Chung, L.W.; Morokuma, K.; Nozaki, K. Mechanistic Studies on the Reversible Hydrogenation of Carbon Dioxide Catalyzed by an Ir-PNP Complex. Organometallics 2011, 30, 6742–6750. [Google Scholar] [CrossRef]
- Gu, X.; Lu, Z.-H.; Jiang, H.-L.; Akita, T.; Xu, Q. Synergistic Catalysis of Metal–Organic Framework-Immobilized Au–Pd Nanoparticles in Dehydrogenation of Formic Acid for Chemical Hydrogen Storage. J. Am. Chem. Soc. 2011, 133, 11822–11825. [Google Scholar] [CrossRef]
- Bi, Q.-Y.; Du, X.-L.; Liu, Y.-M.; Cao, Y.; He, H.-Y.; Fan, K.-N. Efficient Subnanometric Gold-Catalyzed Hydrogen Generation via Formic Acid Decomposition under Ambient Conditions. J. Am. Chem. Soc. 2012, 134, 8926–8933. [Google Scholar] [CrossRef]
- Barnard, J.H.; Wang, C.; Berry, N.G.; Xiao, J. Long-range metal–ligand bifunctional catalysis: Cyclometallated iridium catalysts for the mild and rapid dehydrogenation of formic acid. Chem. Sci. 2013, 4, 1234. [Google Scholar] [CrossRef]
- Boddien, A.; Jackstell, R.; Junge, H.; Spannenberg, A.; Baumann, W.; Ludwig, R.; Beller, M. Ortho-metalation of iron(0) tribenzylphosphine complexes: Homogeneous catalysts for the generation of hydrogen from formic acid. Angew. Chem. Int. Ed. 2010, 49, 8993–8996. [Google Scholar] [CrossRef] [PubMed]
- Boddien, A.; Loges, B.; Gärtner, F.; Torborg, C.; Fumino, K.; Junge, H.; Ludwig, R.; Beller, M. Iron-catalyzed hydrogen production from formic acid. J. Am. Chem. Soc. 2010, 132, 8924–8934. [Google Scholar] [CrossRef] [PubMed]
- Boddien, A.; Mellmann, D.; Gartner, F.; Jackstell, R.; Junge, H.; Dyson, P.J.; Laurenczy, G.; Ludwig, R.; Beller, M. Efficient Dehydrogenation of Formic Acid Using an Iron Catalyst. Science 2011, 333, 1733–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, R.; Iron, M.A.; Konstantinovski, L.; Diskin-Posner, Y.; Leitus, G.; Ben-David, Y.; Milstein, D. Iron borohydride pincer complexes for the efficient hydrogenation of ketones under mild, base-free conditions: Synthesis and mechanistic insight. Chem. A Eur. J. 2012, 18, 7196–7209. [Google Scholar] [CrossRef]
- Zhang, J.; Leitus, G.; Ben-David, Y.; Milstein, D. Efficient homogeneous catalytic hydrogenation of esters to alcohols. Angew. Chem. Int. Ed. 2006, 45, 1113–1115. [Google Scholar] [CrossRef]
- Langer, R.; Diskin-Posner, Y.; Leitus, G.; Shimon, L.J.W.; Ben-David, Y.; Milstein, D. Low-pressure hydrogenation of carbon dioxide catalyzed by an iron pincer complex exhibiting noble metal activity. Angew. Chem. Int. Ed. 2011, 50, 9948–9952. [Google Scholar] [CrossRef] [PubMed]
- Zell, T.; Butschke, B.; Ben-David, Y.; Milstein, D. Efficient hydrogen liberation from formic acid catalyzed by a well-defined iron pincer complex under mild conditions. Chem. A Eur. J. 2013, 19, 8068–8072. [Google Scholar] [CrossRef]
- Bielinski, E.A.; Lagaditis, P.O.; Zhang, Y.; Mercado, B.Q.; Würtele, C.; Bernskoetter, W.H.; Hazari, N.; Schneider, S. Lewis acid-assisted formic acid dehydrogenation using a pincer-supported iron catalyst. J. Am. Chem. Soc. 2014, 136, 10234–10237. [Google Scholar] [CrossRef] [PubMed]
- Kothandaraman, J.; Czaun, M.; Goeppert, A.; Haiges, R.; Jones, J.P.; May, R.B.; Prakash, G.K.S.; Olah, G.A. Amine-free reversible hydrogen storage in formate salts catalyzed by ruthenium pincer complex without pH control or solvent change. ChemSusChem 2015, 8, 1442–1451. [Google Scholar] [CrossRef]
- Mellone, I.; Gorgas, N.; Bertini, F.; Peruzzini, M.; Kirchner, K.; Gonsalvi, L. Selective Formic Acid Dehydrogenation Catalyzed by Fe-PNP Pincer Complexes Based on the 2,6-Diaminopyridine Scaffold. Organometallics 2016, 35, 3344–3349. [Google Scholar] [CrossRef]
- Mellone, I.; Bertini, F.; Peruzzini, M.; Gonsalvi, L. An active, stable and recyclable Ru(II) tetraphosphine-based catalytic system for hydrogen production by selective formic acid dehydrogenation. Catal. Sci. Technol. 2016, 6, 6504–6512. [Google Scholar] [CrossRef]
- Czaun, M.; Kothandaraman, J.; Goeppert, A.; Yang, B.; Greenberg, S.; May, R.B.; Olah, G.A.; Prakash, G.K.S. Iridium-Catalyzed Continuous Hydrogen Generation from Formic Acid and Its Subsequent Utilization in a Fuel Cell: Toward a Carbon Neutral Chemical Energy Storage. ACS Catal. 2016, 6, 7475–7484. [Google Scholar] [CrossRef]
- Cohen, S.; Borin, V.; Schapiro, I.; Musa, S.; De-Botton, S.; Belkova, N.V.; Gelman, D. Ir(III)-PC(sp3)P Bifunctional Catalysts for Production of H2 by Dehydrogenation of Formic Acid: Experimental and Theoretical Study. ACS Catal. 2017, 7, 8139–8146. [Google Scholar] [CrossRef]
- Musa, S.; Fronton, S.; Vaccaro, L.; Gelman, D. Bifunctional ruthenium(II) PCP pincer complexes and their catalytic activity in acceptorless dehydrogenative reactions. Organometallics 2013, 32, 3069–3073. [Google Scholar] [CrossRef]
- Musa, S.; Ghosh, A.; Vaccaro, L.; Ackermann, L.; Gelman, D. Efficient E-Selective Transfer Semihydrogenation of Alkynes by Means of Ligand-Metal Cooperating Ruthenium Catalyst. Adv. Synth. Catal. 2015, 357, 2351–2357. [Google Scholar] [CrossRef]
- Esteruelas, M.A.; García-Yebra, C.; Martín, J.; Oñate, E. Dehydrogenation of Formic Acid Promoted by a Trihydride-Hydroxo-Osmium(IV) Complex: Kinetics and Mechanism. ACS Catal. 2018, 8, 11314–11323. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Pan, C.L.; Zhang, Y.; Li, H.; Min, S.; Guo, X.; Zheng, B.; Chen, H.; Anders, A.; Lai, Z.; et al. Selective hydrogen generation from formic acid with well-defined complexes of ruthenium and phosphorus-nitrogen PN3-pincer ligand. Chem. Asian J. 2016, 11, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- He, L.-P.; Chen, T.; Gong, D.; Lai, Z.; Huang, K.-W. Enhanced Reactivities toward Amines by Introducing an Imine Arm to the Pincer Ligand: Direct Coupling of Two Amines to Form an Imine Without Oxidant. Organometallics 2012, 31, 5208–5211. [Google Scholar] [CrossRef]
- Chen, T.; Li, H.; Qu, S.; Zheng, B.; He, L.; Lai, Z.; Wang, Z.X.; Huang, K.W. Hydrogenation of esters catalyzed by ruthenium PN3-Pincer complexes containing an aminophosphine arm. Organometallics 2014, 33, 4152–4155. [Google Scholar] [CrossRef]
- Min, S.; Rasul, S.; Li, H.; Grills, D.C.; Takanabe, K.; Li, L.J.; Huang, K.W. Electrocatalytic Reduction of Carbon Dioxide with a Well-Defined PN3-Ru Pincer Complex. Chempluschem 2016, 81, 166–171. [Google Scholar] [CrossRef]
- Li, H.; Gonçalves, T.P.; Hu, J.; Zhao, Q.; Gong, D.; Lai, Z.; Wang, Z.; Zheng, J.; Huang, K.W. A Pseudodearomatized PN3P*Ni-H Complex as a Ligand and σ-Nucleophilic Catalyst. J. Org. Chem. 2018, 83, 14969–14977. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ling, E.A.P.; Guan, C.; Zhang, Q.; Wu, W.; Liu, P.; Zheng, N.; Zhang, D.; Lopatin, S.; Lai, Z.; et al. Single-Site Ruthenium Pincer Complex Knitted into Porous Organic Polymers for Dehydrogenation of Formic Acid. ChemSusChem 2018, 11, 3591–3598. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, Y.; Toda, T.; Matsunami, A.; Kayaki, Y.; Kuwata, S. Protic NNN and NCN Pincer-Type Ruthenium Complexes Featuring (Trifluoromethyl) pyrazole Arms: Synthesis and Application to Catalytic Hydrogen Evolution from Formic Acid. Chem. Asian J. 2018, 13, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Curley, J.B.; Smith, N.E.; Bernskoetter, W.H.; Hazari, N.; Mercado, B.Q. Catalytic Formic Acid Dehydrogenation and CO2 Hydrogenation Using Iron PNRP Pincer Complexes with Isonitrile Ligands. Organometallics 2018, 37, 3846–3853. [Google Scholar] [CrossRef]
- Smith, N.E.; Bernskoetter, W.H.; Hazari, N.; Mercado, B.Q. Synthesis and Catalytic Activity of PNP-Supported Iron Complexes with Ancillary Isonitrile Ligands. Organometallics 2017, 36, 3995–4004. [Google Scholar] [CrossRef]
- Prichatz, C.; Trincado, M.; Tan, L.; Casas, F.; Kammer, A.; Junge, H.; Beller, M.; Grützmacher, H. Highly Efficient Base-Free Dehydrogenation of Formic Acid at Low Temperature. ChemSusChem 2018, 11, 3092–3095. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wei, Z.; Spannenberg, A.; Jiao, H.; Junge, K.; Junge, H.; Beller, M. Cobalt-Catalyzed Aqueous Dehydrogenation of Formic Acid. Chem. A Eur. J. 2019, 25, 8459–8464. [Google Scholar] [CrossRef]
- Müller, K.; Stark, K.; Müller, B.; Arlt, W. Amine borane based hydrogen carriers: An evaluation. Energy Fuels 2012, 26, 3691–3696. [Google Scholar] [CrossRef]
- Staubitz, A.; Robertson, A.P.M.; Manners, I. Ammonia-Borane and Related Compounds as Dihydrogen Sources. Chem. Rev. 2010, 110, 4079–4124. [Google Scholar] [CrossRef]
- Staubitz, A.; Robertson, A.P.M.; Sloan, M.E.; Manners, I. Amine– and Phosphine–Borane Adducts: New Interest in Old Molecules. Chem. Rev. 2010, 110, 4023–4078. [Google Scholar] [CrossRef]
- Pagano, J.K.; Stelmach, J.P.W.; Waterman, R. Cobalt-catalyzed ammonia borane dehydrocoupling and transfer hydrogenation under aerobic conditions. Dalt. Trans. 2015, 44, 12074–12077. [Google Scholar] [CrossRef] [PubMed]
- Moury, R.; Demirci, U. Hydrazine Borane and Hydrazinidoboranes as Chemical Hydrogen Storage Materials. Energies 2015, 8, 3118–3141. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, J.; Hamilton, E.J.M.; Chen, X. The continuing story of the diammoniate of diborane. J. Organomet. Chem. 2015, 798, 24–29. [Google Scholar] [CrossRef]
- Jepsen, L.H.; Ley, M.B.; Lee, Y.-S.; Cho, Y.W.; Dornheim, M.; Jensen, J.O.; Filinchuk, Y.; Jørgensen, J.E.; Besenbacher, F.; Jensen, T.R. Boron–nitrogen based hydrides and reactive composites for hydrogen storage. Mater. Today 2014, 17, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhao, J.-C.; Shore, S.G. The Roles of Dihydrogen Bonds in Amine Borane Chemistry. Acc. Chem. Res. 2013, 46, 2666–2675. [Google Scholar] [CrossRef]
- Stubbs, N.E.; Robertson, A.P.M.; Leitao, E.M.; Manners, I. Amine–borane dehydrogenation chemistry: Metal-free hydrogen transfer, new catalysts and mechanisms, and the synthesis of polyaminoboranes. J. Organomet. Chem. 2013, 730, 84–89. [Google Scholar] [CrossRef]
- Peng, B.; Chen, J. Ammonia borane as an efficient and lightweight hydrogen storage medium. Energy Environ. Sci. 2008. [Google Scholar] [CrossRef]
- Davis, B.L.; Dixon, D.A.; Garner, E.B.; Gordon, J.C.; Matus, M.H.; Scott, B.; Stephens, F.H. Efficient Regeneration of Partially Spent Ammonia Borane Fuel. Angew. Chem. Int. Ed. 2009, 48, 6812–6816. [Google Scholar] [CrossRef]
- Sutton, A.D.; Burrell, A.K.; Dixon, D.A.; Garner, E.B.; Gordon, J.C.; Nakagawa, T.; Ott, K.C.; Robinson, J.P.; Vasiliu, M. Regeneration of Ammonia Borane Spent Fuel by Direct Reaction with Hydrazine and Liquid Ammonia. Science 2011, 331, 1426–1429. [Google Scholar] [CrossRef] [Green Version]
- Reller, C.; Mertens, F.O.R.L. A Self-Contained Regeneration Scheme for Spent Ammonia Borane Based on the Catalytic Hydrodechlorination of BCl 3. Angew. Chem. Int. Ed. 2012, 51, 11731–11735. [Google Scholar] [CrossRef]
- Davis, B.L.; Rekken, B.D.; Michalczyk, R.; Garner, E.B., III; Dixon, D.A.; Kalviri, H.; Baker, R.T.; Thorn, D.L. Lewis base assisted B–H bond redistribution in borazine and polyborazylene. Chem. Commun. 2013, 49, 9095. [Google Scholar] [CrossRef] [PubMed]
- Smythe, N.C.; Gordon, J.C. Ammonia Borane as a Hydrogen Carrier: Dehydrogenation and Regeneration. Eur. J. Inorg. Chem. 2010, 2010, 509–521. [Google Scholar] [CrossRef]
- Keaton, R.J.; Blacquiere, J.M.; Baker, R.T. Base Metal Catalyzed Dehydrogenation of Ammonia–Borane for Chemical Hydrogen Storage. J. Am. Chem. Soc. 2007, 129, 1844–1845. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.R.; Robertson, A.P.M.; Lee, K.; Manners, I. Photoactivated, Iron-Catalyzed Dehydrocoupling of Amine-Borane Adducts: Formation of Boron-Nitrogen Oligomers and Polymers. Chem. A Eur. J. 2011, 17, 4099–4103. [Google Scholar] [CrossRef]
- Baker, R.T.; Gordon, J.C.; Hamilton, C.W.; Henson, N.J.; Lin, P.-H.; Maguire, S.; Murugesu, M.; Scott, B.L.; Smythe, N.C. Iron Complex-Catalyzed Ammonia–Borane Dehydrogenation. A Potential Route toward B–N-Containing Polymer Motifs Using Earth-Abundant Metal Catalysts. J. Am. Chem. Soc. 2012, 134, 5598–5609. [Google Scholar] [CrossRef]
- Duman, S.; Metin, Ö.; Özkar, S. B–N Polymer Embedded Iron(0) Nanoparticles as Highly Active and Long Lived Catalyst in the Dehydrogenation of Ammonia Borane. J. Nanosci. Nanotechnol. 2013, 13, 4954–4961. [Google Scholar] [CrossRef]
- Lichtenberg, C.; Adelhardt, M.; Gianetti, T.L.; Meyer, K.; de Bruin, B.; Grützmacher, H. Low-Valent Iron Mono-Diazadiene Compounds: Electronic Structure and Catalytic Application. ACS Catal. 2015, 5, 6230–6240. [Google Scholar] [CrossRef]
- Kawano, Y.; Uruichi, M.; Shimoi, M.; Taki, S.; Kawaguchi, T.; Kakizawa, T.; Ogino, H. Dehydrocoupling reactions of borane-secondary and -primary amine adducts catalyzed by group-6 carbonyl complexes: Formation of aminoboranes and borazines. J. Am. Chem. Soc. 2009, 131, 14946–14957. [Google Scholar] [CrossRef]
- Sloan, M.E.; Staubitz, A.; Clark, T.J.; Russell, C.A.; Lloyd-Jones, G.C.; Manners, I. Homogeneous Catalytic Dehydrocoupling/Dehydrogenation of Amine–Borane Adducts by Early Transition Metal, Group 4 Metallocene Complexes. J. Am. Chem. Soc. 2010, 132, 3831–3841. [Google Scholar] [CrossRef]
- Sonnenberg, J.F.; Morris, R.H. Evidence for Iron Nanoparticles Catalyzing the Rapid Dehydrogenation of Ammonia-Borane. ACS Catal. 2013, 3, 1092–1102. [Google Scholar] [CrossRef]
- Chaplin, A.B.; Weller, A.S. B-H activation at a rhodium(I) center: Isolation of a bimetallic complex relevant to the transition-metal-catalyzed dehydrocoupling of amine-boranes. Angew. Chem. Int. Ed. 2010, 49, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Blaquiere, N.; Diallo-Garcia, S.; Gorelsky, S.I.; Black, D.A.; Fagnou, K. Ruthenium-catalyzed dehydrogenation of ammonia boranes. J. Am. Chem. Soc. 2008, 130, 14034–14035. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, P.; Krause, J.A.; Guan, H. Mechanistic Studies of Ammonia Borane Dehydrogenation Catalyzed by Iron Pincer Complexes. J. Am. Chem. Soc. 2014, 136, 11153–11161. [Google Scholar] [CrossRef] [PubMed]
- Buss, J.A.; Edouard, G.A.; Cheng, C.; Shi, J.; Agapie, T. Molybdenum Catalyzed Ammonia Borane Dehydrogenation: Oxidation State Specific Mechanisms. J. Am. Chem. Soc. 2014, 136, 11272–11275. [Google Scholar] [CrossRef] [Green Version]
- Erickson, K.A.; Stelmach, J.P.W.; Mucha, N.T.; Waterman, R. Zirconium-Catalyzed Amine Borane Dehydrocoupling and Transfer Hydrogenation. Organometallics 2015, 34, 4693–4699. [Google Scholar] [CrossRef]
- Lin, T.-P.; Peters, J.C. Boryl-Mediated Reversible H 2 Activation at Cobalt: Catalytic Hydrogenation, Dehydrogenation, and Transfer Hydrogenation. J. Am. Chem. Soc. 2013, 135, 15310–15313. [Google Scholar] [CrossRef] [Green Version]
- Lichtenberg, C.; Viciu, L.; Adelhardt, M.; Sutter, J.; Meyer, K.; de Bruin, B.; Grützmacher, H. Low-Valent Iron(I) Amido Olefin Complexes as Promotors for Dehydrogenation Reactions. Angew. Chem. Int. Ed. 2015, 54, 5766–5771. [Google Scholar] [CrossRef]
- Conley, B.L.; Guess, D.; Williams, T.J. A Robust, Air-Stable, Reusable Ruthenium Catalyst for Dehydrogenation of Ammonia Borane. J. Am. Chem. Soc. 2011, 133, 14212–14215. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Kam, L.; Trerise, R.; Williams, T.J. Ruthenium-Catalyzed Ammonia Borane Dehydrogenation: Mechanism and Utility. Acc. Chem. Res. 2017, 50, 86–95. [Google Scholar] [CrossRef]
- Rossin, A.; Peruzzini, M. Ammonia-Borane and Amine-Borane Dehydrogenation Mediated by Complex Metal Hydrides. Chem. Rev. 2016, 116, 8848–8872. [Google Scholar] [CrossRef]
- Marziale, A.N.; Friedrich, A.; Klopsch, I.; Drees, M.; Celinski, V.R.; Schmedt Auf Der Günne, J.; Schneider, S. The mechanism of borane-amine dehydrocoupling with bifunctional ruthenium catalysts. J. Am. Chem. Soc. 2013, 135, 13342–13355. [Google Scholar] [CrossRef] [PubMed]
- Glüer, A.; Förster, M.; Celinski, V.R.; Schmedt Auf Der Günne, J.; Holthausen, M.C.; Schneider, S. Highly Active Iron Catalyst for Ammonia Borane Dehydrocoupling at Room Temperature. ACS Catal. 2015, 5, 7214–7217. [Google Scholar] [CrossRef]
- Johnson, H.C.; Hooper, T.N.; Weller, A.S. The Catalytic Dehydrocoupling of Amine–Boranes and Phosphine–Boranes. In Topics in Organometallic Chemistry; Springer: Basel, Switzerland, 2015; pp. 153–220. [Google Scholar]
- Käß, M.; Friedrich, A.; Drees, M.; Schneider, S. Ruthenium complexes with cooperative PNP ligands: Bifunctional catalysts for the dehydrogenation of ammonia-borane. Angew. Chem. Int. Ed. 2009, 48, 905–907. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, A.; Drees, M.; Schneider, S. Ruthenium-catalyzed dimethylamineborane dehydrogenation: Stepwise metal-centered dehydrocyclization. Chem. A Eur. J. 2009, 15, 10339–10342. [Google Scholar] [CrossRef]
- Han, D.; Joksch, M.; Klahn, M.; Spannenberg, A.; Drexler, H.J.; Baumann, W.; Jiao, H.; Knitsch, R.; Hansen, M.R.; Eckert, H.; et al. Iridium(III) hydrido complexes for the catalytic dehydrogenation of hydrazine borane. Dalt. Trans. 2016, 45, 17697–17704. [Google Scholar] [CrossRef]
- Brayton, D.F.; Jensen, C.M. Dehydrogenation of pyrrolidine based liquid organic hydrogen carriers by an iridium pincer catalyst, an isothermal kinetic study. Int. J. Hydrog. Energy 2015, 40, 16266–16270. [Google Scholar] [CrossRef]
- Titova, E.M.; Osipova, E.S.; Pavlov, A.A.; Filippov, O.A.; Safronov, S.V.; Shubina, E.S.; Belkova, N.V. Mechanism of Dimethylamine-Borane Dehydrogenation Catalyzed by an Iridium(III) PCP-Pincer Complex. ACS Catal. 2017, 7, 2325–2333. [Google Scholar] [CrossRef]
- Denney, M.C.; Pons, V.; Hebden, T.J.; Heinekey, D.M.; Goldberg, K.I. Efficient catalysis of ammonia borane dehydrogenation. J. Am. Chem. Soc. 2006, 128, 12048–12049. [Google Scholar] [CrossRef] [PubMed]
- Esteruelas, M.A.; Nolis, P.; Oliván, M.; Oñate, E.; Vallribera, A.; Vélez, A. Ammonia Borane Dehydrogenation Promoted by a Pincer-Square-Planar Rhodium(I) Monohydride: A Stepwise Hydrogen Transfer from the Substrate to the Catalyst. Inorg. Chem. 2016, 55, 7176–7181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, E.H.; Ogawa, H.; Yamashita, M. A Highly Active PBP–Iridium Catalyst for the Dehydrogenation of Dimethylamine–Borane: Catalytic Performance and Mechanism. ChemCatChem 2017, 9, 2457–2462. [Google Scholar] [CrossRef]
- Kwan, E.H.; Kawai, Y.J.; Kamakura, S.; Yamashita, M. A long-tethered (P-B-P)-pincer ligand: Synthesis, complexation, and application to catalytic dehydrogenation of alkanes. Dalt. Trans. 2016, 45, 15931–15941. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Ben-David, Y.; Milstein, D. Rechargeable hydrogen storage system based on the dehydrogenative coupling of ethylenediamine with ethanol. Angew. Chem. Int. Ed. 2016, 55, 1061–1064. [Google Scholar] [CrossRef] [PubMed]
- Kothandaraman, J.; Kar, S.; Sen, R.; Goeppert, A.; Olah, G.A.; Prakash, G.K.S. Efficient Reversible Hydrogen Carrier System Based on Amine Reforming of Methanol. J. Am. Chem. Soc. 2017, 139, 2549–2552. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Hong, S.H. Ruthenium-Catalyzed Urea Synthesis Using Methanol as the C1 Source. Org. Lett. 2016, 18, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Baratta, W.; Ballico, M.; Chelucci, G.; Siega, K.; Rigo, P. Osmium(II) CNN pincer complexes as efficient catalysts for both asymmetric transfer and H2 hydrogenation of ketones. Angew. Chem. Int. Ed. 2008, 47, 4362–4365. [Google Scholar] [CrossRef] [PubMed]
- Abdur-Rashid, K.; Clapham, S.E.; Hadzovic, A.; Harvey, J.N.; Lough, A.J.; Morris, R.H. Mechanism of the hydrogenation of ketones catalyzed by trans-dihydrido(diamine)ruthenium(II) complexes. J. Am. Chem. Soc. 2002, 124, 15104–15118. [Google Scholar] [CrossRef]
- Abdur-Rashid, K.; Faatz, M.; Lough, A.J.; Morris, R.H. Catalytic cycle for the asymmetric hydrogenation of prochiral ketones to chiral alcohols: Direct hydride and proton transfer from chiral catalysts trans-Ru(H)2(diphosphine)(diamine) to ketones and direct addition of dihydrogen to the resulting hydridoamid. J. Am. Chem. Soc. 2001, 123, 7473–7474. [Google Scholar] [CrossRef]
- Rautenstrauch, V.; Hoang-Cong, X.; Churlaud, R.; Abdur-Rashid, K.; Morris, R.H. Hydrogenation versus Transfer Hydrogenation of Ketones: Two Established Ruthenium Systems Catalyze Both. Chem. A Eur. J. 2003, 9, 4954–4967. [Google Scholar] [CrossRef]
- Raja, M.U.; Ramesh, R.; Ahn, K.H. Rhodium(III) NCN pincer complexes catalyzed transfer hydrogenation of ketones. Tetrahedron Lett. 2009, 50, 7014–7017. [Google Scholar] [CrossRef]
- Hou, C.; Li, Y.; Zhao, C.; Ke, Z. A DFT study of Co(i) and Ni(ii) pincer complex-catalyzed hydrogenation of ketones: Intriguing mechanism dichotomy by ligand field variation. Catal. Sci. Technol. 2019, 9, 125–135. [Google Scholar] [CrossRef]
- Filonenko, G.A.; Cosimi, E.; Lefort, L.; Conley, M.P.; Copéret, C.; Lutz, M.; Hensen, E.J.M.; Pidko, E.A. Lutidine-derived Ru-CNC hydrogenation pincer catalysts with versatile coordination properties. ACS Catal. 2014, 4, 2667–2671. [Google Scholar] [CrossRef]
- Saudan, L.A.; Saudan, C.M.; Debieux, C.; Wyss, P. Dihydrogen reduction of carboxylic esters to alcohols under the catalysis of homogeneous ruthenium complexes: High efficiency and unprecedented chemoselectivity. Angew. Chem. Int. Ed. 2007, 46, 7473–7476. [Google Scholar] [CrossRef] [PubMed]
- Yuwen, J.; Chakraborty, S.; Brennessel, W.W.; Jones, W.D. Additive-Free Cobalt-Catalyzed Hydrogenation of Esters to Alcohols. ACS Catal. 2017, 7, 3735–3740. [Google Scholar] [CrossRef]
- Kim, D.; Le, L.; Drance, M.J.; Jensen, K.H.; Bogdanovski, K.; Cervarich, T.N.; Barnard, M.G.; Pudalov, N.J.; Knapp, S.M.M.; Chianese, A.R. Ester Hydrogenation Catalyzed by CNN-Pincer Complexes of Ruthenium. Organometallics 2016, 35, 982–989. [Google Scholar] [CrossRef]
- Le, L.; Liu, J.; He, T.; Malek, J.C.; Cervarich, T.N.; Buttner, J.C.; Pham, J.; Keith, J.M.; Chianese, A.R. Unexpected CNN-to-CC Ligand Rearrangement in Pincer–Ruthenium Precatalysts Leads to a Base-Free Catalyst for Ester Hydrogenation. Organometallics 2019, 38, 3311–3321. [Google Scholar] [CrossRef]
- Filonenko, G.A.; Aguila, M.J.B.; Schulpen, E.N.; Van Putten, R.; Wiecko, J.; Müller, C.; Lefort, L.; Hensen, E.J.M.; Pidko, E.A. Bis-N-heterocyclic Carbene Aminopincer Ligands Enable High Activity in Ru-Catalyzed Ester Hydrogenation. J. Am. Chem. Soc. 2015, 137, 7620–7623. [Google Scholar] [CrossRef]
- He, T.; Buttner, J.C.; Reynolds, E.F.; Pham, J.; Malek, J.C.; Keith, J.M.; Chianese, A.R. Dehydroalkylative Activation of CNN- A nd PNN-Pincer Ruthenium Catalysts for Ester Hydrogenation. J. Am. Chem. Soc. 2019, 141, 17404–17413. [Google Scholar] [CrossRef]
- Acosta-Ramirez, A.; Bertoli, M.; Gusev, D.G.; Schlaf, M. Homogeneous catalytic hydrogenation of long-chain esters by an osmium pincer complex and its potential application in the direct conversion of triglycerides into fatty alcohols. Green Chem. 2012, 14, 1178. [Google Scholar] [CrossRef]
- Chakraborty, S.; Dai, H.; Bhattacharya, P.; Fairweather, N.T.; Gibson, M.S.; Krause, J.A.; Guan, H. Iron-Based Catalysts for the Hydrogenation of Esters to Alcohols. J. Am. Chem. Soc. 2014, 136, 7869–7872. [Google Scholar] [CrossRef]
- Werkmeister, S.; Junge, K.; Wendt, B.; Alberico, E.; Jiao, H.; Baumann, W.; Junge, H.; Gallou, F.; Beller, M. Hydrogenation of esters to alcohols with a well-defined iron complex. Angew. Chem. Int. Ed. 2014, 53, 8722–8726. [Google Scholar] [CrossRef]
- Sun, Y.; Koehler, C.; Tan, R.; Annibale, V.T.; Song, D. Ester hydrogenation catalyzed by Ru-CNN pincer complexes. Chem. Commun. 2011, 47, 8349–8351. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, X.; Liu, B.; Liu, Q.B.; Solan, G.A.; Yang, X.; Sun, W.H. Cooperative interplay between a flexible PNN-Ru(II) complex and a NaBH4 additive in the efficient catalytic hydrogenation of esters. Catal. Sci. Technol. 2017, 7, 1297–1304. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Yang, X. Mechanistic insights into the iridium catalysed hydrogenation of ethyl acetate to ethanol: A DFT study. Dalt. Trans. 2018. [Google Scholar] [CrossRef] [PubMed]
- Spasyuk, D.; Vicent, C.; Gusev, D.G. Chemoselective hydrogenation of carbonyl compounds and acceptorless dehydrogenative coupling of alcohols. J. Am. Chem. Soc. 2015, 137, 3743–3746. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Dai, H.; Dang, Y.; Song, C.; Wang, Z.X.; Guan, H. Computational mechanistic study of Fe-catalyzed hydrogenation of esters to alcohols: Improving catalysis by accelerating precatalyst activation with a lewis base. ACS Catal. 2014, 4, 4377–4388. [Google Scholar] [CrossRef]
- Wei, Z.; Jiao, H. Bifunctional Aliphatic PNP Pincer Catalysts for Hydrogenation: Mechanisms and Scope, 1st ed.; Elsevier: Amsterdam, The Netherland, 2019; ISBN 9780128157282. [Google Scholar]
- Hey, D.A.; Reich, R.M.; Baratta, W.; Kühn, F.E. Current advances on ruthenium(II) N-heterocyclic carbenes in hydrogenation reactions. Coord. Chem. Rev. 2018, 374, 114–132. [Google Scholar] [CrossRef] [Green Version]
- Elangovan, S.; Topf, C.; Fischer, S.; Jiao, H.; Spannenberg, A.; Baumann, W.; Ludwig, R.; Junge, K.; Beller, M. Selective Catalytic Hydrogenations of Nitriles, Ketones, and Aldehydes by Well-Defined Manganese Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 8809–8814. [Google Scholar] [CrossRef]
- Schneck, F.; Assmann, M.; Balmer, M.; Harms, K.; Langer, R. Selective Hydrogenation of Amides to Amines and Alcohols Catalyzed by Improved Iron Pincer Complexes. Organometallics 2016, 35, 1931–1943. [Google Scholar] [CrossRef]
- Artús Suàrez, L.; Culakova, Z.; Balcells, D.; Bernskoetter, W.H.; Eisenstein, O.; Goldberg, K.I.; Hazari, N.; Tilset, M.; Nova, A. The Key Role of the Hemiaminal Intermediate in the Iron-Catalyzed Deaminative Hydrogenation of Amides. ACS Catal. 2018, 8, 8751–8762. [Google Scholar] [CrossRef]
- Jayarathne, U.; Zhang, Y.; Hazari, N.; Bernskoetter, W.H. Selective Iron-Catalyzed Deaminative Hydrogenation of Amides. Organometallics 2017, 36, 409–416. [Google Scholar] [CrossRef]
- Coetzee, J.; Dodds, D.L.; Klankermayer, J.; Brosinski, S.; Leitner, W.; Slawin, A.M.Z.; Cole-Hamilton, D.J. Homogeneous Catalytic Hydrogenation of Amides to Amines. Chem. A Eur. J. 2013, 19, 11039–11050. [Google Scholar] [CrossRef] [PubMed]
- Núñez Magro, A.A.; Eastham, G.R.; Cole-Hamilton, D.J. The synthesis of amines by the homogeneous hydrogenation of secondary and primary amides. Chem. Commun. 2007, 30, 3154–3156. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Juárez, M.; Vaquero, M.; Álvarez, E.; Salazar, V.; Suárez, A. Hydrogenation of imines catalysed by ruthenium(ii) complexes based on lutidine-derived CNC pincer ligands. Dalt. Trans. 2013, 42, 351–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Juárez, M.; López-Serrano, J.; Lara, P.; Morales-Cerón, J.P.; Vaquero, M.; Álvarez, E.; Salazar, V.; Suárez, A. Ruthenium(II) complexes containing lutidine-derived pincer CNC ligands: Synthesis, structure, and catalytic hydrogenation of C=N bonds. Chem. A Eur. J. 2015, 21, 7540–7555. [Google Scholar] [CrossRef]
- Perera, F. Pollution from Fossil-Fuel Combustion is the Leading Environmental Threat to Global Pediatric Health and Equity: Solutions Exist. Int. J. Environ. Res. Public Health 2017, 15, 16. [Google Scholar] [CrossRef] [Green Version]
- Butler, C. Climate Change, Health and Existential Risks to Civilization: A Comprehensive Review (1989–2013). Int. J. Environ. Res. Public Health 2018, 15, 2266. [Google Scholar] [CrossRef] [Green Version]
- Diffenbaugh, N.S.; Singh, D.; Mankin, J.S.; Horton, D.E.; Swain, D.L.; Touma, D.; Charland, A.; Liu, Y.; Haugen, M.; Tsiang, M.; et al. Quantifying the influence of global warming on unprecedented extreme climate events. Proc. Natl. Acad. Sci. USA 2017, 114, 4881–4886. [Google Scholar] [CrossRef] [Green Version]
- Haustein, K.; Allen, M.R.; Forster, P.M.; Otto, F.E.L.; Mitchell, D.M.; Matthews, H.D.; Frame, D.J. A real-time Global Warming Index. Sci. Rep. 2017, 7, 15417. [Google Scholar] [CrossRef] [Green Version]
- Aresta, M. Carbon Dioxide as Chemical Feedstock; John Wiley & Sons: Hoboken, NJ, USA, 2010; ISBN 9783527324750. [Google Scholar]
- Centi, G.; Perathoner, S. Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal. Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- Gibson, D.H. The organometallic chemistry of carbon dioxide. Chem. Rev. 1996, 96, 2063–2095. [Google Scholar] [CrossRef]
- Boot-Handford, M.E.; Abanades, J.C.; Anthony, E.J.; Blunt, M.J.; Brandani, S.; Mac Dowell, N.; Fernández, J.R.; Ferrari, M.-C.; Gross, R.; Hallett, J.P.; et al. Carbon capture and storage update. Energy Environ. Sci. 2014, 7, 130–189. [Google Scholar] [CrossRef]
- Goeppert, A.; Czaun, M.; May, R.B.; Prakash, G.K.S.; Olah, G.A.; Narayanan, S.R. Carbon Dioxide Capture from the Air Using a Polyamine Based Regenerable Solid Adsorbent. J. Am. Chem. Soc. 2011, 133, 20164–20167. [Google Scholar] [CrossRef]
- Gibbins, J.; Chalmers, H. Carbon capture and storage. Energy Policy 2008, 36, 4317–4322. [Google Scholar] [CrossRef] [Green Version]
- Leung, D.Y.C.; Caramanna, G.; Maroto-Valer, M.M. An overview of current status of carbon dioxide capture and storage technologies. Renew. Sustain. Energy Rev. 2014, 39, 426–443. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, S.; Keeling, S.; Malischek, R.; Stanley, T. 20 Years of Carbon Capture and Storage; OECD: Paris, France, 2016; ISBN 9789264267800. [Google Scholar]
- Styring, P.; Jansen, D.; de Coninck, H.; Reith, H.; Armstrong, K. Carbon Capture and Utilisation in the Green Economy; Centre for Low Carbon Futures: New York, NY, USA, 2011. [Google Scholar]
- de Coninck, H.; Benson, S.M. Carbon Dioxide Capture and Storage: Issues and Prospects. Annu. Rev. Environ. Resour. 2014, 39, 243–270. [Google Scholar] [CrossRef]
- Goeppert, A.; Czaun, M.; Surya Prakash, G.K.; Olah, G.A. Air as the renewable carbon source of the future: An overview of CO2 capture from the atmosphere. Energy Environ. Sci. 2012, 5, 7833. [Google Scholar] [CrossRef]
- Cuéllar-Franca, R.M.; Azapagic, A. Carbon capture, storage and utilisation technologies: A critical analysis and comparison of their life cycle environmental impacts. J. CO2 Util. 2015, 9, 82–102. [Google Scholar] [CrossRef]
- Takht Ravanchi, M.; Sahebdelfar, S. Carbon dioxide capture and utilization in petrochemical industry: Potentials and challenges. Appl. Petrochem. Res. 2014, 4, 63–77. [Google Scholar] [CrossRef] [Green Version]
- von der Assen, N.; Jung, J.; Bardow, A. Life-cycle assessment of carbon dioxide capture and utilization: Avoiding the pitfalls. Energy Environ. Sci. 2013, 6, 2721. [Google Scholar] [CrossRef]
- Mac Dowell, N.; Fennell, P.S.; Shah, N.; Maitland, G.C. The role of CO2 capture and utilization in mitigating climate change. Nat. Clim. Chang. 2017, 7, 243–249. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, H.; Aresta, M.; Armor, J.N.; Barteau, M.A.; Beckman, E.J.; Bell, A.T.; Bercaw, J.E.; Creutz, C.; Dinjus, E.; Dixon, D.A.; et al. Catalysis Research of Relevance to Carbon Management: Progress, Challenges, and Opportunities. Chem. Rev. 2001, 101, 953–996. [Google Scholar] [CrossRef] [Green Version]
- Behr, A. Carbon Dioxide as an Alternative C1 Synthetic Unit: Activation by Transition-Metal Complexes. Angew. Chem. Int. Ed. Engl. 1988, 27, 661–678. [Google Scholar] [CrossRef]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Moss, J.R. Recent developments in the activation of carbon dioxide by metal complexes. Coord. Chem. Rev. 1999, 181, 27–59. [Google Scholar] [CrossRef]
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of Carbon Dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef]
- Peters, M.; Köhler, B.; Kuckshinrichs, W.; Leitner, W.; Markewitz, P.; Müller, T.E. Chemical Technologies for Exploiting and Recycling Carbon Dioxide into the Value Chain. ChemSusChem 2011, 4, 1216–1240. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A. Utilisation of CO2 as a chemical feedstock: Opportunities and challenges. Dalt. Trans. 2007, 2975. [Google Scholar] [CrossRef]
- Asinger, F. Methanol—Chemie und Energierohstoff. In Methanol—Chem.-und Energierohstoff; Springer: Berlin/Heidelberg, Germany, 1986. [Google Scholar]
- Leitner, W. Carbon Dioxide as a Raw Material: The Synthesis of Formic Acid and Its Derivatives from CO2. Angew. Chem. Int. Ed. Engl. 1995, 34, 2207–2221. [Google Scholar] [CrossRef]
- Leitner, W. The coordination chemistry of carbon dioxide and its relevance for catalysis: A critical survey. Coord. Chem. Rev. 1996, 153, 257–284. [Google Scholar] [CrossRef]
- Jessop, P.G.; Ikariya, T.; Noyori, R. Homogeneous Hydrogenation of Carbon Dioxide. Chem. Rev. 1995, 95, 259–272. [Google Scholar] [CrossRef]
- Olah, G.A.; Goeppert, A.; Prakash, G.K.S. Methanol-Based Chemicals, Synthetic Hydrocarbons and Materials. In Beyond Oil and Gas: The Methanol Economy; Wiley: Weinheim, Germany, 2009; pp. 375–386. [Google Scholar]
- Goeppert, A.; Czaun, M.; Jones, J.-P.; Surya Prakash, G.K.; Olah, G.A. Recycling of carbon dioxide to methanol and derived products—Closing the loop. Chem. Soc. Rev. 2014, 43, 7995–8048. [Google Scholar] [CrossRef] [PubMed]
- Olah, G.A. Towards Oil Independence Through Renewable Methanol Chemistry. Angew. Chem. Int. Ed. 2013, 52, 104–107. [Google Scholar] [CrossRef]
- Malik, K.; Singh, S.; Basu, S.; Verma, A. Electrochemical reduction of CO2 for synthesis of green fuel. Wiley Interdiscip. Rev. Energy Environ. 2017, 6, e244. [Google Scholar] [CrossRef]
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef]
- Behrens, M. Heterogeneous Catalysis of CO2 Conversion to Methanol on Copper Surfaces. Angew. Chem. Int. Ed. 2014, 53, 12022–12024. [Google Scholar] [CrossRef]
- Dang, S.; Yang, H.; Gao, P.; Wang, H.; Li, X.; Wei, W.; Sun, Y. A review of research progress on heterogeneous catalysts for methanol synthesis from carbon dioxide hydrogenation. Catal. Today 2019, 330, 61–75. [Google Scholar] [CrossRef]
- Jessop, P.G. Homogeneous Hydrogenation of Carbon Dioxide. In The Handbook of Homogeneous Hydrogenation; WILEY-VCH: Weinheim, Germany, 2007; ISBN 9783527311613. [Google Scholar]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Sanz, S.; Azua, A.; Peris, E. ‘(η6-arene)Ru(bis-NHC)’ complexes for the reduction of CO2 to formate with hydrogen and by transfer hydrogenation with iPrOH. Dalt. Trans. 2010, 39, 6339. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, X.; Liu, X.; He, L.N. Ruthenium-promoted reductive transformation of CO2. Sci. China Chem. 2017, 60, 841–852. [Google Scholar] [CrossRef]
- Wiedner, E.S.; Linehan, J.C. Making a Splash in Homogeneous CO2 Hydrogenation: Elucidating the Impact of Solvent on Catalytic Mechanisms. Chem. A Eur. J. 2018, 24, 16964–16971. [Google Scholar] [CrossRef]
- Janes, T.; Yang, Y.; Song, D. Chemical reduction of CO2 facilitated by C-nucleophiles. Chem. Commun. 2017, 53, 11390–11398. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Nichols, A.W.; Machan, C.W. A look at periodic trends in d-block molecular electrocatalysts for CO2 reduction. Dalt. Trans. 2019, 48, 9454–9468. [Google Scholar] [CrossRef]
- Fernández-Alvarez, F.J.; Iglesias, M.; Oro, L.A.; Polo, V. CO2 activation and catalysis driven by iridium complexes. ChemCatChem 2013, 5, 3481–3494. [Google Scholar] [CrossRef]
- Kang, P.; Chen, Z.; Brookhart, M.; Meyer, T.J. Electrocatalytic Reduction of Carbon Dioxide: Let the Molecules Do the Work. Top. Catal. 2015, 58, 30–45. [Google Scholar] [CrossRef]
- Jessop, P.G.; Joó, F.; Tai, C.-C. Recent advances in the homogeneous hydrogenation of carbon dioxide. Coord. Chem. Rev. 2004, 248, 2425–2442. [Google Scholar] [CrossRef]
- Badiei, Y.M.; Wang, W.-H.; Hull, J.F.; Szalda, D.J.; Muckerman, J.T.; Himeda, Y.; Fujita, E. Cp*Co(III) Catalysts with Proton-Responsive Ligands for Carbon Dioxide Hydrogenation in Aqueous Media. Inorg. Chem. 2013, 52, 12576–12586. [Google Scholar] [CrossRef] [PubMed]
- Jeletic, M.S.; Mock, M.T.; Appel, A.M.; Linehan, J.C. A Cobalt-Based Catalyst for the Hydrogenation of CO2 under Ambient Conditions. J. Am. Chem. Soc. 2013, 135, 11533–11536. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Vasudevan, K.V.; Scott, B.L.; Hanson, S.K. Understanding the Mechanisms of Cobalt-Catalyzed Hydrogenation and Dehydrogenation Reactions. J. Am. Chem. Soc. 2013, 135, 8668–8681. [Google Scholar] [CrossRef] [PubMed]
- Glüer, A.; Schneider, S. Iron catalyzed hydrogenation and electrochemical reduction of CO2: The role of functional ligands. J. Organomet. Chem. 2018, 861, 159–173. [Google Scholar] [CrossRef]
- Jeletic, M.S.; Helm, M.L.; Hulley, E.B.; Mock, M.T.; Appel, A.M.; Linehan, J.C. A Cobalt Hydride Catalyst for the Hydrogenation of CO2: Pathways for Catalysis and Deactivation. ACS Catal. 2014, 4, 3755–3762. [Google Scholar] [CrossRef]
- Jeletic, M.S.; Hulley, E.B.; Helm, M.L.; Mock, M.T.; Appel, A.M.; Wiedner, E.S.; Linehan, J.C. Understanding the Relationship Between Kinetics and Thermodynamics in CO2 Hydrogenation Catalysis. ACS Catal. 2017, 7, 6008–6017. [Google Scholar] [CrossRef]
- Shaffer, D.W.; Johnson, S.I.; Rheingold, A.L.; Ziller, J.W.; Goddard, W.A.; Nielsen, R.J.; Yang, J.Y. Reactivity of a Series of Isostructural Cobalt Pincer Complexes with CO2, CO, and H+. Inorg. Chem. 2014, 53, 13031–13041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hojilla Atienza, C.C.; Milsmann, C.; Semproni, S.P.; Turner, Z.R.; Chirik, P.J. Reversible Carbon–Carbon Bond Formation Induced by Oxidation and Reduction at a Redox-Active Cobalt Complex. Inorg. Chem. 2013, 52, 5403–5417. [Google Scholar] [CrossRef] [PubMed]
- Dubey, A.; Nencini, L.; Fayzullin, R.R.; Nervi, C.; Khusnutdinova, J.R. Bio-Inspired Mn(I) Complexes for the Hydrogenation of CO2 to Formate and Formamide. ACS Catal. 2017, 7, 3864–3868. [Google Scholar] [CrossRef]
- Jewess, M.; Crabtree, R.H. Electrocatalytic Nitrogen Fixation for Distributed Fertilizer Production? ACS Sustain. Chem. Eng. 2016, 4, 5855–5858. [Google Scholar] [CrossRef]
- Liu, H. Ammonia Synthesis Catalysts: Innovation and Practice; World Scientific: Beijing, China, 2013; ISBN 9789814355780. [Google Scholar]
- Jacobsen, C.J.H.; Dahl, S.; Clausen, B.S.; Bahn, S.; Logadottir, A.; Nørskov, J.K. Catalyst Design by Interpolation in the Periodic Table: Bimetallic Ammonia Synthesis Catalysts. J. Am. Chem. Soc. 2001, 123, 8404–8405. [Google Scholar] [CrossRef]
- Liu, H. Ammonia synthesis catalyst 100 years: Practice, enlightenment and challenge. Chinese J. Catal. 2014, 35, 1619–1640. [Google Scholar] [CrossRef]
- Levi, P.G.; Cullen, J.M. Mapping Global Flows of Chemicals: From Fossil Fuel Feedstocks to Chemical Products. Environ. Sci. Technol. 2018, 52, 1725–1734. [Google Scholar] [CrossRef]
- Canfield, D.E.; Glazer, A.N.; Falkowski, P.G. The Evolution and Future of Earth’s Nitrogen Cycle. Science 2010, 330, 192–196. [Google Scholar] [CrossRef] [Green Version]
- Galloway, J.N.; Townsend, A.R.; Erisman, J.W.; Bekunda, M.; Cai, Z.; Freney, J.R.; Martinelli, L.A.; Seitzinger, S.P.; Sutton, M.A. Transformation of the Nitrogen Cycle: Recent Trends, Questions, and Potential Solutions. Science 2008, 320, 889–892. [Google Scholar] [CrossRef] [Green Version]
- Gruber, N.; Galloway, J.N. An Earth-system perspective of the global nitrogen cycle. Nature 2008, 451, 293–296. [Google Scholar] [CrossRef]
- U.S. Geological Survay. Mineral Commodity Summaries; U.S. Geological Survay: Reston, VA, USA, 2020; ISBN 9781411343627.
- Lipman, T.; Shah, N. Ammonia as an Alternative Energy Storage Medium for Hydrogen Fuel Cells: Scientific and Technical Review for Near-Term Stationary Power Demonstration Projects, Final Report. Inst. Transp. Stud. Res. Rep. Work. Pap. Proc. 2007. Available online: https://escholarship.org/uc/item/7z69v4wp (accessed on 9 July 2020).
- Lan, R.; Irvine, J.T.S.; Tao, S. Ammonia and related chemicals as potential indirect hydrogen storage materials. Int. J. Hydrog. Energy 2012, 37, 1482–1494. [Google Scholar] [CrossRef]
- Lan, R.; Tao, S. Ammonia as a Suitable Fuel for Fuel Cells. Front. Energy Res. 2014, 2, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Federsel, C.; Jackstell, R.; Beller, M. State-of-the-art catalysts for hydrogenation of carbon dioxide. Angew. Chem. Int. Ed. 2010, 49, 6254–6257. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Goeppert, A.; Prakash, G.K.S. Integrated CO2 Capture and Conversion to Formate and Methanol: Connecting Two Threads. Acc. Chem. Res. 2019, 52, 2892–2903. [Google Scholar] [CrossRef]
- Kar, S.; Kothandaraman, J.; Goeppert, A.; Prakash, G.K.S. Advances in catalytic homogeneous hydrogenation of carbon dioxide to methanol. J. CO2 Util. 2018, 23, 212–218. [Google Scholar] [CrossRef]
- Munshi, P.; Main, A.D.; Linehan, J.C.; Tai, C.C.; Jessop, P.G. Hydrogenation of carbon dioxide catalyzed by ruthenium trimethylphosphine complexes: The accelerating effect of certain alcohols and amines. J. Am. Chem. Soc. 2002, 124, 7963–7971. [Google Scholar] [CrossRef]
- Urakawa, A.; Jutz, F.; Laurenczy, G.; Baiker, A. Carbon dioxide hydrogenation catalyzed by a ruthenium dihydride: A DFT and high-pressure spectroscopic investigation. Chem. A Eur. J. 2007, 13, 3886–3899. [Google Scholar] [CrossRef]
- Jessop, P.G.; Hsiao, Y.; Ikariya, T.; Noyori, R. Homogeneous Catalysis in Supercritical Fluids: Hydrogenation of Supercritical Carbon Dioxide to Formic Acid, Alkyl Formates, and Formamides. J. Am. Chem. Soc. 1996, 118, 344–355. [Google Scholar] [CrossRef]
- Jessop, P.G.; Ikariya, T.; Noyori, R. Homogeneous catalytic hydrogenation of supercritical carbon dioxide. Nature 1994, 368, 231–233. [Google Scholar] [CrossRef]
- Graf, E.; Leitner, W. Direct formation of formic acid from carbon dioxide and dihydrogen using the [{Rh(cod)Cl}2]–Ph2P(CH2)4PPh2 catalyst system. J. Chem. Soc., Chem. Commun. 1992, 623–624. [Google Scholar] [CrossRef]
- Gassner, F.; Leitner, W. Hydrogenation of carbon dioxide to formic acid using water-soluble rhodium catalyststs. J. Chem. Soc. Chem. Commun. 1993, 1465. [Google Scholar] [CrossRef]
- Angermund, K.; Baumann, W.; Dinjus, E.; Fornika, R.; Görls, H.; Kessler, M.; Krüger, C.; Leitner, W.; Lutz, F. Complexes [(P2)Rh(hfacac)] as Model Compounds for the Fragment [(P2)Rh] and as Highly Active Catalysts for CO2 Hydrogenation: The Accessible Molecular Surface (AMS) Model as an Approach to Quantifying the Intrinsic Steric Properties of Chelating Ligands i. Chem. A Eur. J. 1997, 3, 755–764. [Google Scholar] [CrossRef]
- Himeda, Y.; Onozawa-Komatsuzaki, N.; Sugihara, H.; Kasuga, K. Simultaneous tuning of activity and water solubility of complex catalysts by acid-base equilibrium of ligands for conversion of carbon dioxide. Organometallics 2007, 26, 702–712. [Google Scholar] [CrossRef]
- Tanaka, R.; Yamashita, M.; Nozaki, K. Catalytic Hydrogenation of Carbon Dioxide Using Ir(III)-Pincer Complexes RID E-4607-2010 RID E-8779-2010. J. Am. Chem. Soc. 2009, 131, 14168–14169. [Google Scholar] [CrossRef] [PubMed]
- Schmeier, T.J.; Dobereiner, G.E.; Crabtree, R.H.; Hazari, N. Secondary coordination sphere interactions facilitate the insertion step in an iridium(III) CO2 reduction catalyst. J. Am. Chem. Soc. 2011, 133, 9274–9277. [Google Scholar] [CrossRef]
- Federsel, C.; Boddien, A.; Jackstell, R.; Jennerjahn, R.; Dyson, P.J.; Scopelliti, R.; Laurenczy, G.; Beller, M. A well-defined iron catalyst for the reduction of bicarbonates and carbon dioxide to formates, alkyl formates, and formamides. Angew. Chem. Int. Ed. 2010, 49, 9777–9780. [Google Scholar] [CrossRef]
- Balaraman, E.; Gunanathan, C.; Zhang, J.; Shimon, L.J.W.; Milstein, D. Efficient hydrogenation of organic carbonates, carbamates and formates indicates alternative routes to methanol based on CO2 and CO. Nat. Chem. 2011, 3, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Huff, C.A.; Sanford, M.S. Cascade catalysis for the homogeneous hydrogenation of CO2 to methanol. J. Am. Chem. Soc. 2011, 133, 18122–18125. [Google Scholar] [CrossRef]
- Balaraman, E.; Ben-David, Y.; Milstein, D. Unprecedented catalytic hydrogenation of urea derivatives to amines and methanol. Angew. Chem. Int. Ed. 2011, 50, 11702–11705. [Google Scholar] [CrossRef] [PubMed]
- Ostapowicz, T.G.; Hölscher, M.; Leitner, W. CO2 insertion into metal-carbon bonds: A computational study of RhI pincer complexes. Chem. A Eur. J. 2011, 17, 10329–10338. [Google Scholar] [CrossRef]
- Ostapowicz, T.G.; Schmitz, M.; Krystof, M.; Klankermayer, J.; Leitner, W. Carbon dioxide as a C1 building block for the formation of carboxylic acids by formal catalytic hydrocarboxylation. Angew. Chem. Int. Ed. 2013, 52, 12119–12123. [Google Scholar] [CrossRef] [Green Version]
- Wesselbaum, S.; Vom Stein, T.; Klankermayer, J.; Leitner, W. Hydrogenation of carbon dioxide to methanol by using a homogeneous ruthenium-phosphine catalyst. Angew. Chem. Int. Ed. 2012, 51, 7499–7502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geilen, F.M.A.; Engendahl, B.; Hölscher, M.; Klankermayer, J.; Leitner, W. Selective Homogeneous Hydrogenation of Biogenic Carboxylic Acids with [Ru(TriPhos)H] +: A Mechanistic Study. J. Am. Chem. Soc. 2011, 133, 14349–14358. [Google Scholar] [CrossRef] [PubMed]
- Wesselbaum, S.; Moha, V.; Meuresch, M.; Brosinski, S.; Thenert, K.M.; Kothe, J.; vom Stein, T.; Englert, U.; Hölscher, M.; Klankermayer, J.; et al. Hydrogenation of carbon dioxide to methanol using a homogeneous ruthenium-Triphos catalyst: From mechanistic investigations to multiphase catalysis. Chem. Sci. 2015, 6, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Beydoun, K.; Vom Stein, T.; Klankermayer, J.; Leitner, W. Ruthenium-catalyzed direct methylation of primary and secondary aromatic amines using carbon dioxide and molecular hydrogen. Angew. Chem. Int. Ed. 2013, 52, 9554–9557. [Google Scholar] [CrossRef] [Green Version]
- Kang, P.; Cheng, C.; Chen, Z.; Schauer, C.K.; Meyer, T.J.; Brookhart, M. Selective electrocatalytic reduction of CO2 to formate by water-stable iridium dihydride pincer complexes. J. Am. Chem. Soc. 2012, 134, 5500–5503. [Google Scholar] [CrossRef]
- Osadchuk, I.; Tamm, T.; Ahlquist, M.S.G. Reduced State of Iridium PCP Pincer Complexes in Electrochemical CO2 Hydrogenation. ACS Catal. 2016, 6, 3834–3839. [Google Scholar] [CrossRef]
- Elmas, S.; Subhani, M.A.; Vogt, H.; Leitner, W.; Müller, T.E. Facile insertion of CO2 into metal-phenoxide bonds. Green Chem. 2013, 15, 1356–1360. [Google Scholar] [CrossRef] [Green Version]
- Vogt, M.; Rivada-Wheelaghan, O.; Iron, M.A.; Leitus, G.; Diskin-Posner, Y.; Shimon, L.J.W.; Ben-David, Y.; Milstein, D. Anionic nickel(II) complexes with doubly deprotonated PNP pincer-type ligands and their reactivity toward CO2. Organometallics 2013, 32, 300–308. [Google Scholar] [CrossRef]
- Hull, J.F.; Himeda, Y.; Wang, W.-H.; Hashiguchi, B.; Periana, R.; Szalda, D.J.; Muckerman, J.T.; Fujita, E. Reversible hydrogen storage using CO2 and a proton-switchable iridium catalyst in aqueous media under mild temperatures and pressures. Nat. Chem. 2012, 4, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Sordakis, K.; Tsurusaki, A.; Iguchi, M.; Kawanami, H.; Himeda, Y.; Laurenczy, G. Carbon Dioxide to Methanol: The Aqueous Catalytic Way at Room Temperature. Chem. A Eur. J. 2016, 22, 15605–15608. [Google Scholar] [CrossRef] [PubMed]
- Rezayee, N.M.; Huff, C.A.; Sanford, M.S. Tandem amine and ruthenium-catalyzed hydrogenation of CO2 to methanol. J. Am. Chem. Soc. 2015, 137, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Khusnutdinova, J.R.; Garg, J.A.; Milstein, D. Combining Low-Pressure CO2 Capture and Hydrogenation to Form Methanol. ACS Catal. 2015, 5, 2416–2422. [Google Scholar] [CrossRef]
- Shao, R.; Stangeland, A. Amines Used in CO2 Capture-Health and Environmental Impacts. Bellona Found. Norway, Oslo. 2009. Available online: https://network.bellona.org/content/uploads/sites/3/fil_Bellona_report_September_2009_-_Amines_used_in_CO2_capture.pdf (accessed on 9 July 2020).
- Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G.A.; Prakash, G.K.S. Conversion of CO2 from Air into Methanol Using a Polyamine and a Homogeneous Ruthenium Catalyst. J. Am. Chem. Soc. 2016, 138, 778–781. [Google Scholar] [CrossRef]
- Kar, S.; Sen, R.; Goeppert, A.; Prakash, G.K.S. Integrative CO2 Capture and hydrogenation to methanol with reusable catalyst and amine: Toward a carbon neutral methanol economy. J. Am. Chem. Soc. 2018, 140, 1580–1583. [Google Scholar] [CrossRef]
- Kar, S.; Goeppert, A.; Prakash, G.K.S. Combined CO2 Capture and Hydrogenation to Methanol: Amine Immobilization Enables Easy Recycling of Active Elements. ChemSusChem 2019, 12, 3172–3177. [Google Scholar] [CrossRef]
- Kar, S.; Sen, R.; Kothandaraman, J.; Goeppert, A.; Chowdhury, R.; Munoz, S.B.; Haiges, R.; Prakash, G.K.S. Mechanistic Insights into Ruthenium-Pincer-Catalyzed Amine-Assisted Homogeneous Hydrogenation of CO2 to Methanol. J. Am. Chem. Soc. 2019, 141, 3160–3170. [Google Scholar] [CrossRef]
- Rawat, K.S.; Pathak, B. Aliphatic Mn-PNP complexes for the CO2 hydrogenation reaction: A base free mechanism. Catal. Sci. Technol. 2017, 7, 3234–3242. [Google Scholar] [CrossRef]
- Mandal, S.C.; Rawat, K.S.; Nandi, S.; Pathak, B. Theoretical insights into CO2 hydrogenation to methanol by a Mn-PNP complex. Catal. Sci. Technol. 2019, 9, 1867–1878. [Google Scholar] [CrossRef]
- Kar, S.; Goeppert, A.; Kothandaraman, J.; Prakash, G.K.S. Manganese-Catalyzed Sequential Hydrogenation of CO2 to Methanol via Formamide. ACS Catal. 2017, 7, 6347–6351. [Google Scholar] [CrossRef]
- Sen, R.; Goeppert, A.; Kar, S.; Prakash, G.K.S. Hydroxide Based Integrated CO2 Capture from Air and Conversion to Methanol. J. Am. Chem. Soc. 2020, 142, 4544–4549. [Google Scholar] [CrossRef] [PubMed]
- Schneidewind, J.; Adam, R.; Baumann, W.; Jackstell, R.; Beller, M. Low-Temperature Hydrogenation of Carbon Dioxide to Methanol with a Homogeneous Cobalt Catalyst. Angew. Chem. Int. Ed. 2017, 56, 1890–1893. [Google Scholar] [CrossRef] [PubMed]
- Korstanje, T.J.; Ivar van der Vlugt, J.; Elsevier, C.J.; de Bruin, B. Hydrogenation of carboxylic acids with a homogeneous cobalt catalyst. Science 2015, 350, 298–302. [Google Scholar] [CrossRef]
- Scharnagl, F.K.; Hertrich, M.F.; Neitzel, G.; Jackstell, R.; Beller, M. Homogeneous Catalytic Hydrogenation of CO2 to Methanol—Improvements with Tailored Ligands. Adv. Synth. Catal. 2019, 361, 374–379. [Google Scholar]
- Schieweck, B.G.; Klankermayer, J. Tailor-made Molecular Cobalt Catalyst System for the Selective Transformation of Carbon Dioxide to Dialkoxymethane Ethers. Angew. Chem. Int. Ed. 2017, 56, 10854–10857. [Google Scholar] [CrossRef]
- Kumar, A.; Janes, T.; Espinosa-Jalapa, N.A.; Milstein, D. Manganese Catalyzed Hydrogenation of Organic Carbonates to Methanol and Alcohols. Angew. Chem. Int. Ed. 2018, 57, 12076–12080. [Google Scholar] [CrossRef] [PubMed]
- Zubar, V.; Lebedev, Y.; Azofra, L.M.; Cavallo, L.; El-Sepelgy, O.; Rueping, M. Hydrogenation of CO2-Derived Carbonates and Polycarbonates to Methanol and Diols by Metal–Ligand Cooperative Manganese Catalysis. Angew. Chem. Int. Ed. 2018, 57, 13439–13443. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.Q.; Liu, T.; Li, S.; Zhang, J.; Chen, X.; Guan, H. Highly efficient reduction of carbon dioxide with a borane catalyzed by bis(phosphinite) pincer ligated palladium thiolate complexes. Chem. Commun. 2016, 52, 14262–14265. [Google Scholar] [CrossRef]
- Erken, C.; Kaithal, A.; Sen, S.; Weyhermüller, T.; Hölscher, M.; Werlé, C.; Leitner, W. Manganese-catalyzed hydroboration of carbon dioxide and other challenging carbonyl groups. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Motokura, K.; Kashiwame, D.; Takahashi, N.; Miyaji, A.; Baba, T. Highly Active and Selective Catalysis of Copper Diphosphine Complexes for the Transformation of Carbon Dioxide into Silyl Formate. Chem. A Eur. J. 2013, 19, 10030–10037. [Google Scholar] [CrossRef]
- Rit, A.; Zanardi, A.; Spaniol, T.P.; Maron, L.; Okuda, J. A Cationic Zinc Hydride Cluster Stabilized by an N-Heterocyclic Carbene: Synthesis, Reactivity, and Hydrosilylation Catalysis. Angew. Chem. Int. Ed. 2014, 53, 13273–13277. [Google Scholar] [CrossRef]
- Tüchler, M.; Gärtner, L.; Fischer, S.; Boese, A.D.; Belaj, F.; Mösch-Zanetti, N.C. Efficient CO2 Insertion and Reduction Catalyzed by a Terminal Zinc Hydride Complex. Angew. Chem. Int. Ed. 2018, 57, 6906–6909. [Google Scholar] [CrossRef]
- Eisenschmid, T.C.; Eisenberg, R. The iridium complex catalyzed reduction of carbon dioxide to methoxide by alkylsilanes. Organometallics 1989, 8, 1822–1824. [Google Scholar] [CrossRef]
- Riduan, S.N.; Zhang, Y.; Ying, J.Y. Conversion of Carbon Dioxide into Methanol with Silanes over N-Heterocyclic Carbene Catalysts. Angew. Chem. Int. Ed. 2009, 48, 3322–3325. [Google Scholar] [CrossRef]
- Metsänen, T.T.; Oestreich, M. Temperature-Dependent Chemoselective Hydrosilylation of Carbon Dioxide to Formaldehyde or Methanol Oxidation State. Organometallics 2015, 34, 543–546. [Google Scholar] [CrossRef]
- Scheuermann, M.L.; Semproni, S.P.; Pappas, I.; Chirik, P.J. Carbon dioxide hydrosilylation promoted by cobalt pincer complexes. Inorg. Chem. 2014, 53, 9463–9465. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zhang, C.; Jiang, J.; Wang, Z.-X.; Guan, H. How Does the Nickel Pincer Complex Catalyze the Conversion of CO2 to a Methanol Derivative? A Computational Mechanistic Study. Inorg. Chem. 2011, 50, 3816–3825. [Google Scholar] [CrossRef] [PubMed]
- Bertini, F.; Glatz, M.; Stöger, B.; Peruzzini, M.; Veiros, L.F.; Kirchner, K.; Gonsalvi, L. Carbon Dioxide Reduction to Methanol Catalyzed by Mn(I) PNP Pincer Complexes under Mild Reaction Conditions. ACS Catal. 2019, 9, 632–639. [Google Scholar] [CrossRef]
- Mazzotta, M.G.; Xiong, M.; Abu-Omar, M.M. Carbon Dioxide Reduction to Silyl-Protected Methanol Catalyzed by an Oxorhenium Pincer PNN Complex. Organometallics 2017, 36, 1688–1691. [Google Scholar] [CrossRef]
- Ryabchuk, P.; Stier, K.; Junge, K.; Checinski, M.P.; Beller, M. Molecularly Defined Manganese Catalyst for Low-Temperature Hydrogenation of Carbon Monoxide to Methanol. J. Am. Chem. Soc. 2019, 141, 16923–16929. [Google Scholar] [CrossRef]
- Kar, S.; Goeppert, A.; Prakash, G.K.S. Catalytic Homogeneous Hydrogenation of CO to Methanol via Formamide. J. Am. Chem. Soc. 2019, 141, 12518–12521. [Google Scholar] [CrossRef]
- Schneck, F.; Schendzielorz, F.; Hatami, N.; Finger, M.; Würtele, C.; Schneider, S. Photochemically Driven Reverse Water-Gas Shift at Ambient Conditions mediated by a Nickel Pincer Complex. Angew. Chem. Int. Ed. 2018, 57, 14482–14487. [Google Scholar] [CrossRef]
- Schneck, F.; Ahrens, J.; Finger, M.; Stückl, A.C.; Würtele, C.; Schwarzer, D.; Schneider, S. The elusive abnormal CO2 insertion enabled by metal-ligand cooperative photochemical selectivity inversion. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Feller, M.; Ben-Ari, E.; Diskin-Posner, Y.; Milstein, D. CO2 activation by metal–ligand-cooperation mediated by iridium pincer complexes. J. Coord. Chem. 2018, 71, 1679–1689. [Google Scholar] [CrossRef]
- Feller, M.; Gellrich, U.; Anaby, A.; Diskin-Posner, Y.; Milstein, D. Reductive Cleavage of CO2 by Metal-Ligand-Cooperation Mediated by an Iridium Pincer Complex. J. Am. Chem. Soc. 2016, 138, 6445–6454. [Google Scholar] [CrossRef] [PubMed]
- Anaby, A.; Feller, M.; Ben-David, Y.; Leitus, G.; Diskin-Posner, Y.; Shimon, L.J.W.; Milstein, D. Bottom-Up Construction of a CO2-Based Cycle for the Photocarbonylation of Benzene, Promoted by a Rhodium(I) Pincer Complex. J. Am. Chem. Soc. 2016, 138, 9941–9950. [Google Scholar] [CrossRef]
- Krishnan, V.M.; Arman, H.D.; Tonzetich, Z.J. Preparation and reactivity of a square-planar PNP cobalt(II)-hydrido complex: Isolation of the first {Co-NO} 8 -hydride. Dalt. Trans. 2018, 47, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.; Sun, Y.; Heimermann, A.; Menges, F.; Niedner-Schatteburg, G.; van Wüllen, C.; Thiel, W.R. Structure-Reactivity Relationships in the Hydrogenation of Carbon Dioxide with Ruthenium Complexes Bearing Pyridinylazolato Ligands. Chem. A Eur. J. 2013, 19, 7825–7834. [Google Scholar] [CrossRef]
- Thai, T.-T.; Therrien, B.; Süss-Fink, G. Arene ruthenium oxinato complexes: Synthesis, molecular structure and catalytic activity for the hydrogenation of carbon dioxide in aqueous solution. J. Organomet. Chem. 2009, 694, 3973–3981. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Kuncheria, J.K.; Jenkins, H.A.; Puddephatt, R.J.; Yap, G.P.A. The interconversion of formic acid and hydrogen/carbon dioxide using a binuclear ruthenium complex catalyst. J. Chem. Soc. Dalt. Trans. 2000, 3212–3217. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Li, Z.; Wang, H.; Wang, C.Y. Homogeneous catalytic synthesis of formic acid (salts) by hydrogenation of CO2 with H2 in the presence of ruthenium species. J. Mol. Catal. A Chem. 1996, 112, 9–14. [Google Scholar] [CrossRef]
- Wesselbaum, S.; Hintermair, U.; Leitner, W. Continuous-Flow Hydrogenation of Carbon Dioxide to Pure Formic Acid using an Integrated scCO2 Process with Immobilized Catalyst and Base. Angew. Chem. Int. Ed. 2012, 51, 8585–8588. [Google Scholar] [CrossRef] [Green Version]
- Boddien, A.; Federsel, C.; Sponholz, P.; Mellmann, D.; Jackstell, R.; Junge, H.; Laurenczy, G.; Beller, M. Towards the development of a hydrogen battery. Energy Environ. Sci. 2012, 5, 8907. [Google Scholar] [CrossRef]
- Elek, J.; Nádasdi, L.; Papp, G.; Laurenczy, G.; Joó, F. Homogeneous hydrogenation of carbon dioxide and bicarbonate in aqueous solution catalyzed by water-soluble ruthenium(II) phosphine complexes. Appl. Catal. A Gen. 2003, 255, 59–67. [Google Scholar] [CrossRef]
- Jantke, D.; Pardatscher, L.; Drees, M.; Cokoja, M.; Herrmann, W.A.; Kühn, F.E. Hydrogen Production and Storage on a Formic Acid/Bicarbonate Platform using Water-Soluble N -Heterocyclic Carbene Complexes of Late Transition Metals. ChemSusChem 2016, 9, 2849–2854. [Google Scholar] [CrossRef]
- Chatterjee, D.; Sarkar, P. Ru III (edta) catalyzed hydrogenation of bicarbonate to formate. J. Coord. Chem. 2016, 69, 650–655. [Google Scholar] [CrossRef]
- Boddien, A.; Gärtner, F.; Federsel, C.; Sponholz, P.; Mellmann, D.; Jackstell, R.; Junge, H.; Beller, M. CO2-“Neutral” Hydrogen Storage Based on Bicarbonates and Formates. Angew. Chem. Int. Ed. 2011, 50, 6411–6414. [Google Scholar] [CrossRef] [PubMed]
- Federsel, C.; Jackstell, R.; Boddien, A.; Laurenczy, G.; Beller, M. Ruthenium-Catalyzed Hydrogenation of Bicarbonate in Water. ChemSusChem 2010, 3, 1048–1050. [Google Scholar] [CrossRef] [PubMed]
- Ziebart, C.; Federsel, C.; Anbarasan, P.; Jackstell, R.; Baumann, W.; Spannenberg, A.; Beller, M. Well-defined iron catalyst for improved hydrogenation of carbon dioxide and bicarbonate. J. Am. Chem. Soc. 2012, 134, 20701–20704. [Google Scholar] [CrossRef]
- Federsel, C.; Ziebart, C.; Jackstell, R.; Baumann, W.; Beller, M. Catalytic hydrogenation of carbon dioxide and bicarbonates with a well-defined cobalt dihydrogen complex. Chem. A Eur. J. 2012, 18, 72–75. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, L.; Gülak, S.; Rockstroh, N.; Jackstell, R.; Beller, M. Towards a sustainable synthesis of formate salts: Combined catalytic methanol dehydrogenation and bicarbonate hydrogenation. Angew. Chem. Int. Ed. 2014, 53, 7085–7088. [Google Scholar] [CrossRef] [PubMed]
- Huff, C.A.; Sanford, M.S. Catalytic CO2 hydrogenation to formate by a ruthenium pincer complex. ACS Catal. 2013. [Google Scholar] [CrossRef]
- Filonenko, G.A.; van Putten, R.; Schulpen, E.N.; Hensen, E.J.M.; Pidko, E.A. Highly Efficient Reversible Hydrogenation of Carbon Dioxide to Formates Using a Ruthenium PNP-Pincer Catalyst. ChemCatChem 2014, 6, 1526–1530. [Google Scholar] [CrossRef]
- Zhang, Y.; MacIntosh, A.D.; Wong, J.L.; Bielinski, E.A.; Williard, P.G.; Mercado, B.Q.; Hazari, N.; Bernskoetter, W.H. Iron catalyzed CO2 hydrogenation to formate enhanced by Lewis acid co-catalysts. Chem. Sci. 2015, 6, 4291–4299. [Google Scholar] [CrossRef] [Green Version]
- Bernskoetter, W.H.; Hazari, N. Reversible Hydrogenation of Carbon Dioxide to Formic Acid and Methanol: Lewis Acid Enhancement of Base Metal Catalysts. Acc. Chem. Res. 2017, 50, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Spentzos, A.Z.; Barnes, C.L.; Bernskoetter, W.H. Effective Pincer Cobalt Precatalysts for Lewis Acid Assisted CO2 Hydrogenation. Inorg. Chem. 2016, 55, 8225–8233. [Google Scholar] [CrossRef]
- Mills, M.R.; Barnes, C.L.; Bernskoetter, W.H. Influences of Bifunctional PNP-Pincer Ligands on Low Valent Cobalt Complexes Relevant to CO2 Hydrogenation. Inorg. Chem. 2018, 57, 1590–1597. [Google Scholar] [CrossRef]
- Rivada-Wheelaghan, O.; Dauth, A.; Leitus, G.; Diskin-Posner, Y.; Milstein, D. Synthesis and reactivity of iron complexes with a new pyrazine-based pincer ligand, and application in catalytic low-pressure hydrogenation of carbon dioxide. Inorg. Chem. 2015, 54, 4526–4538. [Google Scholar] [CrossRef]
- Bertini, F.; Gorgas, N.; Stöger, B.; Peruzzini, M.; Veiros, L.F.; Kirchner, K.; Gonsalvi, L. Efficient and Mild Carbon Dioxide Hydrogenation to Formate Catalyzed by Fe(II) Hydrido Carbonyl Complexes Bearing 2,6-(Diaminopyridyl)diphosphine Pincer Ligands. ACS Catal. 2016, 6, 2889–2893. [Google Scholar] [CrossRef]
- Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G.A.; Surya Prakash, G.K. CO2 capture by amines in aqueous media and its subsequent conversion to formate with reusable ruthenium and iron catalysts. Green Chem. 2016, 18, 5831–5838. [Google Scholar] [CrossRef]
- Kar, S.; Goeppert, A.; Galvan, V.; Chowdhury, R.; Olah, J.; Prakash, G.K.S. A Carbon-Neutral CO2 Capture, Conversion, and Utilization Cycle with Low-Temperature Regeneration of Sodium Hydroxide. J. Am. Chem. Soc. 2018, 140, 16873–16876. [Google Scholar] [CrossRef] [PubMed]
- Treigerman, Z.; Sasson, Y. Generation and Quantification of Formate Ion Produced from Aqueous Sodium Bicarbonate in the Presence of Homogeneous Ruthenium Catalyst. ACS Omega 2018, 3, 12797–12801. [Google Scholar] [CrossRef]
- Dai, Z.; Luo, Q.; Cong, H.; Zhang, J.; Peng, T. New Ru(ii) N′NN′-type pincer complexes: Synthesis, characterization and the catalytic hydrogenation of CO2 or bicarbonates to formate salts. New J. Chem. 2017, 41, 3055–3060. [Google Scholar] [CrossRef]
- Bertini, F.; Glatz, M.; Gorgas, N.; Stöger, B.; Peruzzini, M.; Veiros, L.F.; Kirchner, K.; Gonsalvi, L. Carbon dioxide hydrogenation catalysed by well-defined Mn(i) PNP pincer hydride complexes. Chem. Sci. 2017, 8, 5024–5029. [Google Scholar] [CrossRef] [Green Version]
- Ramaraj, A.; Nethaji, M.; Jagirdar, B.R. Hydrogenation of CO2, carbonyl and imine substrates catalyzed by [IrH3(PhPNHP)] complex. J. Organomet. Chem. 2019, 883, 25–34. [Google Scholar] [CrossRef]
- Lo, H.K.; Copéret, C. CO2 Hydrogenation to Formate with Immobilized Ru-Catalysts Based on Hybrid Organo-Silica Mesostructured Materials. ChemCatChem 2019, 11, 430–434. [Google Scholar] [CrossRef] [Green Version]
- Foster, S.L.; Bakovic, S.I.P.; Duda, R.D.; Maheshwari, S.; Milton, R.D.; Minteer, S.D.; Janik, M.J.; Renner, J.N.; Greenlee, L.F. Catalysts for nitrogen reduction to ammonia. Nat. Catal. 2018, 1, 490–500. [Google Scholar] [CrossRef]
- Hölscher, M.; Prechtl, M.H.G.; Leitner, W. Can [M(H)2(H2)(PXP)] pincer complexes (M = Fe, Ru, Os; X = N, O, S) serve as catalyst lead structures for NH3 synthesis from N2 and H2? Chem. A Eur. J. 2007, 13, 6636–6643. [Google Scholar] [CrossRef] [Green Version]
- Stucke, N.; Flöser, B.M.; Weyrich, T.; Tuczek, F. Nitrogen Fixation Catalyzed by Transition Metal Complexes: Recent Developments. Eur. J. Inorg. Chem. 2018, 2018, 1337–1355. [Google Scholar] [CrossRef]
- Nishibayashi, Y. Development of catalytic nitrogen fixation using transition metal-dinitrogen complexes under mild reaction conditions. Dalt. Trans. 2018, 47, 11290–11297. [Google Scholar] [CrossRef]
- Nishibayashi, Y. Recent Progress in Transition-Metal-Catalyzed Reduction of Molecular Dinitrogen under Ambient Reaction Conditions. Inorg. Chem. 2015, 54, 9234–9247. [Google Scholar] [CrossRef]
- Tanabe, Y.; Nishibayashi, Y. Catalytic Dinitrogen Fixation to Form Ammonia at Ambient Reaction Conditions Using Transition Metal-Dinitrogen Complexes. Chem. Rec. 2016, 16, 1549–1577. [Google Scholar] [CrossRef]
- Nishibayashi, Y. Molybdenum-catalyzed reduction of molecular dinitrogen into ammonia under ambient reaction conditions. Comptes Rendus Chim. 2015, 18, 776–784. [Google Scholar] [CrossRef]
- Burford, R.J.; Fryzuk, M.D. Examining the relationship between coordination mode and reactivity of dinitrogen. Nat. Rev. Chem. 2017, 1, 0026. [Google Scholar] [CrossRef]
- Shipman, M.A.; Symes, M.D. Recent progress towards the electrosynthesis of ammonia from sustainable resources. Catal. Today 2017, 286, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Cherkasov, N.; Ibhadon, A.O.; Fitzpatrick, P. A review of the existing and alternative methods for greener nitrogen fixation. Chem. Eng. Process Process Intensif. 2015, 90, 24–33. [Google Scholar] [CrossRef]
- Giddey, S.; Badwal, S.P.S.; Kulkarni, A. Review of electrochemical ammonia production technologies and materials. Int. J. Hydrog. Energy 2013, 38, 14576–14594. [Google Scholar] [CrossRef]
- Cui, X.; Tang, C.; Zhang, Q. A Review of Electrocatalytic Reduction of Dinitrogen to Ammonia under Ambient Conditions. Adv. Energy Mater. 2018, 8, 1800369. [Google Scholar] [CrossRef]
- Roux, Y.; Duboc, C.; Gennari, M. Molecular Catalysts for N2 Reduction: State of the Art, Mechanism, and Challenges. ChemPhysChem 2017, 18, 2606–2617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, H.; Nishibayashi, Y.; Yoshizawa, K. Interplay between Theory and Experiment for Ammonia Synthesis Catalyzed by Transition Metal Complexes. Acc. Chem. Res. 2016, 49, 987–995. [Google Scholar] [CrossRef] [PubMed]
- van der Ham, C.J.M.; Koper, M.T.M.; Hetterscheid, D.G.H. Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 2014, 43, 5183–5191. [Google Scholar] [CrossRef] [PubMed]
- Renner, J.N.; Greenlee, L.F.; Ayres, K.E.; Herring, A.M. Electrochemical Synthesis of Ammonia: A Low Pressure, Low Temperature Approach. Interface Mag. 2015, 24, 51–57. [Google Scholar] [CrossRef]
- Allen, A.D.; Senoff, C.V. Nitrogenopentammineruthenium(II) complexes. Chem. Commun. 1965, 24, 621–622. [Google Scholar] [CrossRef]
- Benedek, Z.; Papp, M.; Oláh, J.; Szilvási, T. Identifying the Rate-Limiting Elementary Steps of Nitrogen Fixation with Single-Site Fe Model Complexes. Inorg. Chem. 2018, 57, 8499–8508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanabe, Y.; Nishibayashi, Y. Recent advances in nitrogen fixation upon vanadium complexes. Coord. Chem. Rev. 2019, 381, 135–150. [Google Scholar] [CrossRef]
- Clentsmith, G.K.B.; Bates, V.M.E.; Hitchcock, P.B.; Cloke, F.G.N. Reductive Cleavage of Dinitrogen by a Vanadium Diamidoamine Complex: The Molecular Structures of [V(Me3SiN{CH2CH2NSiMe3}2)(μ-N)]2 and K[V(Me3SiN{CH2CH2NSiMe3}2)(μ-N)]2. J. Am. Chem. Soc. 1999, 121, 10444–10445. [Google Scholar] [CrossRef]
- Gradert, C.; Stucke, N.; Krahmer, J.; Näther, C.; Tuczek, F. Molybdenum complexes supported by mixed NHC/phosphine ligands: Activation of N2 and reaction with P(OMe)3 to the first meta-phosphite complex. Chem. A Eur. J. 2015, 21, 1130–1137. [Google Scholar] [CrossRef]
- Husch, T.; Reiher, M. Mechanistic Consequences of Chelate Ligand Stabilization on Nitrogen Fixation by Yandulov-Schrock-Type Complexes. ACS Sustain. Chem. Eng. 2017, 5, 10527–10537. [Google Scholar] [CrossRef]
- Hidai, M.; Tominari, K.; Uchida, Y. Preparation and properties of dinitrogen-molybdenum complexes. J. Am. Chem. Soc. 1972, 94, 110–114. [Google Scholar] [CrossRef]
- Hidai, M.; Mizobe, Y. Research inspired by the chemistry of nitrogenase Novel metal complexes and their reactivity toward dinitrogen, nitriles, and alkynes. Can. J. Chem. 2005, 83, 358–374. [Google Scholar] [CrossRef]
- Studt, F.; Lehnert, N.; Wiesler, B.E.; Scherer, A.; Beckhaus, R.; Tuczek, F. Spectroscopic Comparison of Dinuclear Ti+ and Ti2+ μ-η1:η1 Dinitrogen Complexes with Cp*/Pentafulvene and Amine/Amide Ligation: Moderate versus Strong Activation of N2. Eur. J. Inorg. Chem. 2006, 2006, 291–297. [Google Scholar] [CrossRef]
- Fryzuk, M.D. Transformation of Coordinated Dinitrogen by Reaction with Dihydrogen and Primary Silanes. Science 1997, 275, 1445–1447. [Google Scholar] [CrossRef]
- Bobadova-Parvanova, P.; Wang, Q.; Quinonero-Santiago, D.; Morokuma, K.; Musaev, D.G. Does Dinitrogen Hydrogenation Follow Different Mechanisms for [(η5-C5Me4H)2Zr]2(μ2,η2,η2-N2) and {[PhP(CH2SiMe2NSiMe2CH2)PPh]Zr}2(μ2,η2,η2-N2) Complexes? A Computational Study. J. Am. Chem. Soc. 2006, 128, 11391–11403. [Google Scholar] [CrossRef]
- Hoffman, B.M.; Lukoyanov, D.; Yang, Z.-Y.; Dean, D.R.; Seefeldt, L.C. Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem. Rev. 2014, 114, 4041–4062. [Google Scholar] [CrossRef]
- Hu, Y.; Ribbe, M.W. Nitrogenases-A Tale of Carbon Atom(s). Angew. Chem. Int. Ed. 2016, 55, 8216–8226. [Google Scholar] [CrossRef] [PubMed]
- Solari, E.; Da Silva, C.; Iacono, B.; Hesschenbrouck, J.; Rizzoli, C.; Scopelliti, R.; Floriani, C. Photochemical activation of the N≡N bond in a dimolybdenum-dinitrogen complex: Formation of a molybdenum nitride. Angew. Chem. Int. Ed. 2001, 40, 3907–3909. [Google Scholar] [CrossRef]
- Reiher, M.; Kirchner, B.; Hutter, J.; Sellmann, D.; Hess, B.A. A Photochemical Activation Scheme of Inert Dinitrogen by Dinuclear RuII and FeII Complexes. Chem. A Eur. J. 2004, 10, 4443–4453. [Google Scholar] [CrossRef] [PubMed]
- Kunkely, H.; Vogler, A. Photolysis of Aqueous [(NH3)5Os(μ-N2)Os(NH3)5]5+: Cleavage of Dinitrogen by an Intramolecular Photoredox Reaction. Angew. Chem. Int. Ed. 2010, 49, 1591–1593. [Google Scholar] [CrossRef] [PubMed]
- Huss, A.S.; Curley, J.J.; Cummins, C.C.; Blank, D.A. Relaxation and Dissociation Following Photoexcitation of the (μ-N2)[Mo(N[t-Bu]Ar)3]2 Dinitrogen Cleavage Intermediate. J. Phys. Chem. B 2013, 117, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Fieser, M.E.; Bates, J.E.; Ziller, J.W.; Furche, F.; Evans, W.J. Dinitrogen Reduction via Photochemical Activation of Heteroleptic Tris(cyclopentadienyl) Rare-Earth Complexes. J. Am. Chem. Soc. 2013, 135, 3804–3807. [Google Scholar] [CrossRef] [PubMed]
- Fieser, M.E.; Johnson, C.W.; Bates, J.E.; Ziller, J.W.; Furche, F.; Evans, W.J. Dinitrogen Reduction, Sulfur Reduction, and Isoprene Polymerization via Photochemical Activation of Trivalent Bis(cyclopentadienyl) Rare-Earth-Metal Allyl Complexes. Organometallics 2015, 34, 4387–4393. [Google Scholar] [CrossRef]
- Miyazaki, T.; Tanaka, H.; Tanabe, Y.; Yuki, M.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Cleavage and Formation of Molecular Dinitrogen in a Single System Assisted by Molybdenum Complexes Bearing Ferrocenyldiphosphine. Angew. Chem. Int. Ed. 2014, 53, 11488–11492. [Google Scholar] [CrossRef]
- Rebreyend, C.; de Bruin, B. Photolytic N2 Splitting: A Road to Sustainable NH3 Production? Angew. Chem. Int. Ed. 2015, 54, 42–44. [Google Scholar] [CrossRef]
- Schrock, R.R. Catalytic reduction of dinitrogen to ammonia by molybdenum: Theory versus experiment. Angew. Chem. Int. Ed. 2008, 47, 5512–5522. [Google Scholar] [CrossRef]
- Yandulov, D.V.; Schrock, R. Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center. Science 2003, 301, 76–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arashiba, K.; Miyake, Y.; Nishibayashi, Y. A molybdenum complex bearing PNP-type pincer ligands leads to the catalytic reduction of dinitrogen into ammonia. Nat. Chem. 2011, 3, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Arashiba, K.; Kuriyama, S.; Sasada, A.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Unique behaviour of dinitrogen-bridged dimolybdenum complexes bearing pincer ligand towards catalytic formation of ammonia. Nat. Commun. 2014, 5, 3737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuriyama, S.; Arashiba, K.; Nakajima, K.; Tanaka, H.; Kamaru, N.; Yoshizawa, K.; Nishibayashi, Y. Catalytic Formation of Ammonia from Molecular Dinitrogen by Use of Dinitrogen-Bridged Dimolybdenum–Dinitrogen Complexes Bearing PNP-Pincer Ligands: Remarkable Effect of Substituent at PNP-Pincer Ligand. J. Am. Chem. Soc. 2014, 136, 9719–9731. [Google Scholar] [CrossRef]
- Kuriyama, S.; Arashiba, K.; Nakajima, K.; Tanaka, H.; Yoshizawa, K.; Nishibayashi, Y. Nitrogen fixation catalyzed by ferrocene-substituted dinitrogen-bridged dimolybdenum-dinitrogen complexes: Unique behavior of ferrocene moiety as redox active site. Chem. Sci. 2015, 6, 3940–3951. [Google Scholar] [CrossRef] [Green Version]
- Kuriyama, S.; Arashiba, K.; Nakajima, K.; Tanaka, H.; Yoshizawa, K.; Nishibayashi, Y. Azaferrocene-Based PNP-Type Pincer Ligand: Synthesis of Molybdenum, Chromium, and Iron Complexes and Reactivity toward Nitrogen Fixation. Eur. J. Inorg. Chem. 2016, 2016, 4856–4861. [Google Scholar] [CrossRef]
- Klopsch, I.; Yuzik-Klimova, E.Y.; Schneider, S. Functionalization of N2 by Mid to Late Transition Metals via N–N Bond Cleavage. In Topics in Organometallic Chemistry; Springer: Basel, Switzerland, 2017; pp. 71–112. [Google Scholar]
- Silantyev, G.A.; Förster, M.; Schluschaß, B.; Abbenseth, J.; Würtele, C.; Volkmann, C.; Holthausen, M.C.; Schneider, S. Dinitrogen Splitting Coupled to Protonation. Angew. Chem. Int. Ed. 2017, 56, 5872–5876. [Google Scholar] [CrossRef]
- Klopsch, I.; Finger, M.; Würtele, C.; Milde, B.; Werz, D.B.; Schneider, S. Dinitrogen Splitting and Functionalization in the Coordination Sphere of Rhenium. J. Am. Chem. Soc. 2014, 136, 6881–6883. [Google Scholar] [CrossRef]
- Lindley, B.M.; Van Alten, R.S.; Finger, M.; Schendzielorz, F.; Würtele, C.; Miller, A.J.M.; Siewert, I.; Schneider, S. Mechanism of Chemical and Electrochemical N2 Splitting by a Rhenium Pincer Complex. J. Am. Chem. Soc. 2018, 140, 7922–7935. [Google Scholar] [CrossRef]
- Klopsch, I.; Kinauer, M.; Finger, M.; Würtele, C.; Schneider, S. Conversion of Dinitrogen into Acetonitrile under Ambient Conditions. Angew. Chem. Int. Ed. 2016, 55, 4786–4789. [Google Scholar] [CrossRef]
- Arashiba, K.; Kinoshita, E.; Kuriyama, S.; Eizawa, A.; Nakajima, K.; Tanaka, H.; Yoshizawa, K.; Nishibayashi, Y. Catalytic Reduction of Dinitrogen to Ammonia by Use of Molybdenum–Nitride Complexes Bearing a Tridentate Triphosphine as Catalysts. J. Am. Chem. Soc. 2015, 137, 5666–5669. [Google Scholar] [CrossRef] [PubMed]
- Eizawa, A.; Arashiba, K.; Tanaka, H.; Kuriyama, S.; Matsuo, Y.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Remarkable catalytic activity of dinitrogen-bridged dimolybdenum complexes bearing NHC-based PCP-pincer ligands toward nitrogen fixation. Nat. Commun. 2017, 8, 14874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matoba, K.; Eizawa, A.; Nishimura, S.; Arashiba, K.; Nakajima, K.; Nishibayashi, Y. Practical Synthesis of a PCP-Type Pincer Ligand and Its Metal Complexes. Synthesis 2018, 50, 1015–1019. [Google Scholar] [CrossRef]
- Kinoshita, E.; Arashiba, K.; Kuriyama, S.; Eizawa, A.; Nakajima, K.; Nishibayashi, Y. Synthesis and Catalytic Activity of Molybdenum-Nitride Complexes Bearing Pincer Ligands. Eur. J. Inorg. Chem. 2015, 2015, 1789–1794. [Google Scholar] [CrossRef]
- Sheng, X.L.; Batista, E.R.; Duan, Y.X.; Tian, Y.H. Dimension and bridging ligand effects on Mo-mediated catalytic transformation of dinitrogen to ammonia: Chain-like extended models of Nishibayashi’s catalyst. Comput. Theor. Chem. 2016, 1095, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.S.; Rittle, J.; Peters, J.C. Catalytic conversion of nitrogen to ammonia by an iron model complex. Nature 2013, 501, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Chalkley, M.J.; Del Castillo, T.J.; Matson, B.D.; Roddy, J.P.; Peters, J.C. Catalytic N2-to-NH3 Conversion by Fe at Lower Driving Force: A Proposed Role for Metallocene-Mediated PCET. ACS Cent. Sci. 2017, 3, 217–223. [Google Scholar] [CrossRef]
- Chalkley, M.J.; Del Castillo, T.J.; Matson, B.D.; Peters, J.C. Fe-Mediated Nitrogen Fixation with a Metallocene Mediator: Exploring pKa Effects and Demonstrating Electrocatalysis. J. Am. Chem. Soc. 2018, 140, 6122–6129. [Google Scholar] [CrossRef] [Green Version]
- Arashiba, K.; Eizawa, A.; Tanaka, H.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Catalytic nitrogen fixation via direct cleavage of nitrogen-nitrogen triple bond of molecular dinitrogen under ambient reaction conditions. Bull. Chem. Soc. Jpn. 2017, 90, 1111–1118. [Google Scholar] [CrossRef]
- Itabashi, T.; Mori, I.; Arashiba, K.; Eizawa, A.; Nakajima, K.; Nishibayashi, Y. Effect of substituents on molybdenum triiodide complexes bearing PNP-type pincer ligands toward catalytic nitrogen fixation. Dalt. Trans. 2019, 48, 3182–3186. [Google Scholar] [CrossRef]
- Fajardo, J.; Peters, J.C. Catalytic Nitrogen-to-Ammonia Conversion by Osmium and Ruthenium Complexes. J. Am. Chem. Soc. 2017, 139, 16105–16108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Castillo, T.J.; Thompson, N.B.; Suess, D.L.M.; Ung, G.; Peters, J.C. Evaluating Molecular Cobalt Complexes for the Conversion of N2 to NH3. Inorg. Chem. 2015, 54, 9256–9262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuriyama, S.; Arashiba, K.; Nakajima, K.; Matsuo, Y.; Tanaka, H.; Ishii, K.; Yoshizawa, K.; Nishibayashi, Y. Catalytic transformation of dinitrogen into ammonia and hydrazine by iron-dinitrogen complexes bearing pincer ligand. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, Y.; Kuriyama, S.; Eizawa, A.; Arashiba, K.; Nakajima, K.; Nishibayashi, Y. Synthesis and reactivity of iron-dinitrogen complexes bearing anionic methyl- and phenyl-substituted pyrrole-based PNP-type pincer ligands toward catalytic nitrogen fixation. Chem. Commun. 2017, 53, 12040–12043. [Google Scholar] [CrossRef] [PubMed]
- Kuriyama, S.; Arashiba, K.; Tanaka, H.; Matsuo, Y.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Direct Transformation of Molecular Dinitrogen into Ammonia Catalyzed by Cobalt Dinitrogen Complexes Bearing Anionic PNP Pincer Ligands. Angew. Chem. Int. Ed. 2016, 55, 14291–14295. [Google Scholar] [CrossRef]
- Sekiguchi, Y.; Arashiba, K.; Tanaka, H.; Eizawa, A.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Catalytic Reduction of Molecular Dinitrogen to Ammonia and Hydrazine Using Vanadium Complexes. Angew. Chem. Int. Ed. 2018, 57, 9064–9068. [Google Scholar] [CrossRef]
- Sekiguchi, Y.; Meng, F.; Tanaka, H.; Eizawa, A.; Arashiba, K.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Synthesis and reactivity of titanium- and zirconium-dinitrogen complexes bearing anionic pyrrole-based PNP-type pincer ligands. Dalt. Trans. 2018, 47, 11322–11326. [Google Scholar] [CrossRef]
- Doyle, L.R.; Wooles, A.J.; Jenkins, L.C.; Tuna, F.; McInnes, E.J.L.; Liddle, S.T. Catalytic Dinitrogen Reduction to Ammonia at a Triamidoamine-Titanium Complex. Angew. Chem. Int. Ed. 2018, 57, 6314–6318. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, Y.; Arashiba, K.; Nakajima, K.; Nishibayashi, Y. Catalytic Conversion of Dinitrogen into Ammonia under Ambient Reaction Conditions by Using Proton Source from Water. Chem. An Asian J. 2017, 12, 2544–2548. [Google Scholar] [CrossRef]
- Higuchi, J.; Kuriyama, S.; Eizawa, A.; Arashiba, K.; Nakajima, K.; Nishibayashi, Y. Preparation and reactivity of iron complexes bearing anionic carbazole-based PNP-type pincer ligands toward catalytic nitrogen fixation. Dalt. Trans. 2018, 47, 1117–1121. [Google Scholar] [CrossRef]
- Wickramasinghe, L.A.; Ogawa, T.; Schrock, R.R.; Müller, P. Reduction of Dinitrogen to Ammonia Catalyzed by Molybdenum Diamido Complexes. J. Am. Chem. Soc. 2017, 139, 9132–9135. [Google Scholar] [CrossRef] [PubMed]
- Kiernicki, J.J.; Zeller, M.; Szymczak, N.K. Hydrazine Capture and N-N Bond Cleavage at Iron Enabled by Flexible Appended Lewis Acids. J. Am. Chem. Soc. 2017, 139, 18194–18197. [Google Scholar] [CrossRef] [PubMed]
- Connor, G.P.; Lease, N.; Casuras, A.; Goldman, A.S.; Holland, P.L.; Mayer, J.M. Protonation and electrochemical reduction of rhodium- and iridium-dinitrogen complexes in organic solution. Dalt. Trans. 2017, 46, 14325–14330. [Google Scholar] [CrossRef]
- Stucke, N.; Krahmer, J.; Näther, C.; Tuczek, F. Molybdenum Complexes Supported by PN3P Pincer Ligands: Synthesis, Characterization, and Application to Synthetic Nitrogen Fixation. Eur. J. Inorg. Chem. 2018, 2018, 5108–5116. [Google Scholar] [CrossRef]
- Shiina, K. Reductive silylation of molecular nitrogen via fixation to tris (trialkylsilyl) amine. J. Am. Chem. Soc. 1972, 94, 9266–9267. [Google Scholar] [CrossRef]
- Komori, K.; Oshita, H.; Mizobe, Y.; Hidai, M. Preparation and properties of molybdenum and tungsten dinitrogen complexes. 25. Catalytic conversion of molecular nitrogen into silylamines using molybdenum and tungsten dinitrogen complexes. J. Am. Chem. Soc. 1989, 111, 1939–1940. [Google Scholar] [CrossRef]
- Tanaka, H.; Sasada, A.; Kouno, T.; Yuki, M.; Miyake, Y.; Nakanishi, H.; Nishibayashi, Y.; Yoshizawa, K. Molybdenum-Catalyzed Transformation of Molecular Dinitrogen into Silylamine: Experimental and DFT Study on the Remarkable Role of Ferrocenyldiphosphine Ligands. J. Am. Chem. Soc. 2011, 133, 3498–3506. [Google Scholar] [CrossRef] [PubMed]
- Yuki, M.; Tanaka, H.; Sasaki, K.; Miyake, Y.; Yoshizawa, K.; Nishibayashi, Y. Iron-catalysed transformation of molecular dinitrogen into silylamine under ambient conditions. Nat. Commun. 2012, 3, 1254. [Google Scholar] [CrossRef] [Green Version]
- Araake, R.; Sakadani, K.; Tada, M.; Sakai, Y.; Ohki, Y. [Fe4] and [Fe6] Hydride Clusters Supported by Phosphines: Synthesis, Characterization, and Application in N2 Reduction. J. Am. Chem. Soc. 2017, 139, 5596–5606. [Google Scholar] [CrossRef]
- Imayoshi, R.; Nakajima, K.; Nishibayashi, Y. Vanadium-catalyzed Reduction of Molecular Dinitrogen into Silylamine under Ambient Reaction Conditions. Chem. Lett. 2017, 46, 466–468. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Shin-ichi, H.; Mori, M. Incorporation of molecular nitrogen into organic compounds. Titanium catalyzed nitrogenation. Tetrahedron Lett. 1993, 34, 6907–6910. [Google Scholar] [CrossRef]
- Ghana, P.; van Krüchten, F.D.; Spaniol, T.P.; van Leusen, J.; Kögerler, P.; Okuda, J. Conversion of dinitrogen to tris(trimethylsilyl)amine catalyzed by titanium triamido-amine complexes. Chem. Commun. 2019, 55, 3231–3234. [Google Scholar] [CrossRef] [PubMed]
- Siedschlag, R.B.; Bernales, V.; Vogiatzis, K.D.; Planas, N.; Clouston, L.J.; Bill, E.; Gagliardi, L.; Lu, C.C. Catalytic Silylation of Dinitrogen with a Dicobalt Complex. J. Am. Chem. Soc. 2015, 137, 4638–4641. [Google Scholar] [CrossRef] [PubMed]
- Imayoshi, R.; Tanaka, H.; Matsuo, Y.; Yuki, M.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Cobalt-Catalyzed Transformation of Molecular Dinitrogen into Silylamine under Ambient Reaction Conditions. Chem. A Eur. J. 2015, 21, 8905–8909. [Google Scholar] [CrossRef] [PubMed]
- Dzik, W. Silylation of Dinitrogen Catalyzed by Hydridodinitrogentris(Triphenylphosphine)Cobalt(I). Inorganics 2016, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Li, G.; Deng, L. Bis(dinitrogen)cobalt(−1) Complexes with NHC Ligation: Synthesis, Characterization, and Their Dinitrogen Functionalization Reactions Affording Side-on Bound Diazene Complexes. J. Am. Chem. Soc. 2018, 140, 2239–2250. [Google Scholar] [CrossRef]
- Suzuki, T.; Fujimoto, K.; Takemoto, Y.; Wasada-Tsutsui, Y.; Ozawa, T.; Inomata, T.; Fryzuk, M.D.; Masuda, H. Efficient Catalytic Conversion of Dinitrogen to N(SiMe3)3 Using a Homogeneous Mononuclear Cobalt Complex. ACS Catal. 2018, 8, 3011–3015. [Google Scholar] [CrossRef]
- Tanabe, Y.; Nishibayashi, Y. Recent advances in catalytic silylation of dinitrogen using transition metal complexes. Coord. Chem. Rev. 2019, 389, 73–93. [Google Scholar] [CrossRef]
- Liao, Q.; Cavaillé, A.; Saffon-Merceron, N.; Mézailles, N. Direct Synthesis of Silylamine from N2 and a Silane: Mediated by a Tridentate Phosphine Molybdenum Fragment. Angew. Chem. Int. Ed. 2016, 128, 11378–11382. [Google Scholar] [CrossRef]
- Imayoshi, R.; Nakajima, K.; Takaya, J.; Iwasawa, N.; Nishibayashi, Y. Synthesis and Reactivity of Iron– and Cobalt–Dinitrogen Complexes Bearing PSiP-Type Pincer Ligands toward Nitrogen Fixation. Eur. J. Inorg. Chem. 2017, 2017, 3768. [Google Scholar] [CrossRef] [Green Version]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts, and Engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chheda, J.N.; Huber, G.W.; Dumesic, J.A. Liquid-Phase Catalytic Processing of Biomass-Derived Oxygenated Hydrocarbons to Fuels and Chemicals. Angew. Chem. Int. Ed. 2007, 46, 7164–7183. [Google Scholar] [CrossRef] [PubMed]
- Corma, A.; Iborra, S.; Velty, A. Chemical Routes for the Transformation of Biomass into Chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.-L.; Alaimo, P.J. Useful Products from Complex Starting Materials: Common Chemicals from Biomass Feedstocks. Chem. A Eur. J. 2010, 16, 4970–4980. [Google Scholar] [CrossRef] [PubMed]
- Werpy, T.A.; Holladay, J.E.; White, J.F. Top Value Added Chemicals from Biomass: I. Results of Screening for Potential Candidates from Sugars and Synthesis Gas; Pacific Northwest National Lab.: Richland, WA, USA, 2004. [Google Scholar]
- Takagaki, A.; Nishimura, S.; Ebitani, K. Catalytic Transformations of Biomass-Derived Materials into Value-Added Chemicals. Catal. Surv. Asia 2012, 16, 164–182. [Google Scholar] [CrossRef]
- Zhou, C.-H.; Xia, X.; Lin, C.-X.; Tong, D.-S.; Beltramini, J. Catalytic conversion of lignocellulosic biomass to fine chemicals and fuels. Chem. Soc. Rev. 2011, 40, 5588. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Yu, I.K.M.; Liu, Y.; Ruan, X.; Tsang, D.C.W.; Hunt, A.J.; Ok, Y.S.; Song, H.; Zhang, S. Lignin valorization for the production of renewable chemicals: State-of-the-art review and future prospects. Bioresour. Technol. 2018, 269, 465–475. [Google Scholar] [CrossRef]
- Azadi, P.; Inderwildi, O.R.; Farnood, R.; King, D.A. Liquid fuels, hydrogen and chemicals from lignin: A critical review. Renew. Sustain. Energy Rev. 2013, 21, 506–523. [Google Scholar] [CrossRef]
- van Putten, R.-J.; van der Waal, J.C.; de Jong, E.; Rasrendra, C.B.; Heeres, H.J.; de Vries, J.G. Hydroxymethylfurfural, A Versatile Platform Chemical Made from Renewable Resources. Chem. Rev. 2013, 113, 1499–1597. [Google Scholar] [CrossRef]
- Rosatella, A.A.; Simeonov, S.P.; Frade, R.F.M.; Afonso, C.A.M. 5-Hydroxymethylfurfural (HMF) as a building block platform: Biological properties, synthesis and synthetic applications. Green Chem. 2011, 13, 754. [Google Scholar] [CrossRef]
- Wright, W.R.H.; Palkovits, R. Development of Heterogeneous Catalysts for the Conversion of Levulinic Acid to γ-Valerolactone. ChemSusChem 2012, 5, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Liguori, F.; Moreno-Marrodan, C.; Barbaro, P. Environmentally Friendly Synthesis of γ-Valerolactone by Direct Catalytic Conversion of Renewable Sources. ACS Catal. 2015, 5, 1882–1894. [Google Scholar] [CrossRef]
- Démolis, A.; Essayem, N.; Rataboul, F. Synthesis and Applications of Alkyl Levulinates. ACS Sustain. Chem. Eng. 2014, 2, 1338–1352. [Google Scholar] [CrossRef]
- Raspolli Galletti, A.M.; Antonetti, C.; Ribechini, E.; Colombini, M.P.; Nassi o Di Nasso, N.; Bonari, E. From giant reed to levulinic acid and gamma-valerolactone: A high yield catalytic route to valeric biofuels. Appl. Energy 2013, 102, 157–162. [Google Scholar] [CrossRef]
- Bond, J.Q.; Alonso, D.M.; Wang, D.; West, R.M.; Dumesic, J.A. Integrated Catalytic Conversion of -Valerolactone to Liquid Alkenes for Transportation Fuels. Science 2010, 327, 1110–1114. [Google Scholar] [CrossRef]
- Yan, K.; Yang, Y.; Chai, J.; Lu, Y. Catalytic reactions of gamma-valerolactone: A platform to fuels and value-added chemicals. Appl. Catal. B Environ. 2015, 179, 292–304. [Google Scholar] [CrossRef]
- Mika, L.T.; Cséfalvay, E.; Horváth, I.T. The role of water in catalytic biomass-based technologies to produce chemicals and fuels. Catal. Today 2015, 247, 33–46. [Google Scholar] [CrossRef]
- Horváth, I.T. Solvents from nature. Green Chem. 2008, 10, 1024. [Google Scholar] [CrossRef]
- Portillo Perez, G.; Mukherjee, A.; Dumont, M.J. Insights into HMF catalysis. J. Ind. Eng. Chem. 2019, 70, 1–34. [Google Scholar] [CrossRef]
- Omoruyi, U.; Page, S.; Hallett, J.; Miller, P.W. Homogeneous catalyzed reactions of levulinic acid: To Γ-valerolactone and beyond. ChemSusChem 2016, 9, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Li, J.; Lai, D.-M.; Fu, Y.; Guo, Q.-X. Catalytic Conversion of Biomass-Derived Carbohydrates into γ-Valerolactone without Using an External H 2 Supply. Angew. Chem. Int. Ed. 2009, 48, 6529–6532. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Horváth, I.T. Catalytic Conversion of Fructose to γ-Valerolactone in γ-Valerolactone. ACS Catal. 2012, 2, 2247–2249. [Google Scholar] [CrossRef]
- Tukacs, J.M.; Király, D.; Strádi, A.; Novodarszki, G.; Eke, Z.; Dibó, G.; Kégl, T.; Mika, L.T. Efficient catalytic hydrogenation of levulinic acid: A key step in biomass conversion. Green Chem. 2012, 14, 2057. [Google Scholar] [CrossRef]
- Tukacs, J.M.; Novák, M.; Dibó, G.; Mika, L.T. An improved catalytic system for the reduction of levulinic acid to γ-valerolactone. Catal. Sci. Technol. 2014, 4, 2908–2912. [Google Scholar] [CrossRef]
- Fábos, V.; Mika, L.T.; Horváth, I.T. Selective Conversion of Levulinic and Formic Acids to γ-Valerolactone with the Shvo Catalyst. Organometallics 2014, 33, 181–187. [Google Scholar] [CrossRef]
- Geilen, F.M.A.; Engendahl, B.; Harwardt, A.; Marquardt, W.; Klankermayer, J.; Leitner, W. Selective and Flexible Transformation of Biomass-Derived Platform Chemicals by a Multifunctional Catalytic System. Angew. Chem. Int. Ed. 2010, 49, 5510–5514. [Google Scholar] [CrossRef]
- vom Stein, T.; Meuresch, M.; Limper, D.; Schmitz, M.; Hölscher, M.; Coetzee, J.; Cole-Hamilton, D.J.; Klankermayer, J.; Leitner, W. Highly Versatile Catalytic Hydrogenation of Carboxylic and Carbonic Acid Derivatives using a Ru-Triphos Complex: Molecular Control over Selectivity and Substrate Scope. J. Am. Chem. Soc. 2014, 136, 13217–13225. [Google Scholar] [CrossRef]
- Meuresch, M.; Westhues, S.; Leitner, W.; Klankermayer, J. Tailor-Made Ruthenium-Triphos Catalysts for the Selective Homogeneous Hydrogenation of Lactams. Angew. Chem. Int. Ed. 2016, 55, 1392–1395. [Google Scholar] [CrossRef]
- vom Stein, T.; Weigand, T.; Merkens, C.; Klankermayer, J.; Leitner, W. Trimethylenemethane-Ruthenium(II)-Triphos Complexes as Highly Active Catalysts for Catalytic C–O Bond Cleavage Reactions of Lignin Model Compounds. ChemCatChem 2013, 5, 439–441. [Google Scholar] [CrossRef]
- Cui, X.; Li, Y.; Topf, C.; Junge, K.; Beller, M. Direct Ruthenium-Catalyzed Hydrogenation of Carboxylic Acids to Alcohols. Angew. Chem. Int. Ed. 2015, 54, 10596–10599. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Topf, C.; Cui, X.; Junge, K.; Beller, M. Lewis Acid Promoted Ruthenium(II)-Catalyzed Etherifications by Selective Hydrogenation of Carboxylic Acids/Esters. Angew. Chem. Int. Ed. 2015, 54, 5196–5200. [Google Scholar] [CrossRef]
- Dutta Chowdhury, A.; Jackstell, R.; Beller, M. Towards the Efficient Development of Homogeneous Catalytic Transformation to γ-Valerolactone from Biomass-Derived Platform Chemicals. ChemCatChem 2014, 6, 3360–3365. [Google Scholar] [CrossRef]
- Balaraman, E.; Fogler, E.; Milstein, D. Efficient hydrogenation of biomass-derived cyclic di-esters to 1,2-diols. Chem. Commun. 2012, 48, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, Y.; Pan, T.; Xu, Q.; Guo, Q.-X.; Fu, Y. Conversion of Carbohydrate Biomass to γ-Valerolactone by using Water-Soluble and Reusable Iridium Complexes in Acidic Aqueous Media. ChemSusChem 2013, 6, 1163–1167. [Google Scholar] [CrossRef]
- Wang, S.; Huang, H.; Dorcet, V.; Roisnel, T.; Bruneau, C.; Fischmeister, C. Efficient Iridium Catalysts for Base-Free Hydrogenation of Levulinic Acid. Organometallics 2017, 36, 3152–3162. [Google Scholar] [CrossRef]
- Ortiz-Cervantes, C.; Flores-Alamo, M.; García, J.J. Hydrogenation of Biomass-Derived Levulinic Acid into γ-Valerolactone Catalyzed by Palladium Complexes. ACS Catal. 2015, 5, 1424–1431. [Google Scholar] [CrossRef]
- Dwivedi, A.D.; Gupta, K.; Tyagi, D.; Rai, R.K.; Mobin, S.M.; Singh, S.K. Ruthenium and Formic Acid Based Tandem Catalytic Transformation of Bioderived Furans to Levulinic Acid and Diketones in Water. ChemCatChem 2015, 7, 4050–4058. [Google Scholar] [CrossRef]
- Li, W.; Xie, J.-H.; Lin, H.; Zhou, Q.-L. Highly efficient hydrogenation of biomass-derived levulinic acid to γ-valerolactone catalyzed by iridium pincer complexes. Green Chem. 2012, 14, 2388. [Google Scholar] [CrossRef]
- Gao, H.; Chen, J. Hydrogenation of biomass-derived levulinic acid to γ-valerolactone catalyzed by PNP-Ir pincer complexes: A computational study. J. Organomet. Chem. 2015, 797, 165–170. [Google Scholar] [CrossRef]
- Phanopoulos, A.; White, A.J.P.; Long, N.J.; Miller, P.W. Catalytic Transformation of Levulinic Acid to 2-Methyltetrahydrofuran Using Ruthenium-N-Triphos Complexes. ACS Catal. 2015, 5, 2500–2512. [Google Scholar] [CrossRef]
- Deng, L.; Kang, B.; Englert, U.; Klankermayer, J.; Palkovits, R. Direct hydrogenation of biobased carboxylic acids mediated by a nitrogen-centered tridentate phosphine ligand. ChemSusChem 2016, 9, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.J.; Kim, J.; Hoyt, C.; Silks, L.A.; Schlaf, M. Ruthenium-8-quinolinethiolate-phenylterpyridine versus ruthenium-bipyridine-phenyl-terpyridine complexes as homogeneous water and high temperature stable hydrogenation catalysts for biomass-derived substrates. Polyhedron 2016, 108, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Wozniak, B.; Spannenberg, A.; Li, Y.; Hinze, S.; de Vries, J.G. Cyclopentanone Derivatives from 5-Hydroxymethylfurfural via 1-Hydroxyhexane-2,5-dione as Intermediate. ChemSusChem 2018, 11, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, B.; Tin, S.; de Vries, J.G. Bio-based building blocks from 5-hydroxymethylfurfural via 1-hydroxyhexane-2,5-dione as intermediate. Chem. Sci. 2019, 10, 6024–6034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, B.M.; Puylaert, P.; Diekamp, J.; van Heck, R.; Fan, Y.; Spannenberg, A.; Hinze, S.; de Vries, J.G. Inexpensive Ruthenium NNS-Complexes as Efficient Ester Hydrogenation Catalysts with High C=O vs. C=C Selectivities. Adv. Synth. Catal. 2018, 360, 1151–1158. [Google Scholar] [CrossRef]
- Yi, Y.; Liu, H.; Xiao, L.P.; Wang, B.; Song, G. Highly Efficient Hydrogenation of Levulinic Acid into γ-Valerolactone using an Iron Pincer Complex. ChemSusChem 2018, 11, 1474–1478. [Google Scholar] [CrossRef]
- Padilla, R.; Jørgensen, M.S.B.; Paixão, M.W.; Nielsen, M. Efficient catalytic hydrogenation of alkyl levulinates to γ-valerolactone. Green Chem. 2019, 21, 5195–5200. [Google Scholar] [CrossRef]
- Irrgang, T.; Kempe, R. 3d-Metal Catalyzed N- and C-Alkylation Reactions via Borrowing Hydrogen or Hydrogen Autotransfer. Chem. Rev. 2019, 119, 2524–2549. [Google Scholar] [CrossRef]
- Corma, A.; Navas, J.; Sabater, M.J. Advances in One-Pot Synthesis through Borrowing Hydrogen Catalysis. Chem. Rev. 2018, 118, 1410–1459. [Google Scholar] [CrossRef]
- Matsunami, A.; Kayaki, Y. Upgrading and expanding the scope of homogeneous transfer hydrogenation. Tetrahedron Lett. 2018, 59, 504–513. [Google Scholar] [CrossRef]
- Morris, R.H. Mechanisms of the H2- and transfer hydrogenation of polar bonds catalyzed by iron group hydrides. Dalt. Trans. 2018, 47, 10809–10826. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, S.; Fujii, A.; Takehara, J.; Ikariya, T.; Noyori, R. Asymmetric Transfer Hydrogenation of Aromatic Ketones Catalyzed by Chiral Ruthenium(II) Complexes. J. Am. Chem. Soc. 1995, 117, 7562–7563. [Google Scholar] [CrossRef]
- Noyori, R.; Hashiguchi, S. Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes. Acc. Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Ikariya, T. Bifunctional Transition Metal-Based Molecular Catalysts for Asymmetric Syntheses. In Topics in Organometallic Chemistry; Springer: Berlin/Heidelberg, Germany, 2011; pp. 31–53. ISBN 9783642207303. [Google Scholar]
- Haack, K.-J.; Hashiguchi, S.; Fujii, A.; Ikariya, T.; Noyori, R. The Catalyst Precursor, Catalyst, and Intermediate in the RuII-Promoted Asymmetric Hydrogen Transfer between Alcohols and Ketones. Angew. Chem. Int. Ed. Engl. 1997, 36, 285–288. [Google Scholar] [CrossRef]
- Zweifel, T.; Naubron, J.-V.; Büttner, T.; Ott, T.; Grützmacher, H. Ethanol as Hydrogen Donor: Highly Efficient Transfer Hydrogenations with Rhodium(I) Amides. Angew. Chem. Int. Ed. 2008, 47, 3245–3249. [Google Scholar] [CrossRef]
- Samec, J.S.M.; Bäckvall, J.-E.; Andersson, P.G.; Brandt, P. Mechanistic aspects of transition metal-catalyzed hydrogen transfer reactions. Chem. Soc. Rev. 2006, 35, 237. [Google Scholar] [CrossRef]
- Foubelo, F.; Nájera, C.; Yus, M. Catalytic asymmetric transfer hydrogenation of ketones: Recent advances. Tetrahedron Asymmetry 2015, 26, 769–790. [Google Scholar] [CrossRef] [Green Version]
- Mai, V.H.; Lee, S.-H.; Nikonov, G.I. Transfer Hydrogenation of Unsaturated Substrates by Half-sandwich Ruthenium Catalysts using Ammonium Formate as Reducing Reagent. ChemistrySelect 2017, 2, 7751–7757. [Google Scholar] [CrossRef]
- Kayan, C.; Meriç, N.; Aydemir, M.; Ocak, Y.S.; Baysal, A.; Temel, H. Novel cyclohexyl-based aminophosphine ligands and use of their Ru(II) complexes in transfer hydrogenation of ketones. Appl. Organomet. Chem. 2014, 28, 127–133. [Google Scholar] [CrossRef]
- Ikariya, T.; Blacker, A.J. Asymmetric Transfer Hydrogenation of Ketones with Bifunctional Transition Metal-Based Molecular Catalysts. Acc. Chem. Res. 2007, 40, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, H.; Adolfsson, H. Ruthenium-catalyzed asymmetric transfer hydrogenation of ketones in ethanol. Tetrahedron Lett. 2011, 52, 2754–2758. [Google Scholar] [CrossRef]
- Castellanos-Blanco, N.; Arévalo, A.; García, J.J. Nickel-catalyzed transfer hydrogenation of ketones using ethanol as a solvent and a hydrogen donor. Dalt. Trans. 2016, 45, 13604–13614. [Google Scholar] [CrossRef] [PubMed]
- Sanz, S.; Benítez, M.; Peris, E. A New Approach to the Reduction of Carbon Dioxide: CO2 Reduction to Formate by Transfer Hydrogenation in i PrOH. Organometallics 2010, 29, 275–277. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, C.; Jiang, X.; Xue, D.; Li, J.; Xiao, J. Highly efficient transformation of levulinic acid into pyrrolidinones by iridium catalysed transfer hydrogenation. Chem. Commun. 2013, 49, 5408. [Google Scholar] [CrossRef]
- Crabtree, R.H. Transfer Hydrogenation with Glycerol as H-Donor: Catalyst Activation, Deactivation and Homogeneity. ACS Sustain. Chem. Eng. 2019, 7, 15845–15853. [Google Scholar] [CrossRef]
- Chen, T.; He, L.-P.; Gong, D.; Yang, L.; Miao, X.; Eppinger, J.; Huang, K.-W. Ruthenium(II) pincer complexes with oxazoline arms for efficient transfer hydrogenation reactions. Tetrahedron Lett. 2012, 53, 4409–4412. [Google Scholar] [CrossRef]
- He, L.-P.; Chen, T.; Xue, D.-X.; Eddaoudi, M.; Huang, K.-W. Efficient transfer hydrogenation reaction Catalyzed by a dearomatized PN3P ruthenium pincer complex under base-free Conditions. J. Organomet. Chem. 2012, 700, 202–206. [Google Scholar] [CrossRef]
- Gladiali, S.; Alberico, E. Asymmetric transfer hydrogenation: Chiral ligands and applications. Chem. Soc. Rev. 2006, 35, 226–236. [Google Scholar] [CrossRef]
- Baldino, S.; Facchetti, S.; Zanotti-Gerosa, A.; Nedden, H.G.; Baratta, W. Transfer Hydrogenation and Hydrogenation of Commercial-Grade Aldehydes to Primary Alcohols Catalyzed by 2-(Aminomethyl)pyridine and Pincer Benzo[h]quinoline Ruthenium Complexes. ChemCatChem 2016, 8, 2279–2288. [Google Scholar] [CrossRef]
- Liu, W.-P.; Yuan, M.-L.; Yang, X.-H.; Li, K.; Xie, J.-H.; Zhou, Q.-L. Efficient asymmetric transfer hydrogenation of ketones in ethanol with chiral iridium complexes of spiroPAP ligands as catalysts. Chem. Commun. 2015, 51, 6123–6125. [Google Scholar] [CrossRef] [PubMed]
- Dubey, A.; Khaskin, E. Catalytic Ester Metathesis Reaction and Its Application to Transfer Hydrogenation of Esters. ACS Catal. 2016, 6, 3998–4002. [Google Scholar] [CrossRef]
- Weingart, P.; Thiel, W.R. Applying Le Chatelier’s Principle for a Highly Efficient Catalytic Transfer Hydrogenation with Ethanol as the Hydrogen Source. ChemCatChem 2018, 10, 4858–4862. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, Z.; Leng, X.; Zhu, H.; Liu, G.; Huang, Z. Transfer Hydrogenation of Alkenes Using Ethanol Catalyzed by a NCP Pincer Iridium Complex: Scope and Mechanism. J. Am. Chem. Soc. 2018, 140, 4417–4429. [Google Scholar] [CrossRef] [PubMed]
- Farrar-Tobar, R.A.; Wozniak, B.; Savini, A.; Hinze, S.; Tin, S.; de Vries, J.G. Base-Free Iron Catalyzed Transfer Hydrogenation of Esters Using EtOH as Hydrogen Source. Angew. Chem. Int. Ed. 2019, 58, 1129–1133. [Google Scholar] [CrossRef]
- Krall, E.M.; Klein, T.W.; Andersen, R.J.; Nett, A.J.; Glasgow, R.W.; Reader, D.S.; Dauphinais, B.C.; Mc Ilrath, S.P.; Fischer, A.A.; Carney, M.J.; et al. Controlled hydrogenative depolymerization of polyesters and polycarbonates catalyzed by ruthenium(ii) PNN pincer complexes. Chem. Commun. 2014, 50, 4884. [Google Scholar] [CrossRef]
- Sun, J.; Wang, Y. Recent advances in catalytic conversion of ethanol to chemicals. ACS Catal. 2014, 4, 1078–1090. [Google Scholar] [CrossRef]
- Ndou, A.S.; Plint, N.; Coville, N.J. Dimerisation of ethanol to butanol over solid-base catalysts. Appl. Catal. A Gen. 2003, 251, 337–345. [Google Scholar] [CrossRef]
- Chakraborty, S.; Piszel, P.E.; Hayes, C.E.; Baker, R.T.; Jones, W.D. Highly Selective Formation of n-Butanol from Ethanol through the Guerbet Process: A Tandem Catalytic Approach. J. Am. Chem. Soc. 2015, 137, 14264–14267. [Google Scholar] [CrossRef]
- Aitchison, H.; Wingad, R.L.; Wass, D.F. Homogeneous Ethanol to Butanol Catalysi-Guerbet Renewed. ACS Catal. 2016, 6, 7125–7132. [Google Scholar] [CrossRef] [Green Version]
- Sheehan, J.; Aden, A.; Paustian, K.; Killian, K.; Brenner, J.; Walsh, M.; Nelson, R. Energy and Environmental Aspects of Using Corn Stover for Fuel Ethanol. J. Ind. Ecol. 2003, 7, 117–146. [Google Scholar] [CrossRef]
- Kim, S.; Dale, B.E. Life cycle assessment of various cropping systems utilized for producing biofuels: Bioethanol and biodiesel. Biomass Bioenergy 2005, 29, 426–439. [Google Scholar] [CrossRef]
- Wu, M.; Wang, M.; Huo, H. Fuel-Cycle Assessment of Selected Bioethanol Production Pathways in the United States. Renew. Energy 2006, 120. [Google Scholar]
- Atsumi, S.; Cann, A.F.; Connor, M.R.; Shen, C.R.; Smith, K.M.; Brynildsen, M.P.; Chou, K.J.Y.; Hanai, T.; Liao, J.C. Metabolic engineering of Escherichia coli for 1-butanol production. Metab. Eng. 2008, 10, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Zhao, X.-Q.; Liu, C.-G.; Chen, L.-J.; Bai, F.-W. Prospective and development of butanol as an advanced biofuel. Biotechnol. Adv. 2013, 31, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Dürre, P. Biobutanol: An attractive biofuel. Biotechnol. J. 2007, 2, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Yao, M.; Liu, H.; Lee, C.F.; Ji, J. Progress in the production and application of n-butanol as a biofuel. Renew. Sustain. Energy Rev. 2011, 15, 4080–4106. [Google Scholar] [CrossRef]
- Veibel, S.; Nielsen, J.I. On the mechanism of the Guerbet reaction. Tetrahedron 1967, 23, 1723–1733. [Google Scholar] [CrossRef]
- Xie, Y.; Ben-David, Y.; Shimon, L.J.W.; Milstein, D. Highly Efficient Process for Production of Biofuel from Ethanol Catalyzed by Ruthenium Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 9077–9080. [Google Scholar] [CrossRef]
- Tseng, K.N.T.; Lin, S.; Kampf, J.W.; Szymczak, N.K. Upgrading ethanol to 1-butanol with a homogeneous air-stable ruthenium catalyst. Chem. Commun. 2016, 52, 2901–2904. [Google Scholar] [CrossRef]
- Tseng, K.N.T.; Kampf, J.W.; Szymczak, N.K. Base-free, acceptorless, and chemoselective alcohol dehydrogenation catalyzed by an amide-derived NNN-ruthenium(II) hydride complex. Organometallics 2013, 32, 2046–2049. [Google Scholar] [CrossRef]
- Tseng, K.N.T.; Kampf, J.W.; Szymczak, N.K. Mechanism of N,N,N-Amide Ruthenium(II) Hydride Mediated Acceptorless Alcohol Dehydrogenation: Inner-Sphere β-H Elimination versus Outer-Sphere Bifunctional Metal-Ligand Cooperativity. ACS Catal. 2015, 5, 5468–5485. [Google Scholar] [CrossRef]
- Wingad, R.L.; Gates, P.J.; Street, S.T.G.; Wass, D.F. Catalytic Conversion of Ethanol to n-Butanol Using Ruthenium P-N Ligand Complexes. ACS Catal. 2015, 5, 5822–5826. [Google Scholar] [CrossRef]
- Everett, M.; Pellow, K.J.; Wass, D.F. Catalytic conversion of methanol/ethanol to isobutanol—A highly selective route to an advanced biofuel. Chem. Commun. 2016, 52, 5202–5204. [Google Scholar]
- Pellow, K.J.; Wingad, R.L.; Wass, D.F. Towards the upgrading of fermentation broths to advanced biofuels: A water tolerant catalyst for the conversion of ethanol to isobutanol. Catal. Sci. Technol. 2017, 7, 5128–5134. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Shao, Z.; Wang, Y.; Liu, Q. Manganese-Catalyzed Upgrading of Ethanol into 1-Butanol. J. Am. Chem. Soc. 2017, 139, 11941–11948. [Google Scholar] [CrossRef]
- Kulkarni, N.V.; Brennessel, W.W.; Jones, W.D. Catalytic Upgrading of Ethanol to n-Butanol via Manganese-Mediated Guerbet Reaction. ACS Catal. 2018, 8, 997–1002. [Google Scholar] [CrossRef]


























































































































{kind=link}
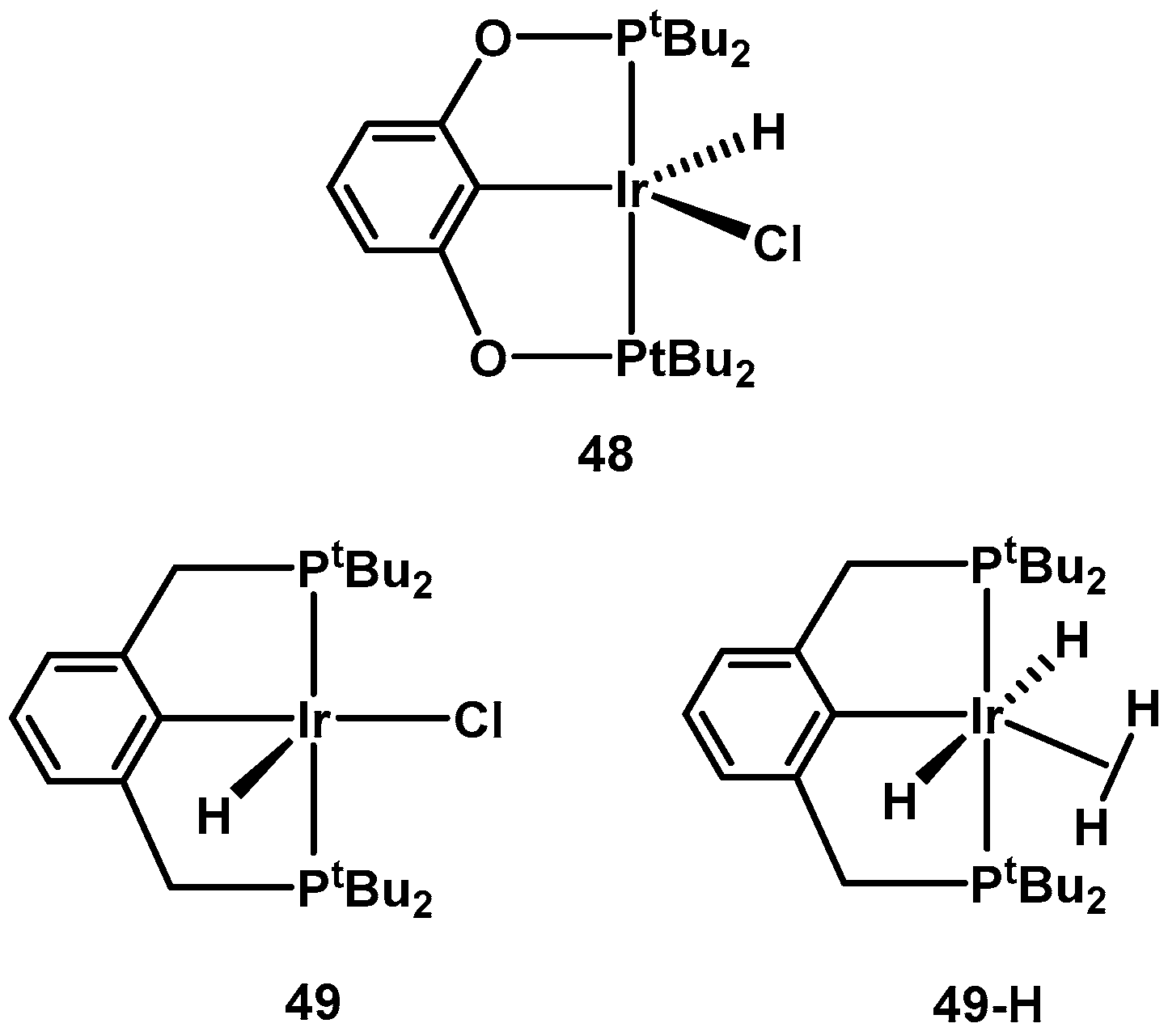{kind=link}
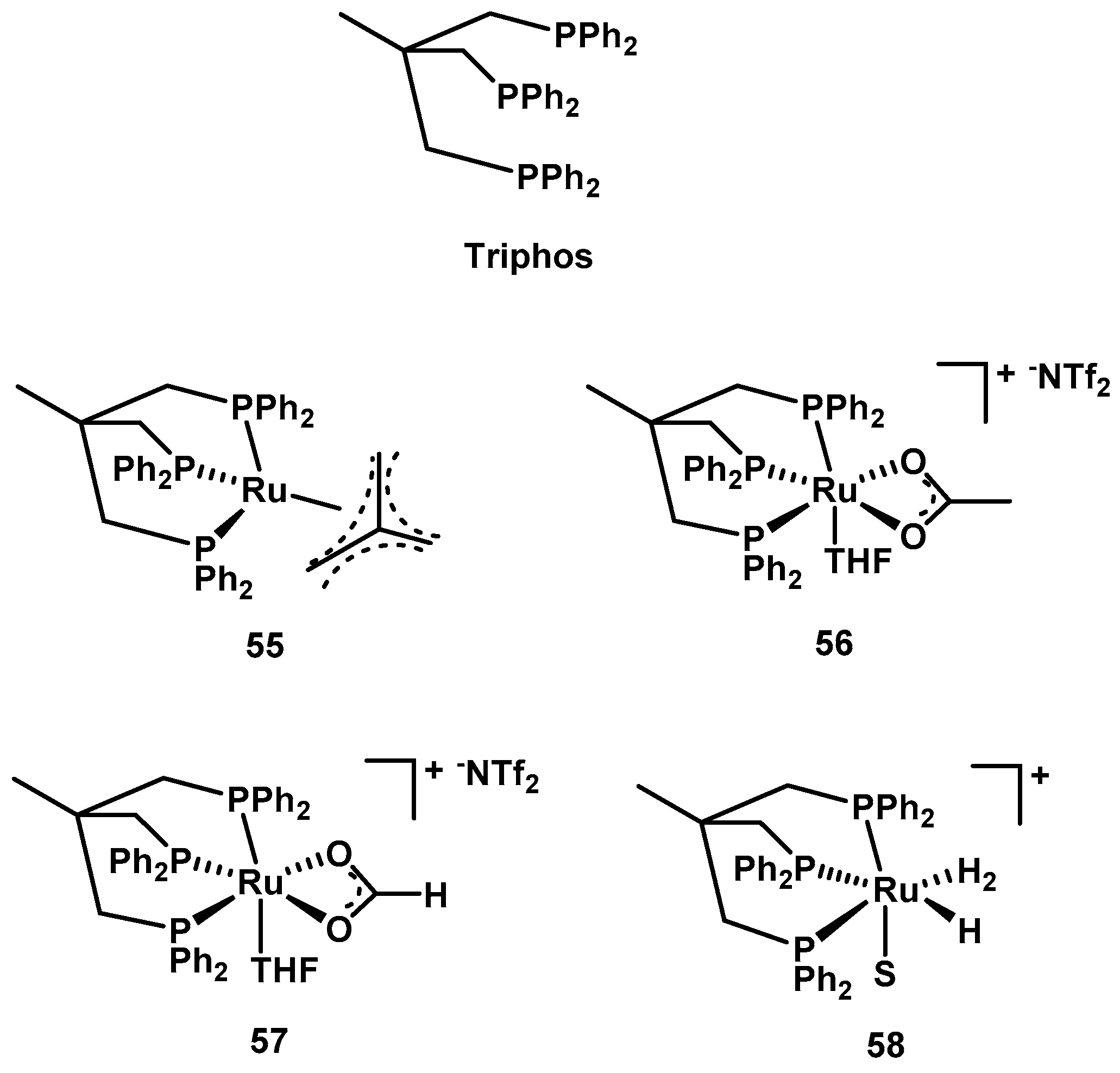{kind=link}
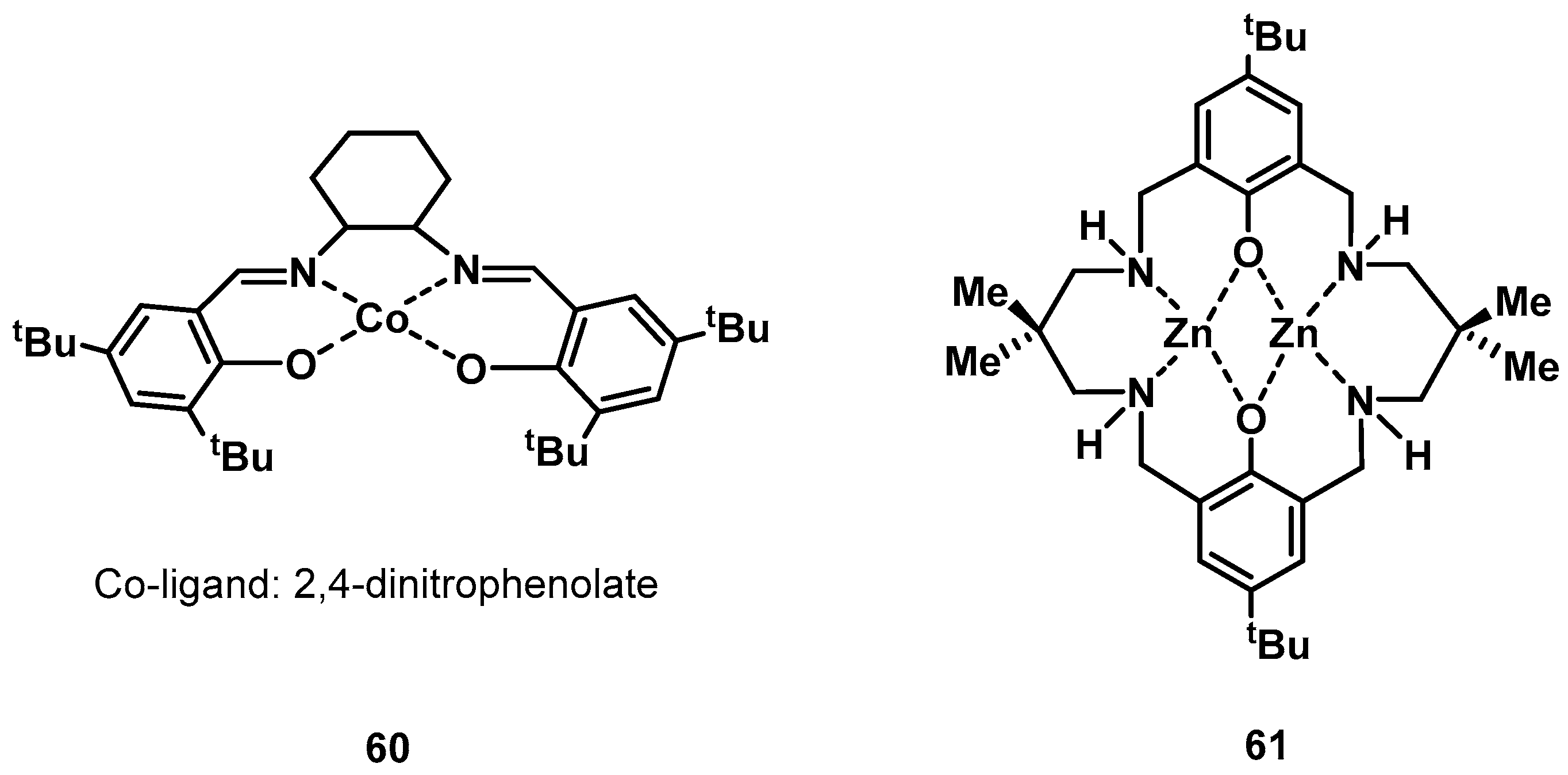{kind=link}
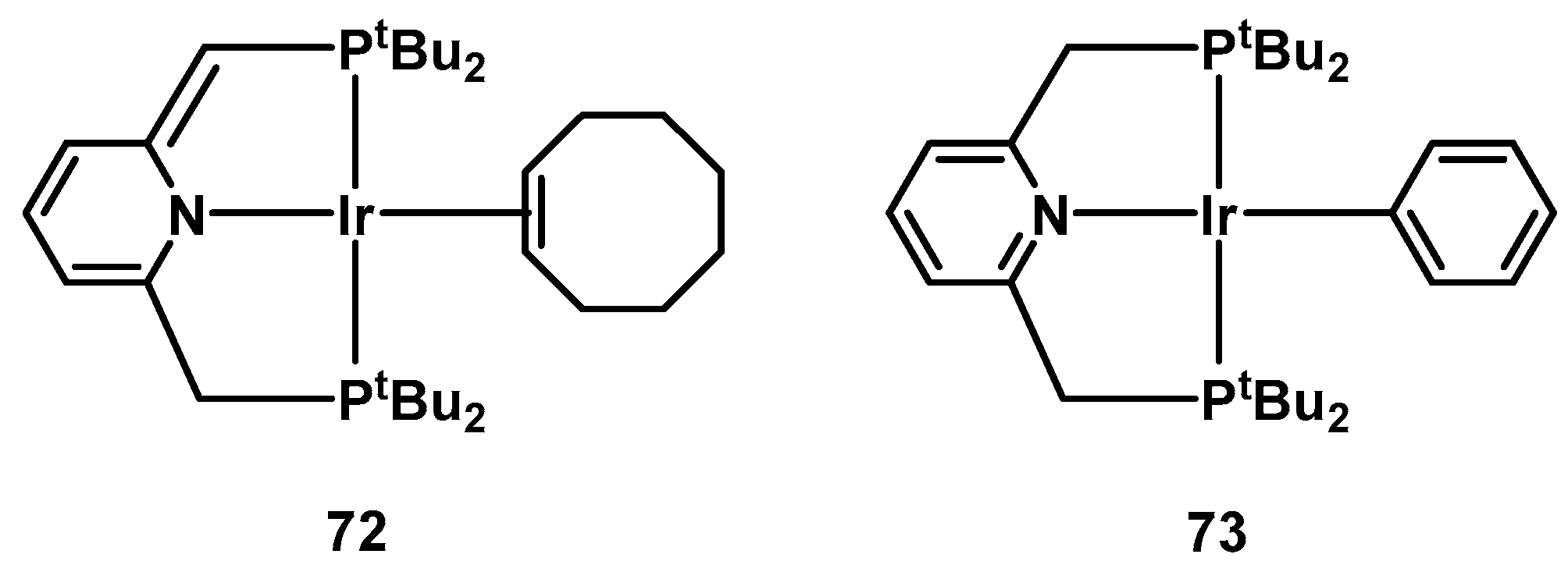{kind=link}
{kind=link}
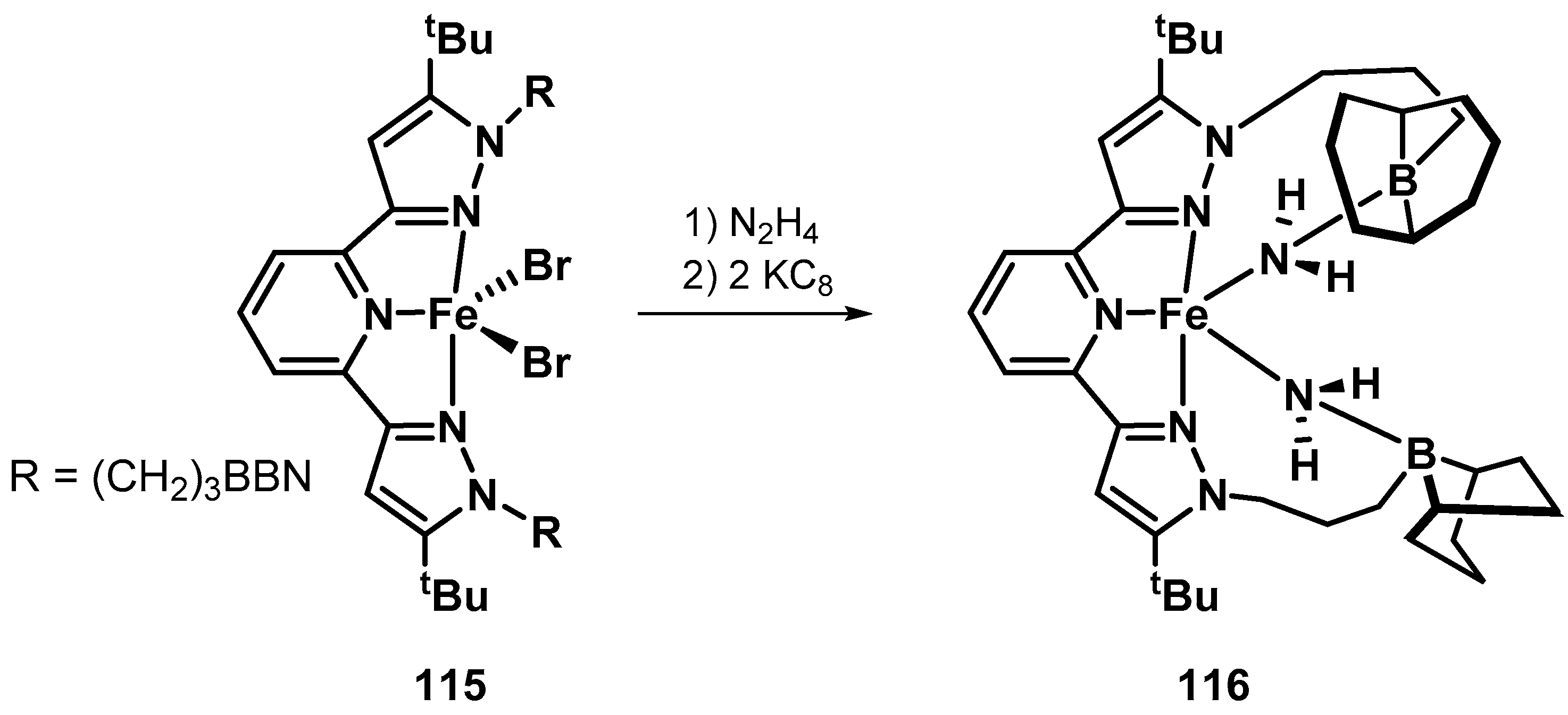{kind=link}
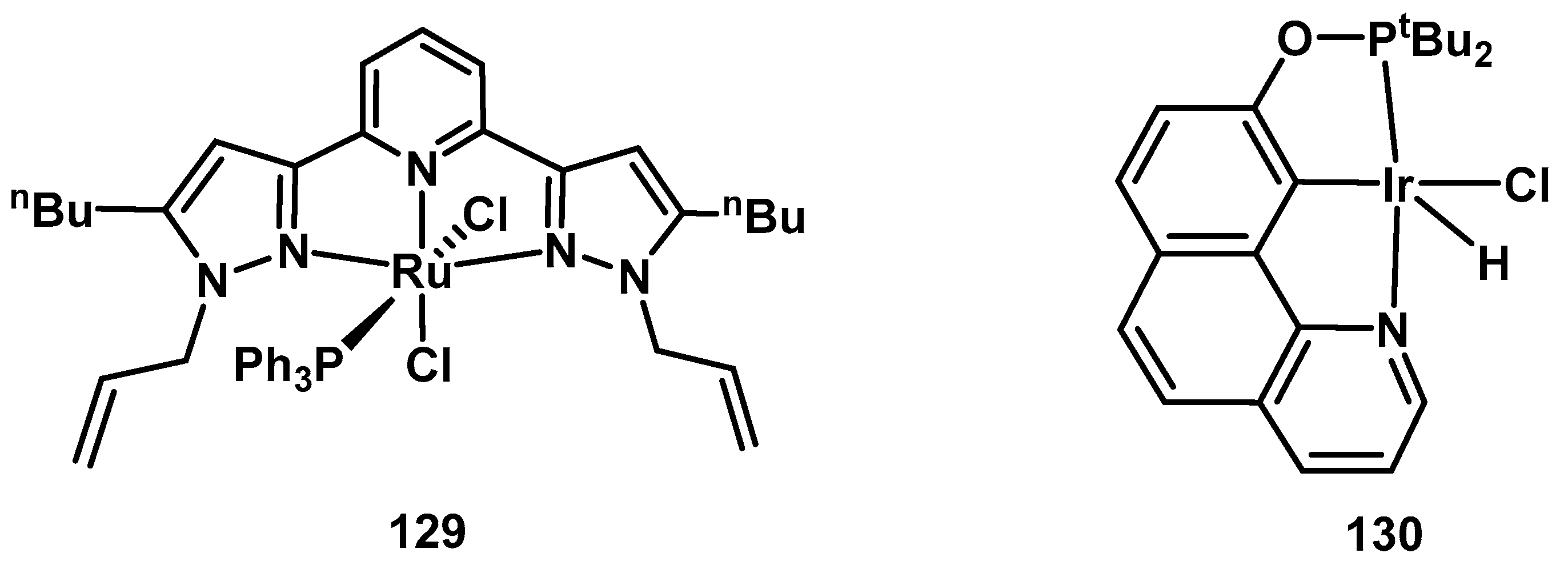{kind=link}
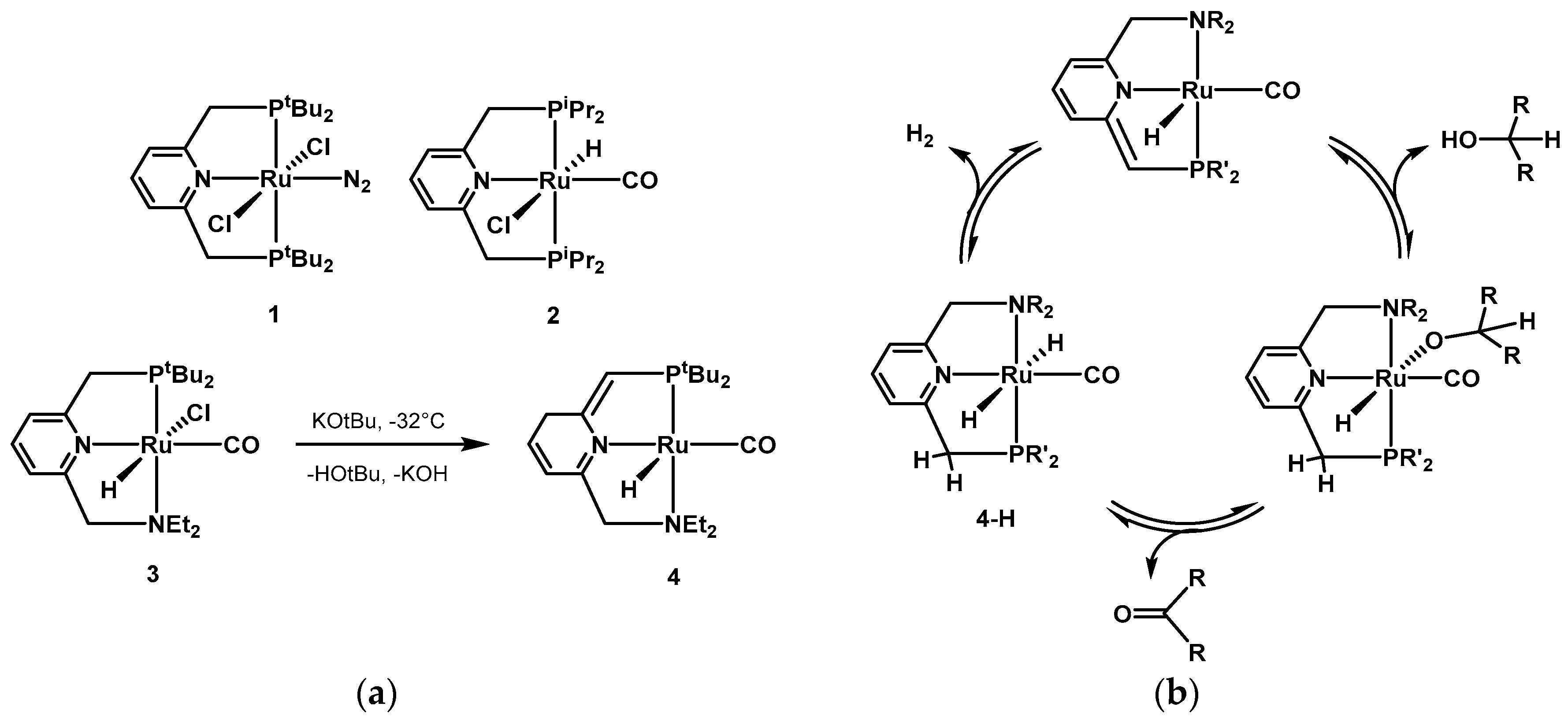{kind=link}
{kind=link}
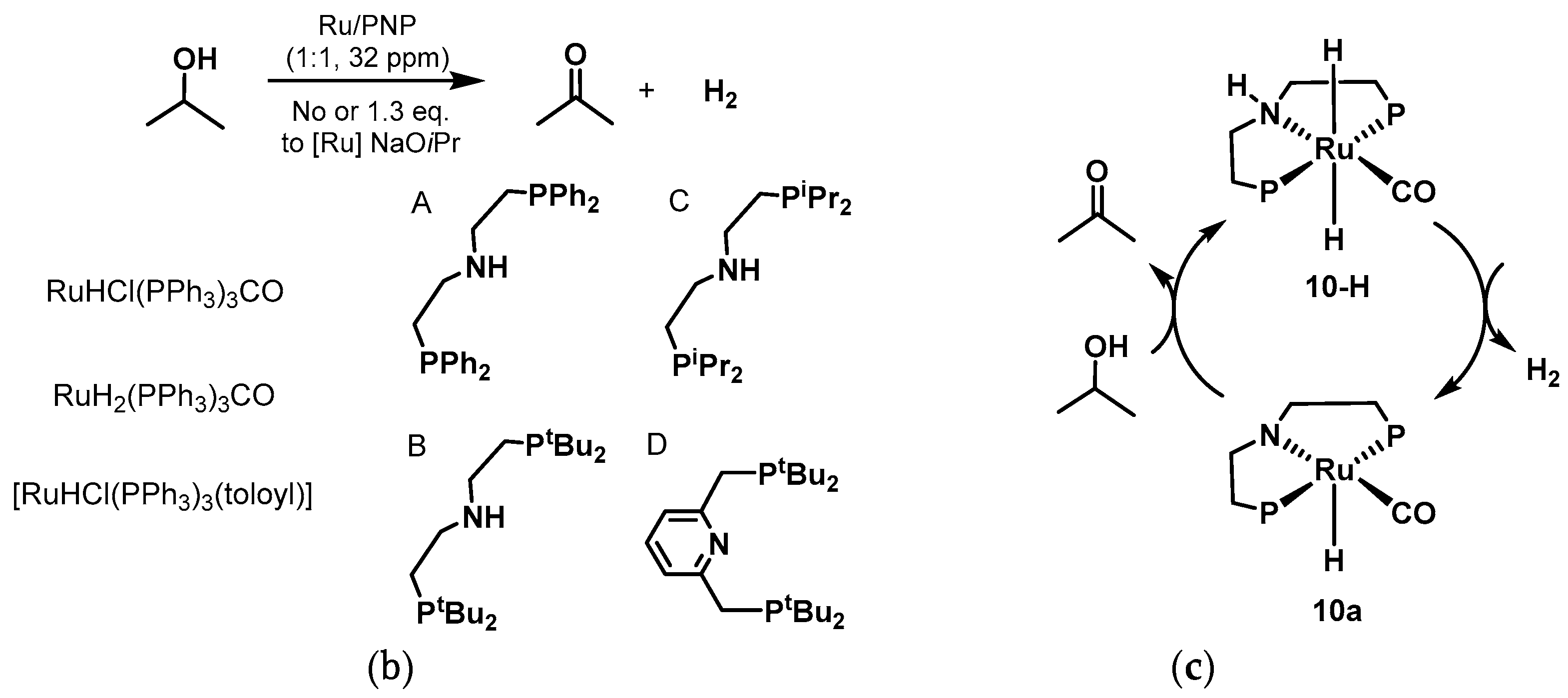{kind=link}
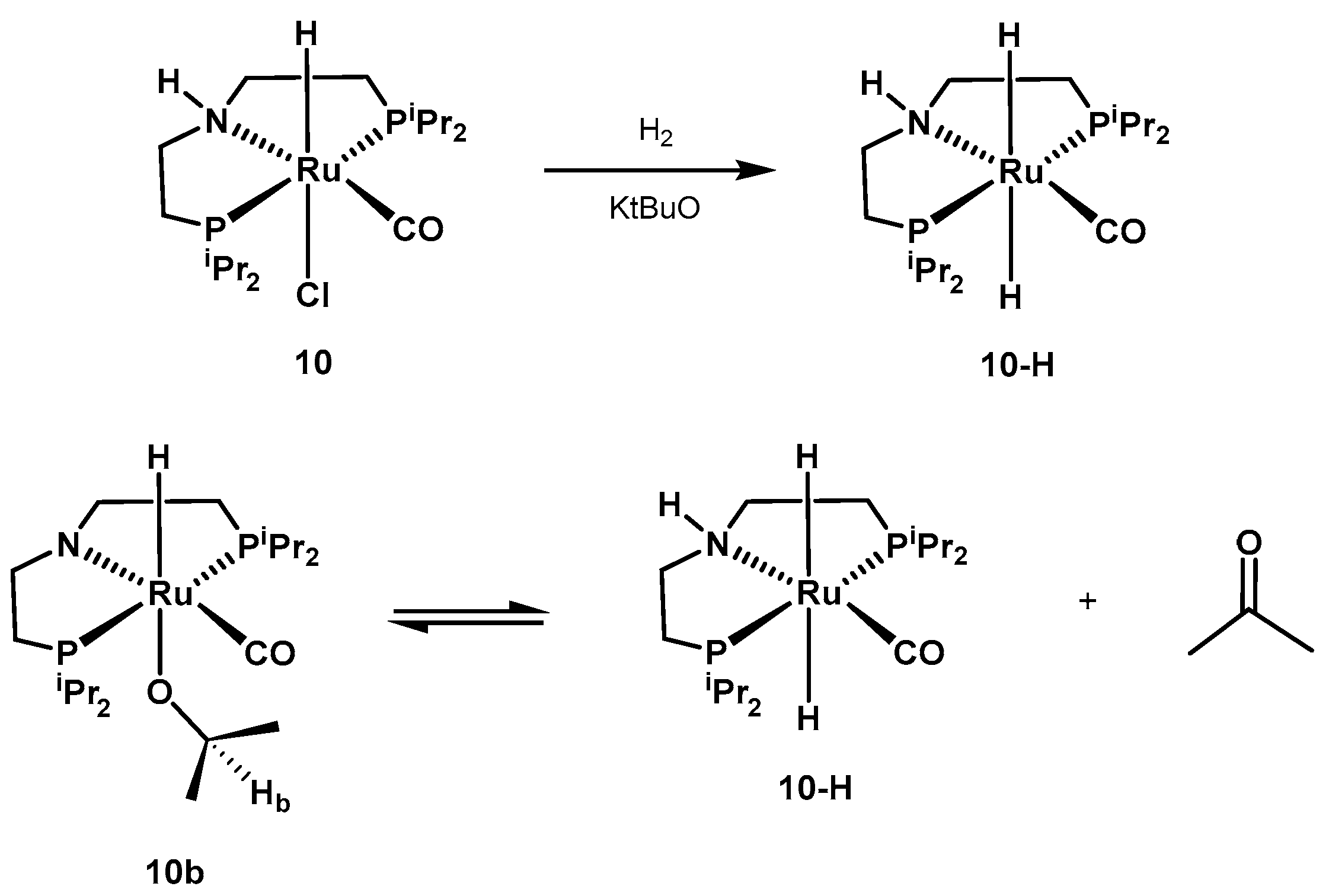{kind=link}
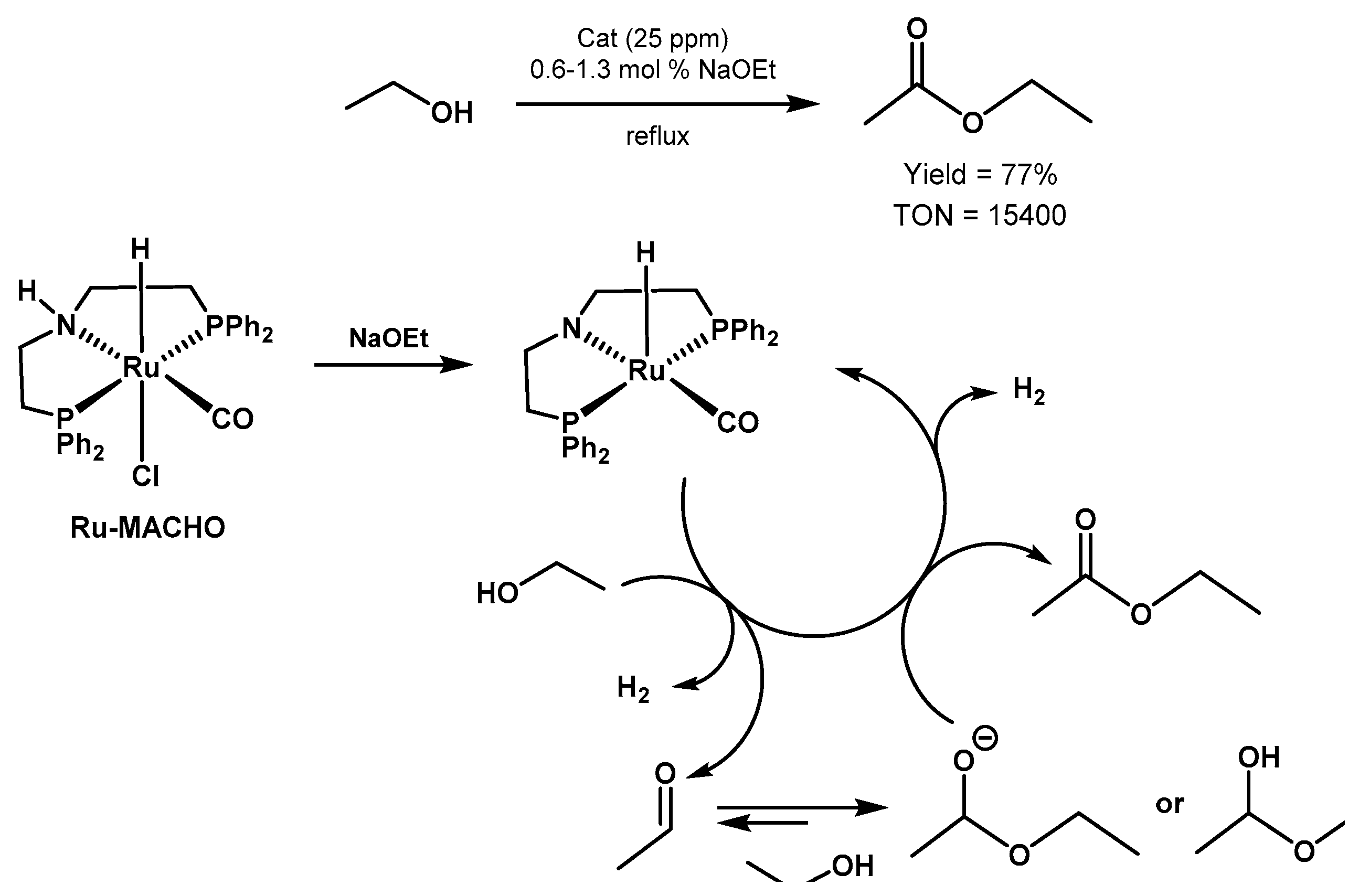{kind=link}
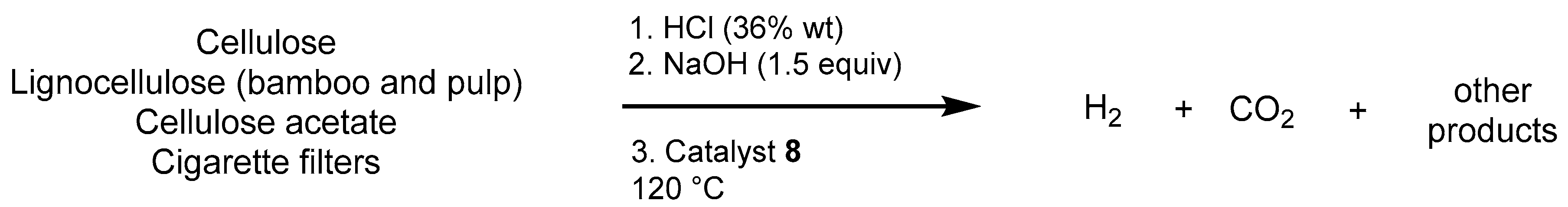{kind=link}
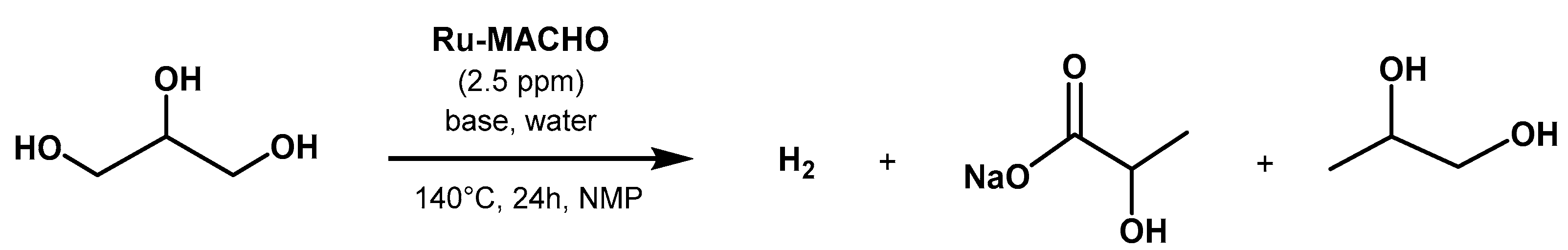{kind=link}
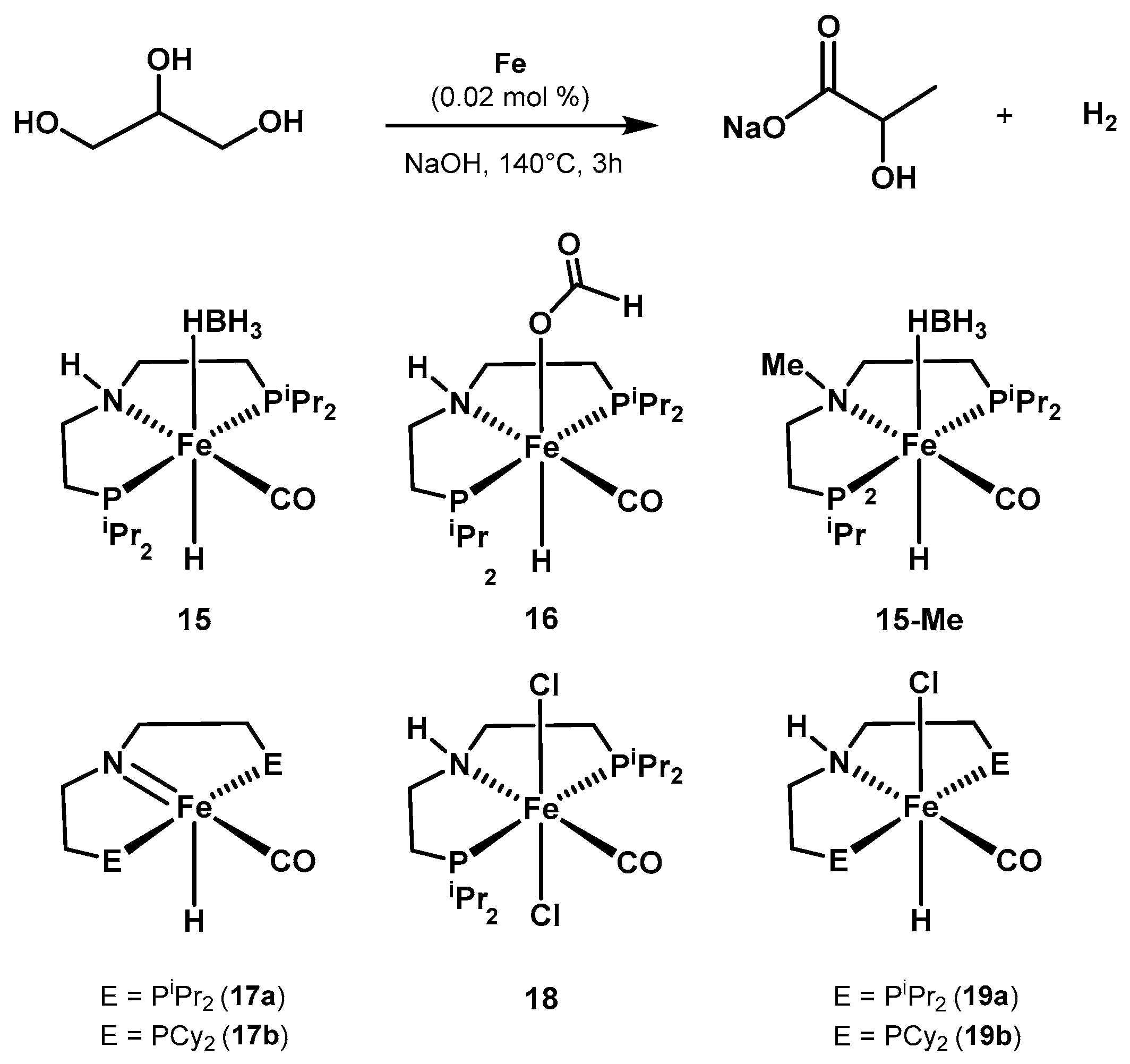{kind=link}
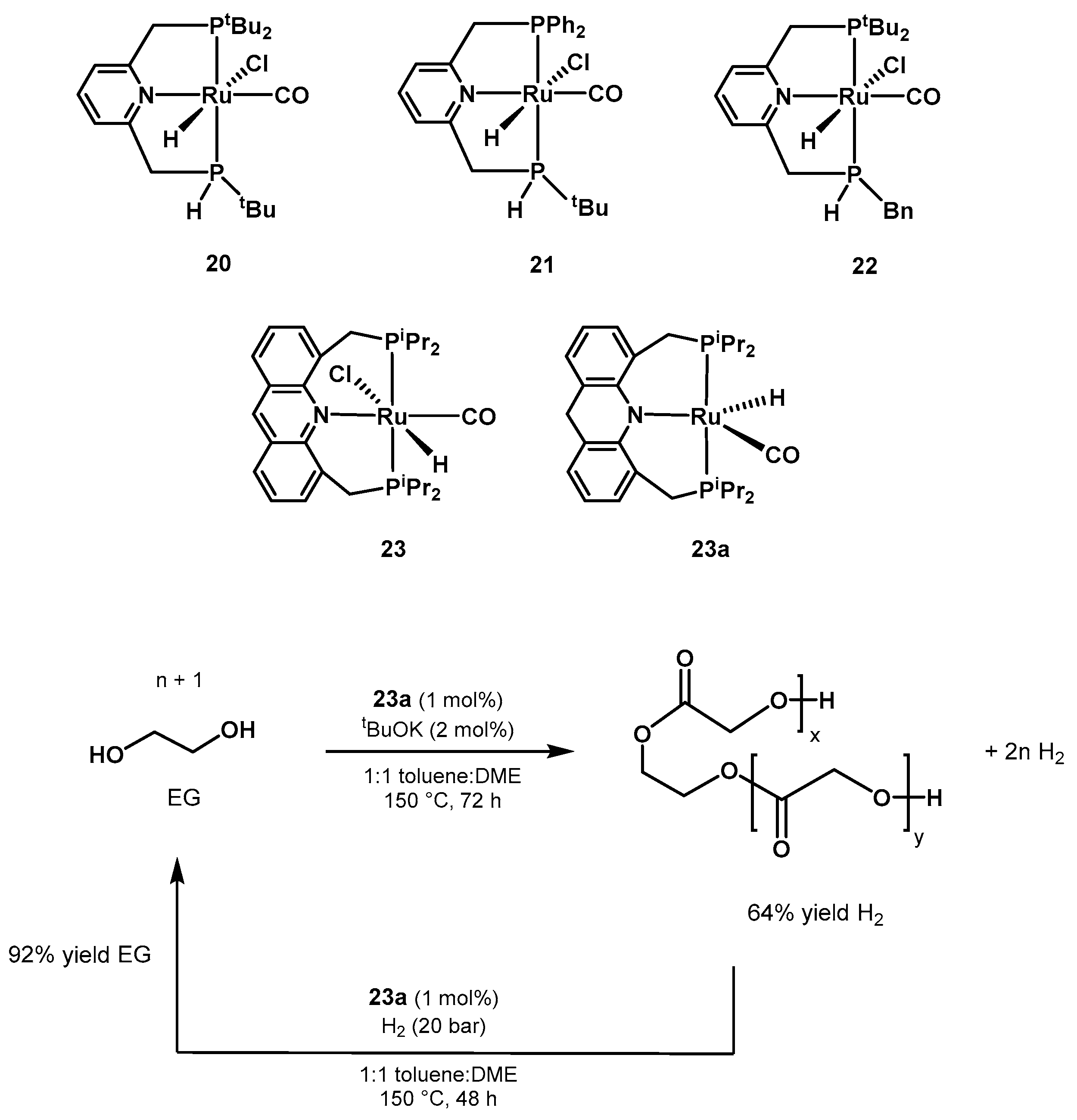{kind=link}
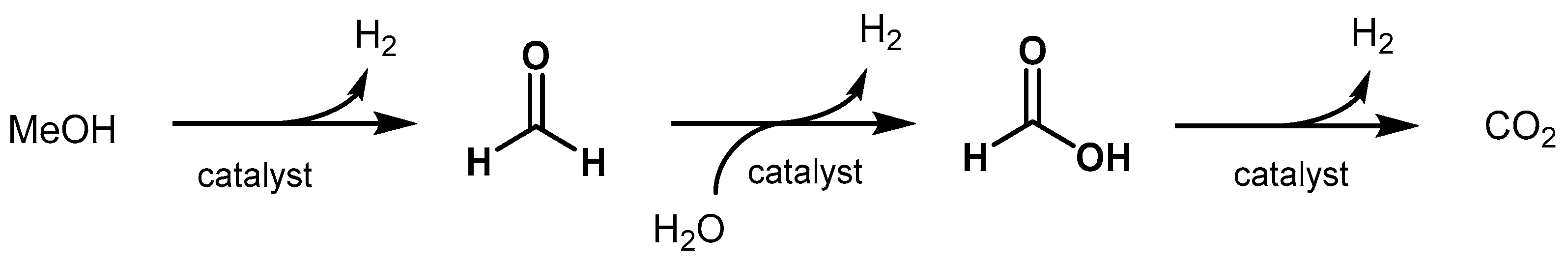{kind=link}
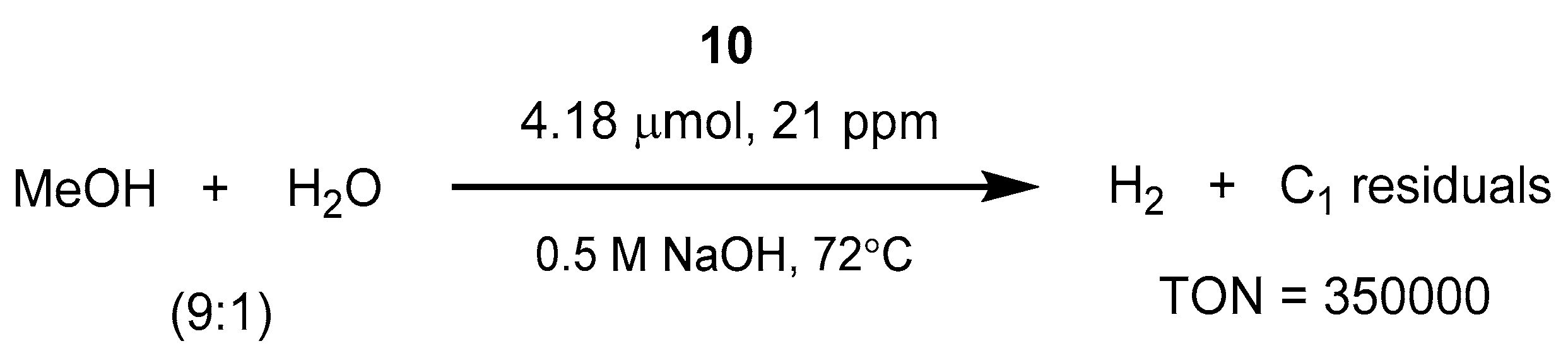{kind=link}
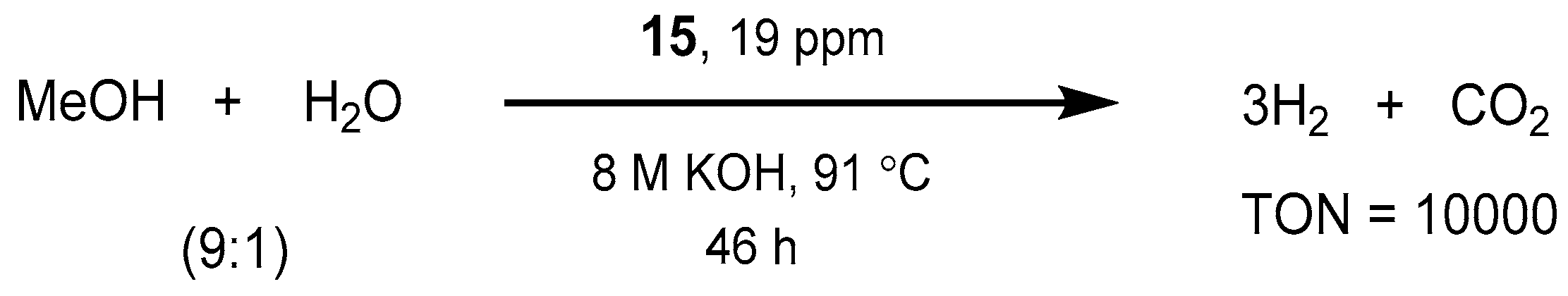{kind=link}
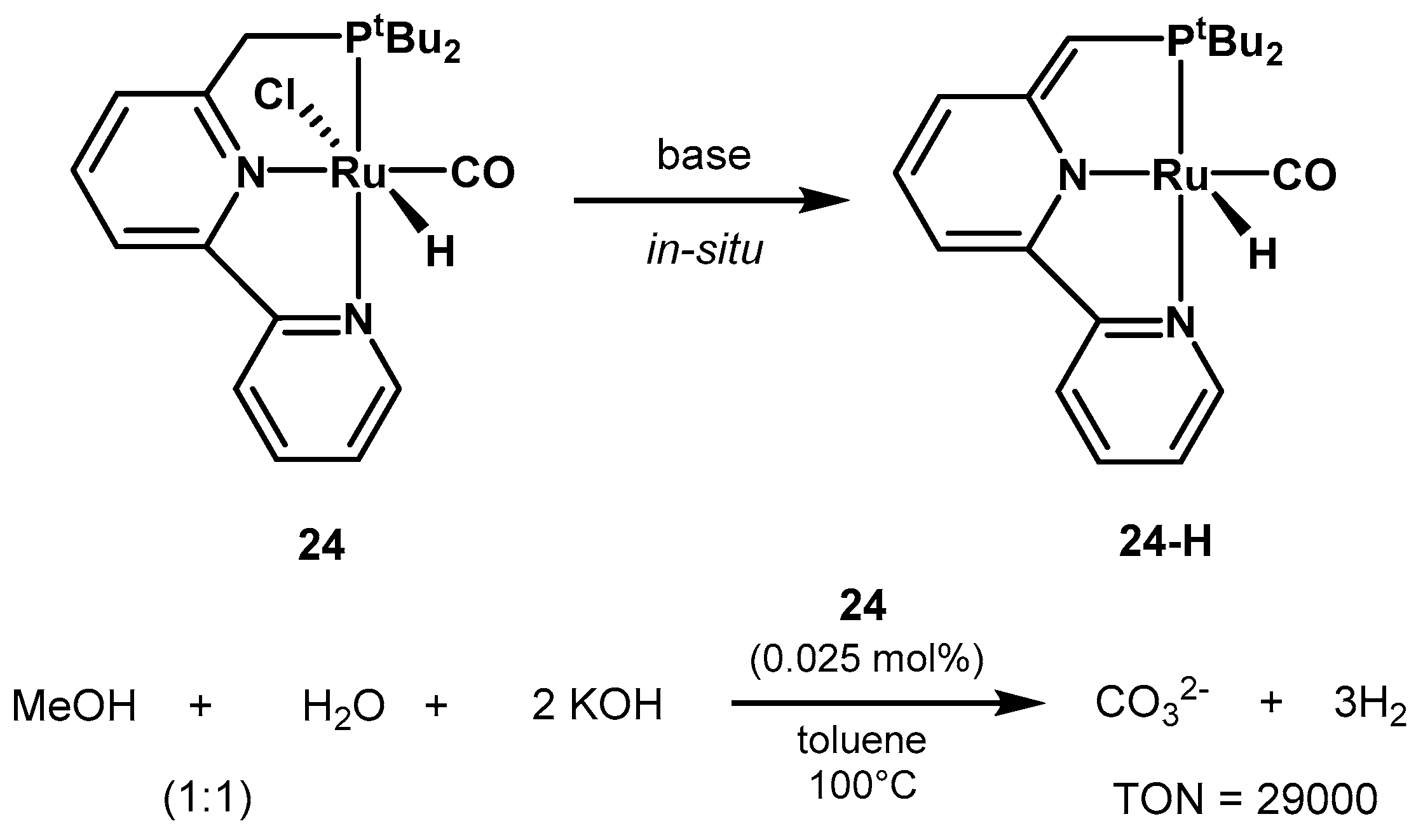{kind=link}
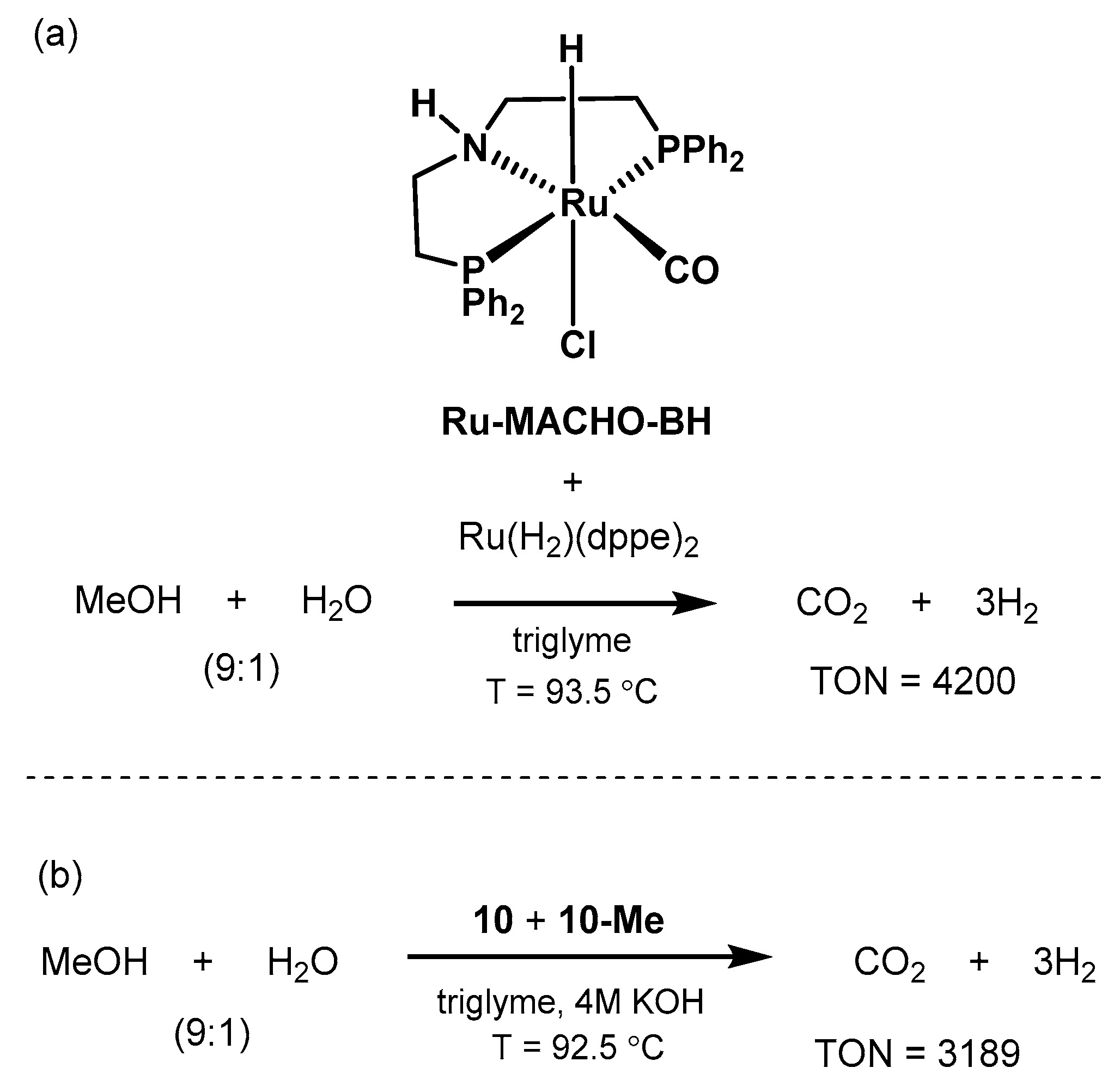{kind=link}
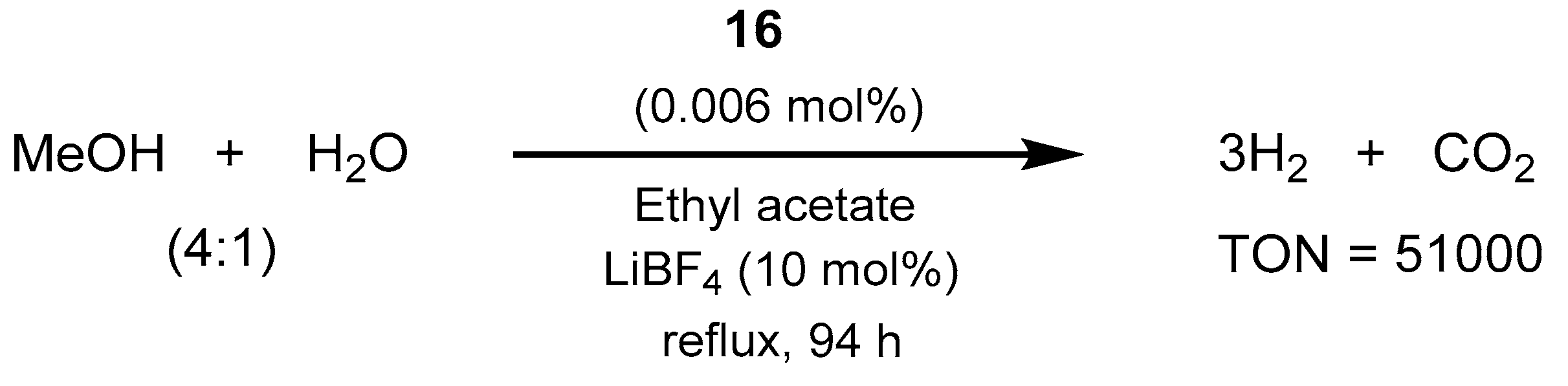{kind=link}
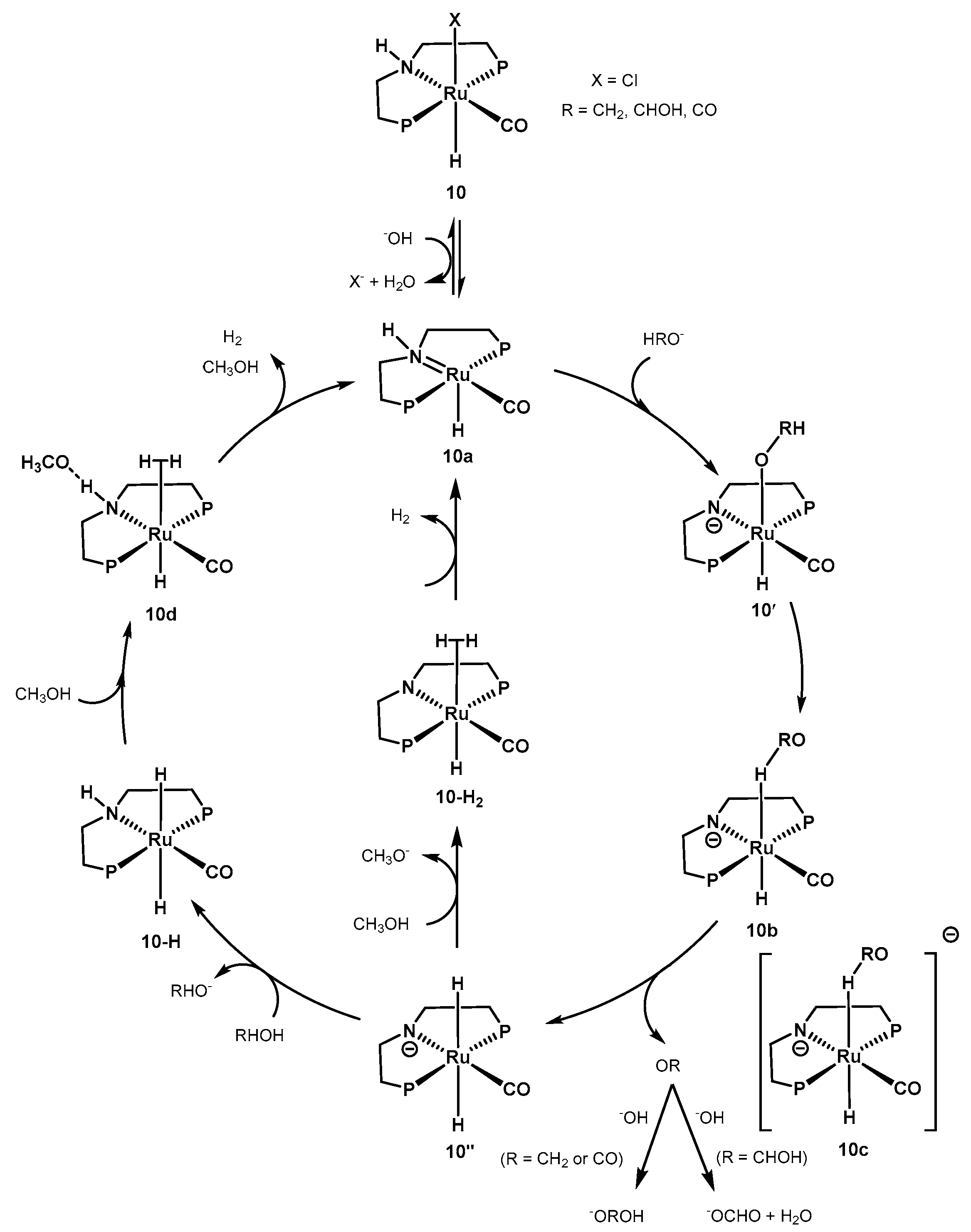{kind=link}
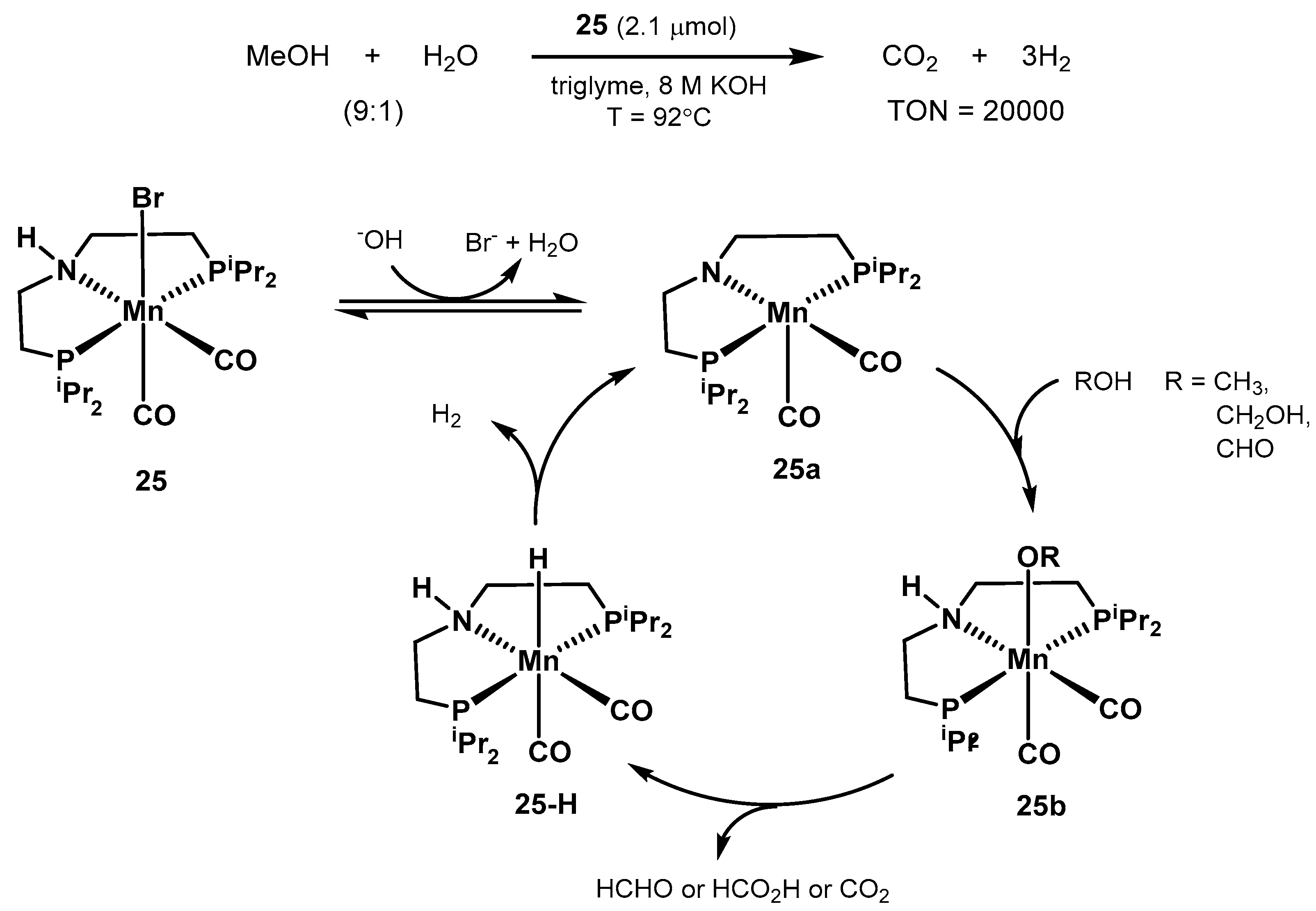{kind=link}
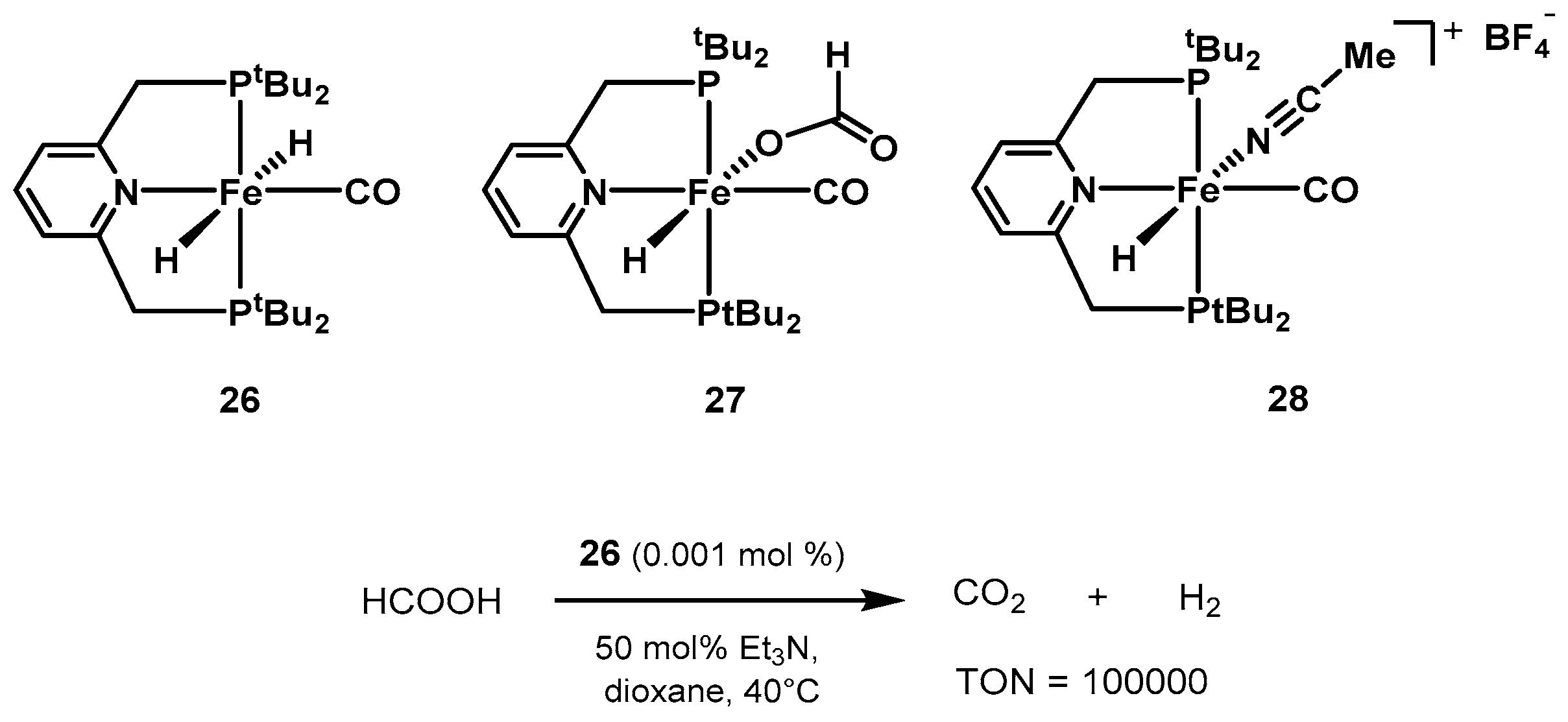{kind=link}
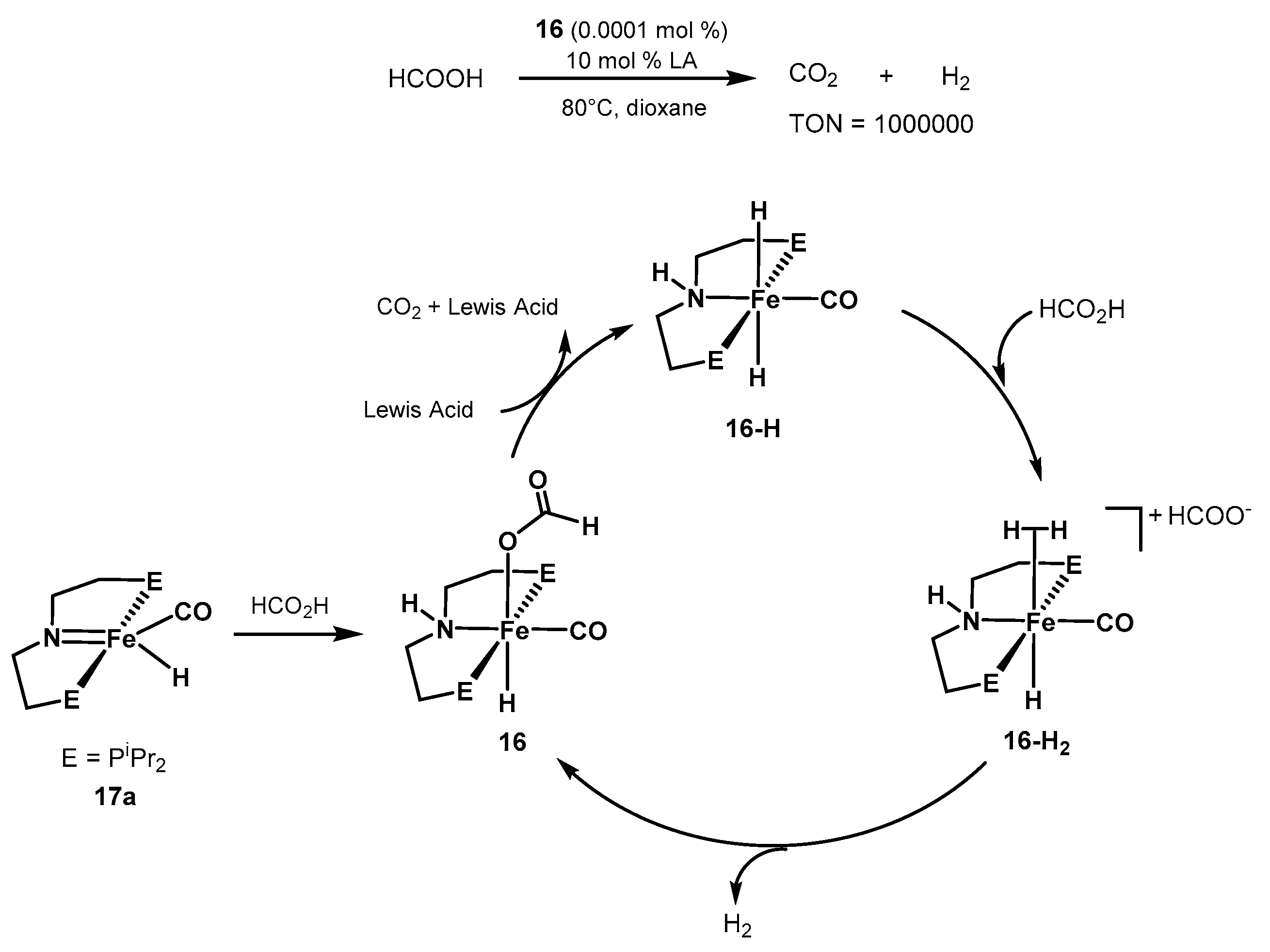{kind=link}
{kind=link}
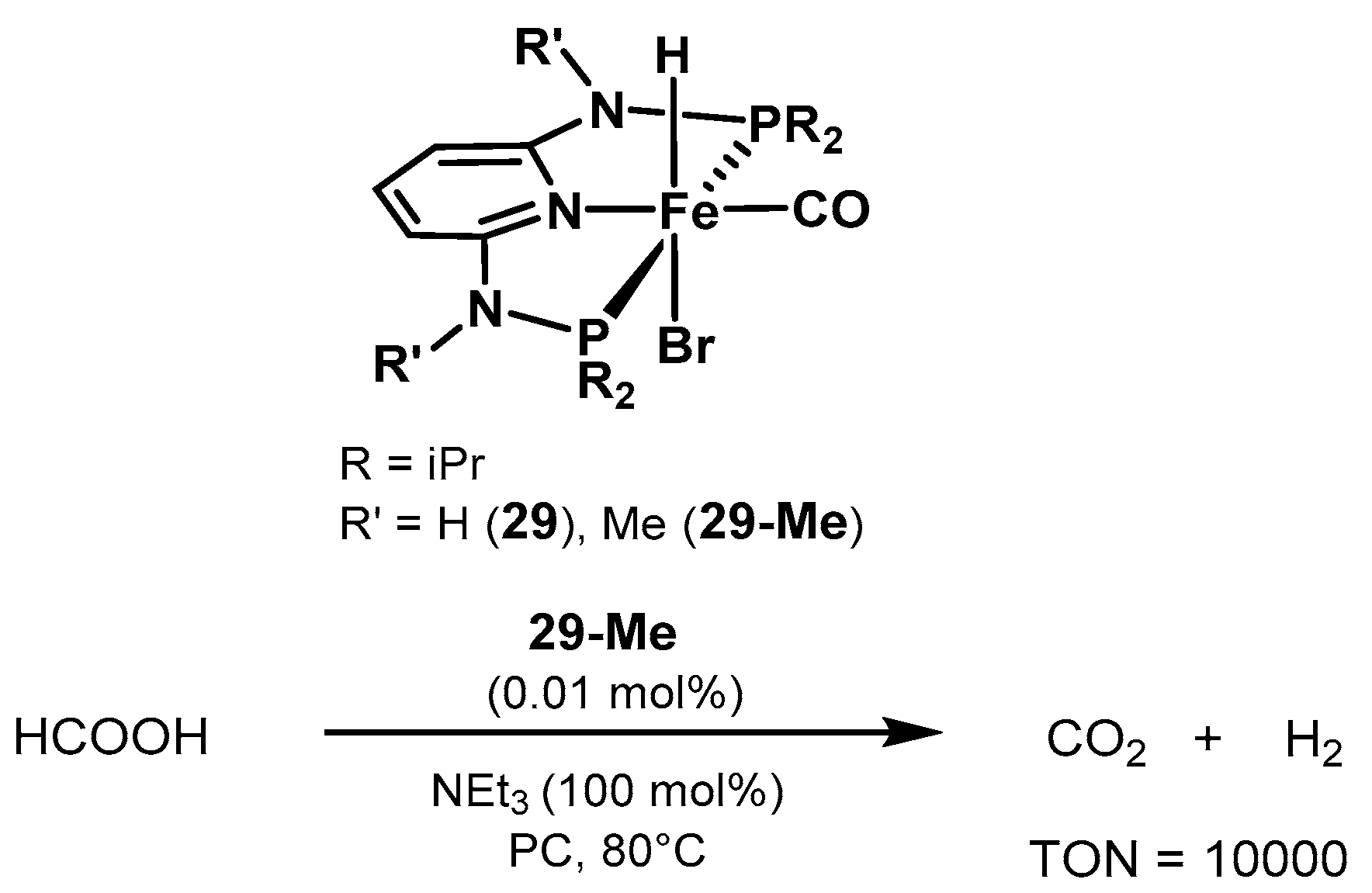{kind=link}
{kind=link}
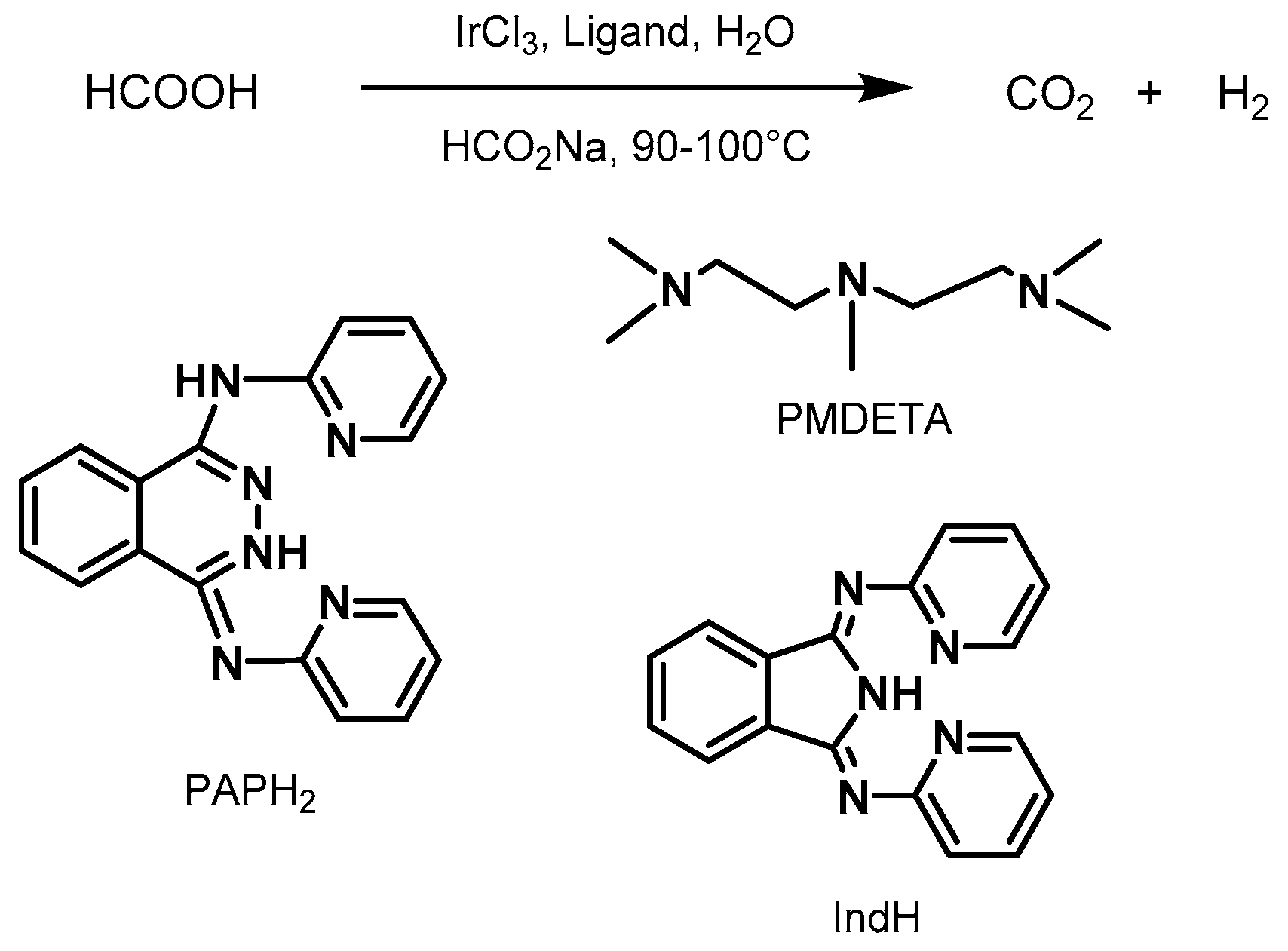{kind=link}
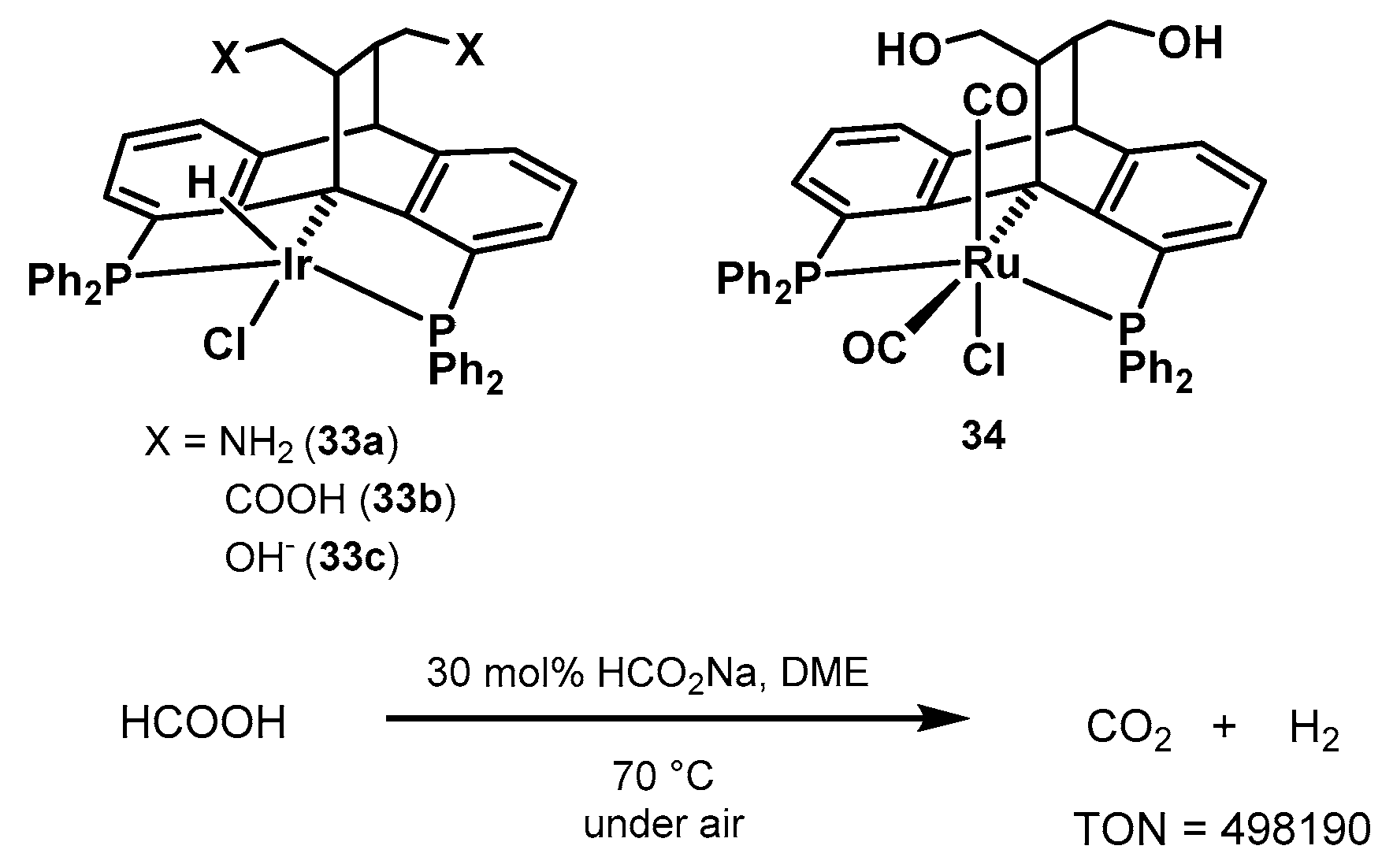{kind=link}
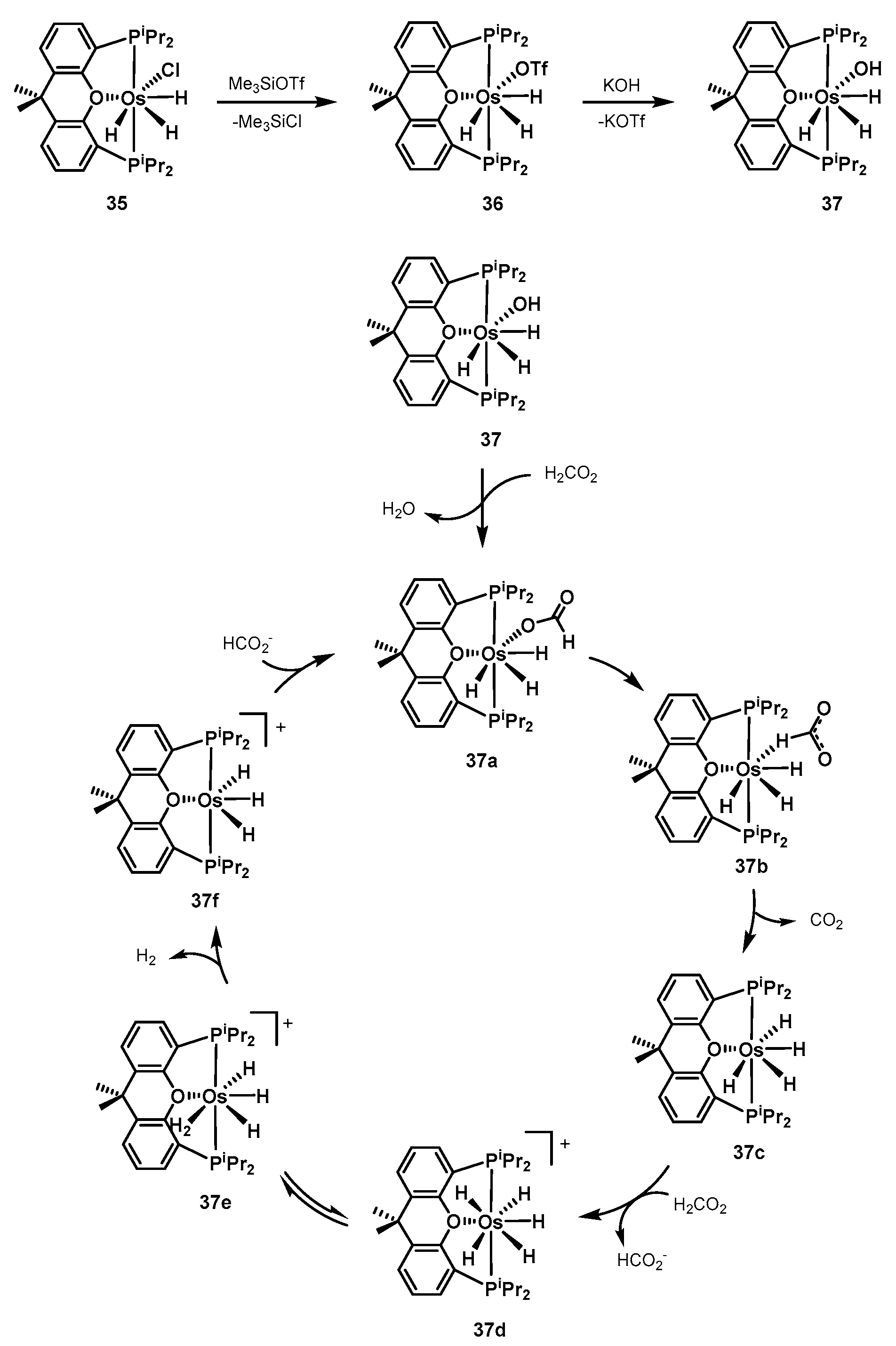{kind=link}
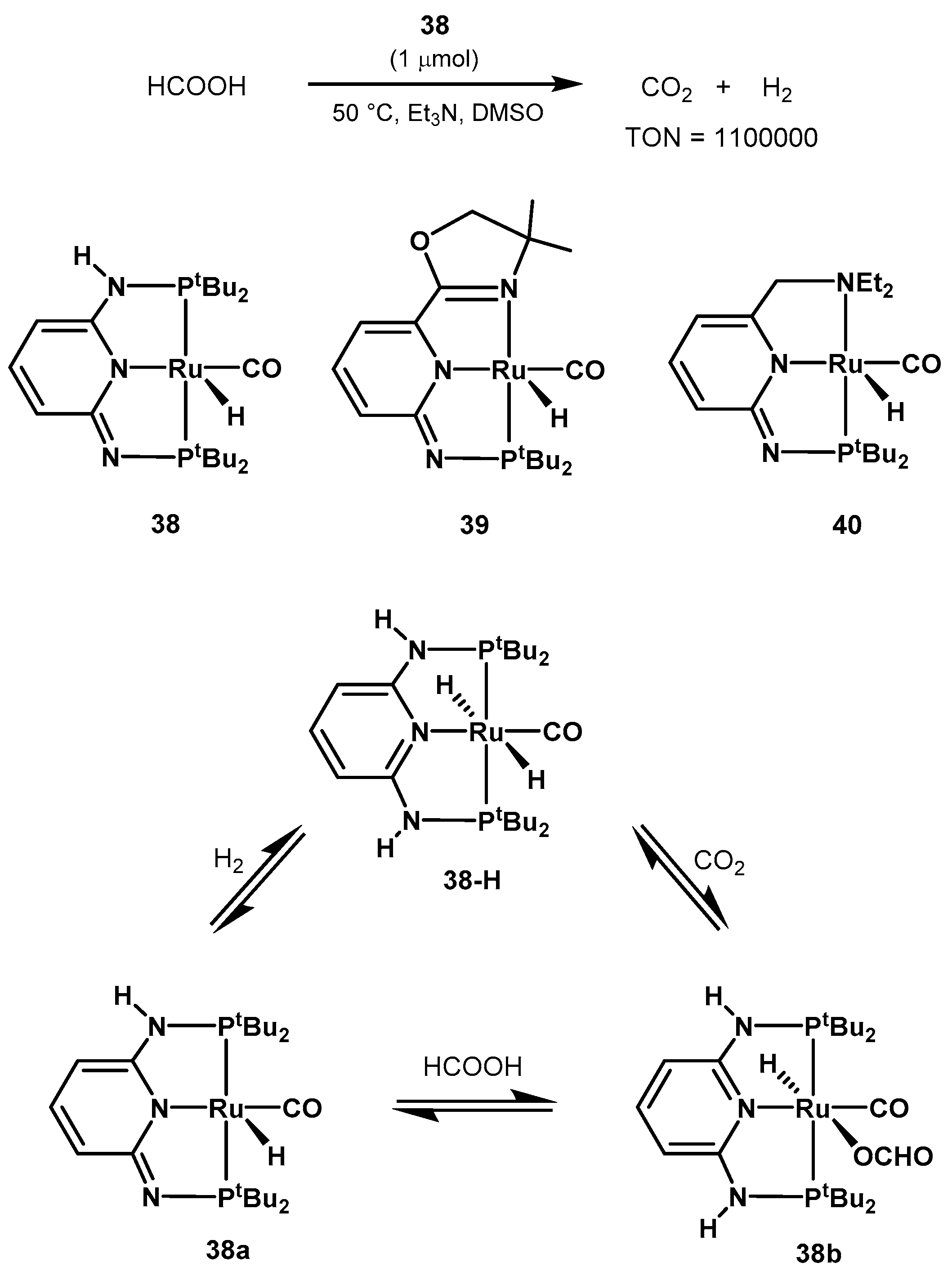{kind=link}
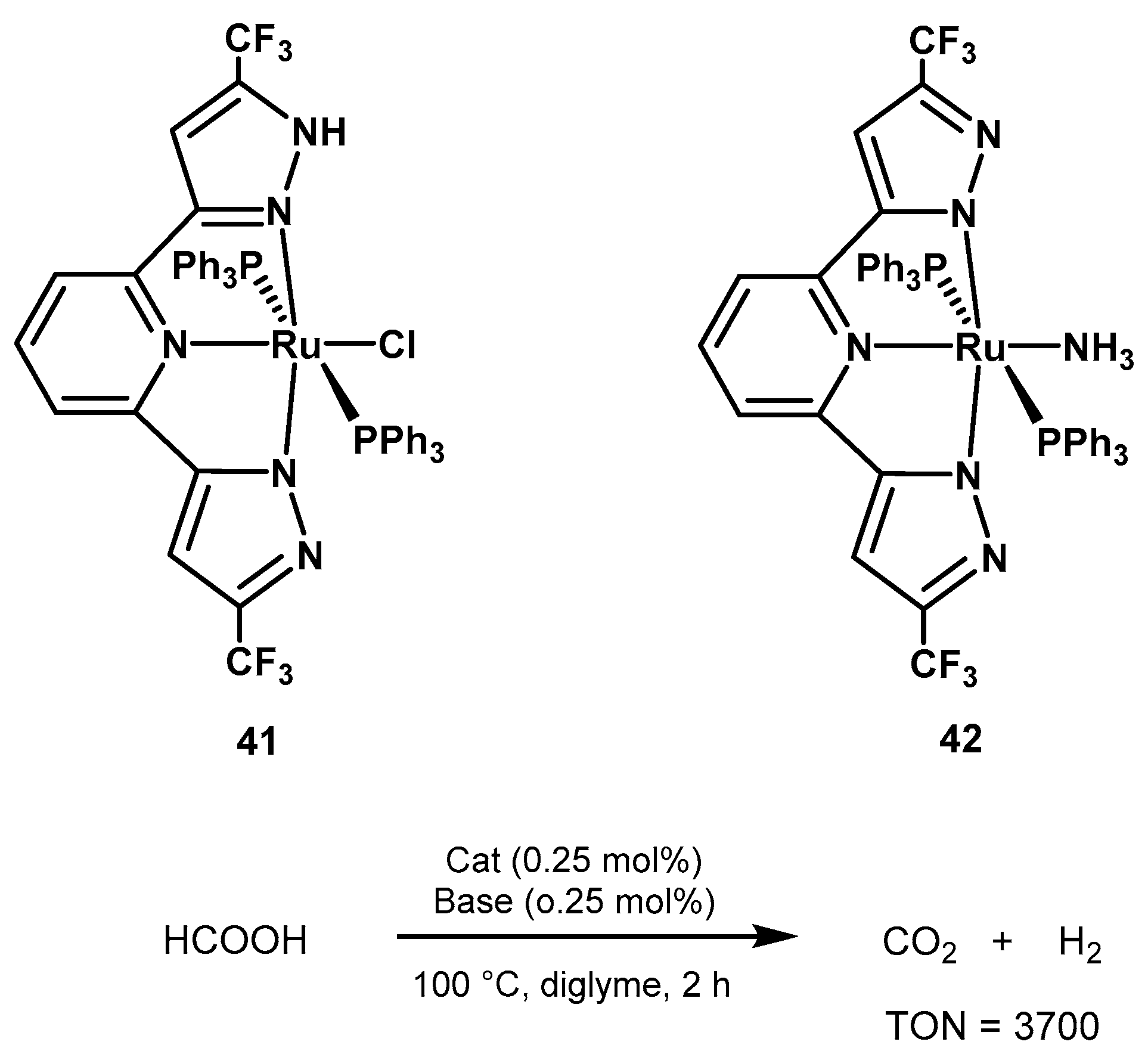{kind=link}
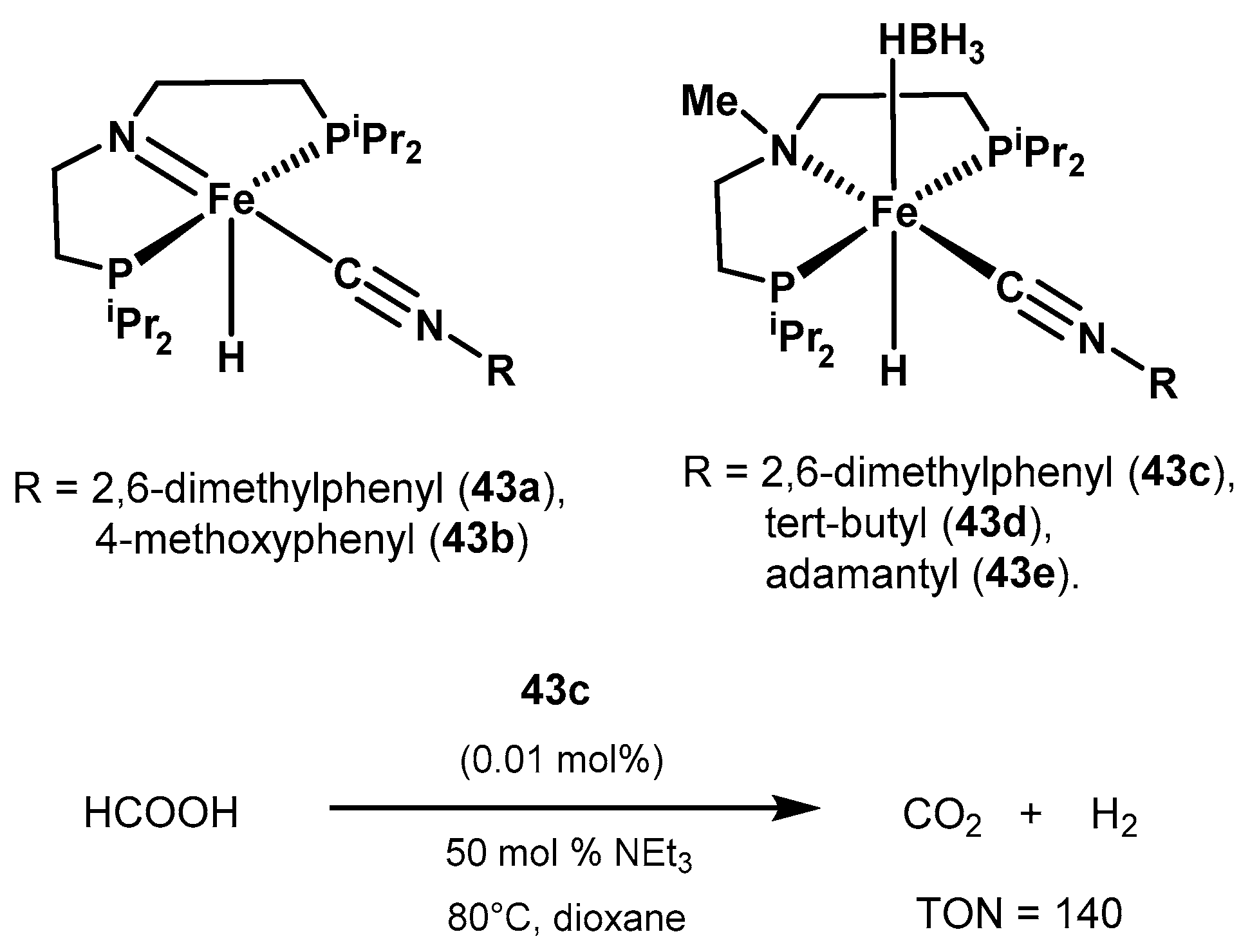{kind=link}
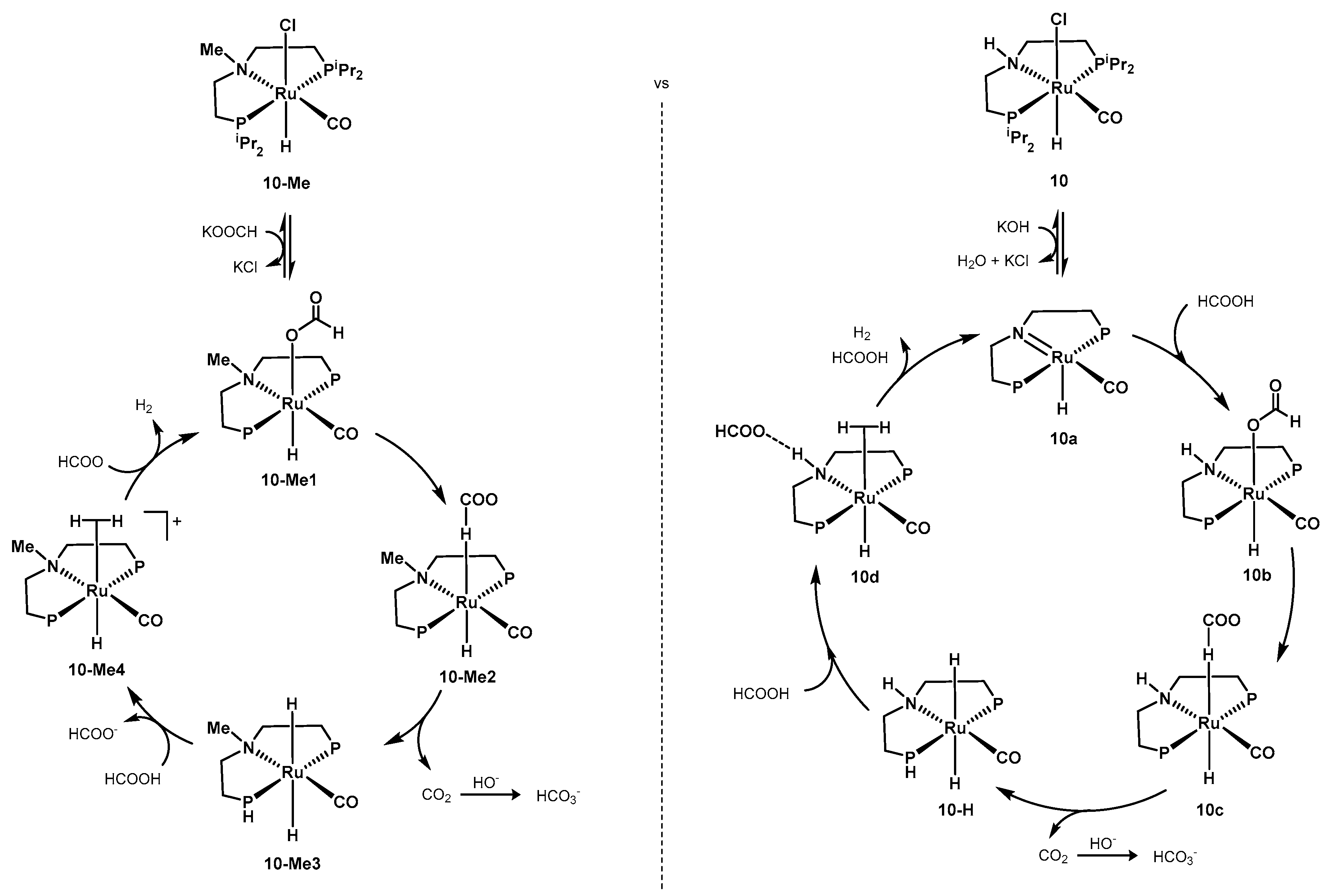{kind=link}
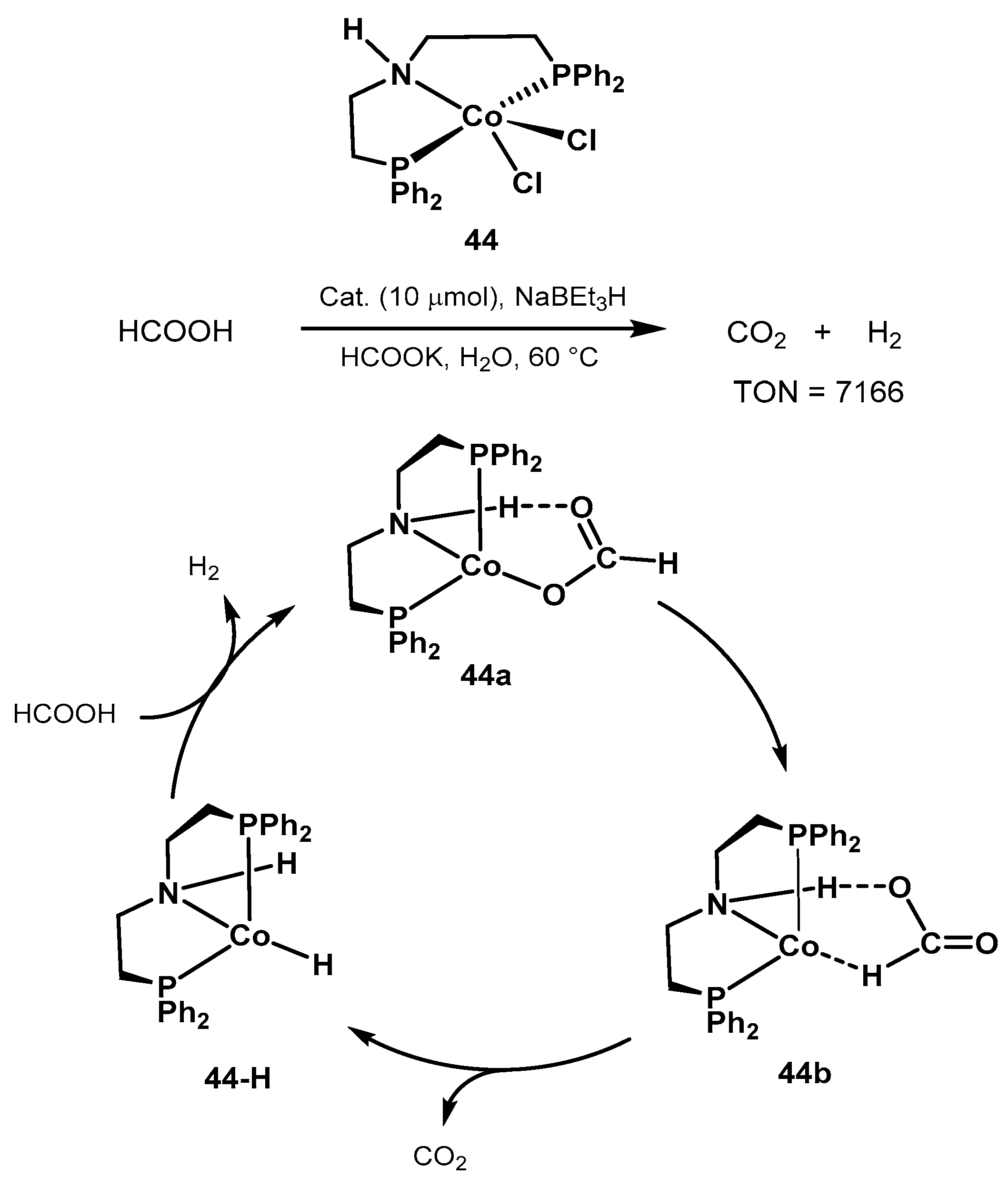{kind=link}
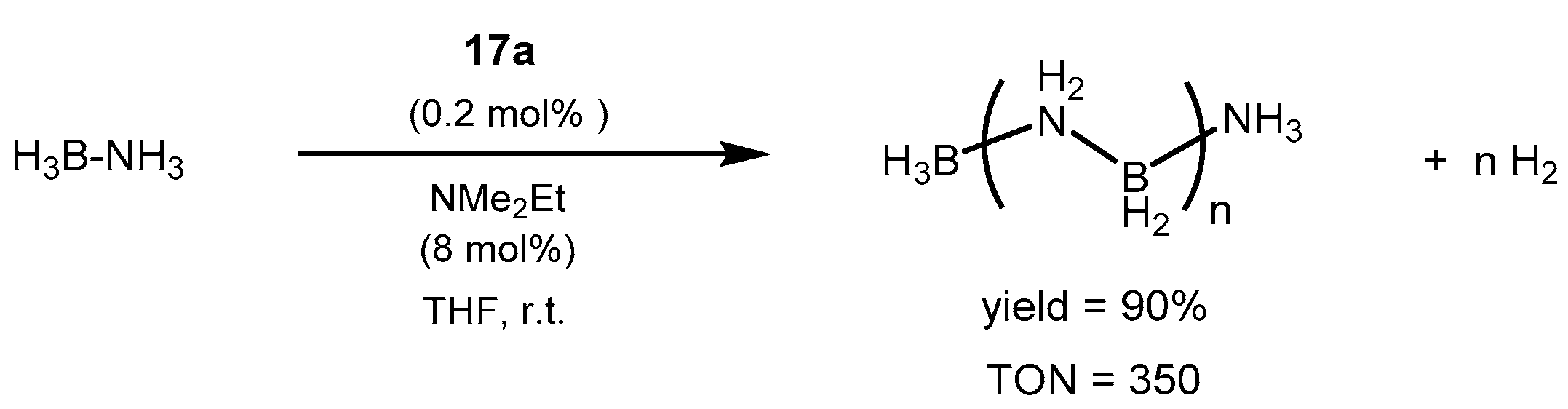{kind=link}
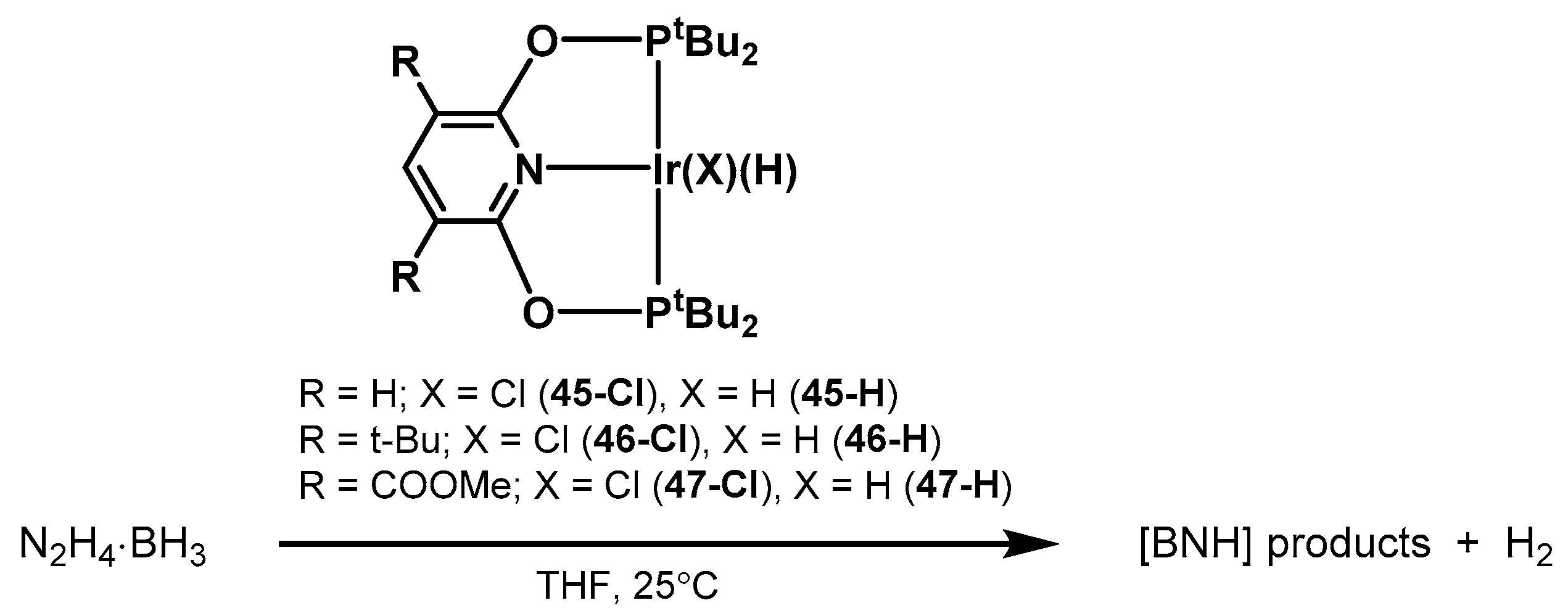{kind=link}
{kind=link}
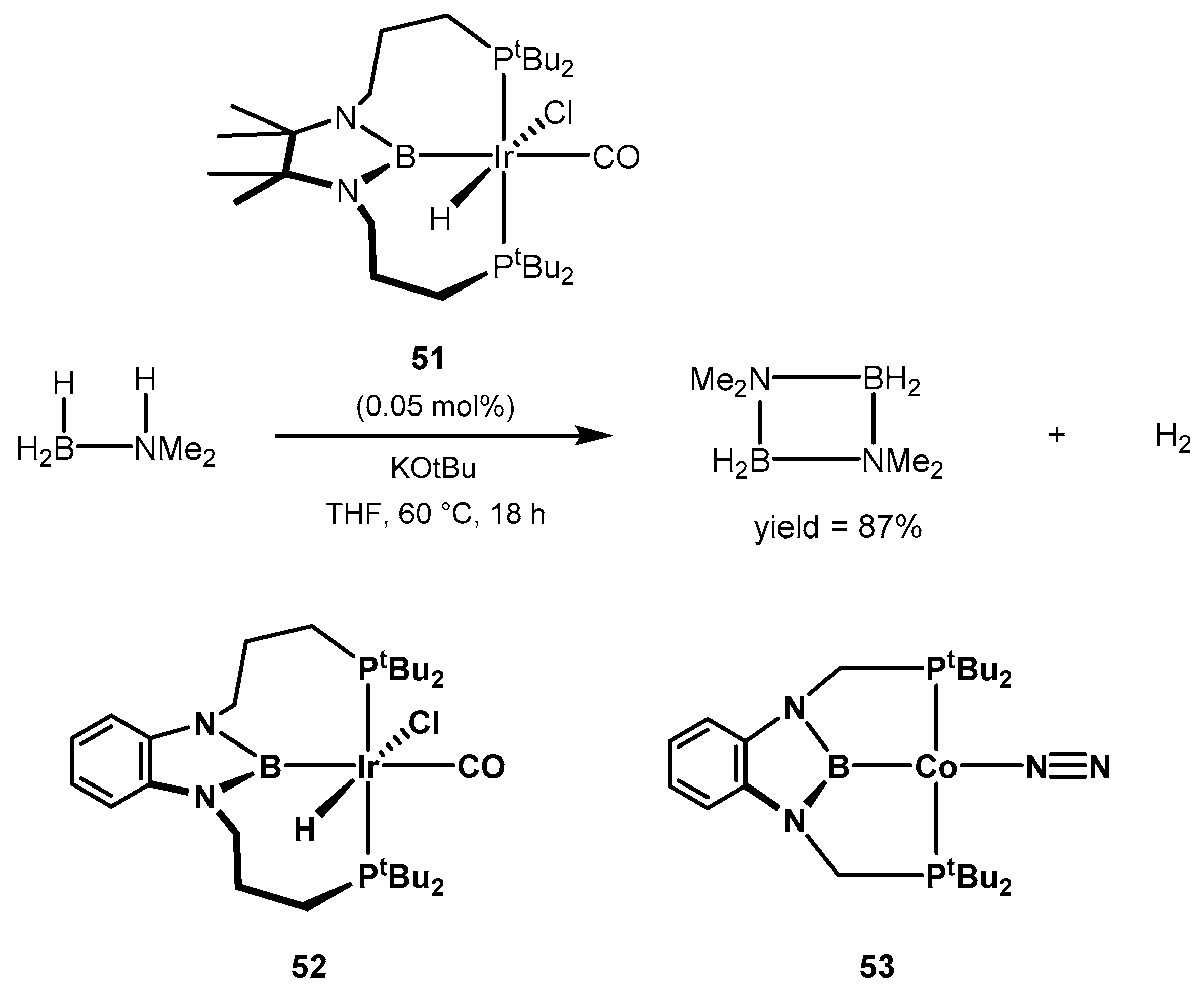{kind=link}
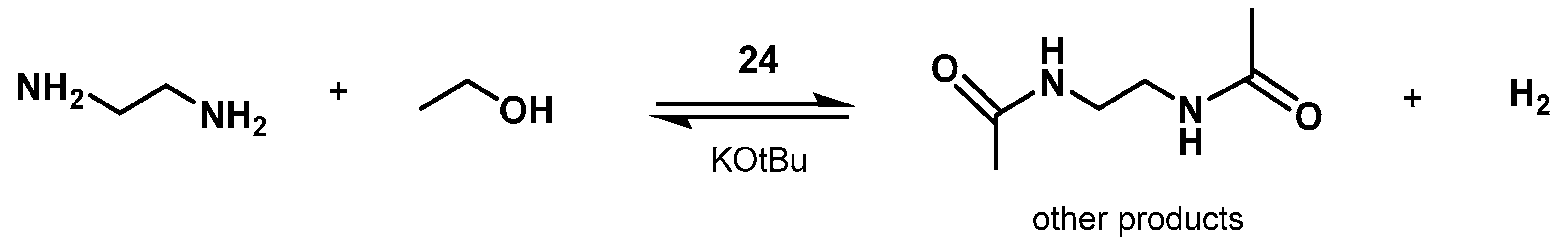{kind=link}
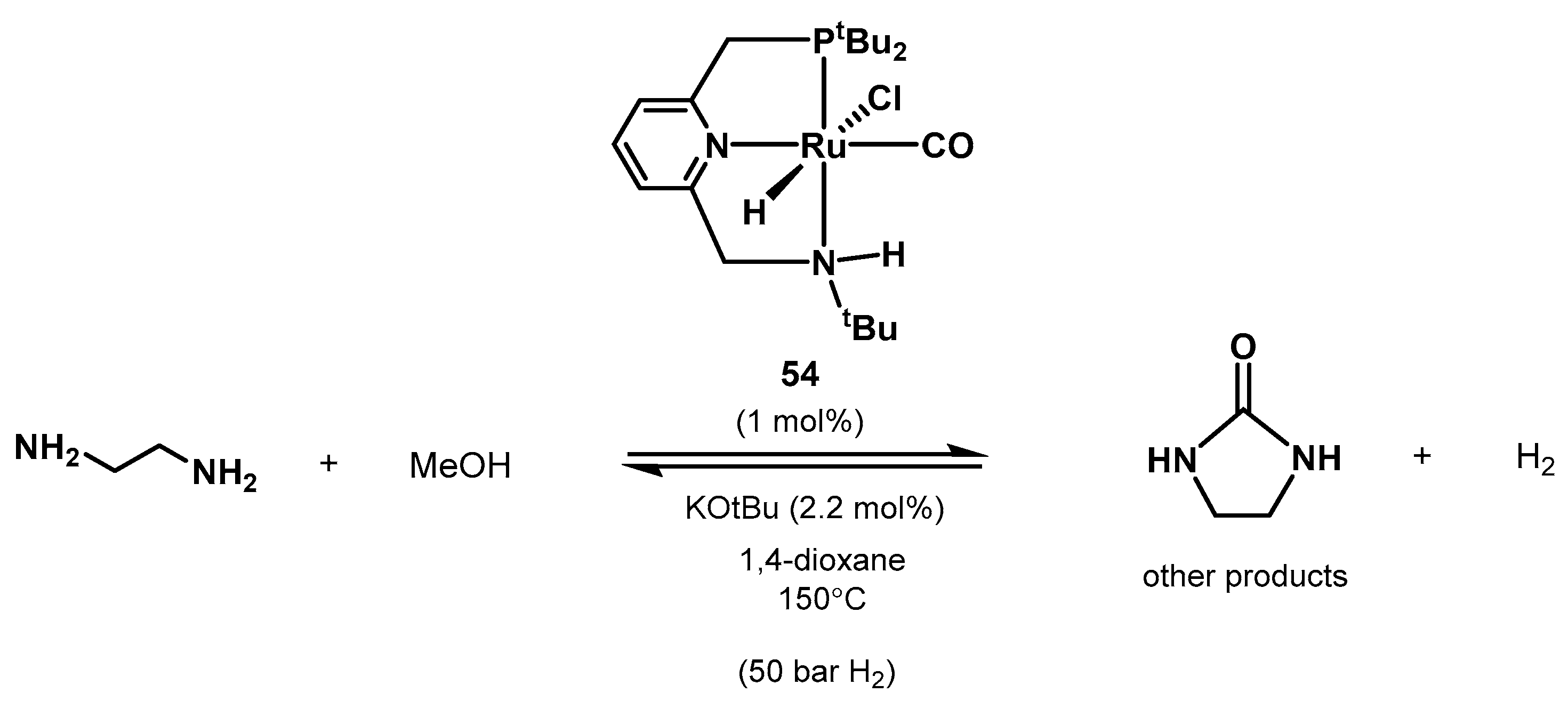{kind=link}
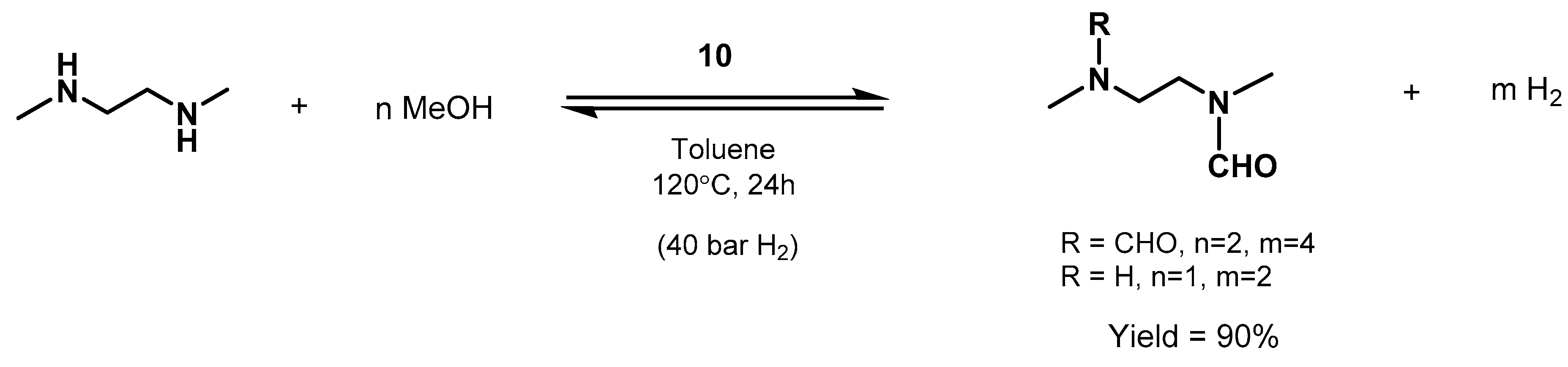{kind=link}
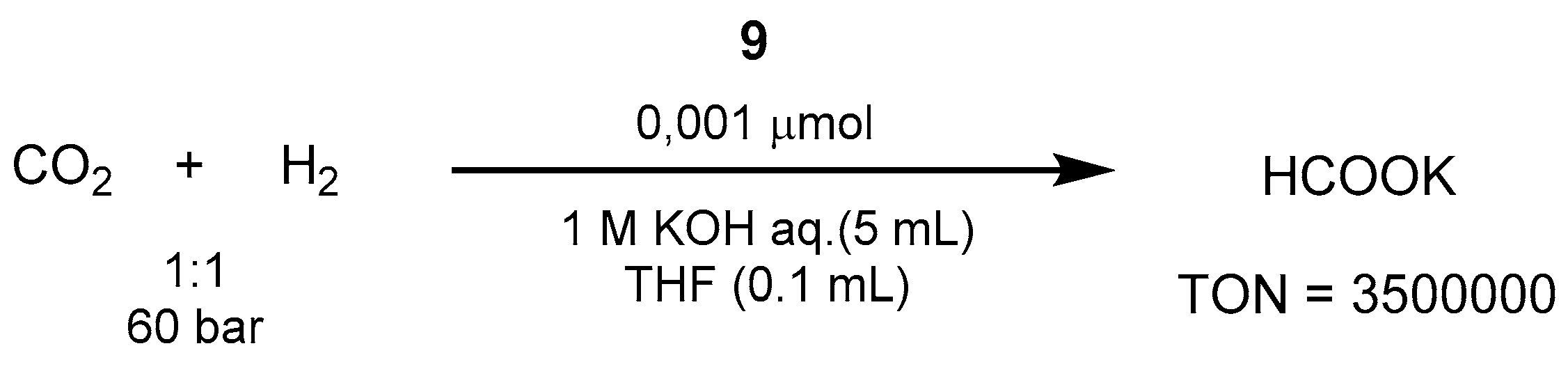{kind=link}
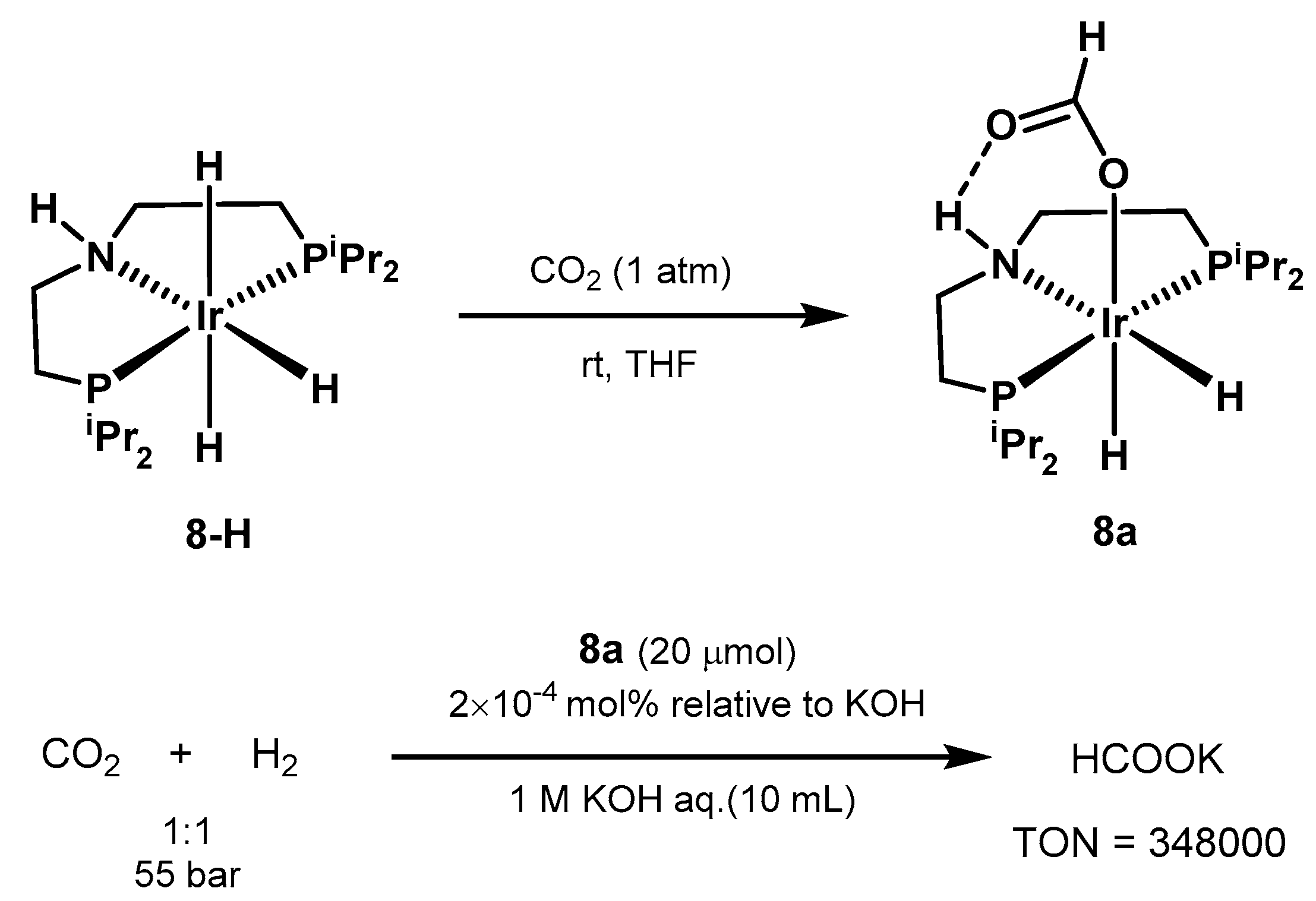{kind=link}
{kind=link}
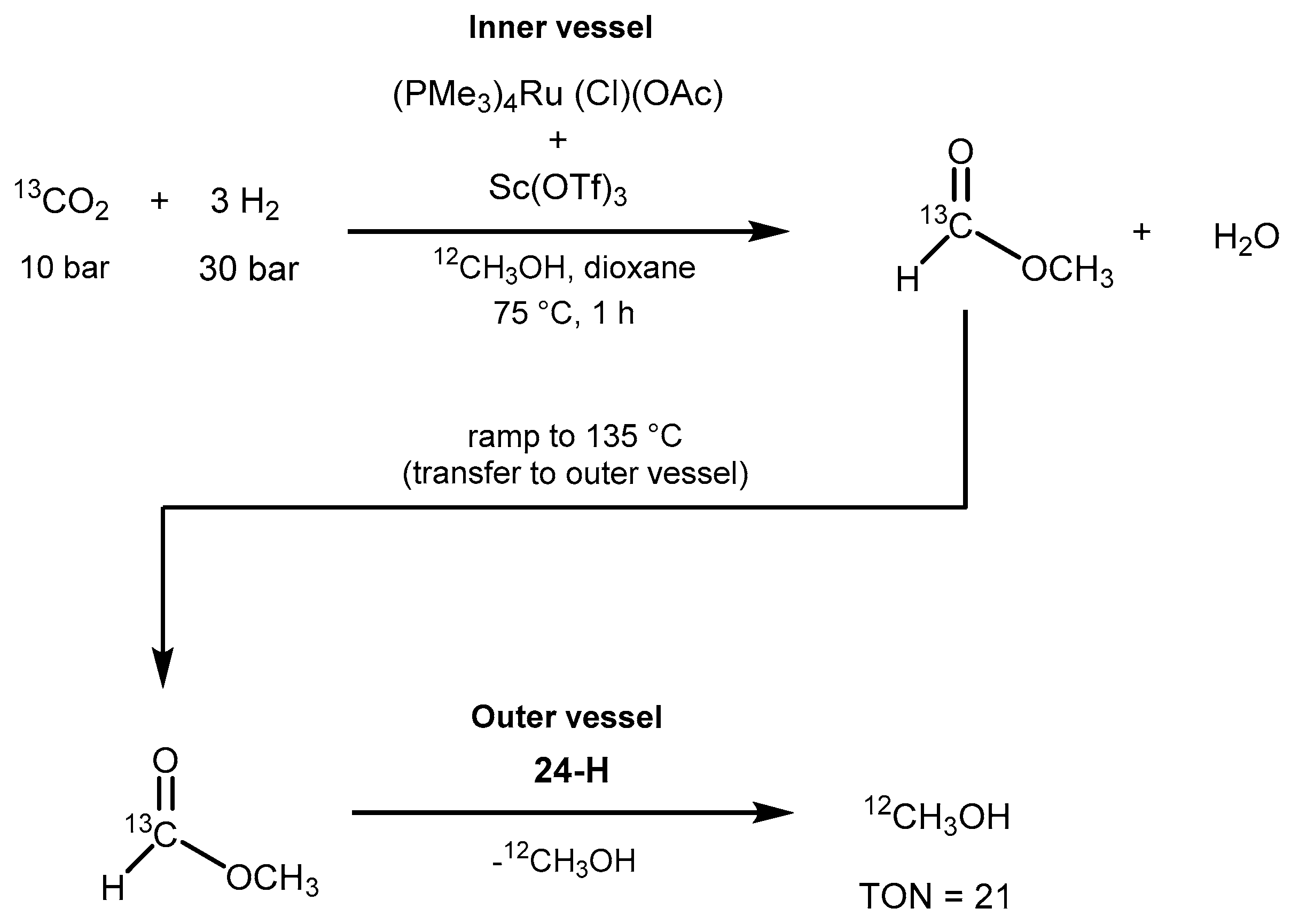{kind=link}
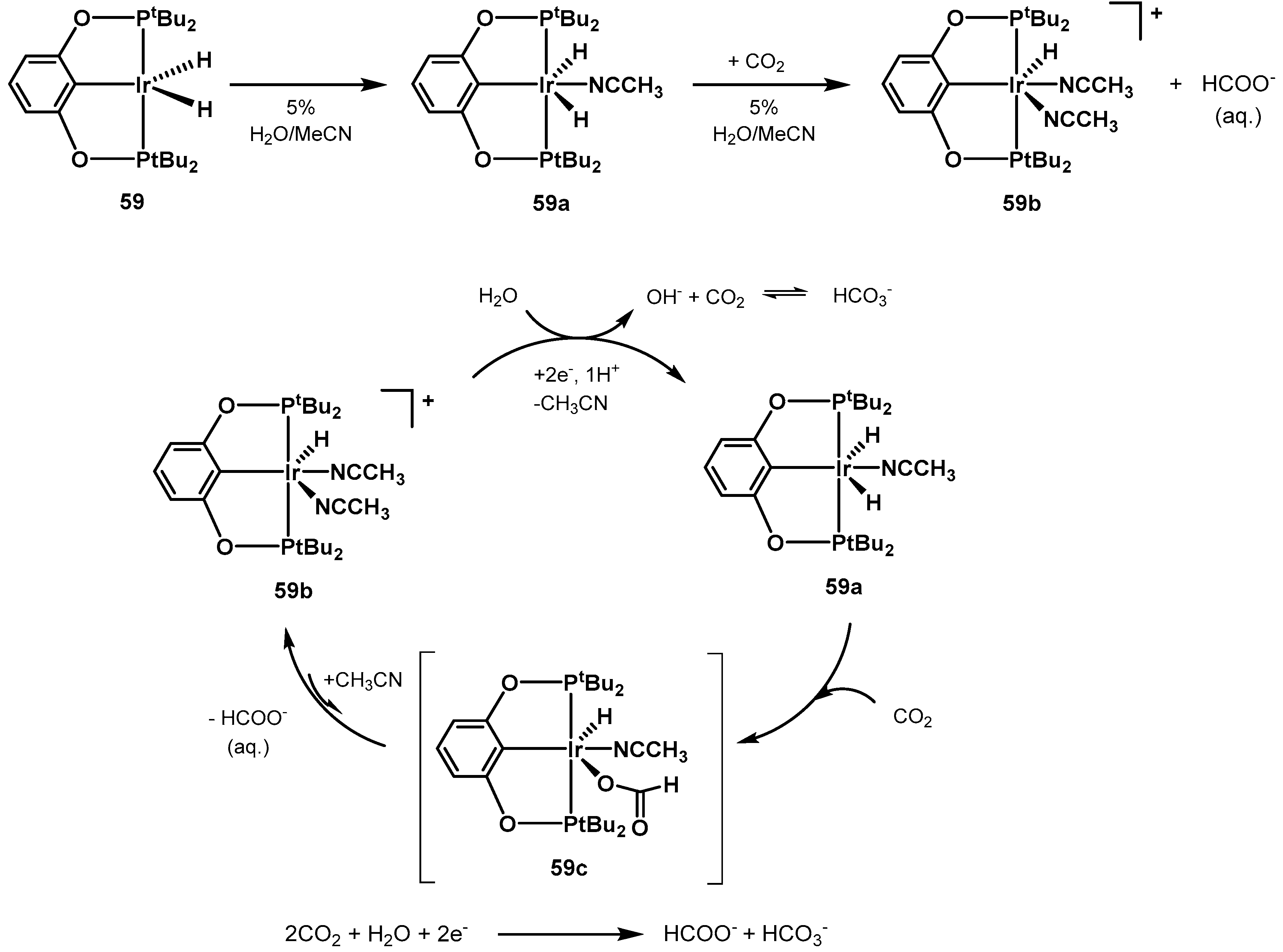{kind=link}
{kind=link}
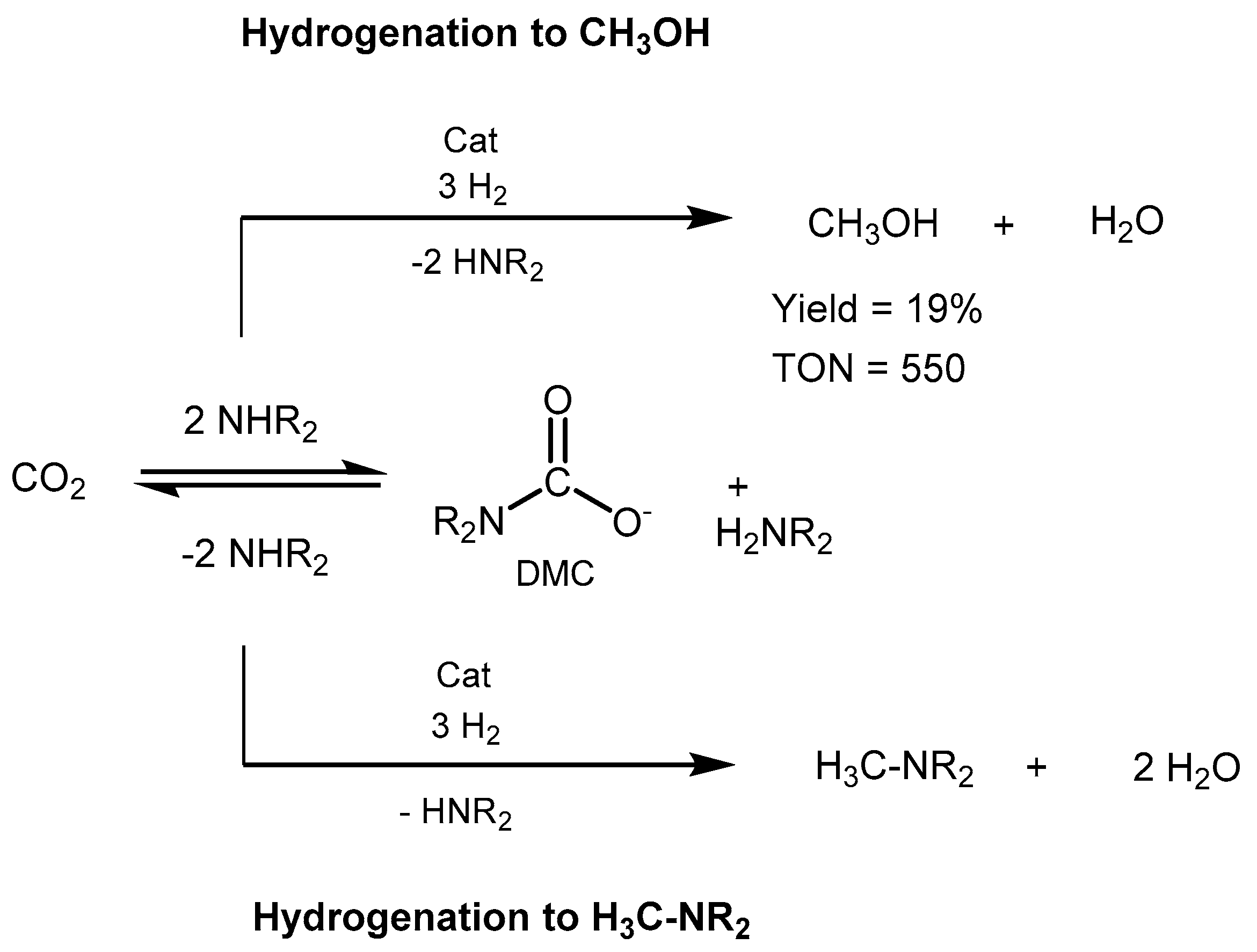{kind=link}
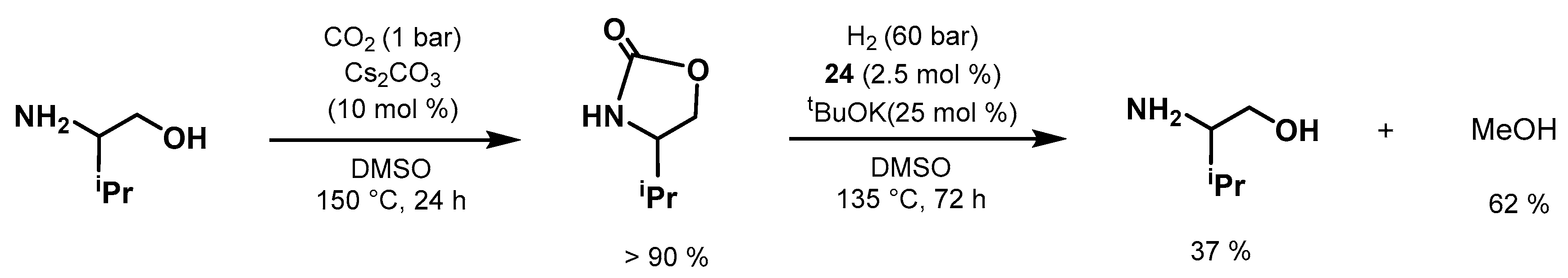{kind=link}
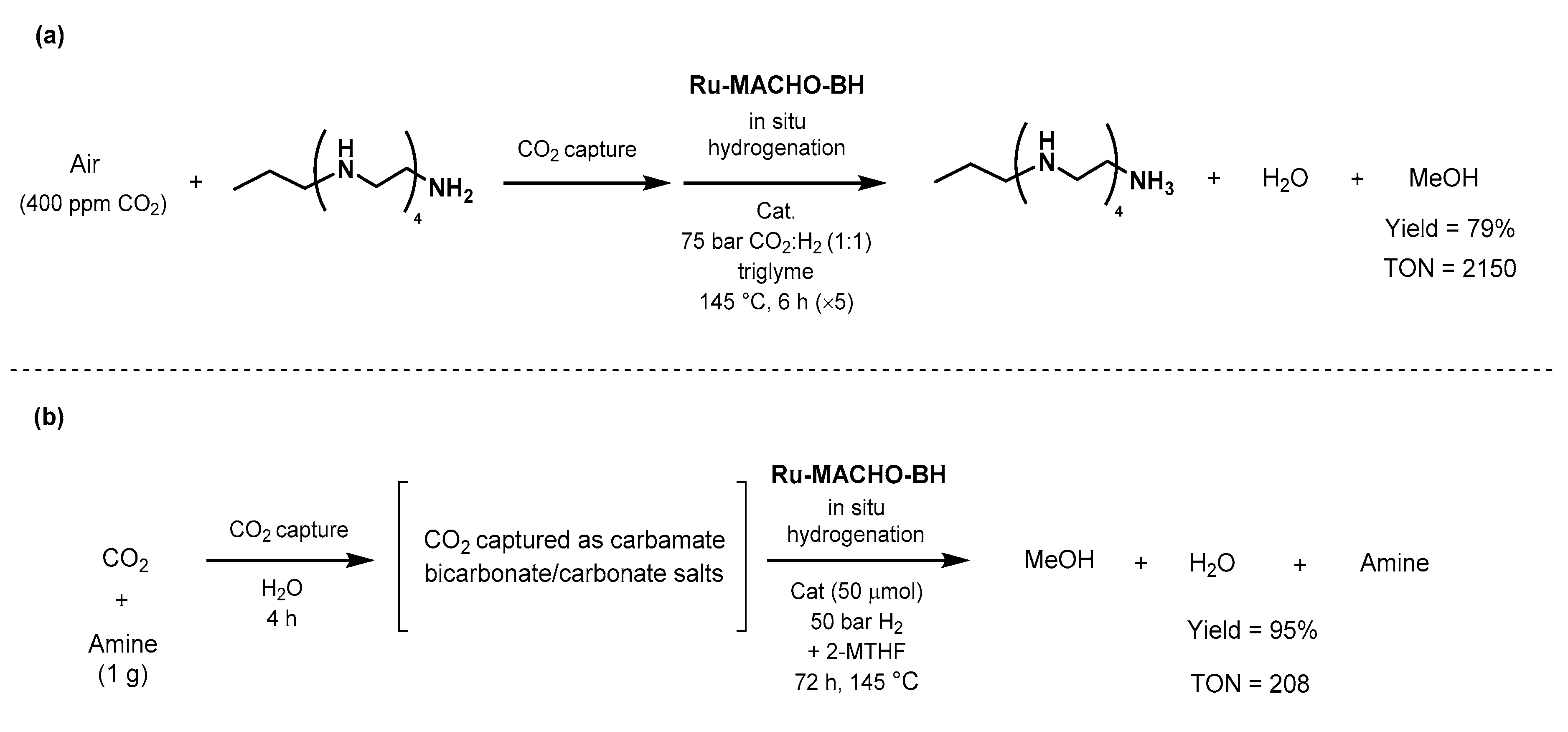{kind=link}
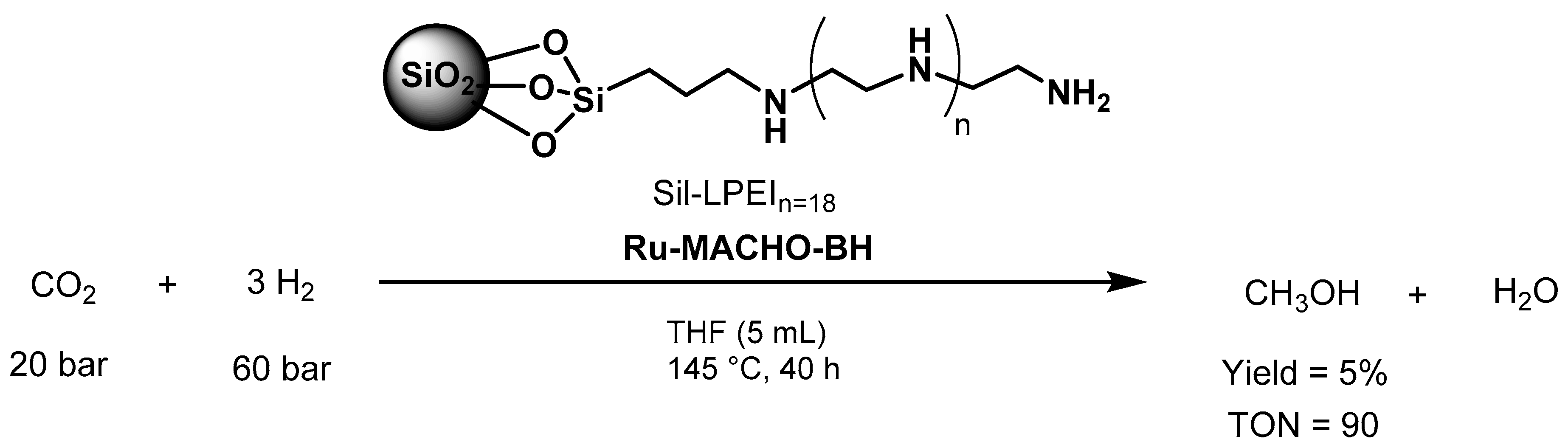{kind=link}
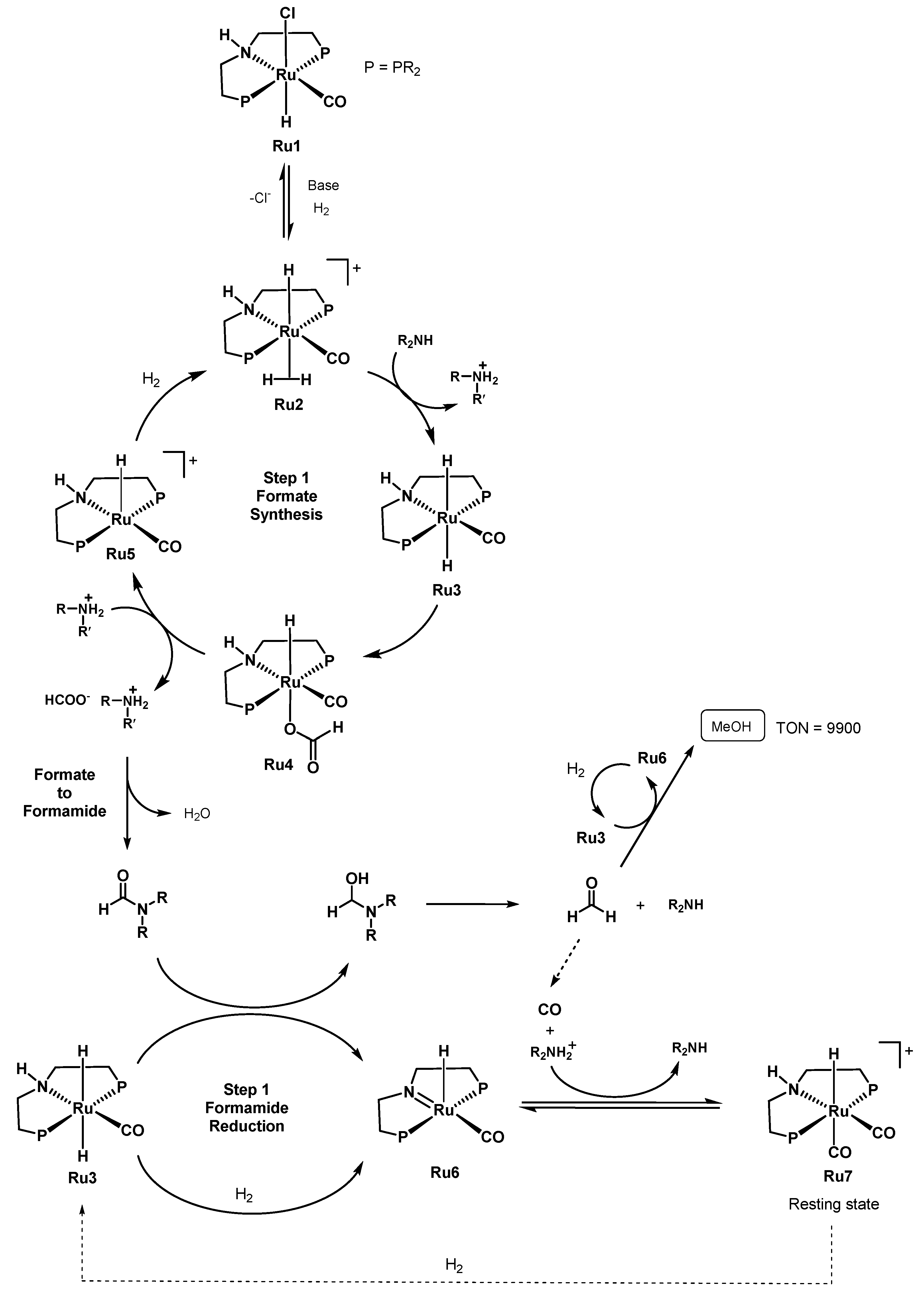{kind=link}
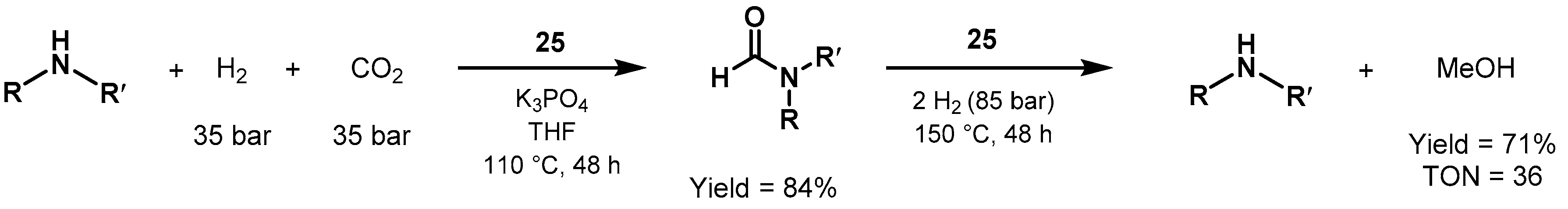{kind=link}
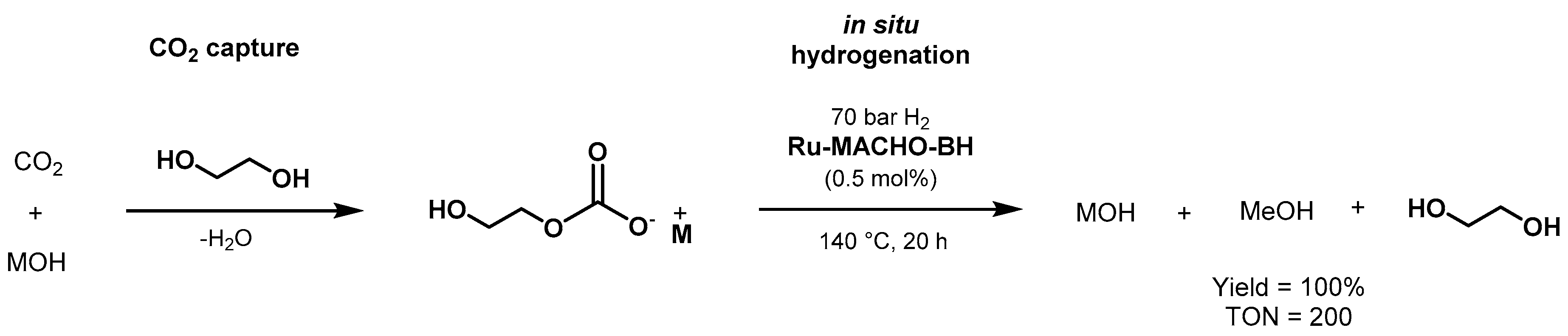{kind=link}
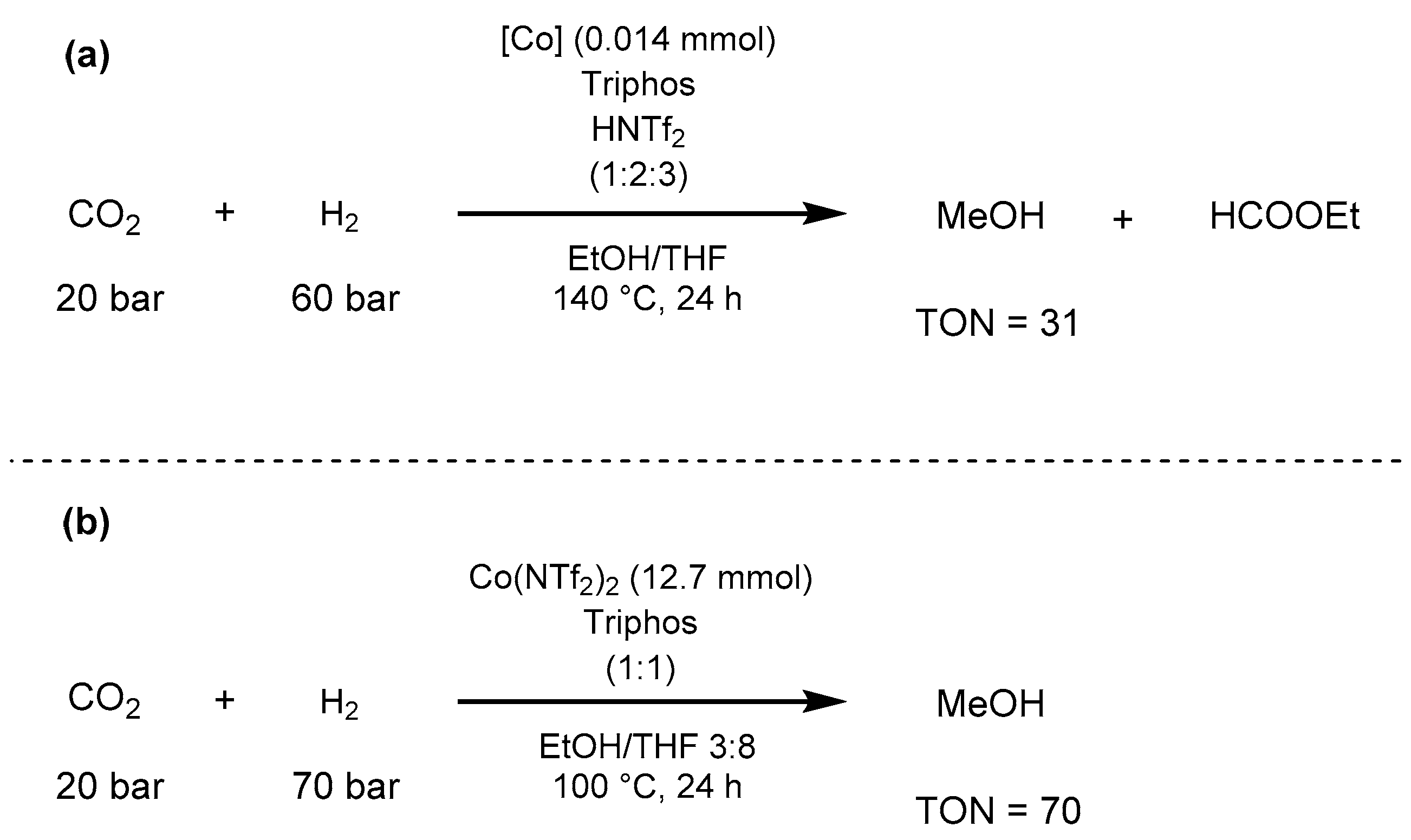{kind=link}
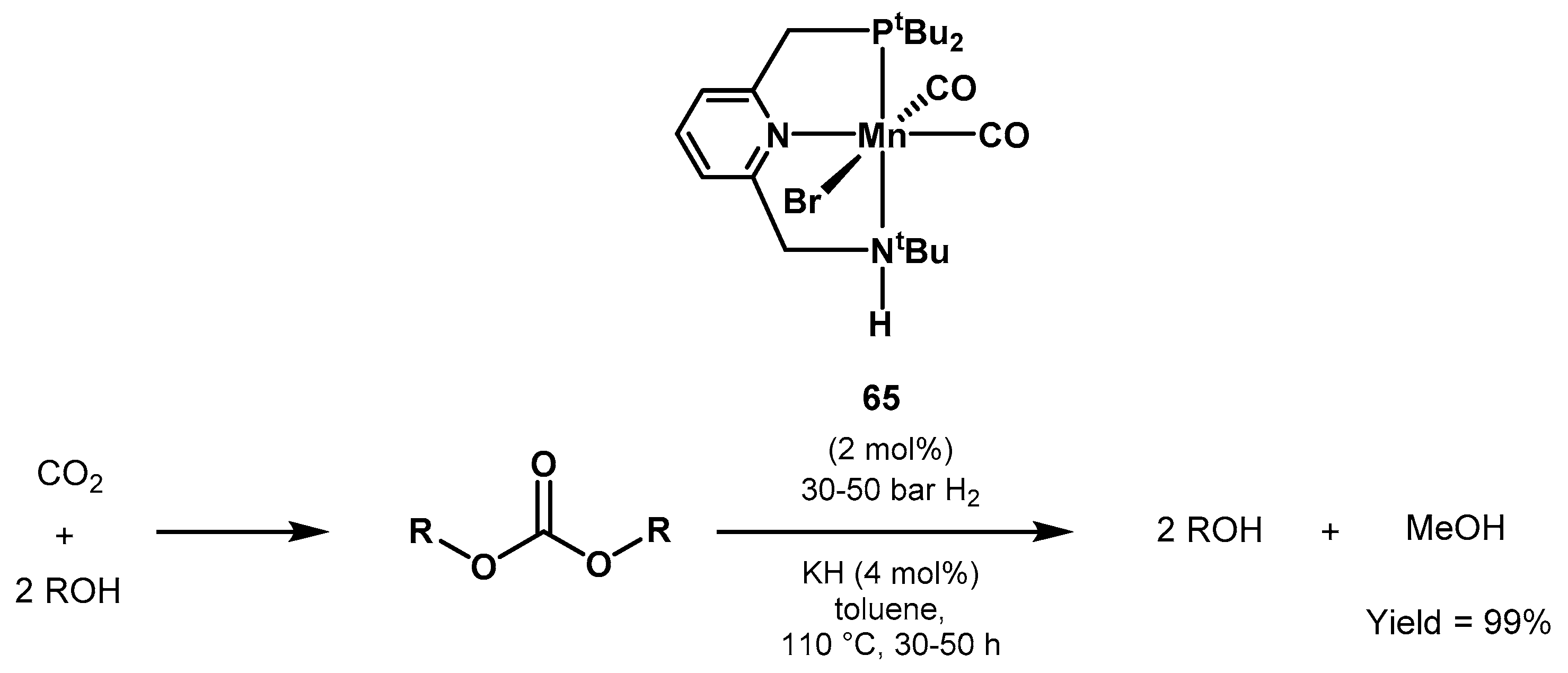{kind=link}
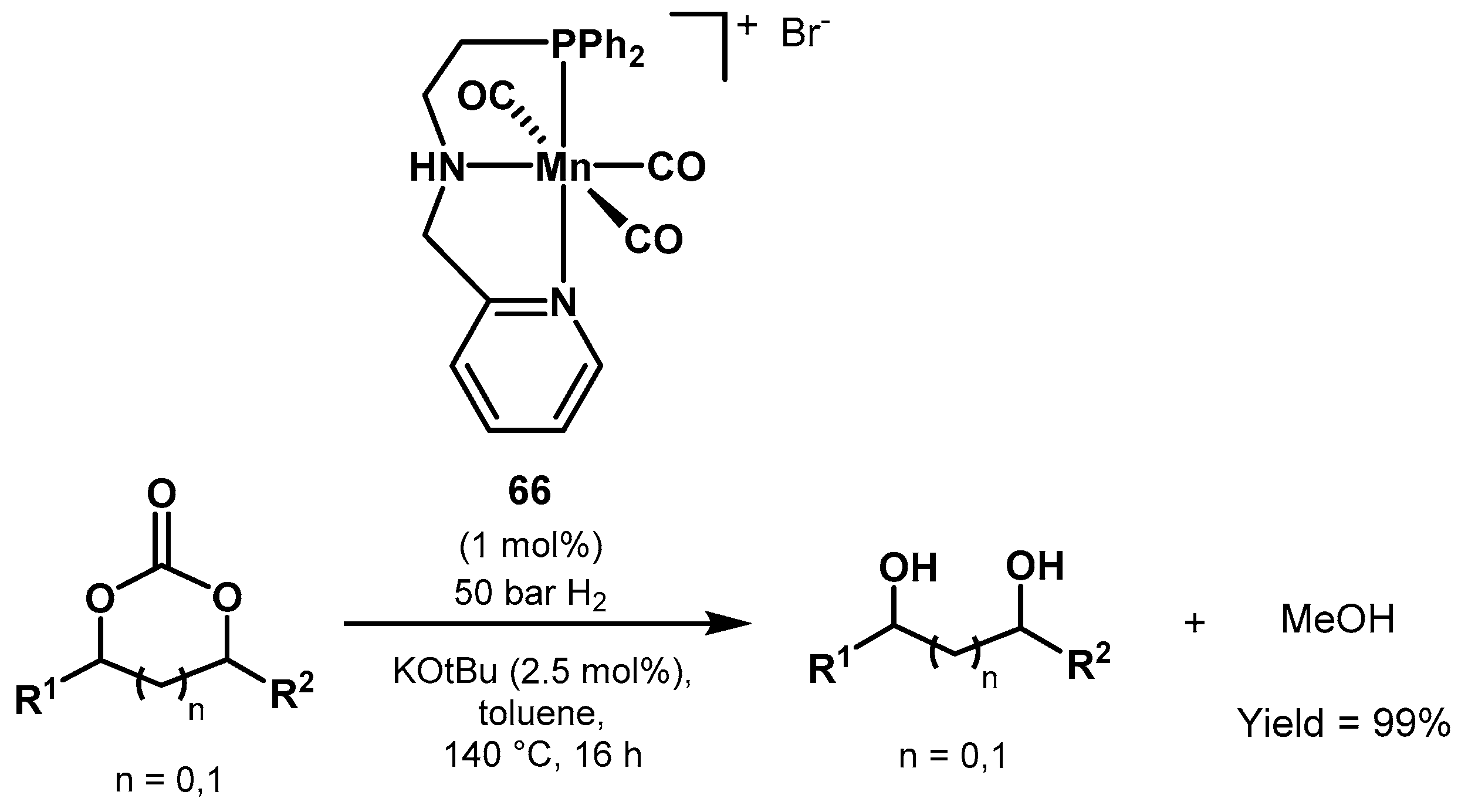{kind=link}
{kind=link}
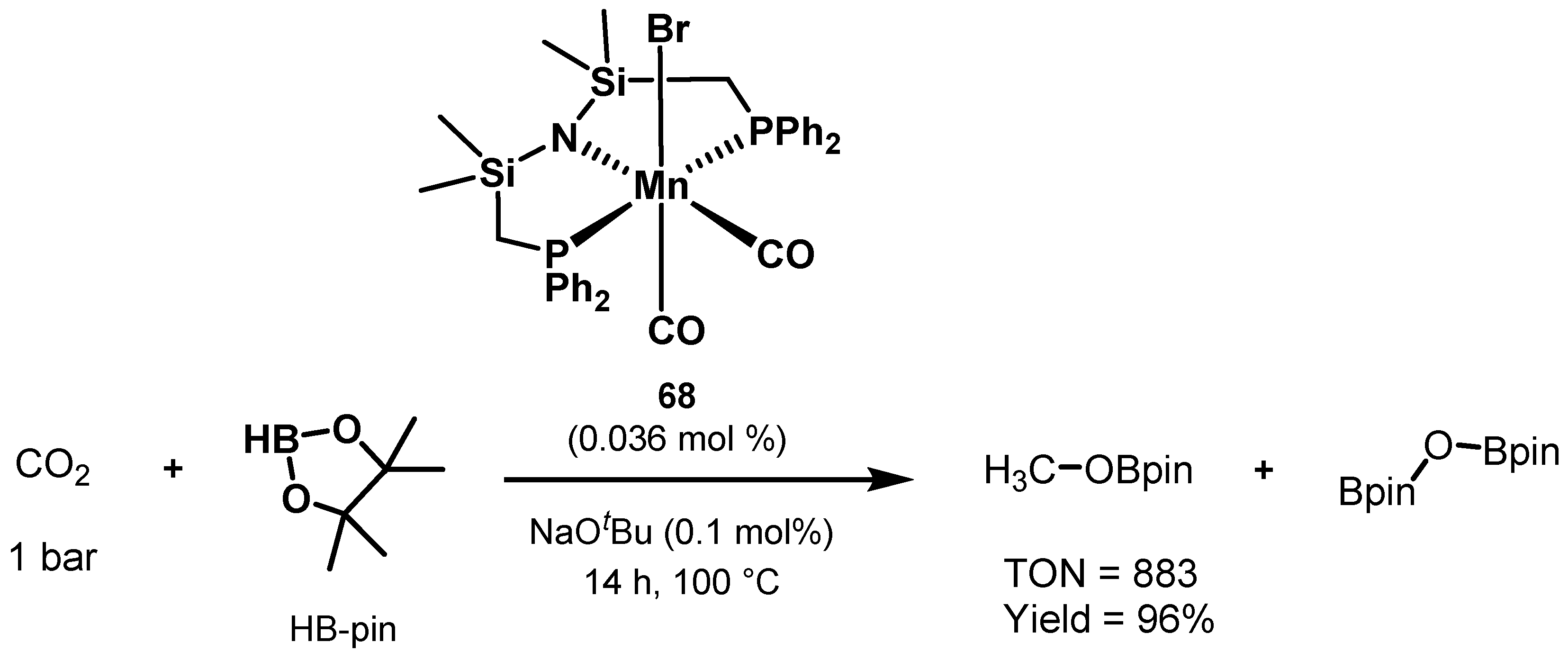{kind=link}
{kind=link}
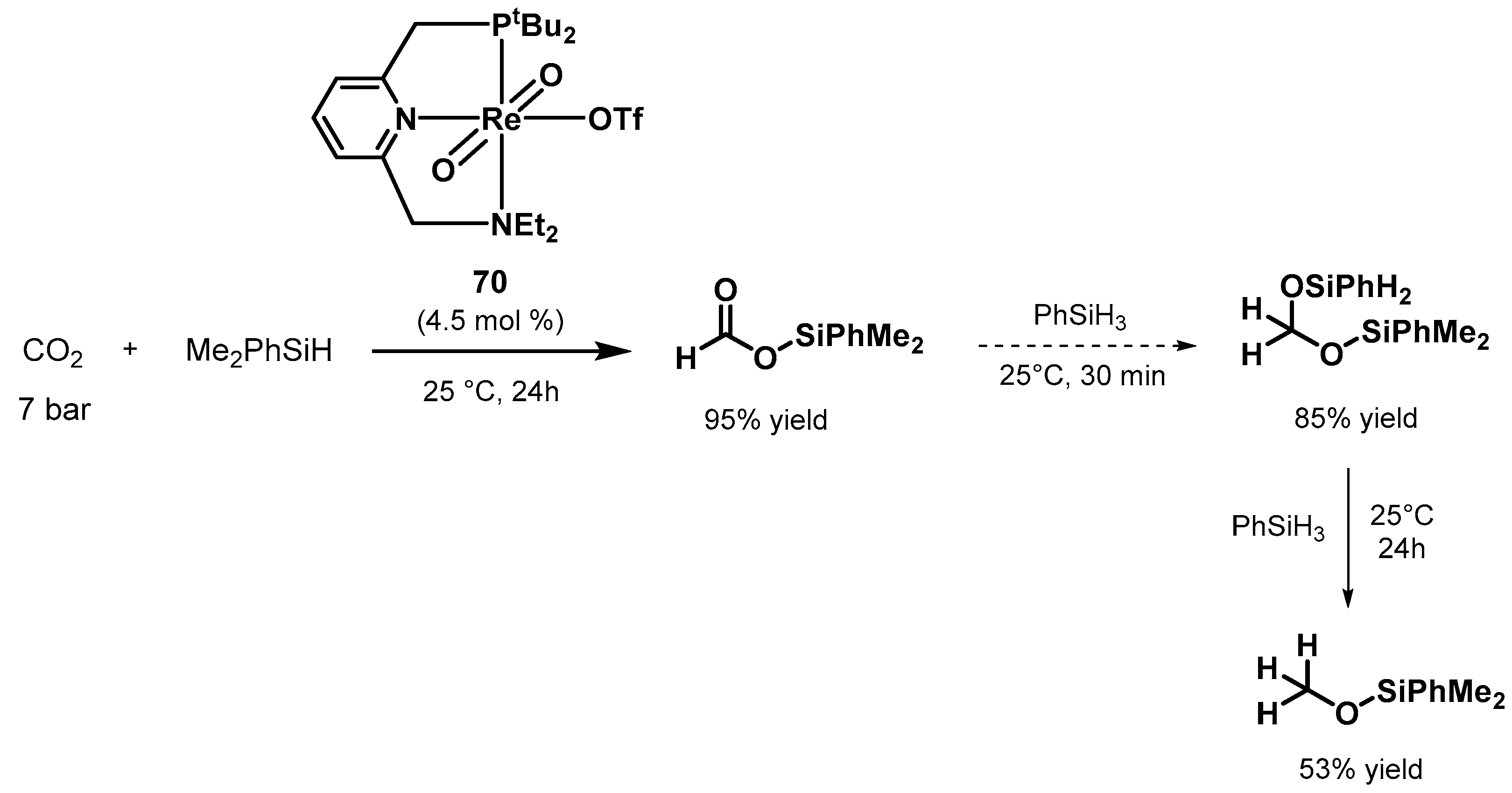{kind=link}
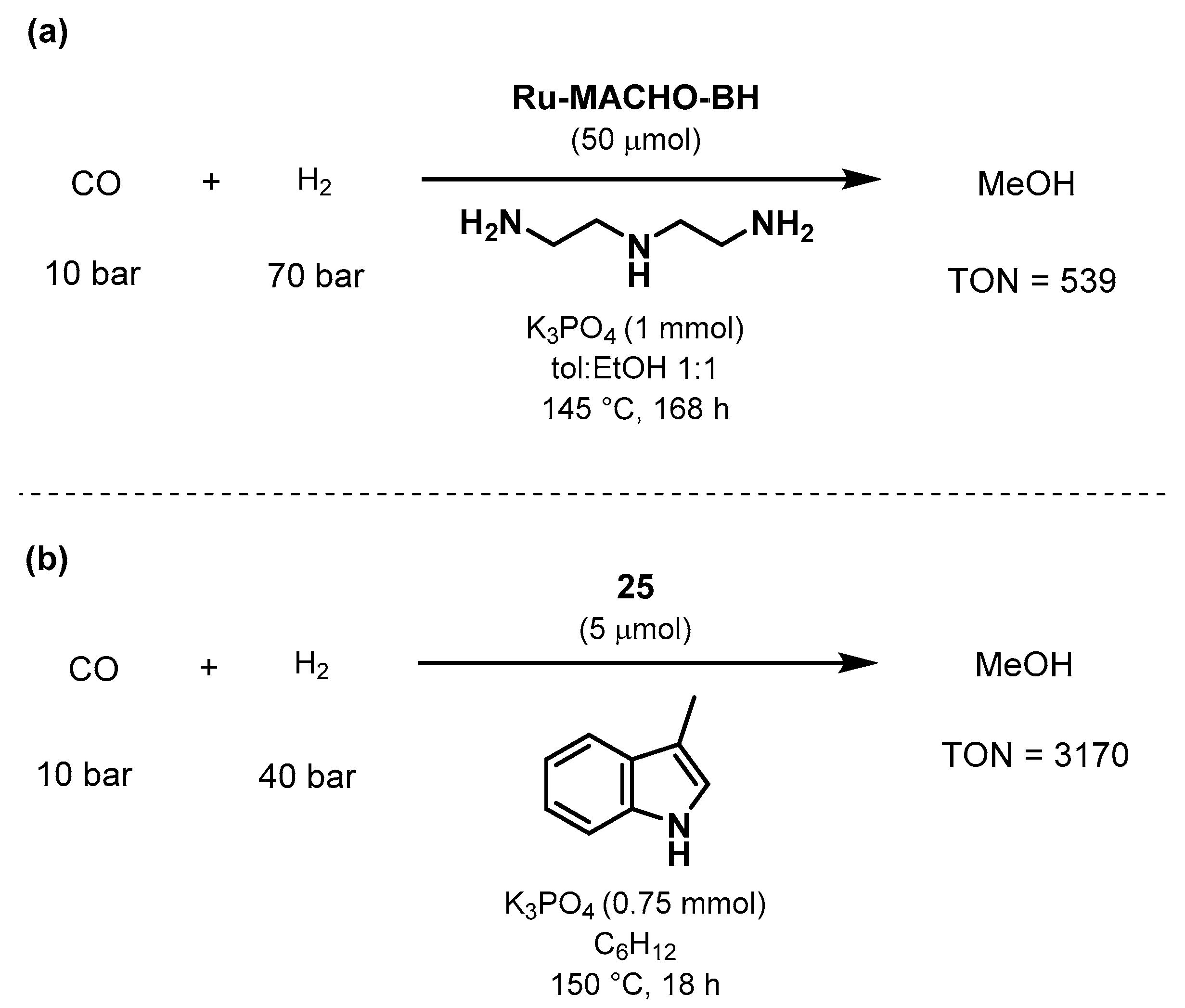{kind=link}
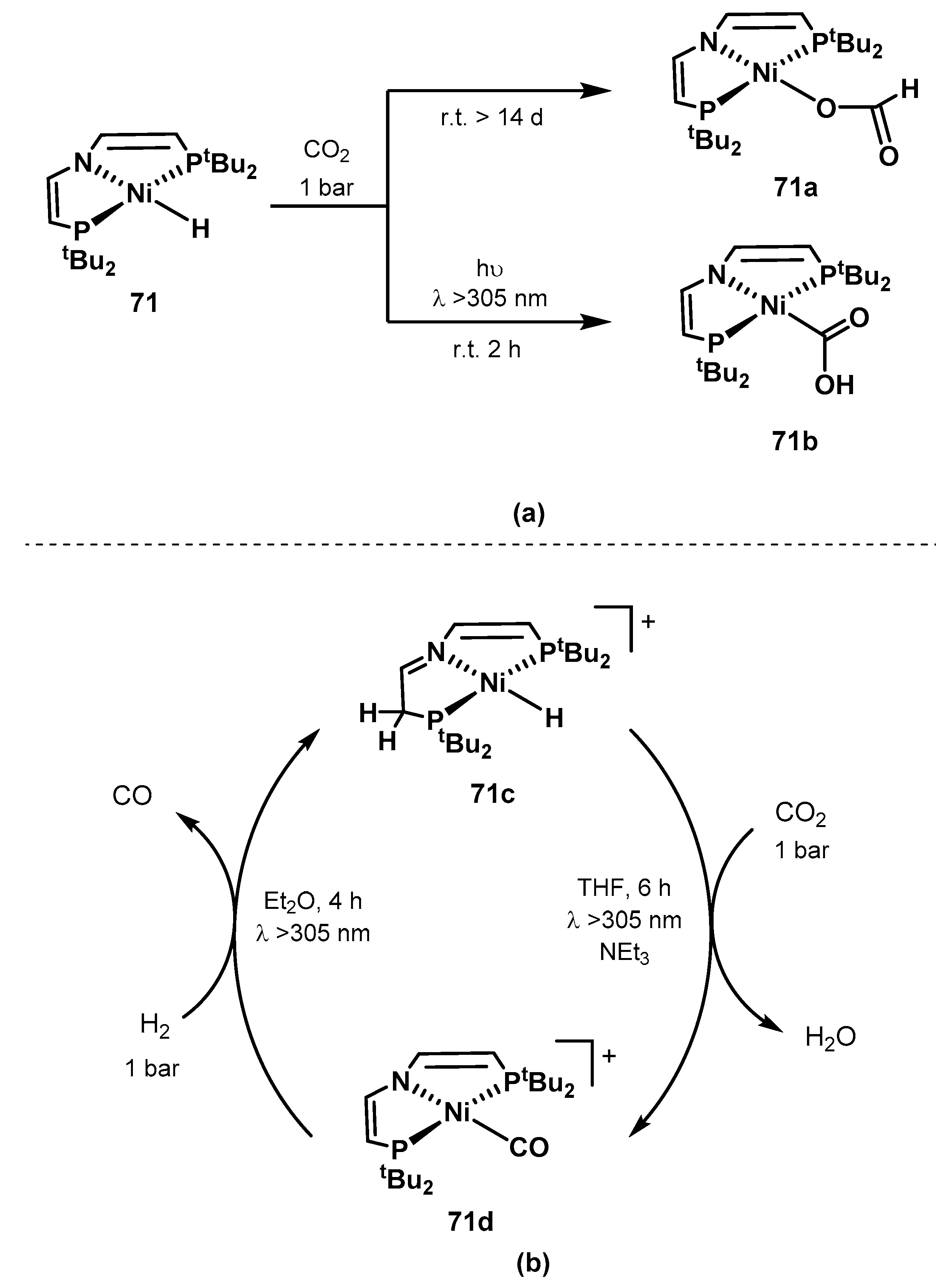{kind=link}
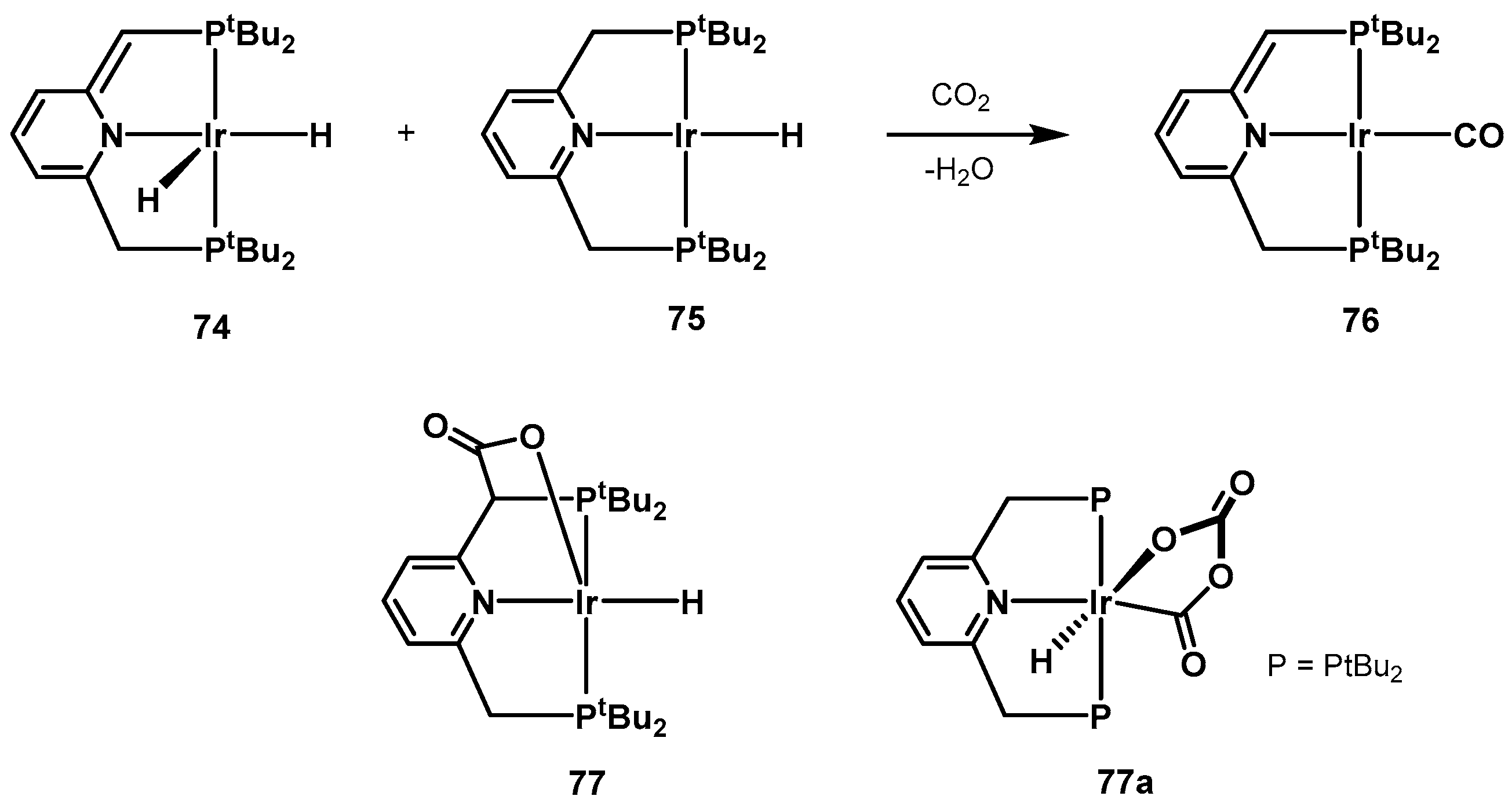{kind=link}
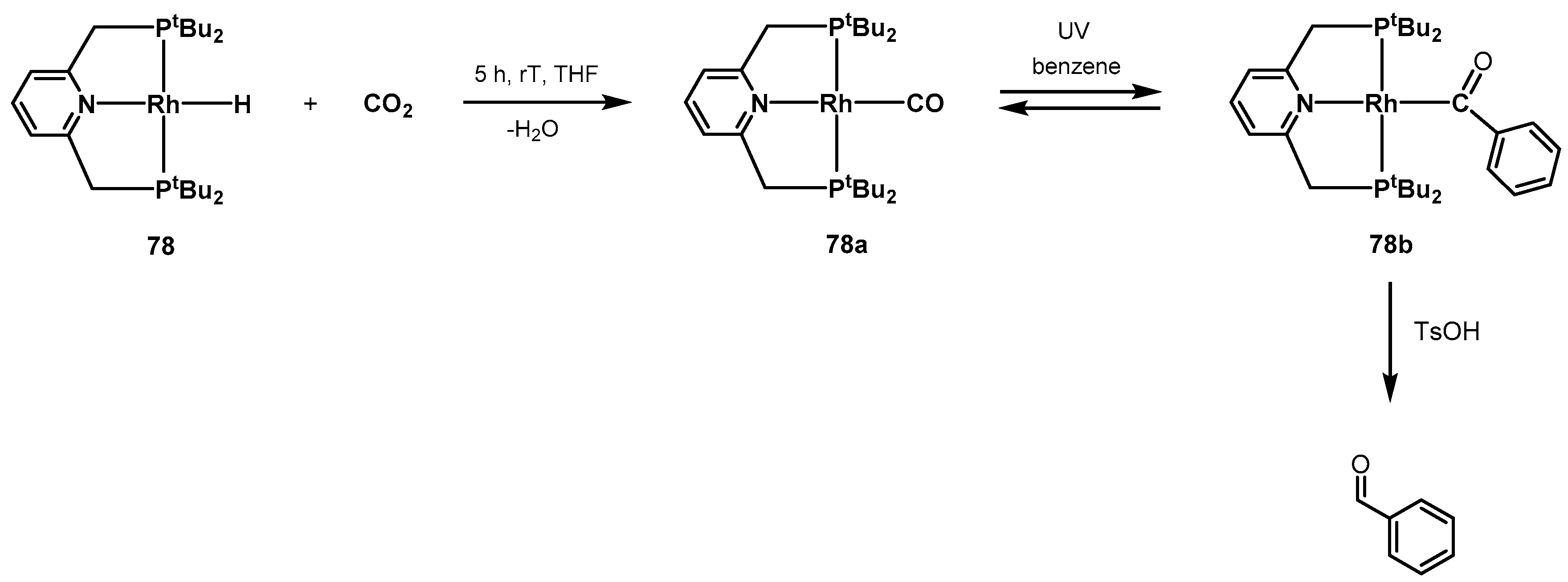{kind=link}
{kind=link}
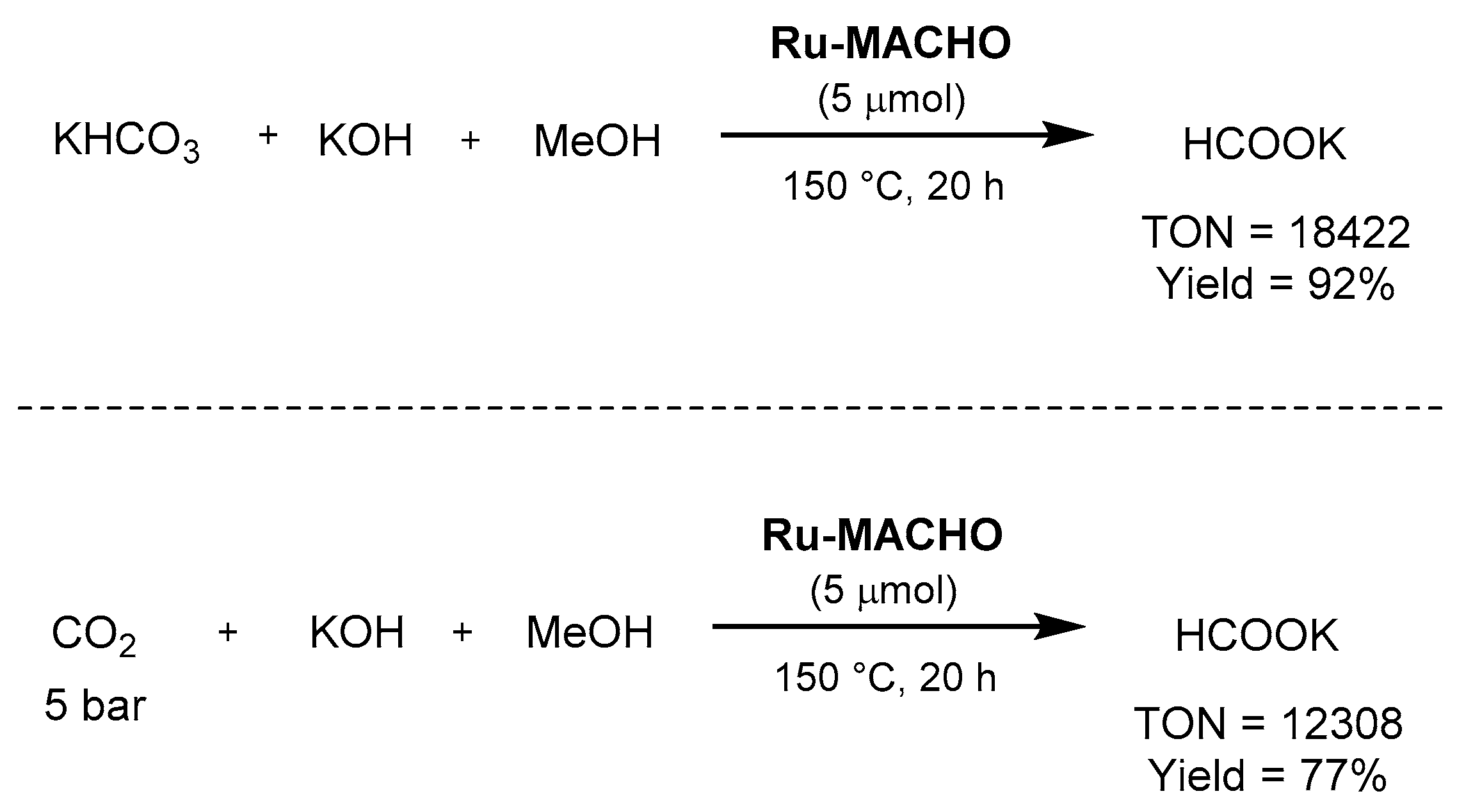{kind=link}
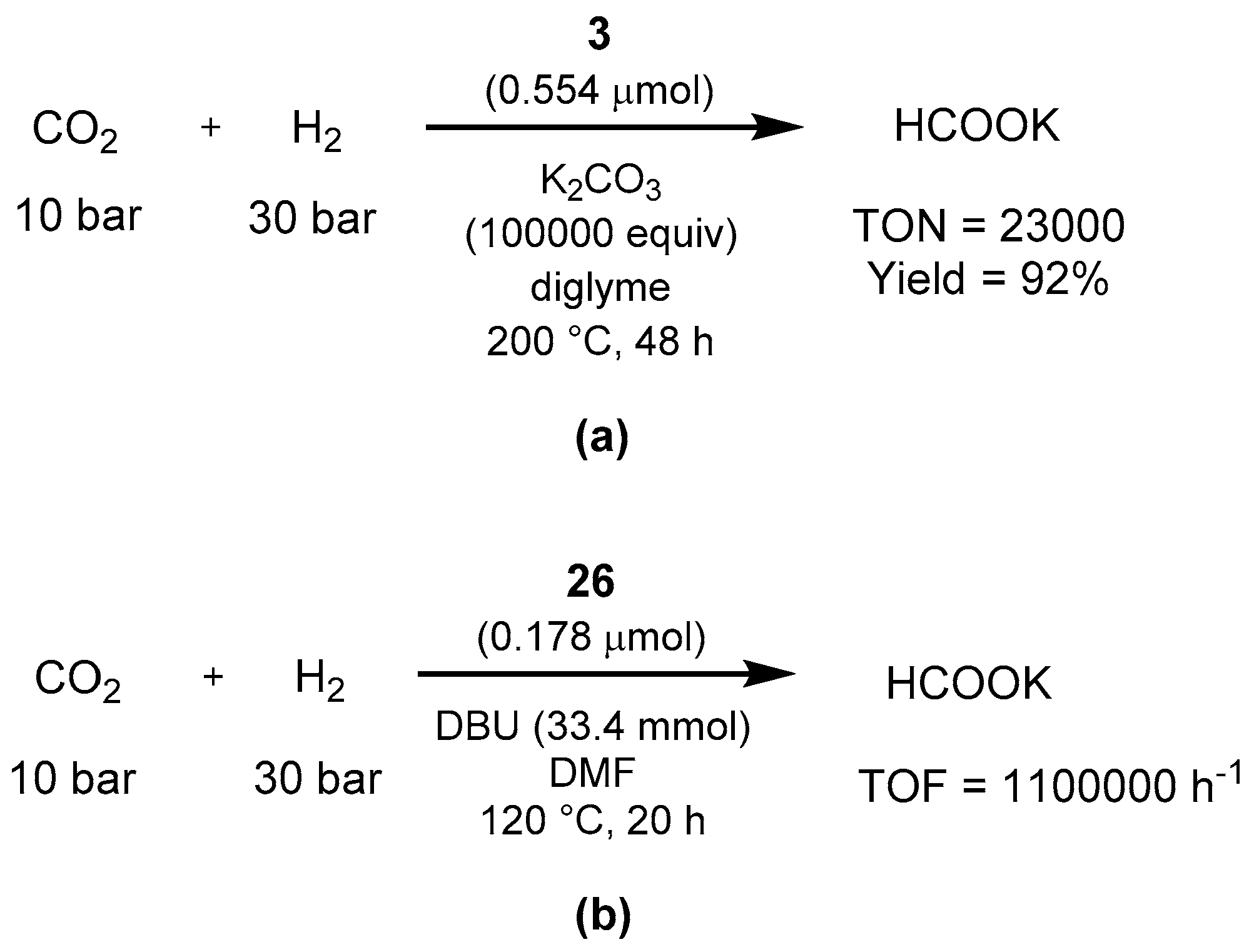{kind=link}
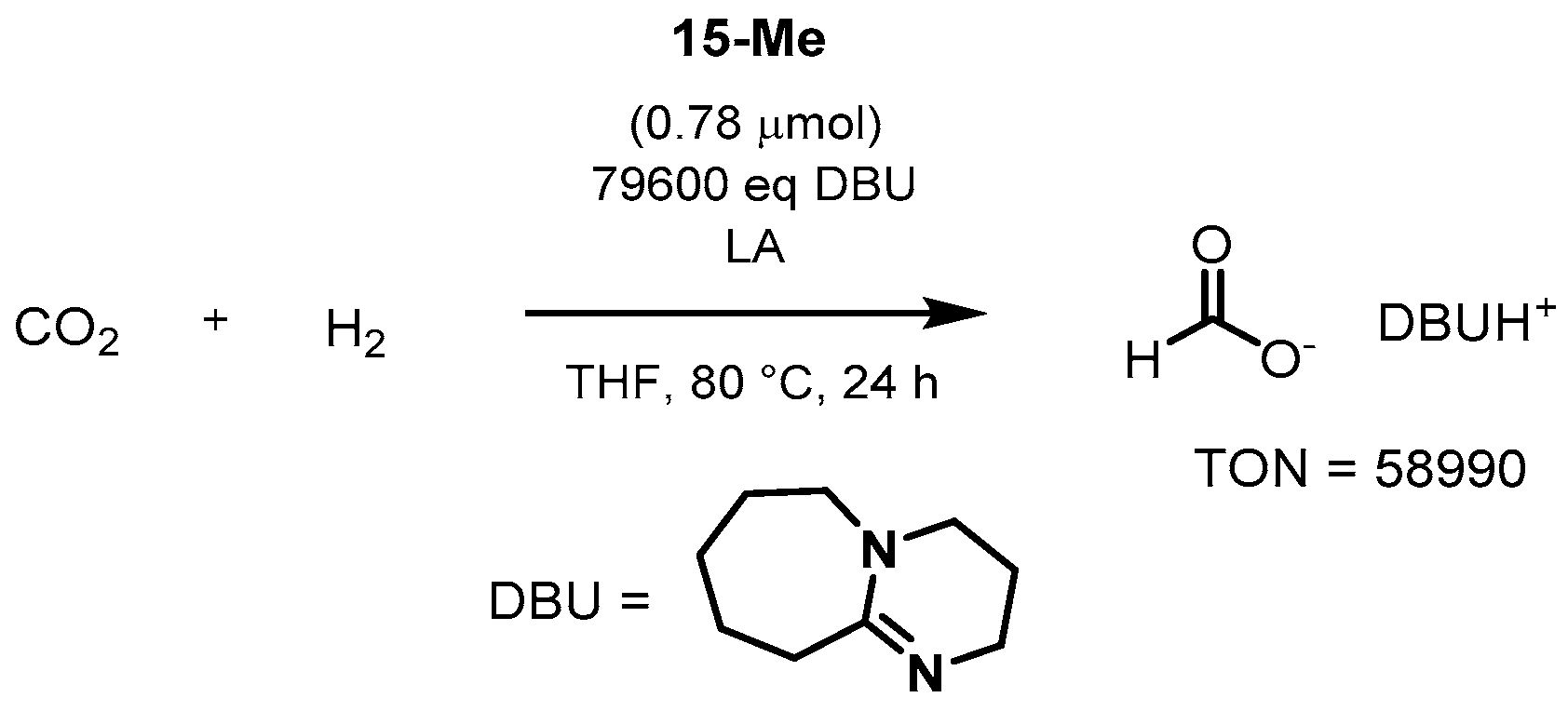{kind=link}
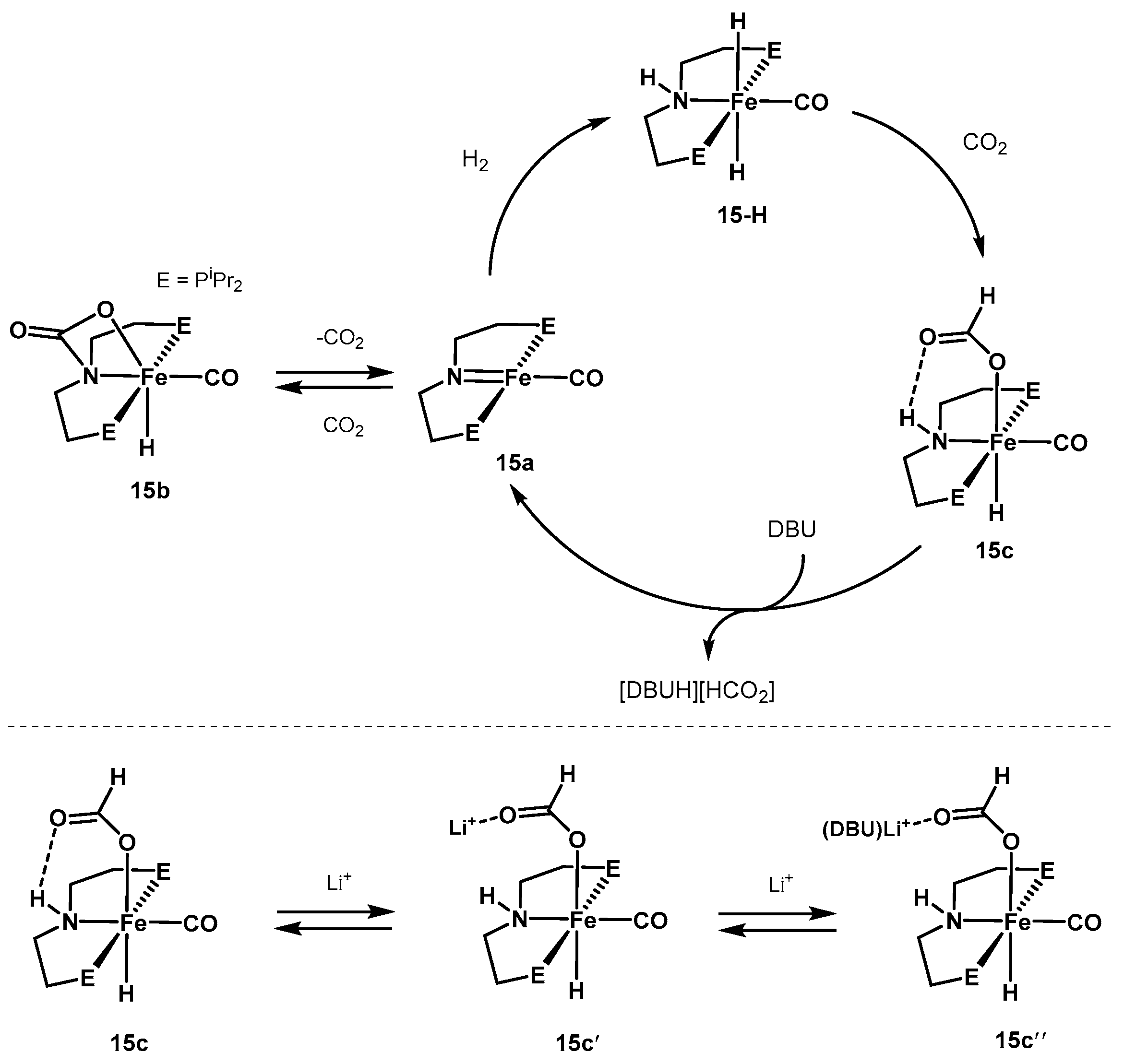{kind=link}
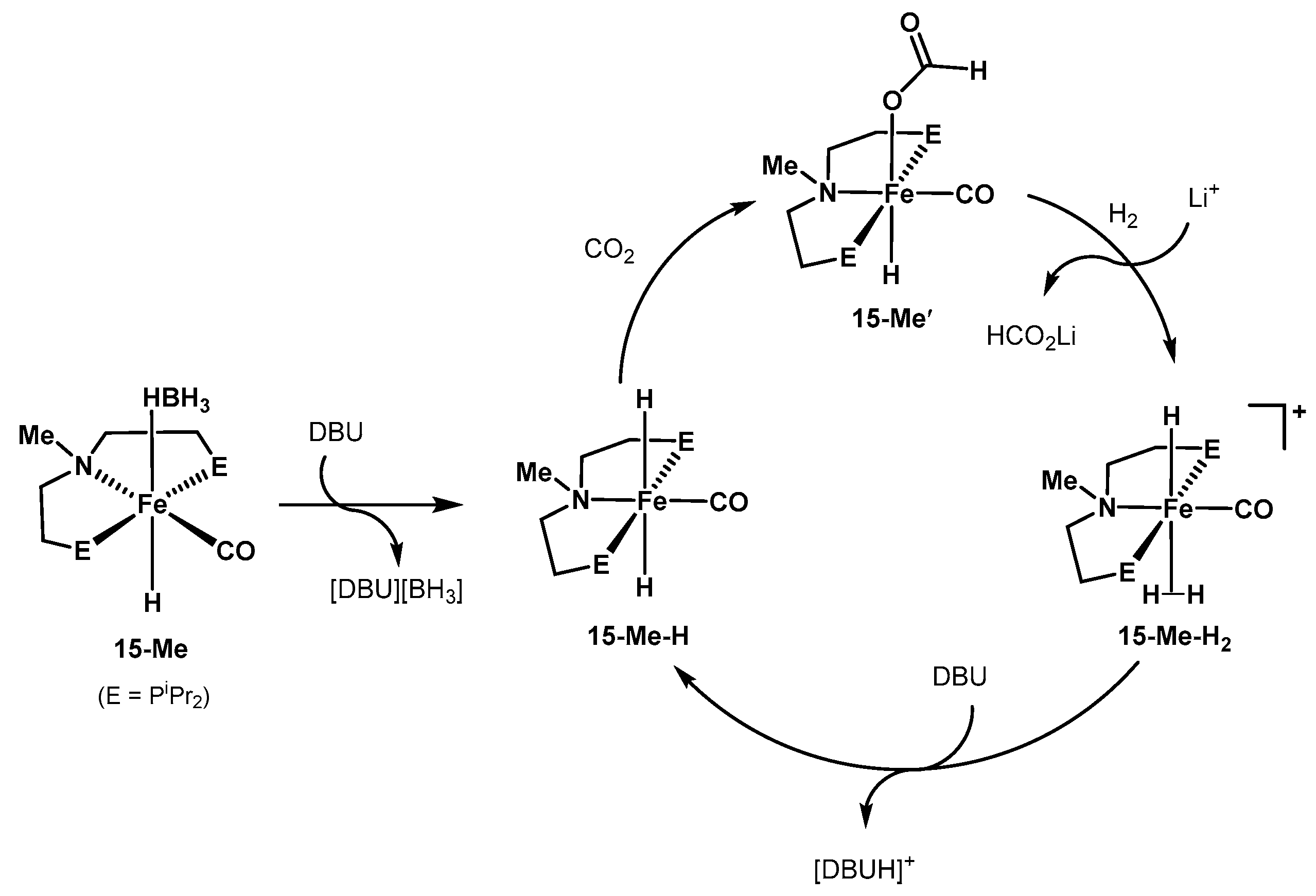{kind=link}
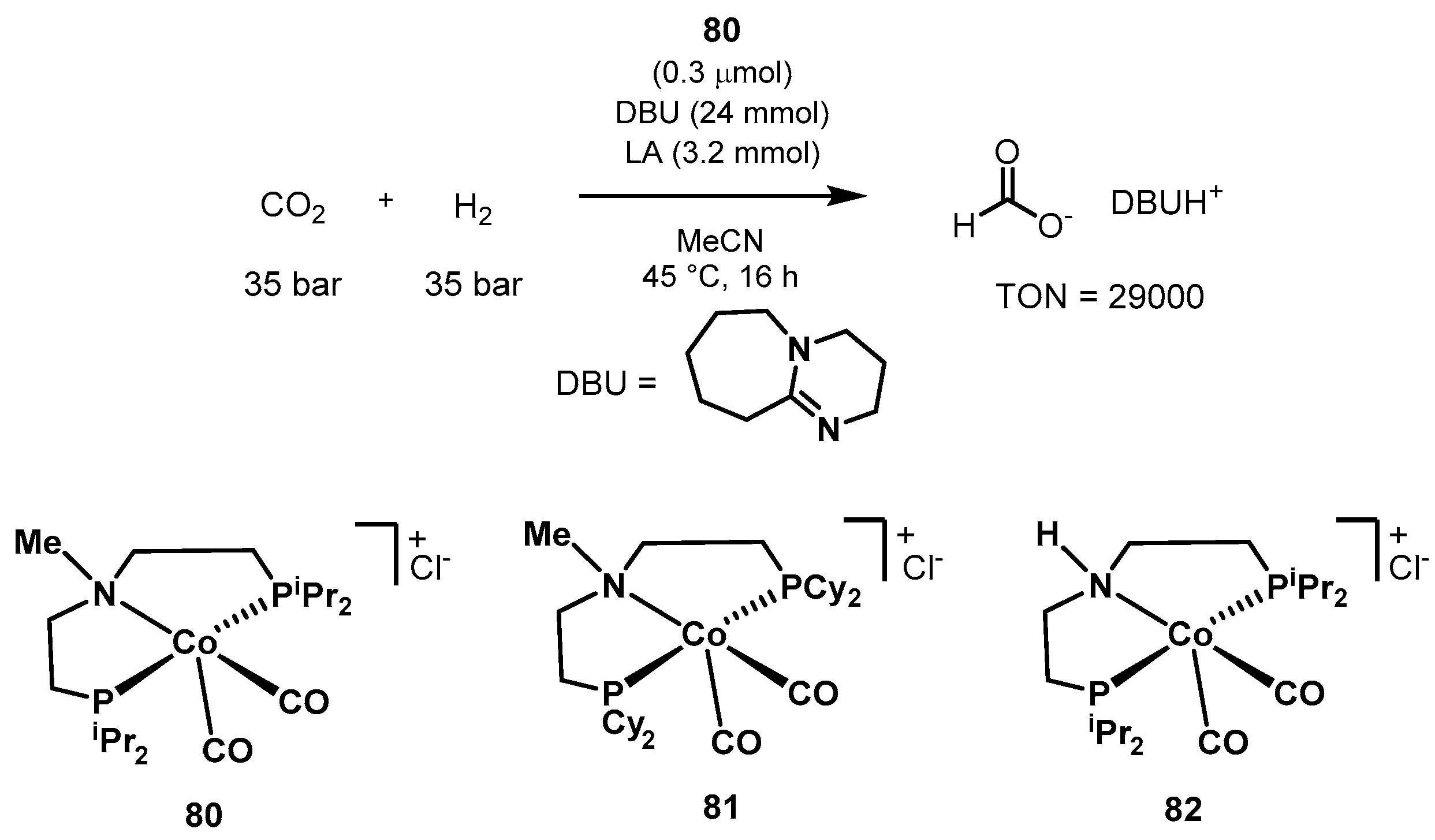{kind=link}
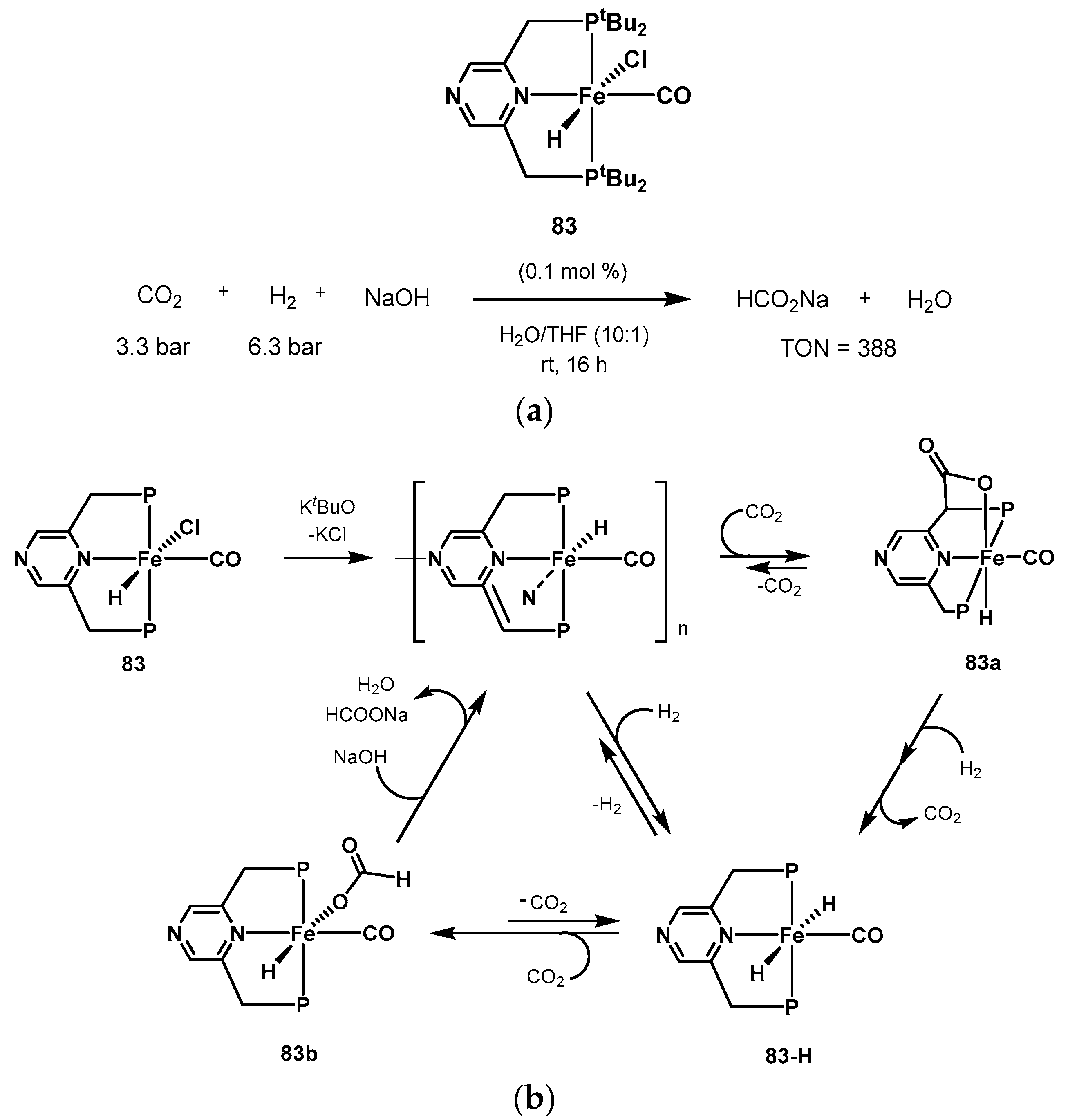{kind=link}
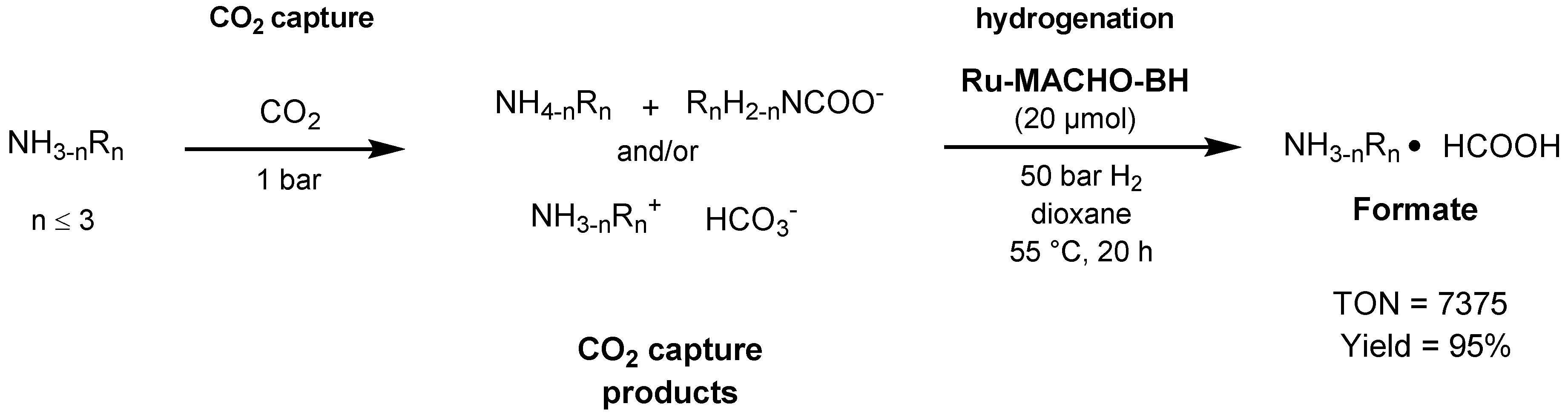{kind=link}
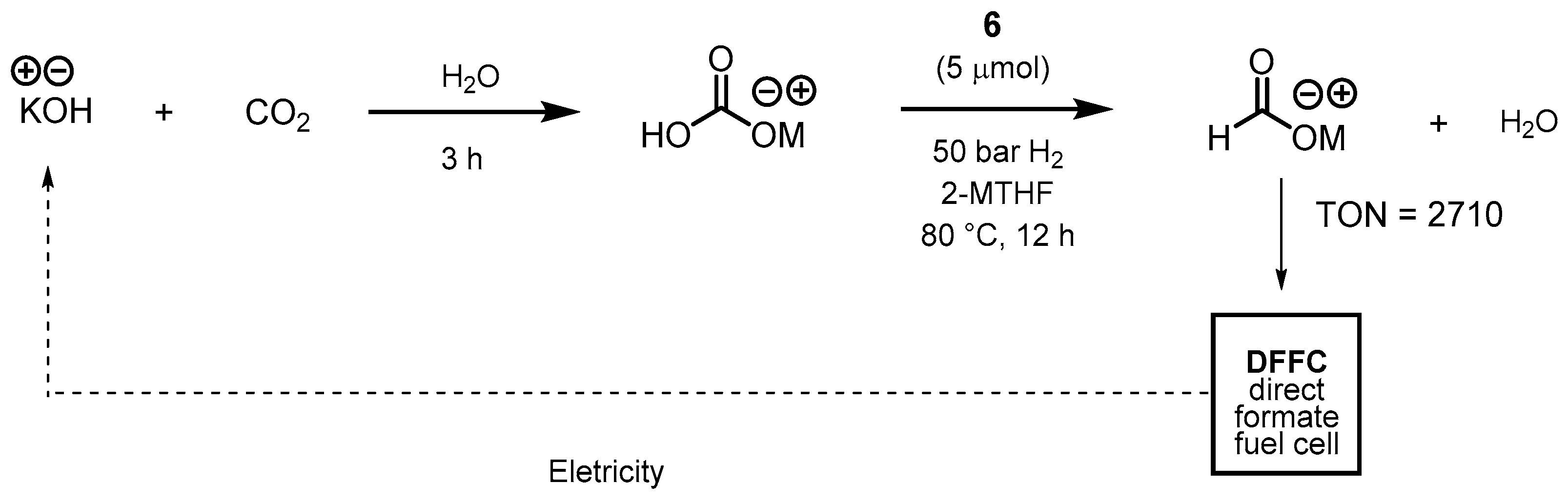{kind=link}
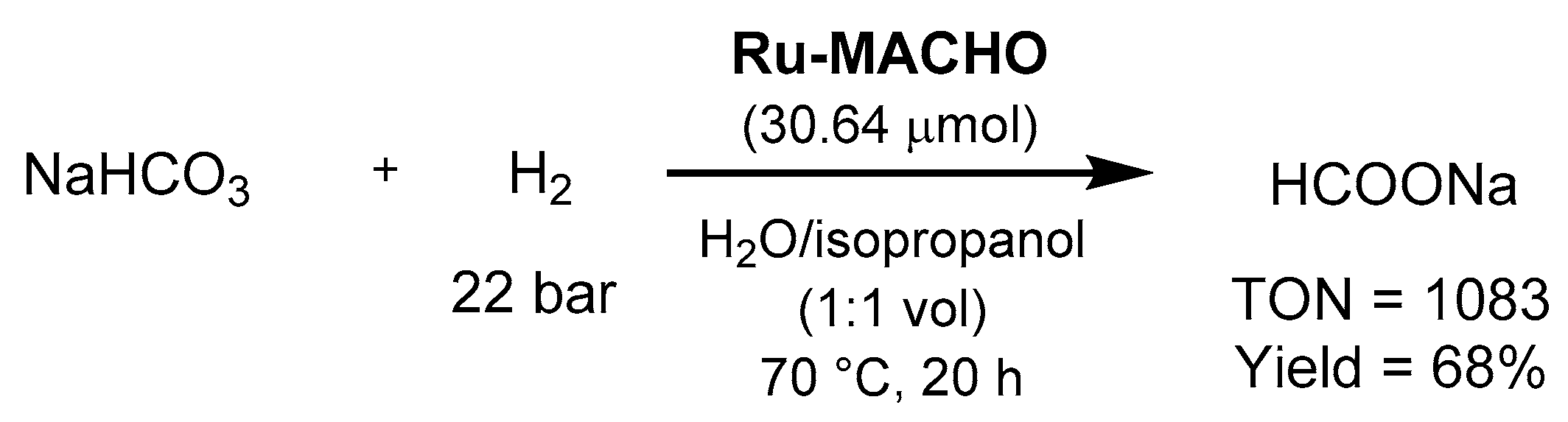{kind=link}
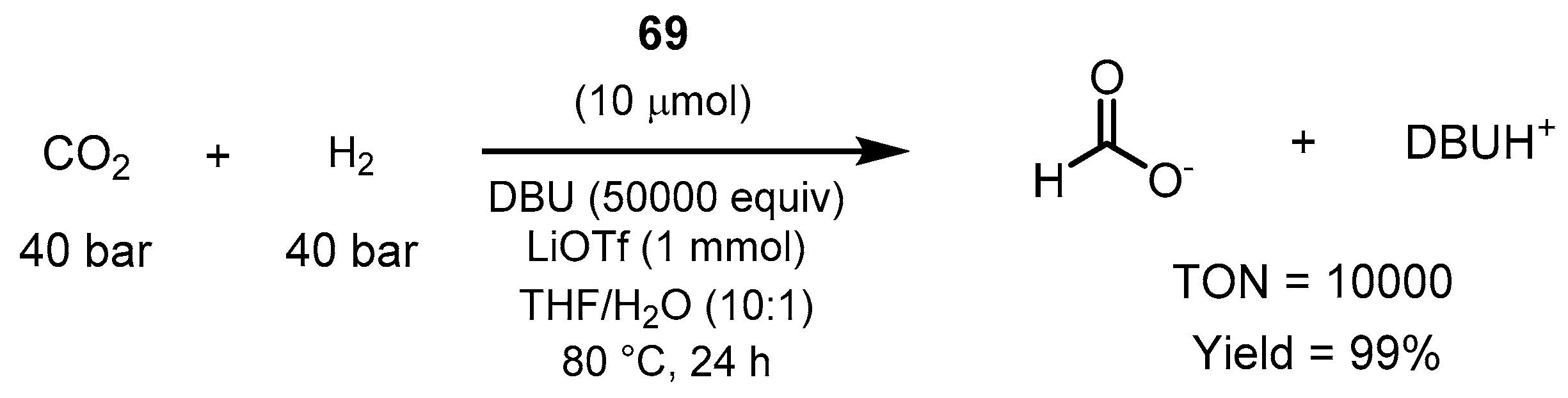{kind=link}
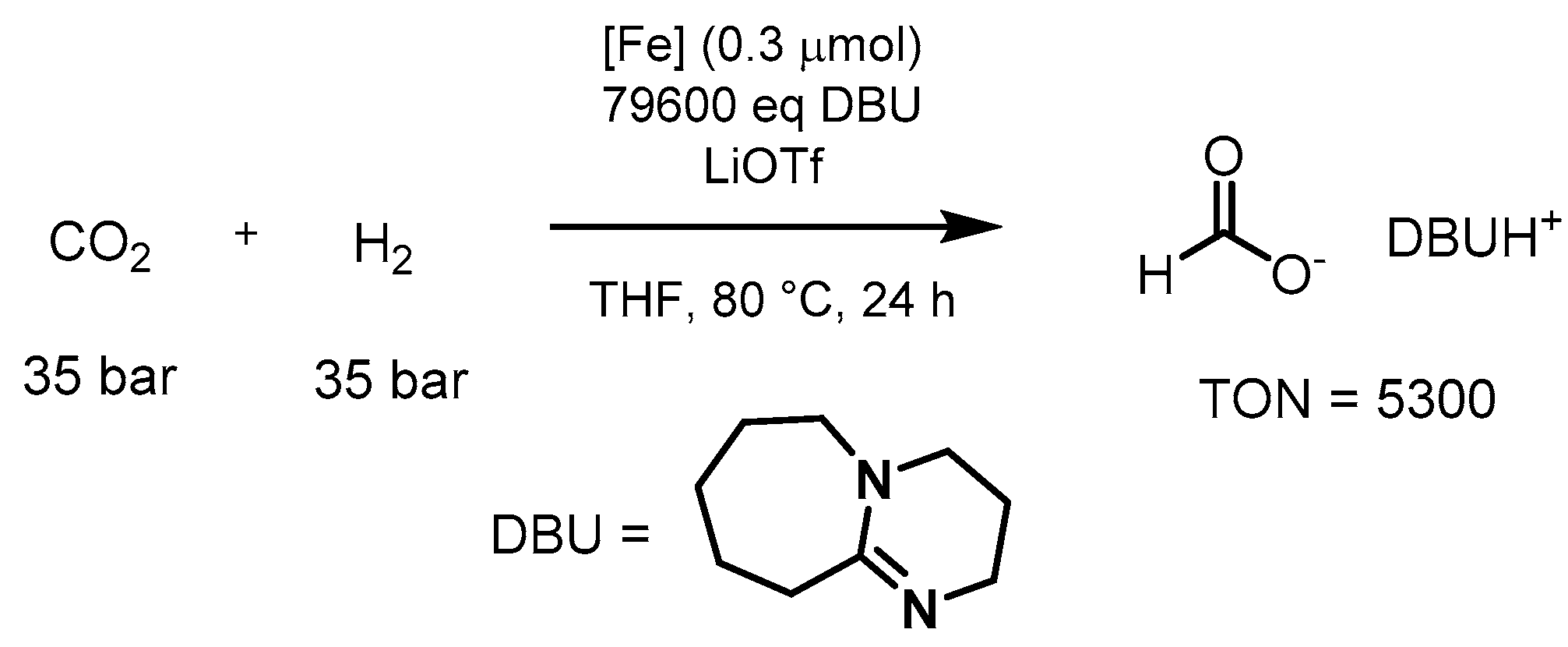{kind=link}
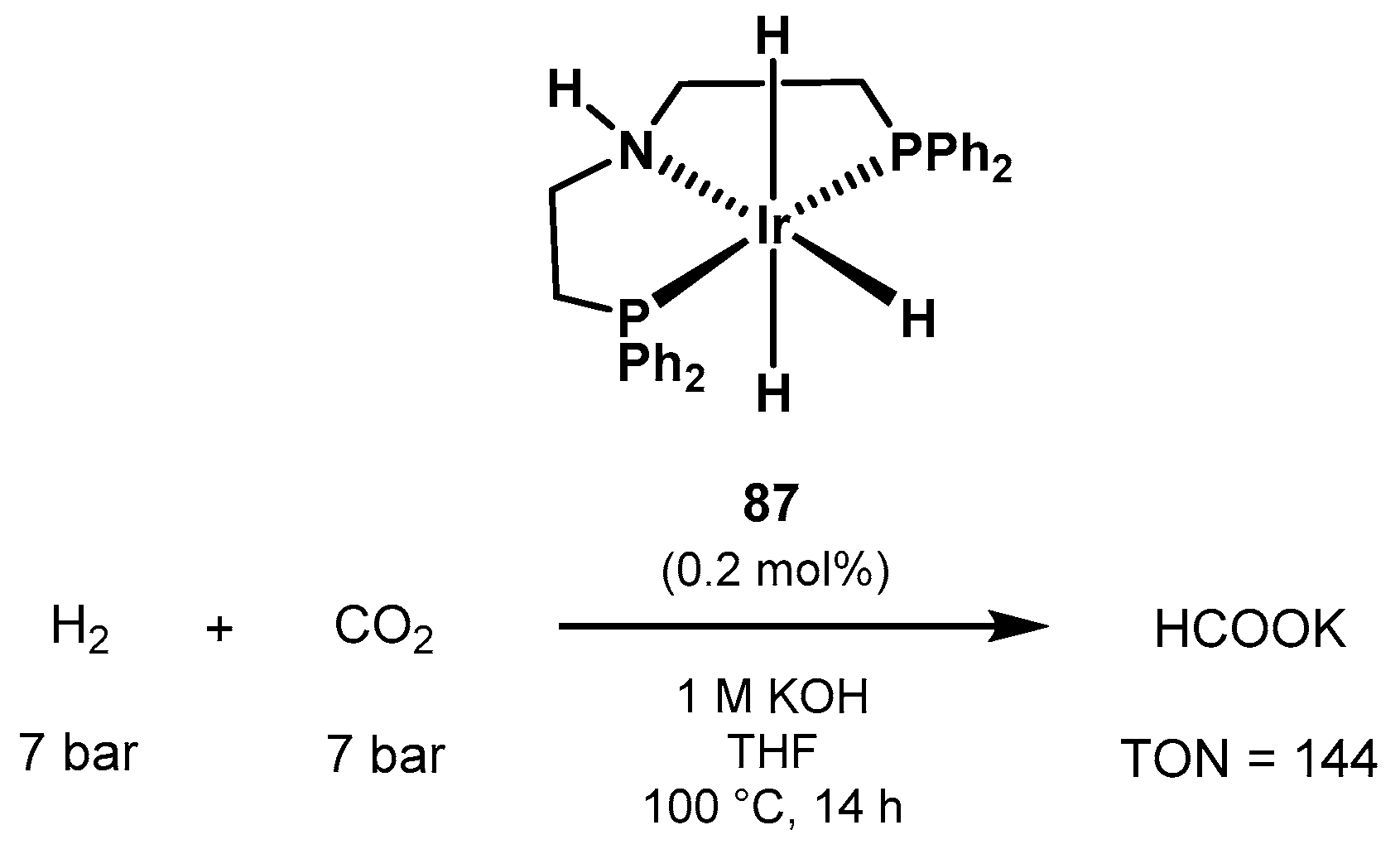{kind=link}
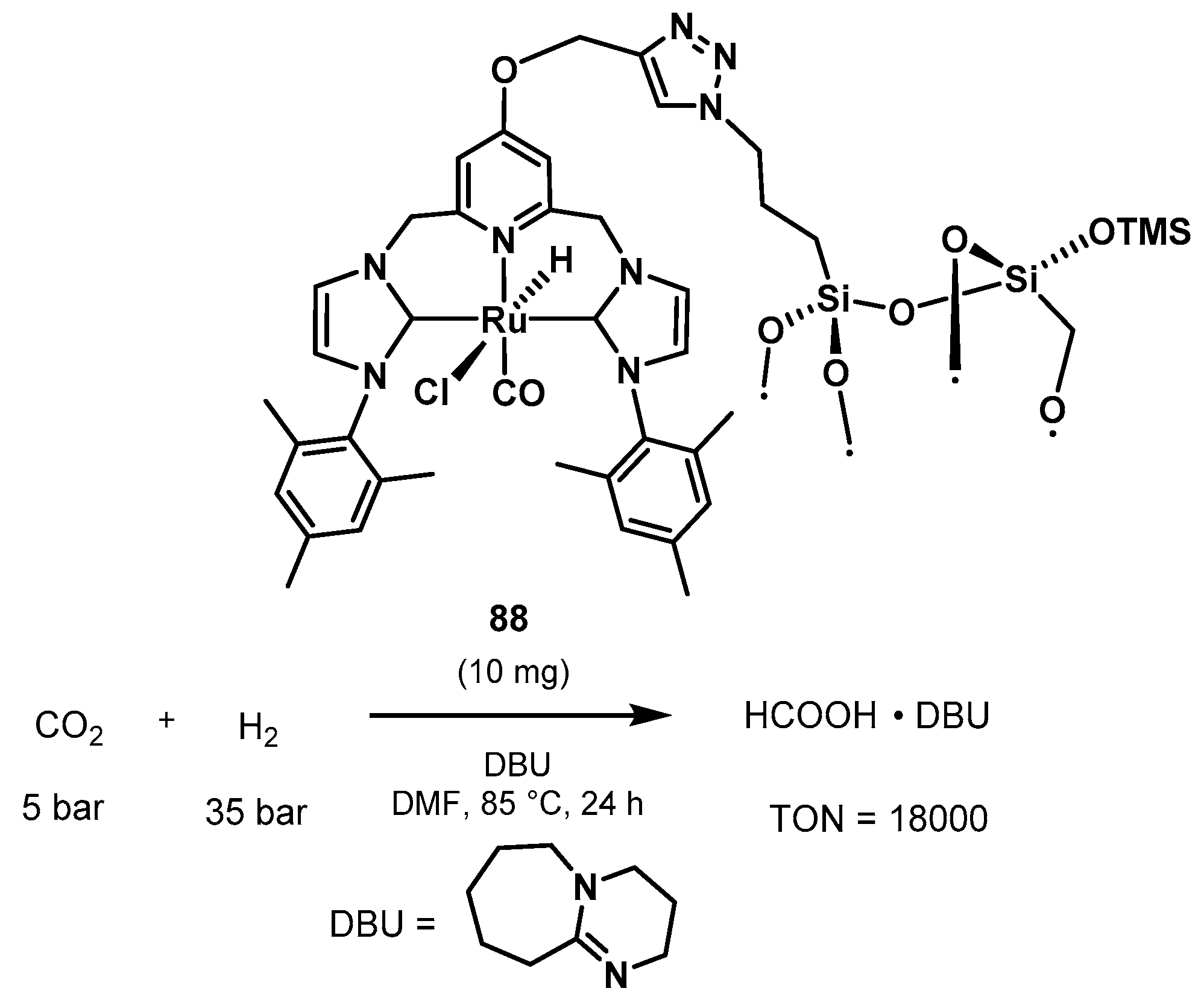{kind=link}
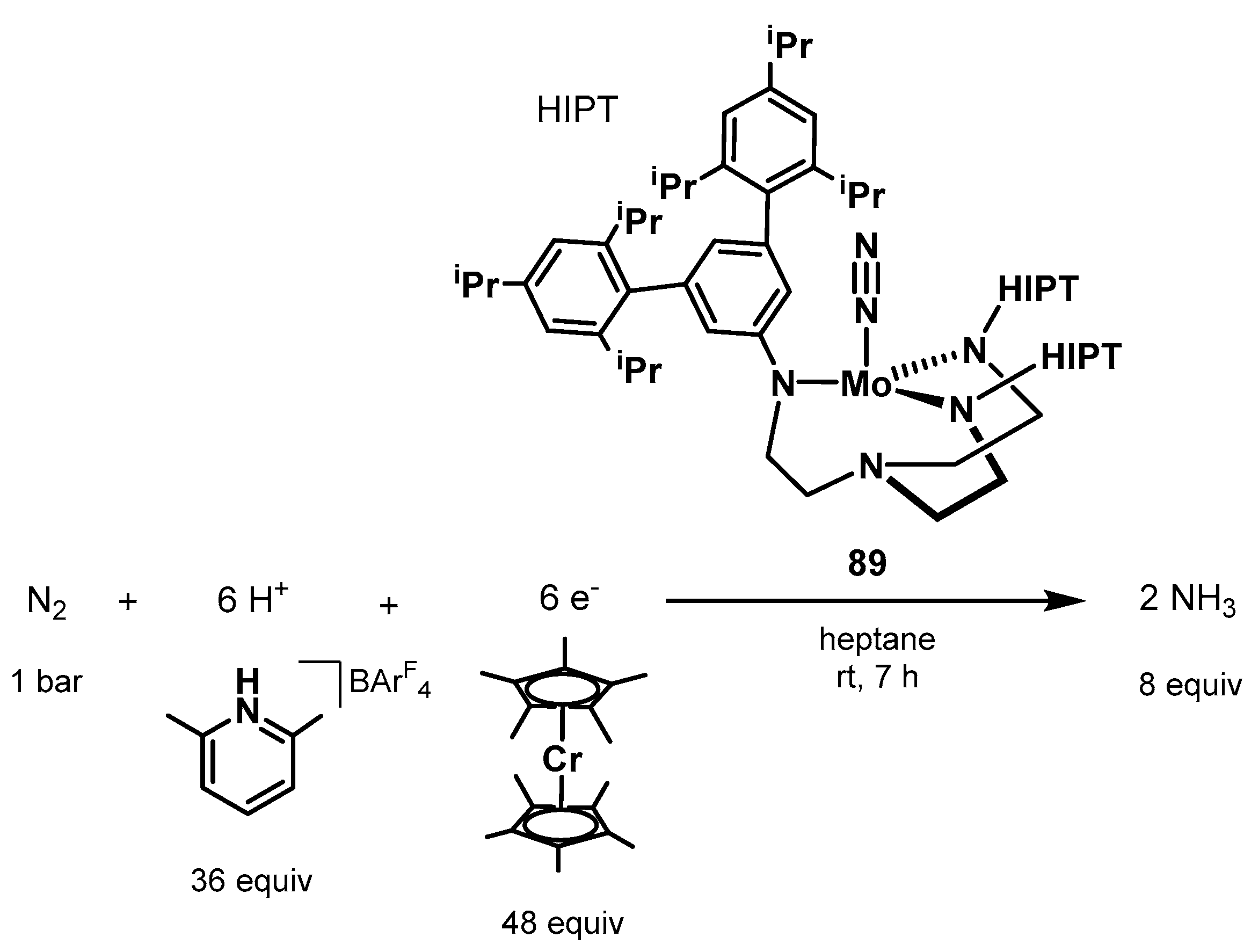{kind=link}
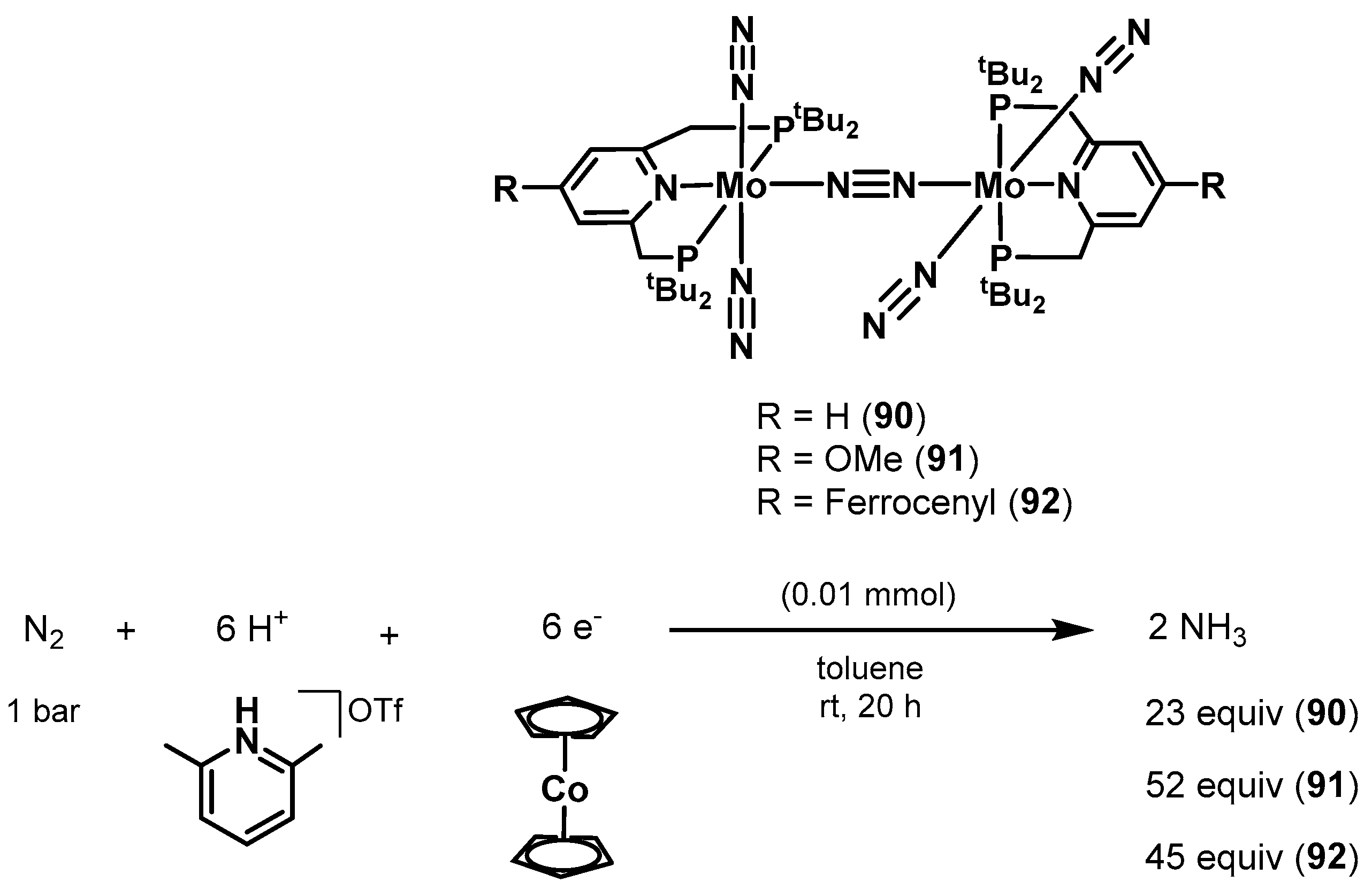{kind=link}
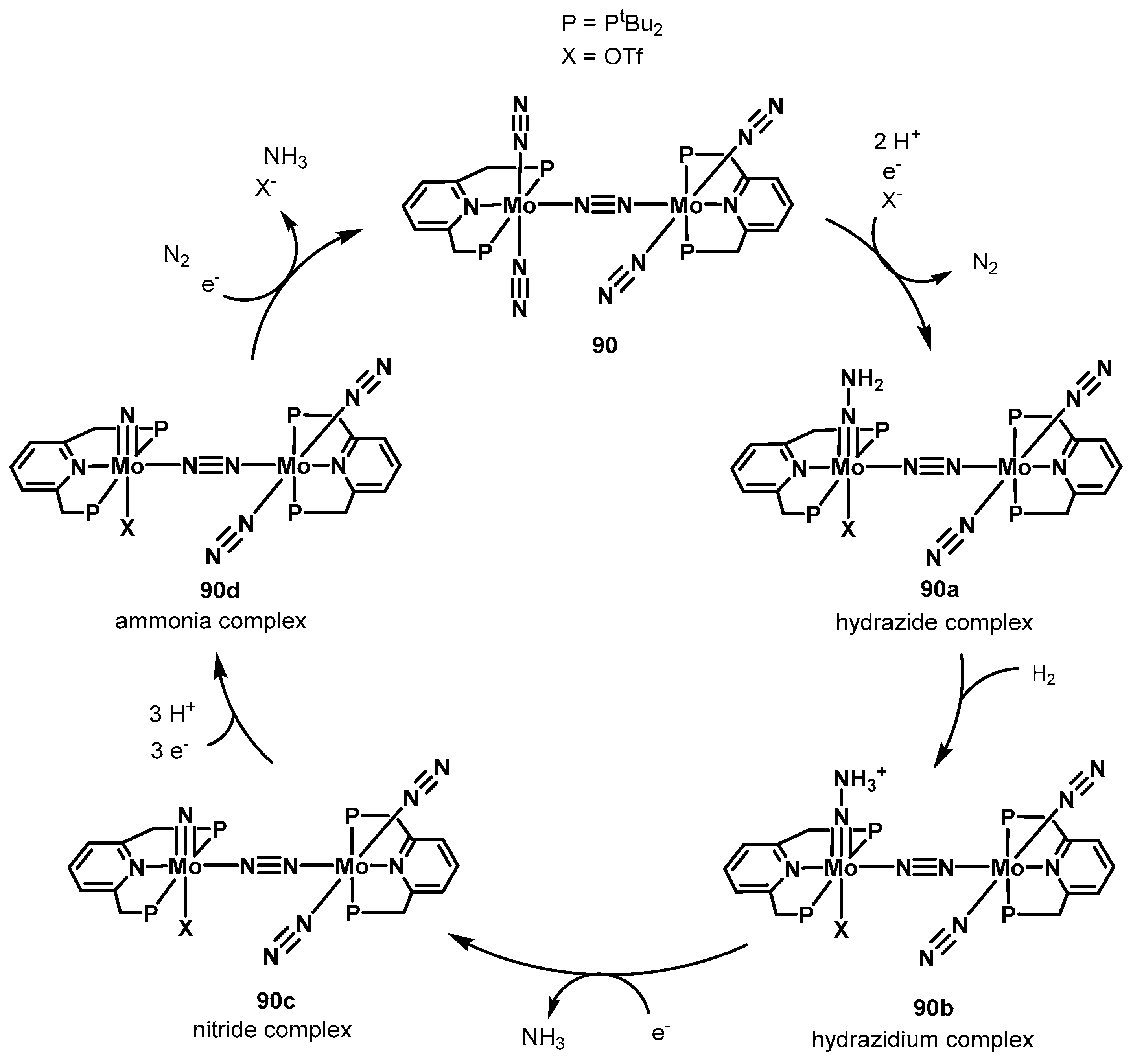{kind=link}
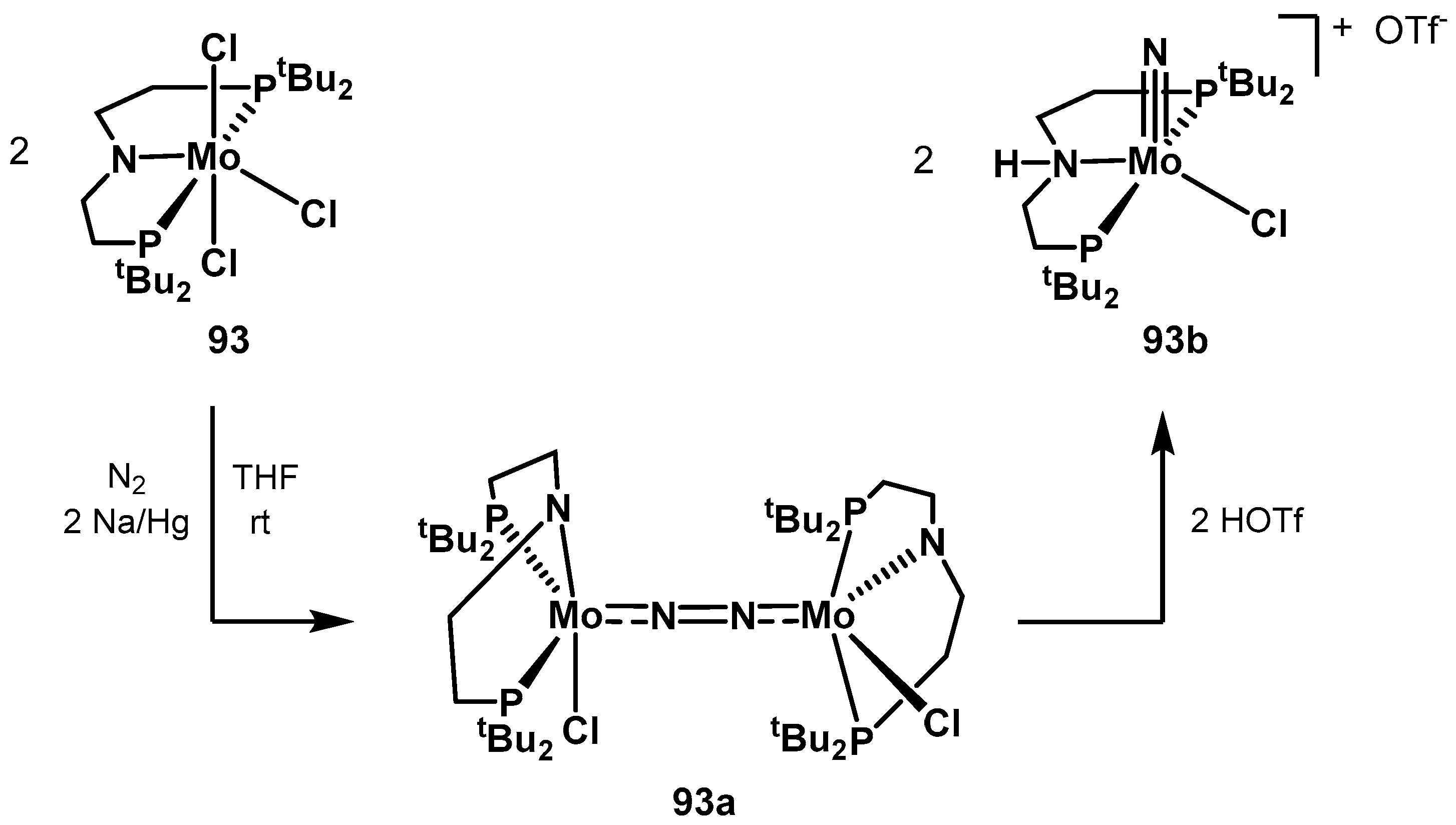{kind=link}
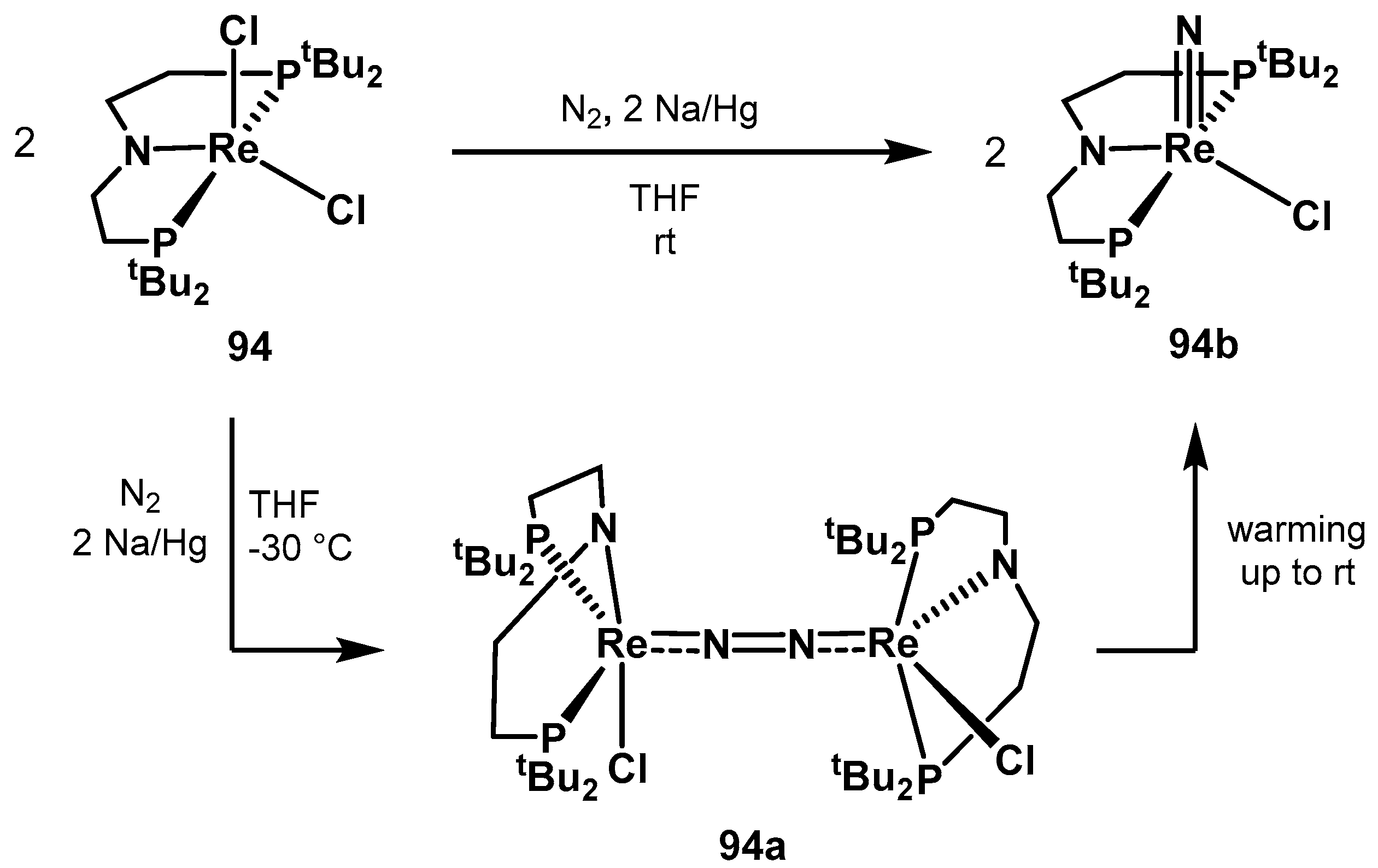{kind=link}
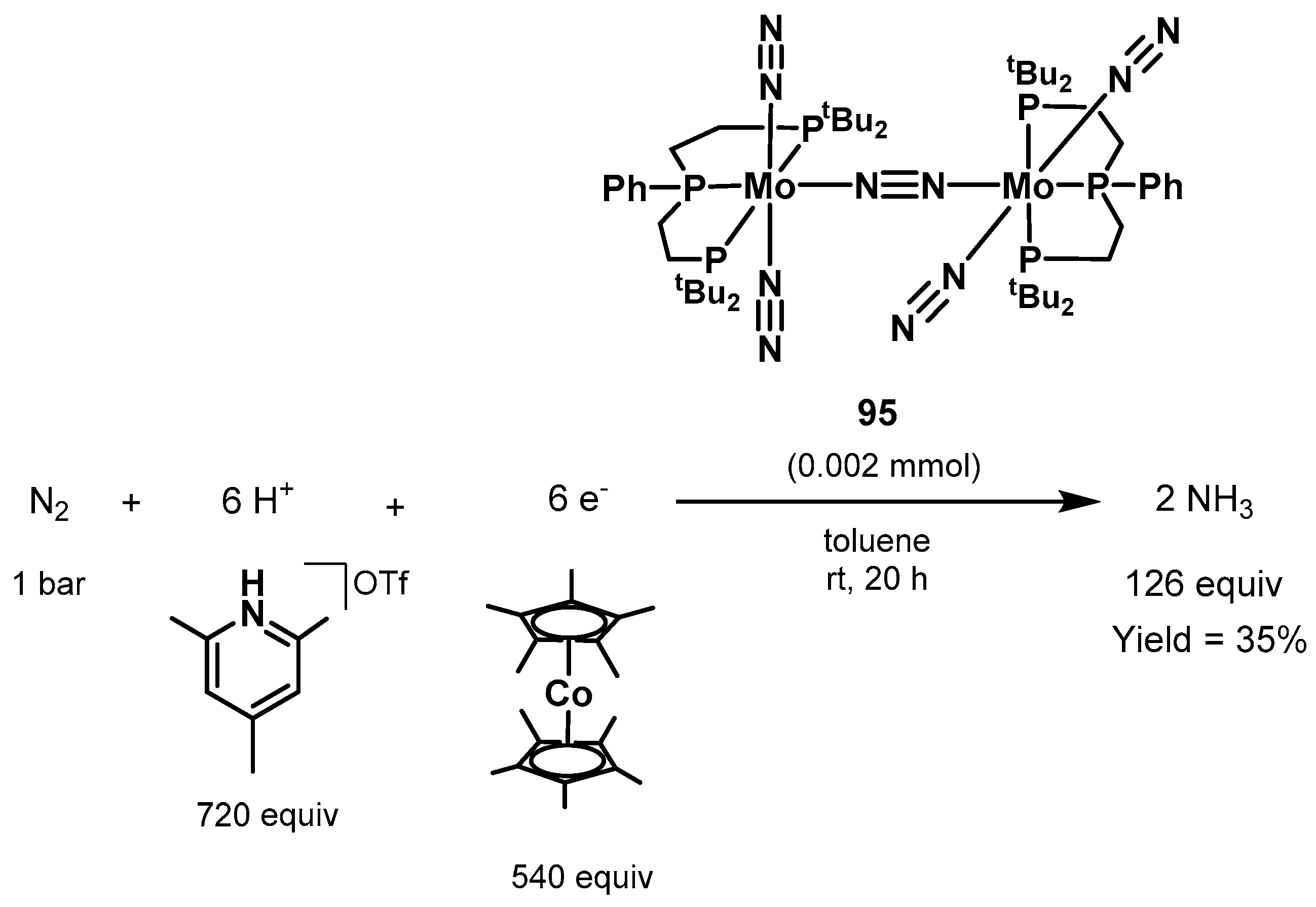{kind=link}
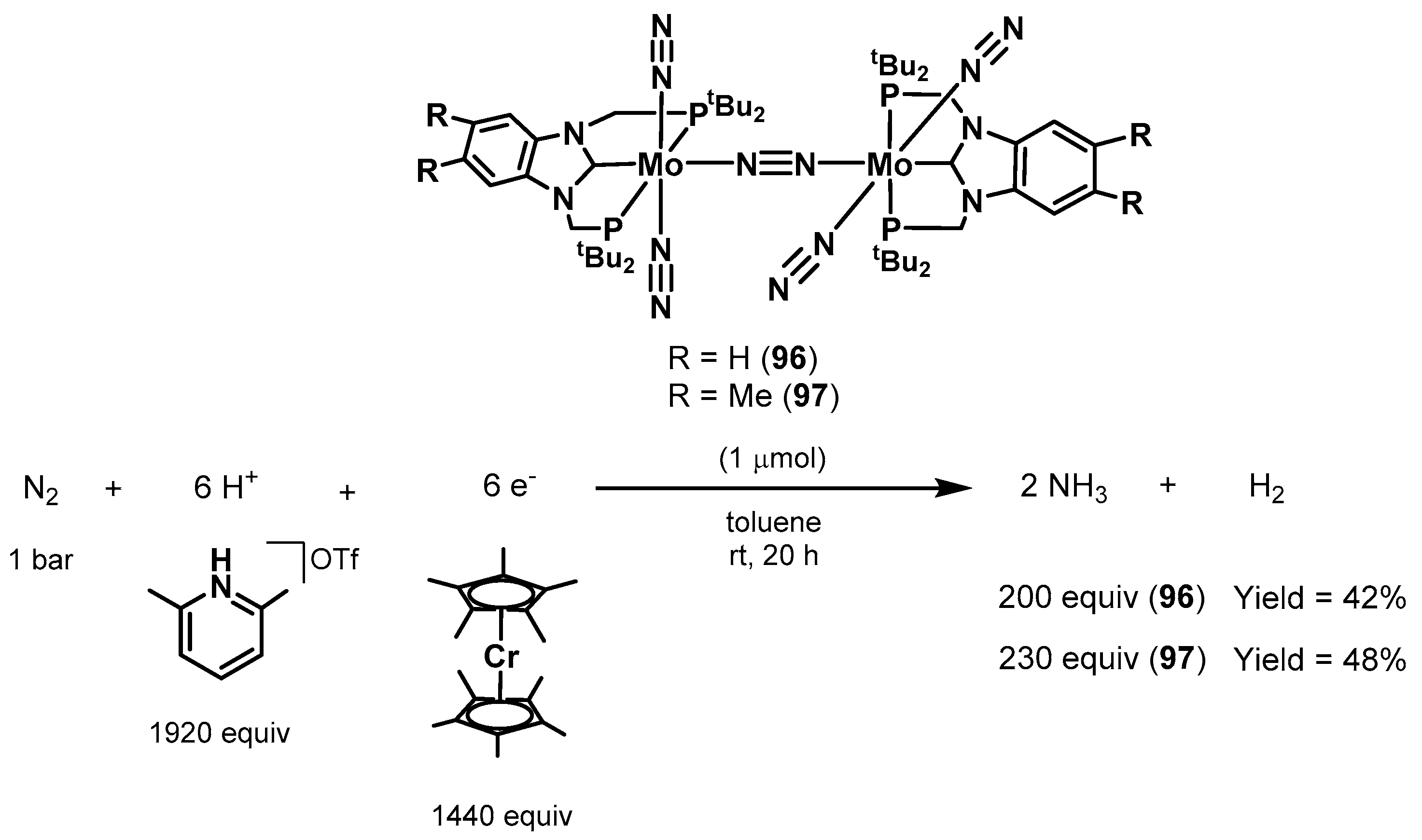{kind=link}
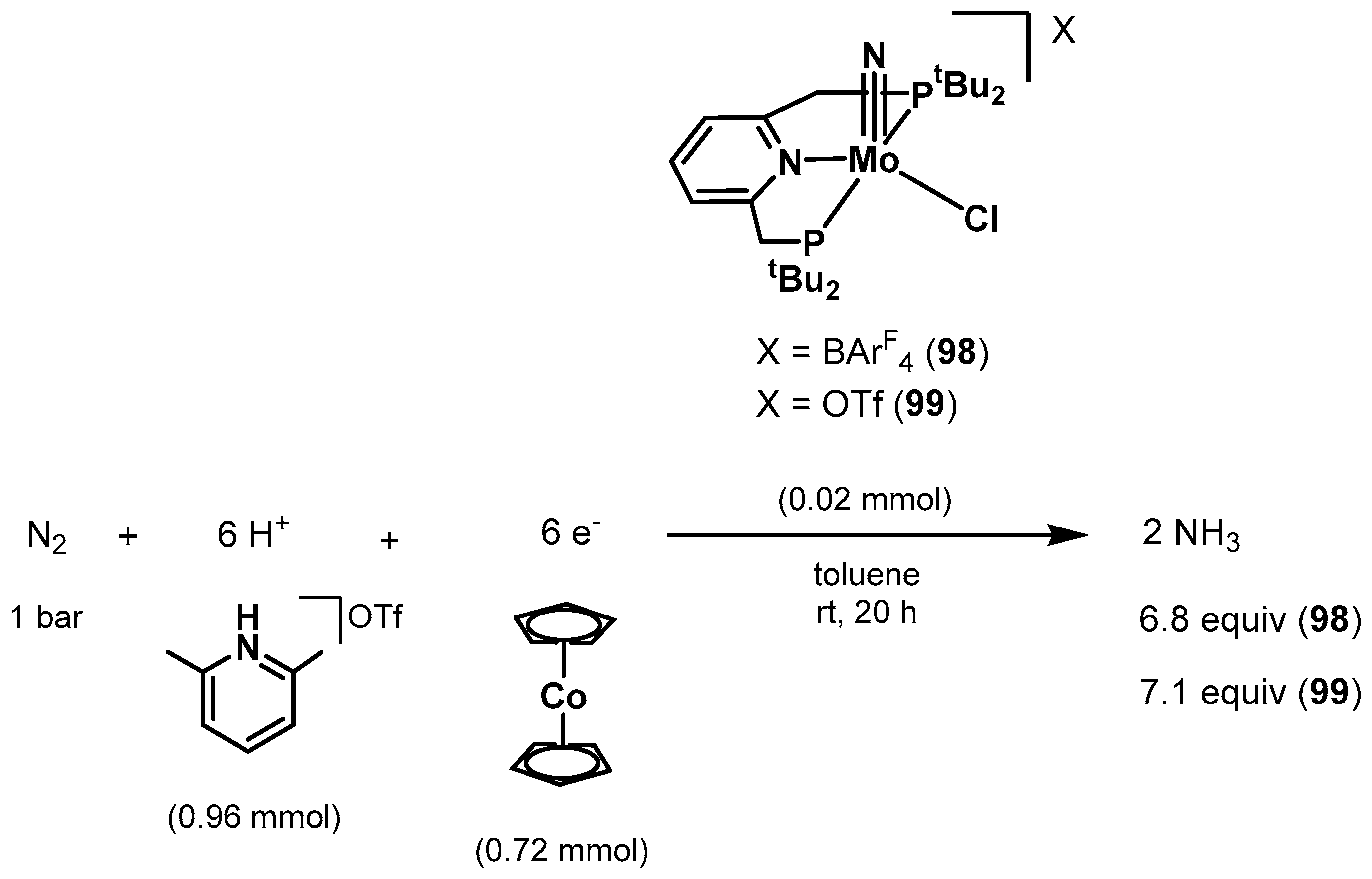{kind=link}
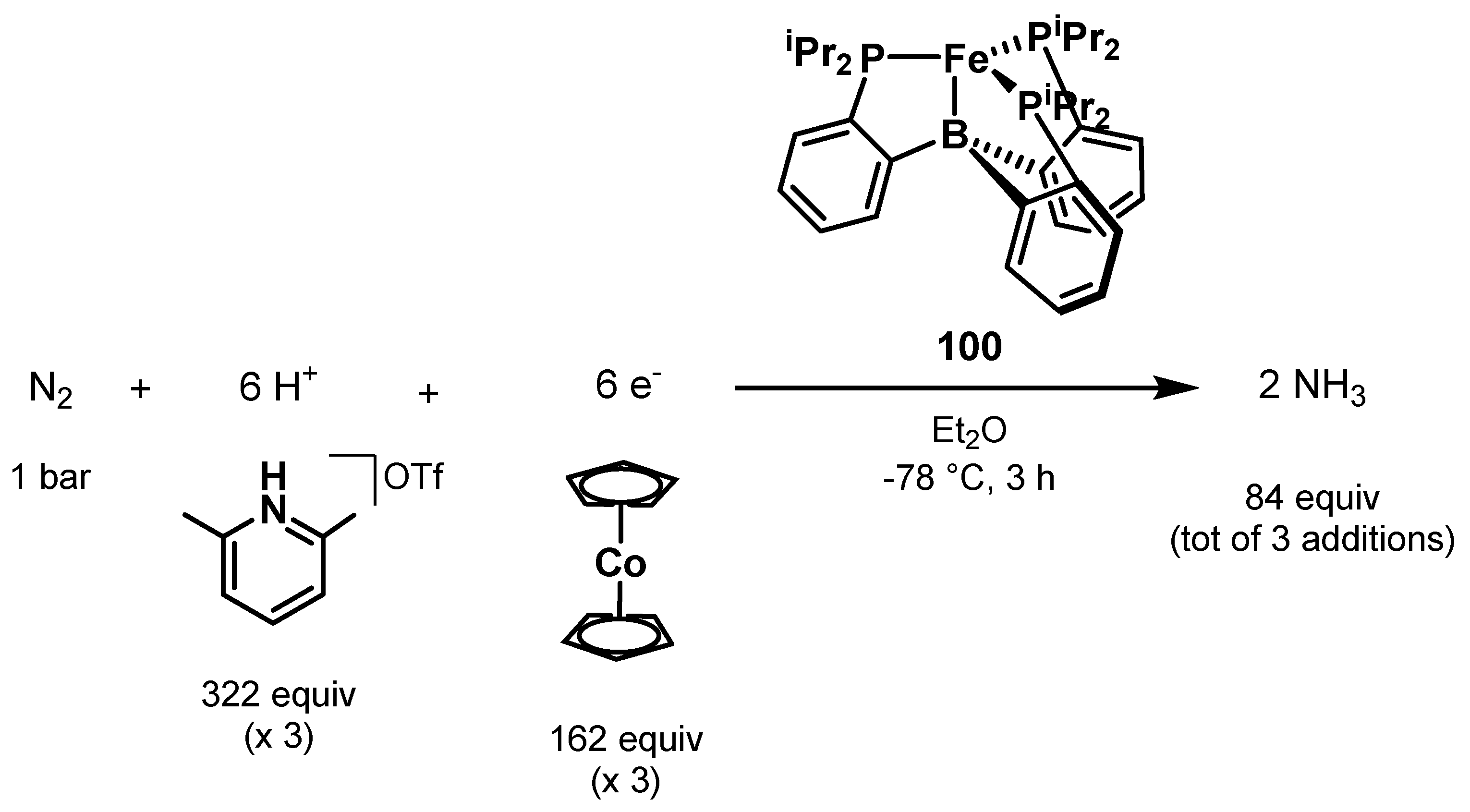{kind=link}
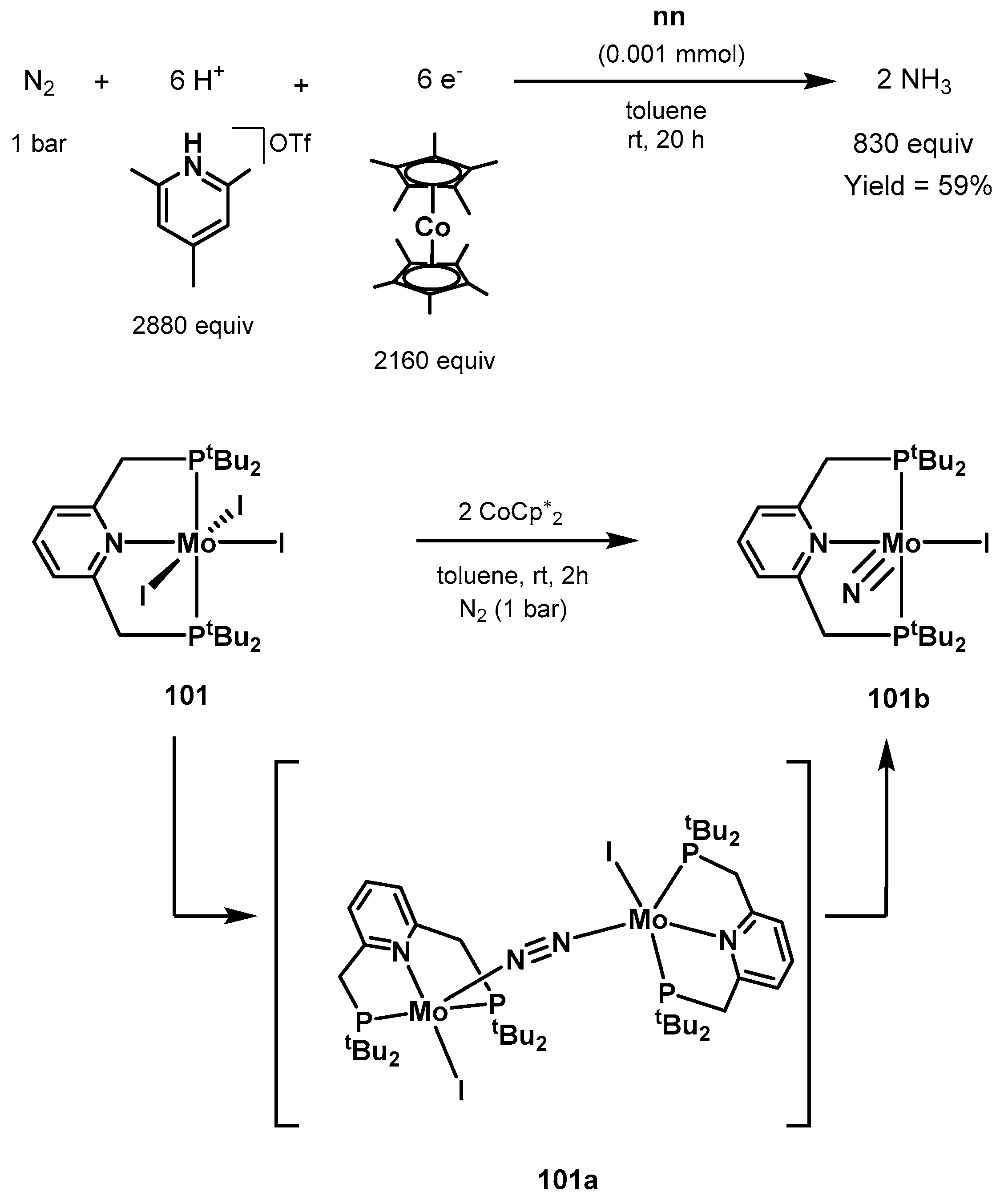{kind=link}
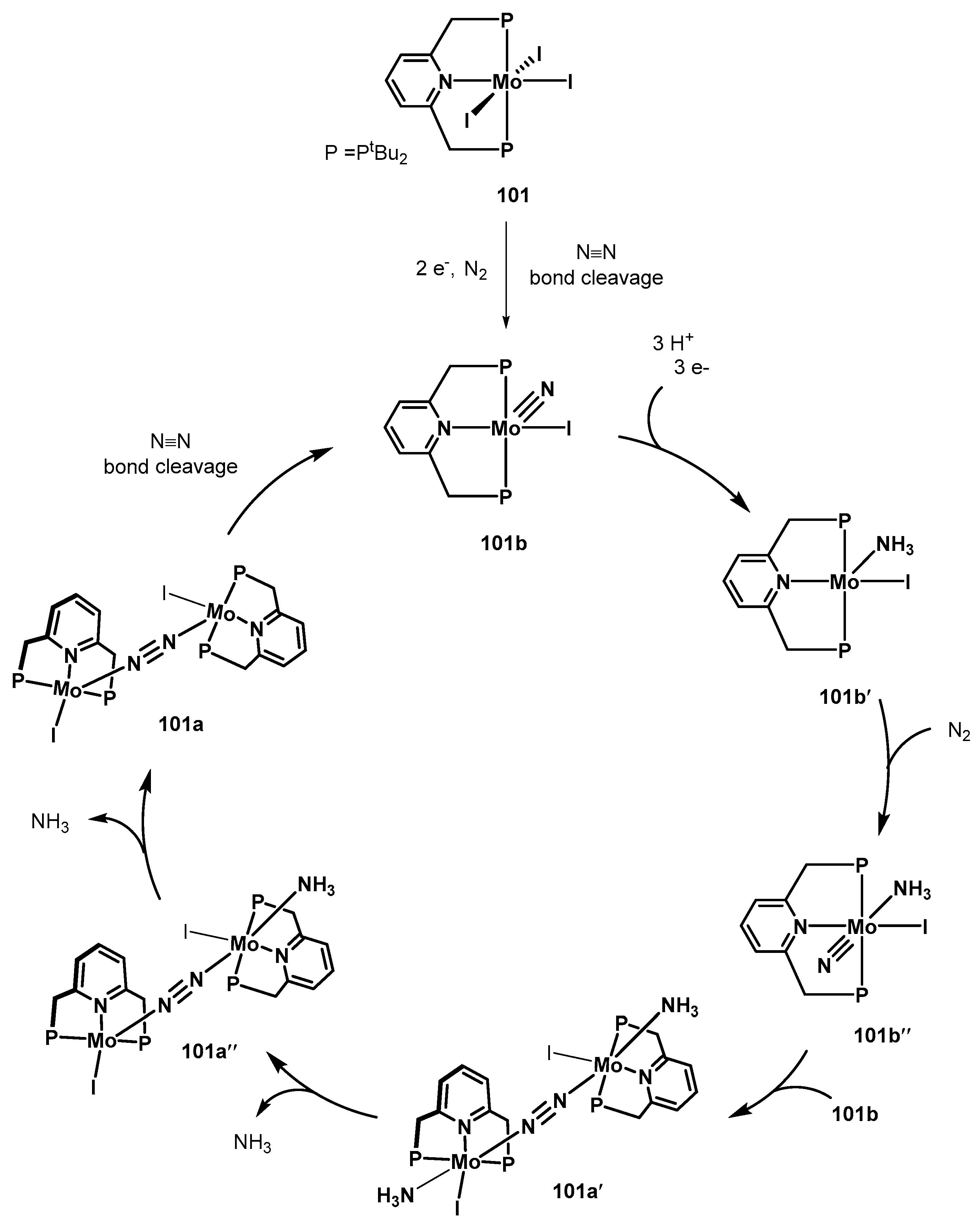{kind=link}
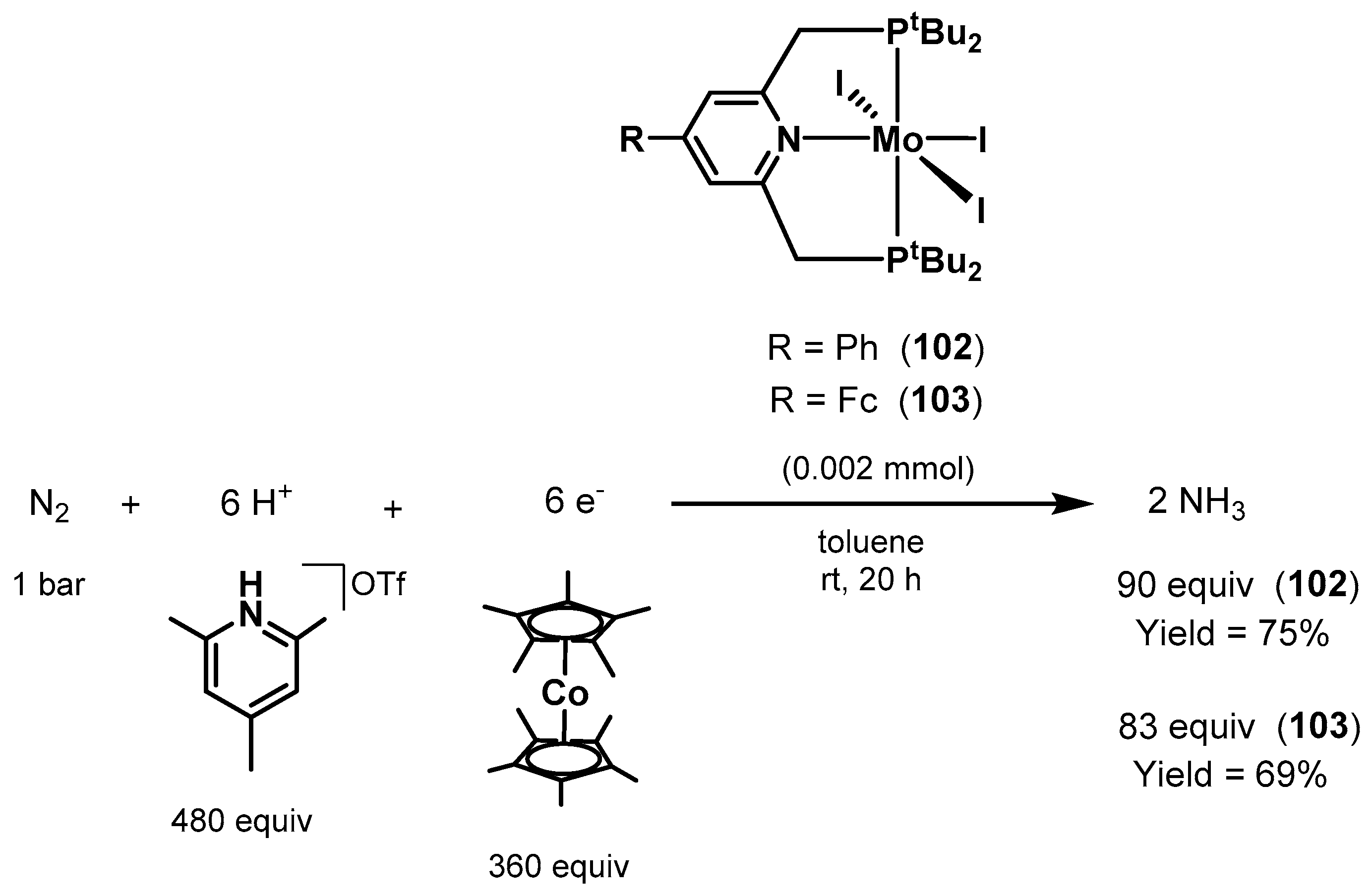{kind=link}
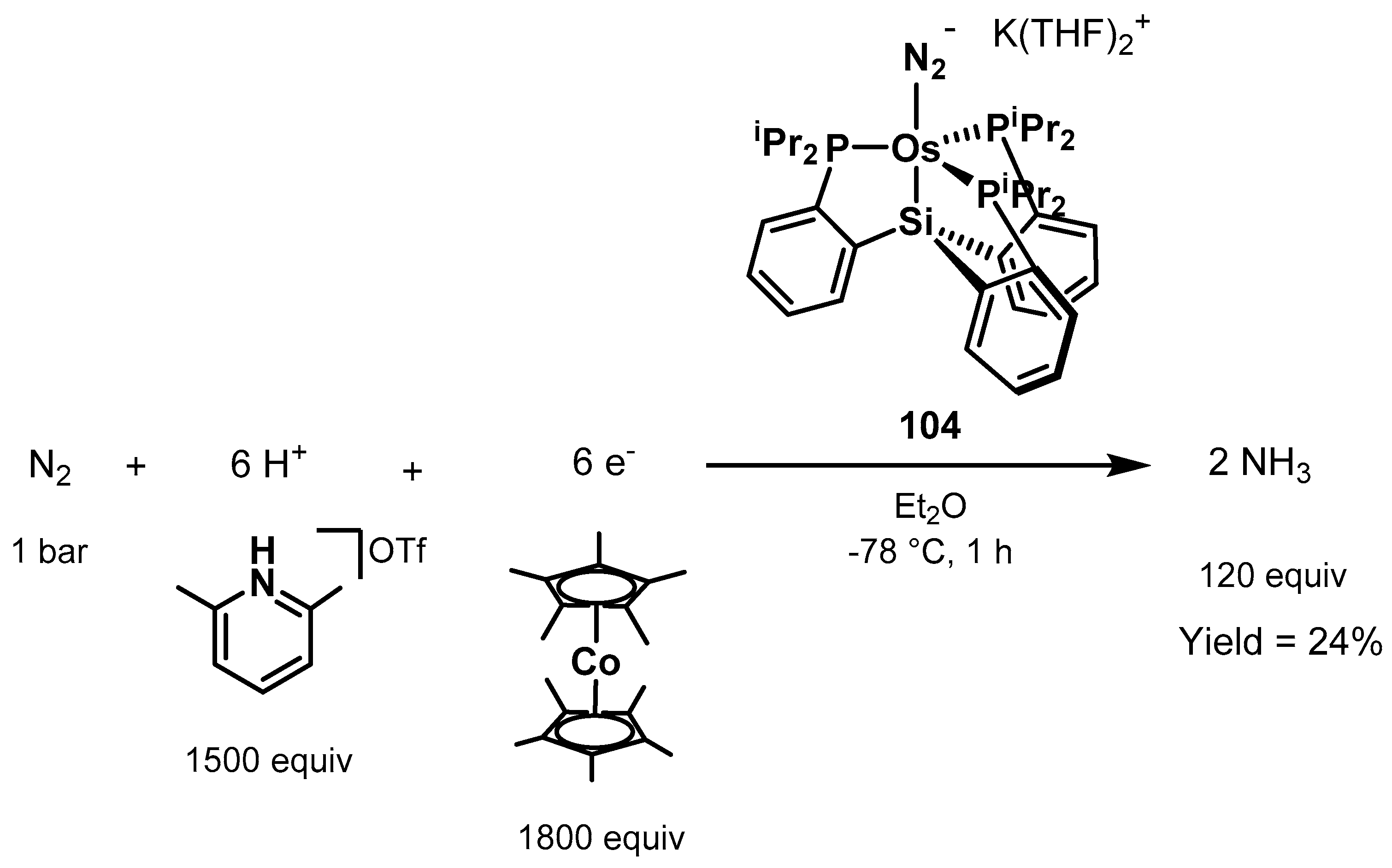{kind=link}
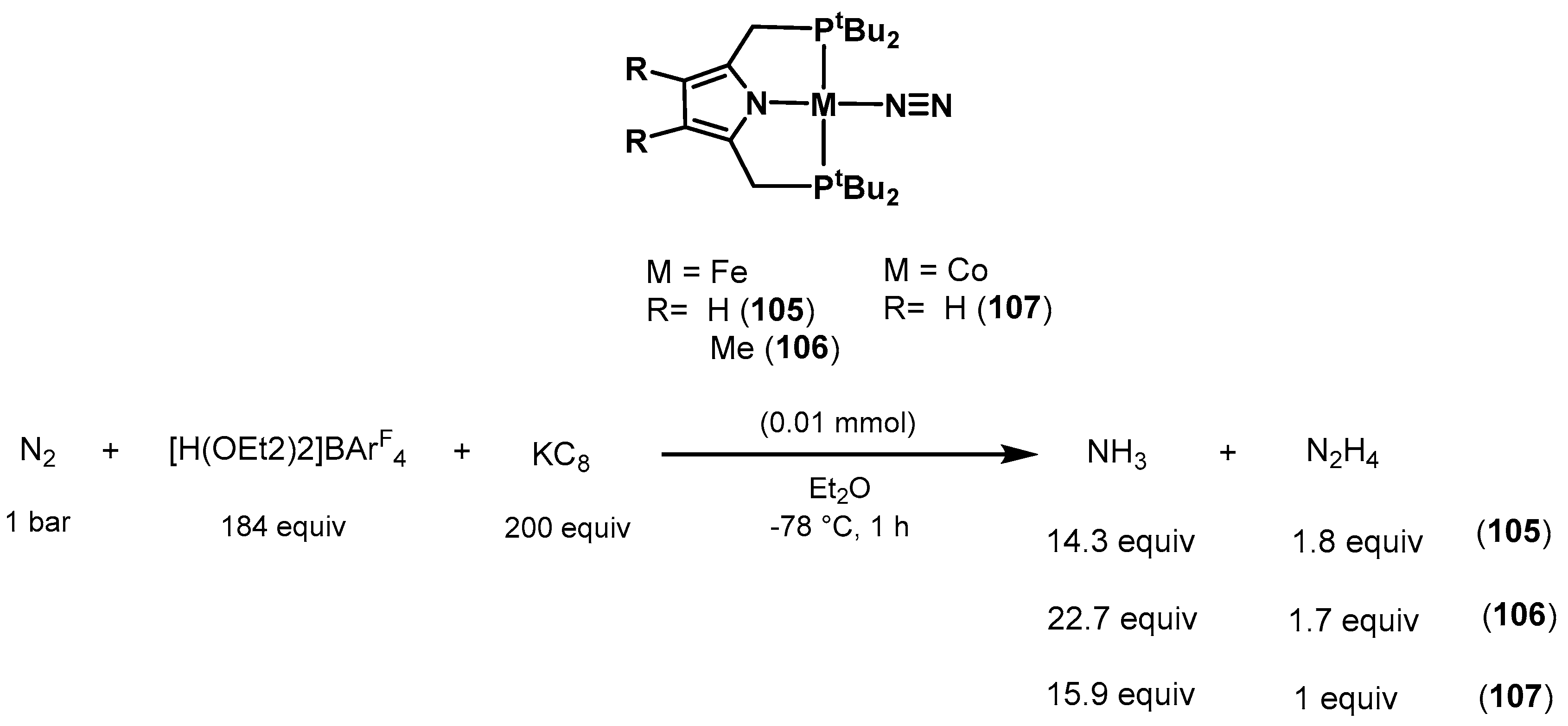{kind=link}
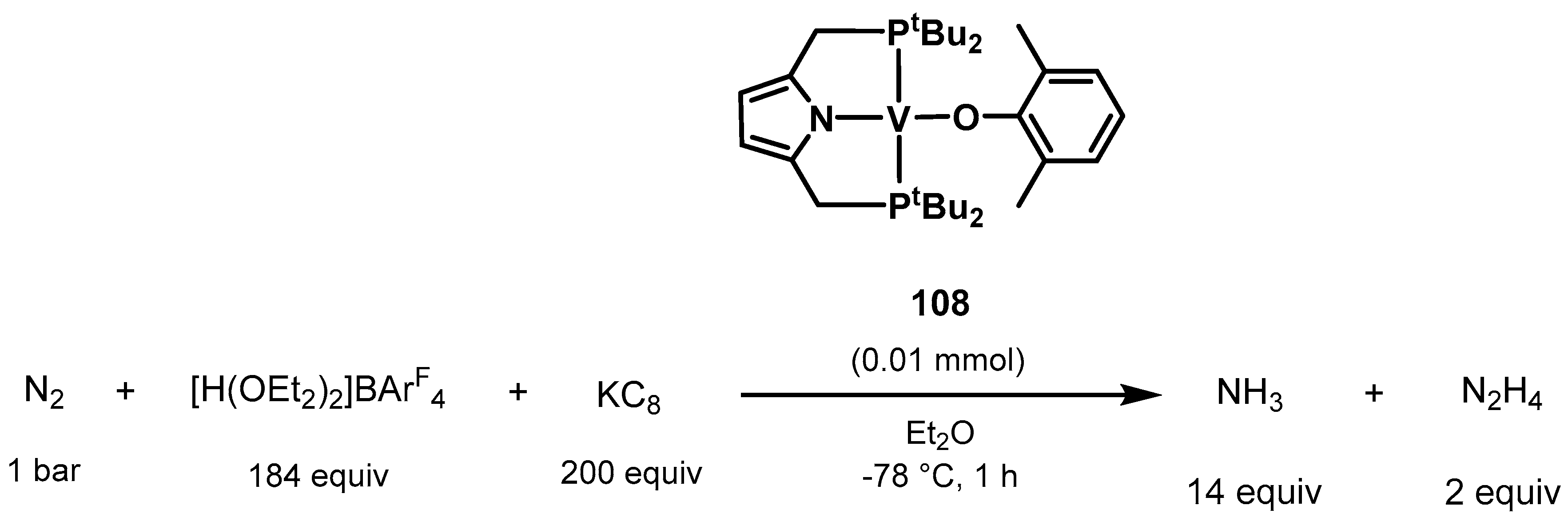{kind=link}
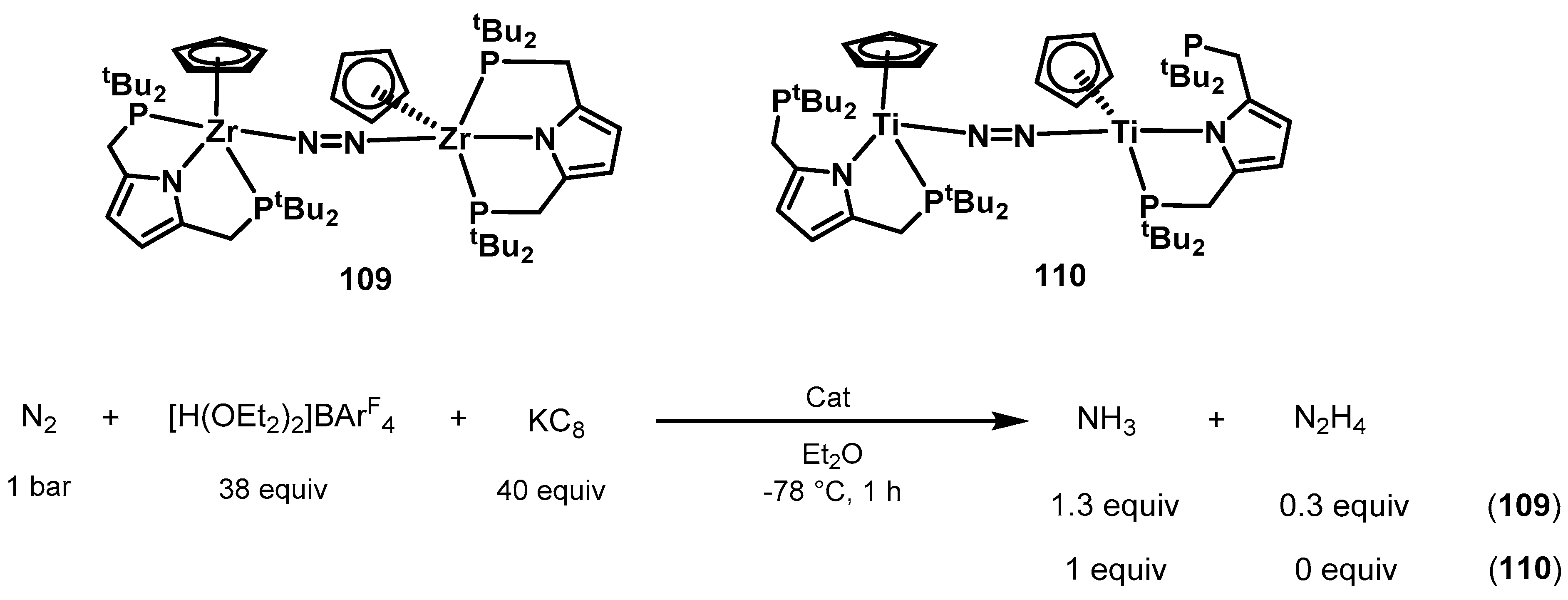{kind=link}
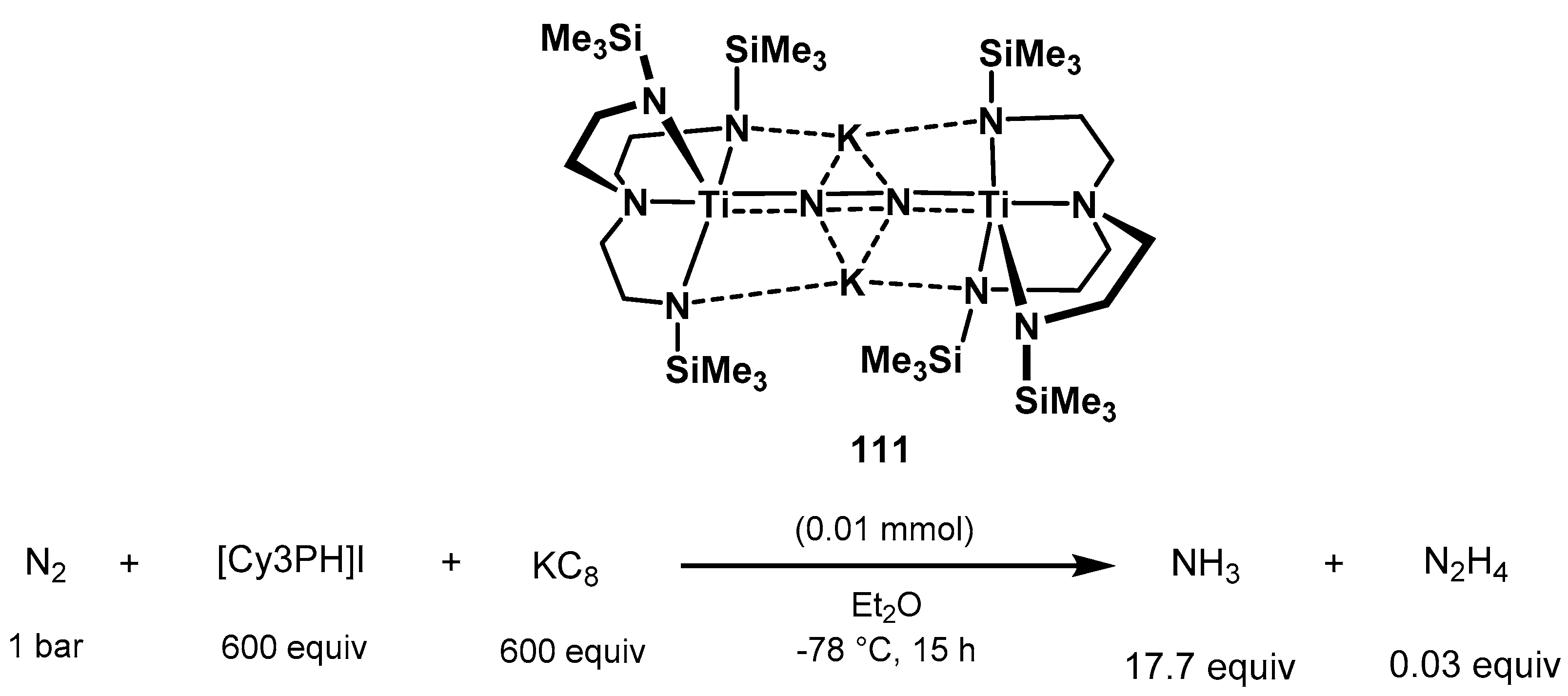{kind=link}
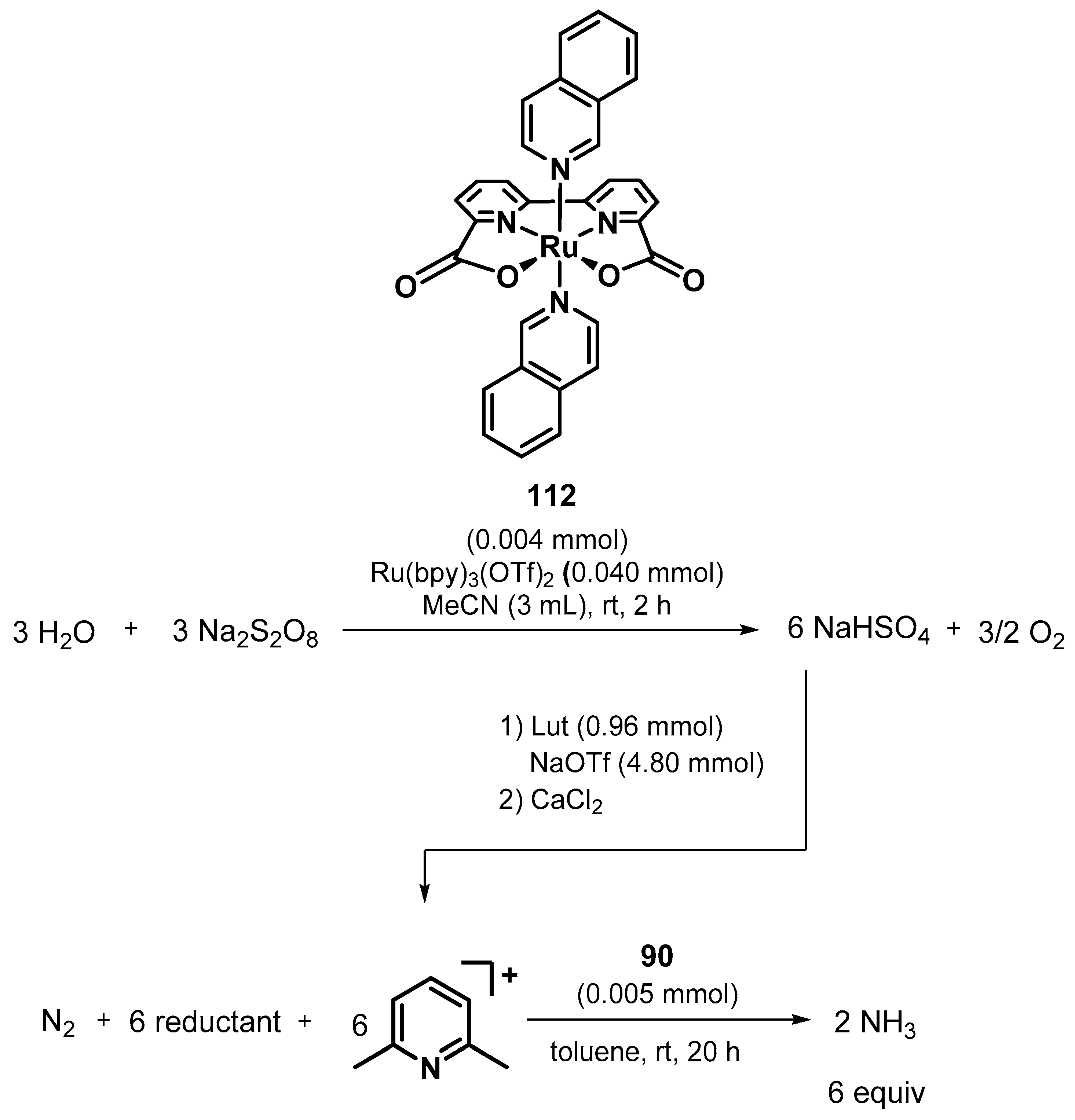{kind=link}
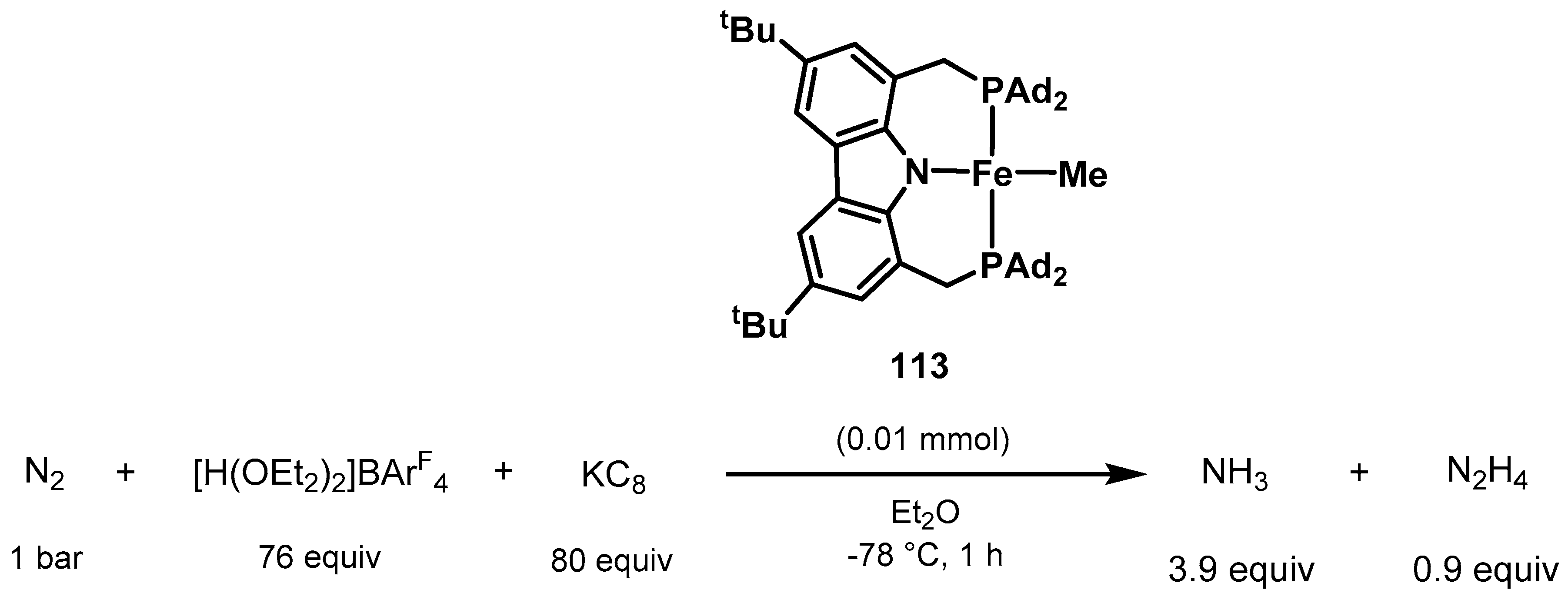{kind=link}
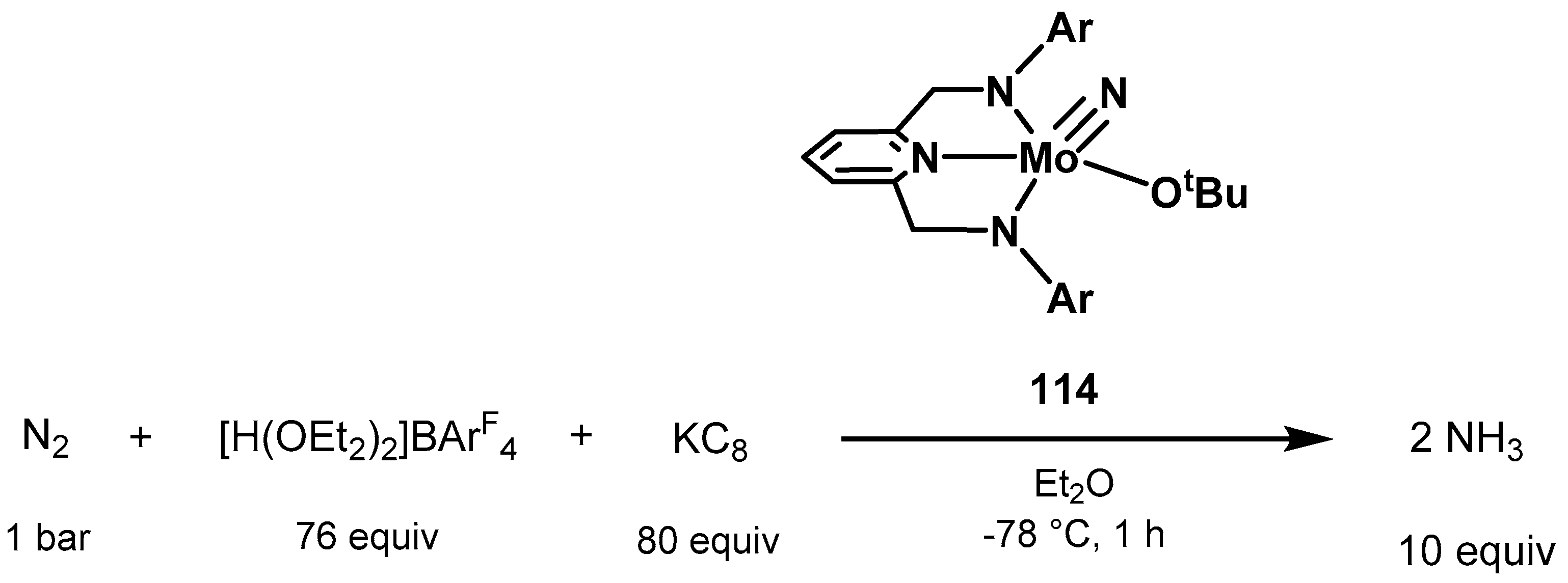{kind=link}
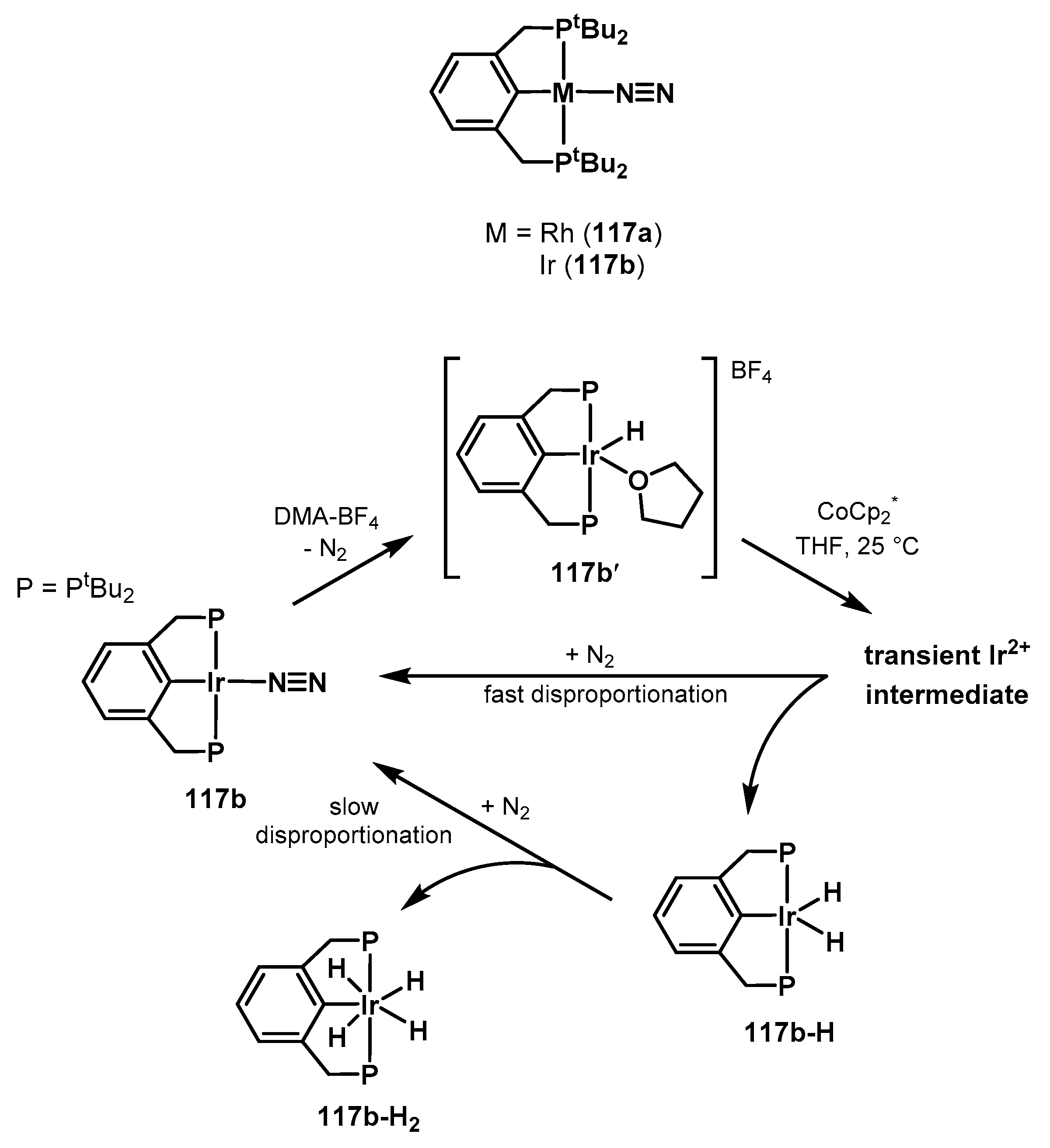{kind=link}
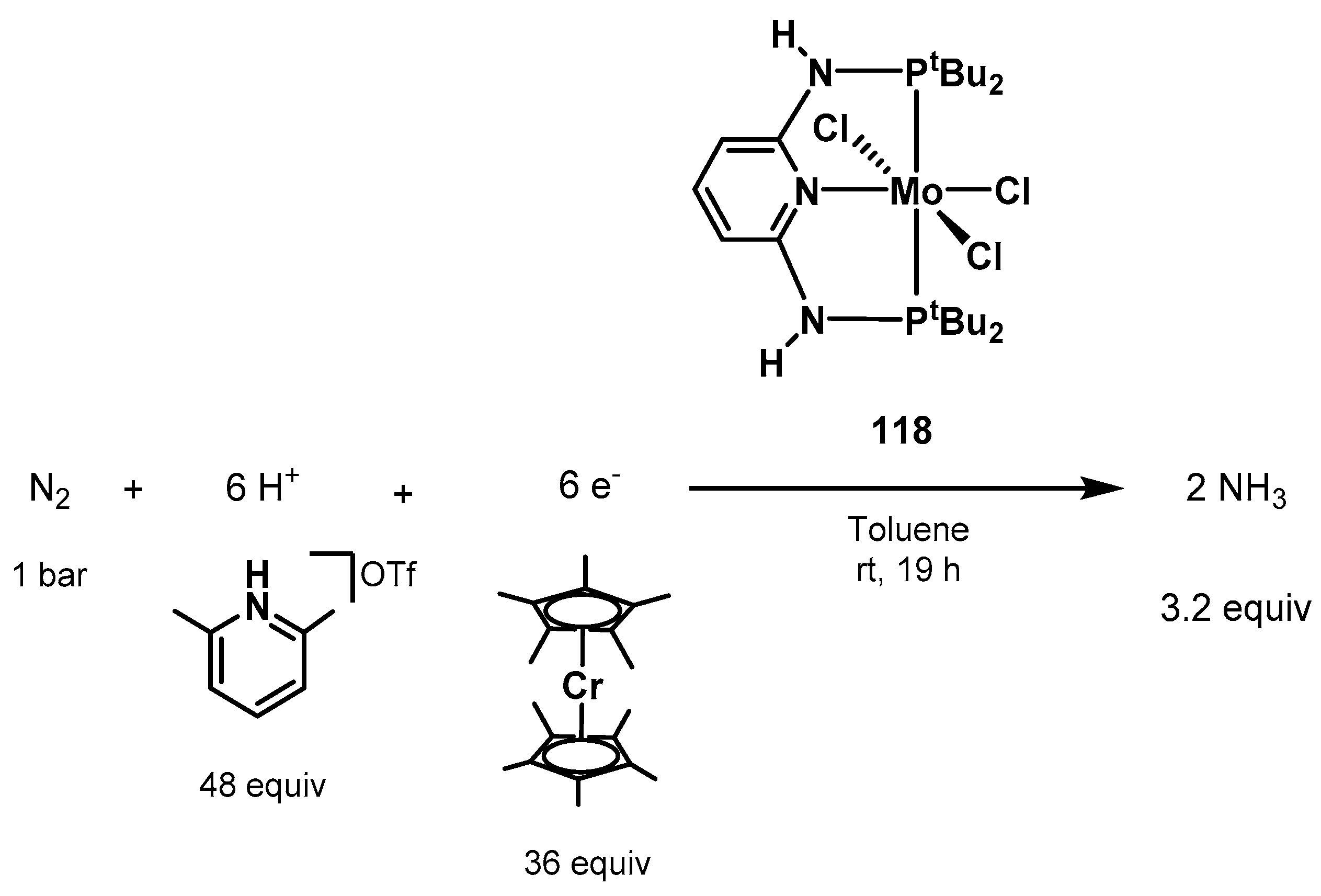{kind=link}
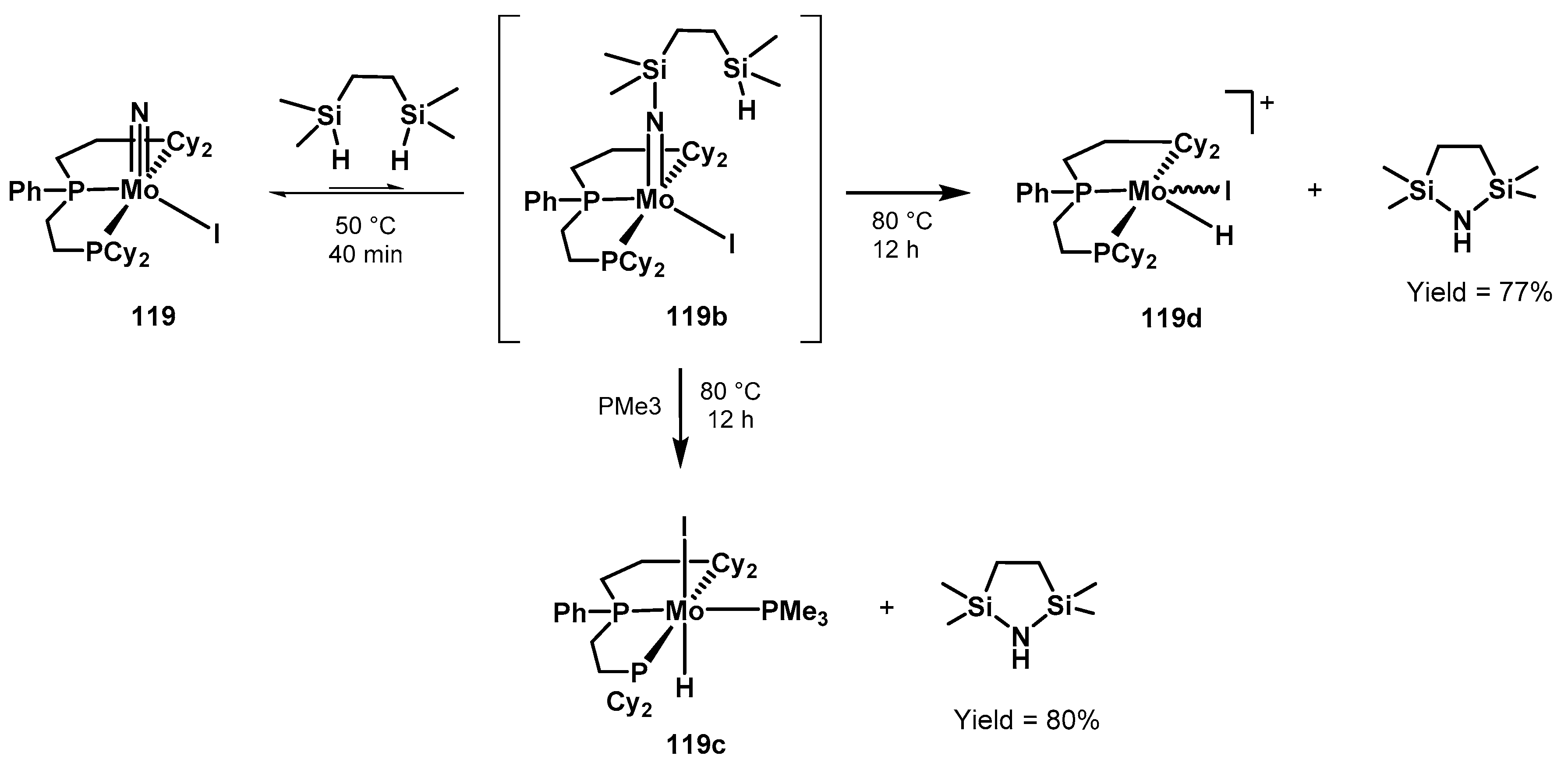{kind=link}
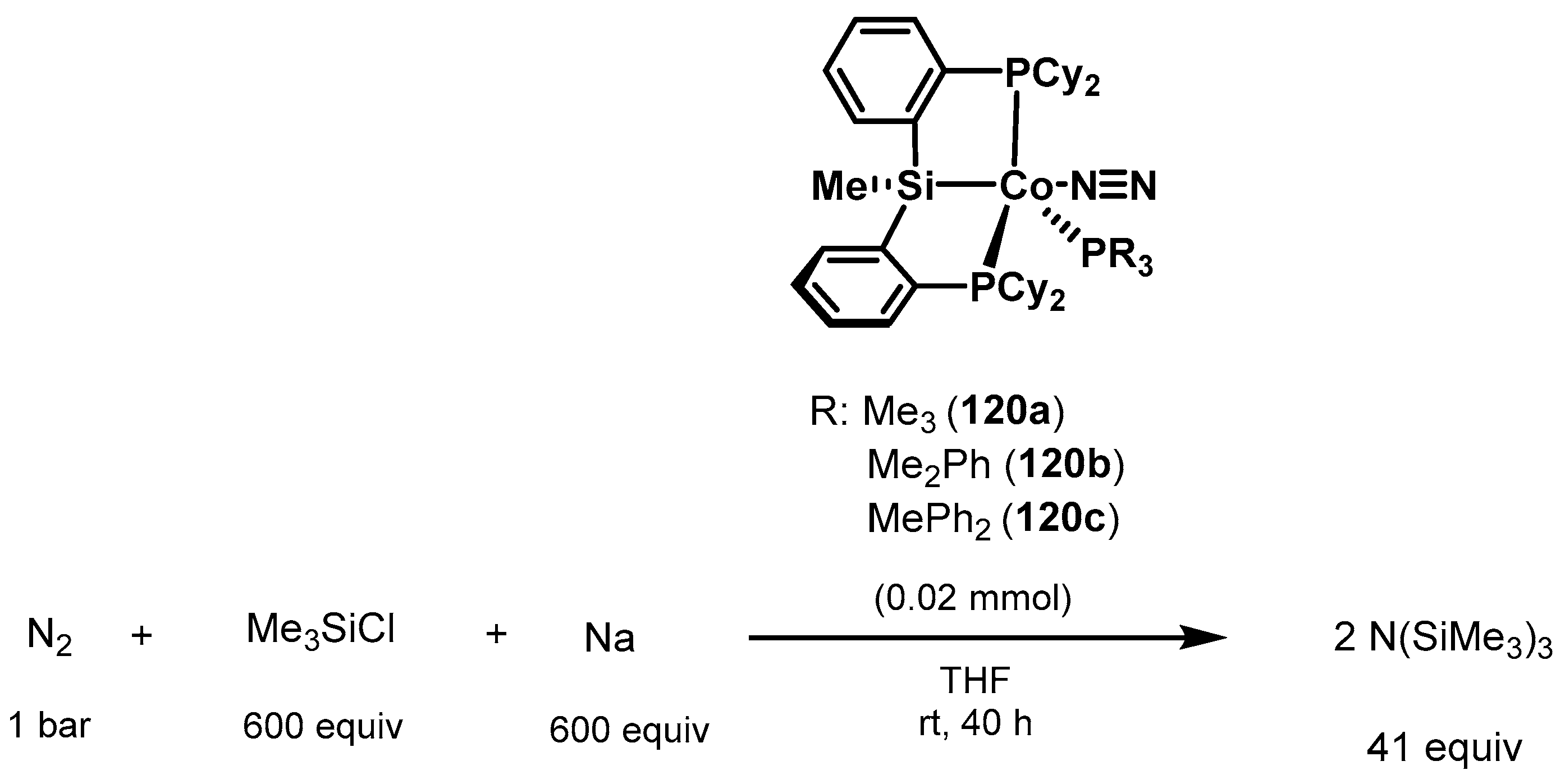{kind=link}
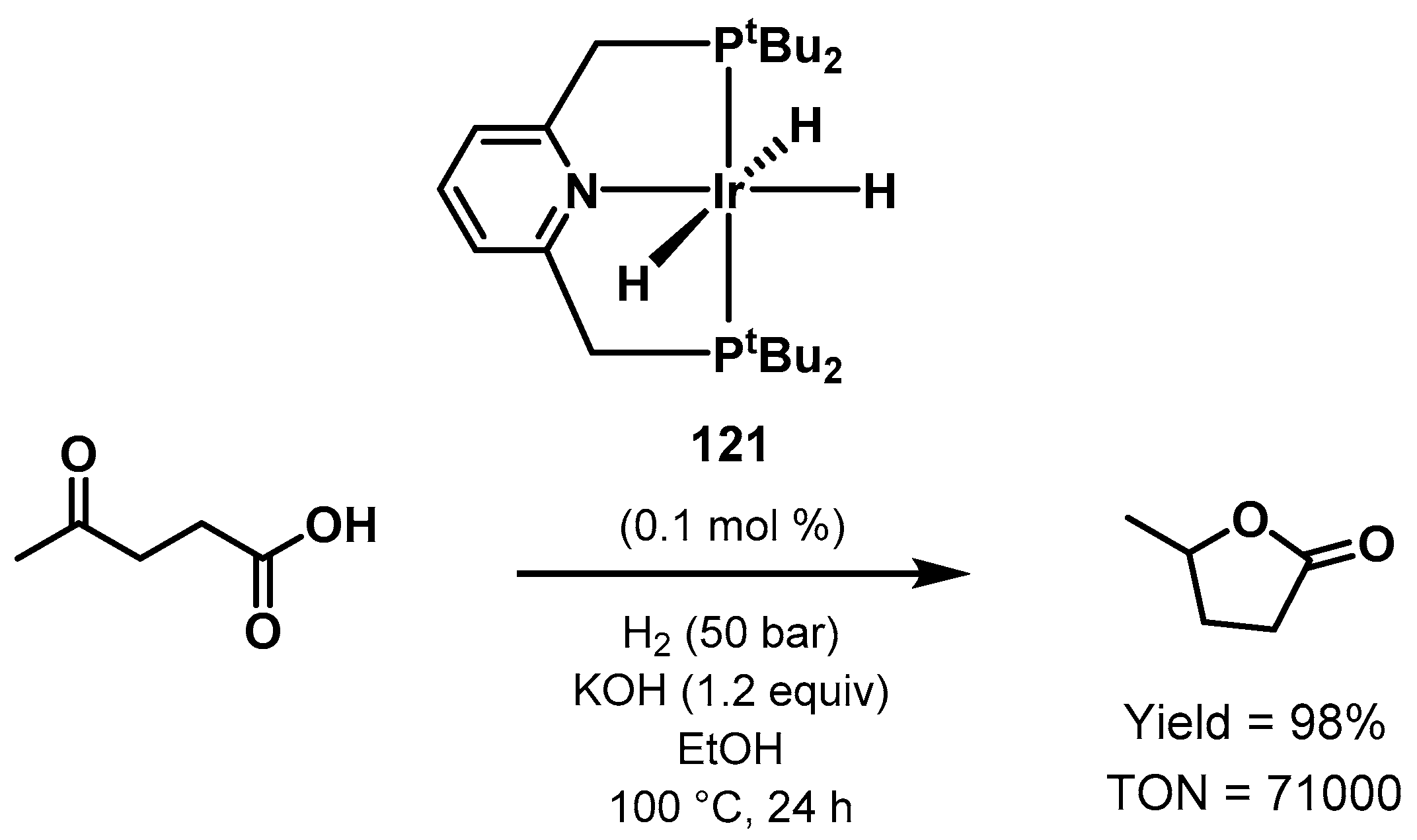{kind=link}
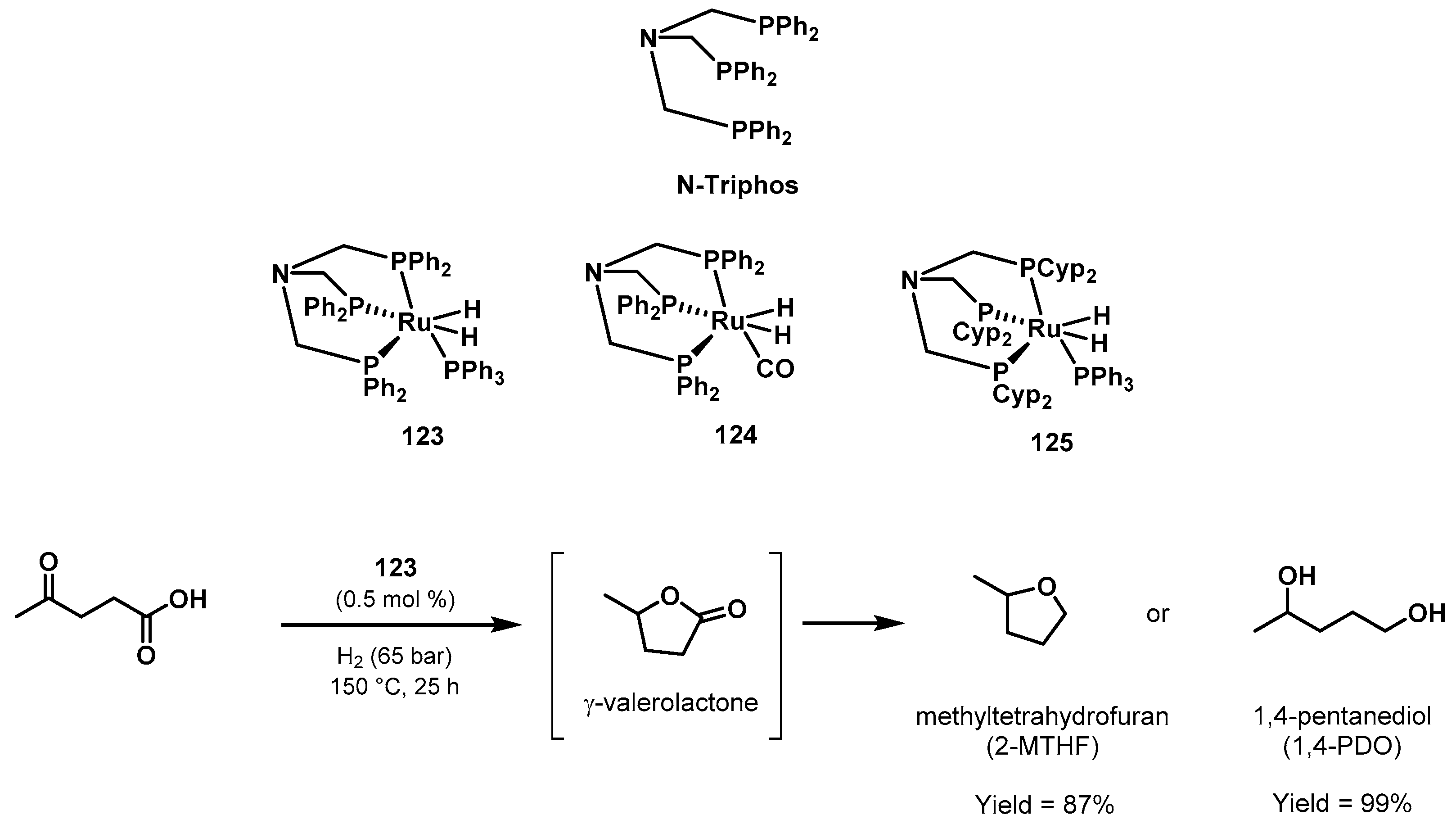{kind=link}
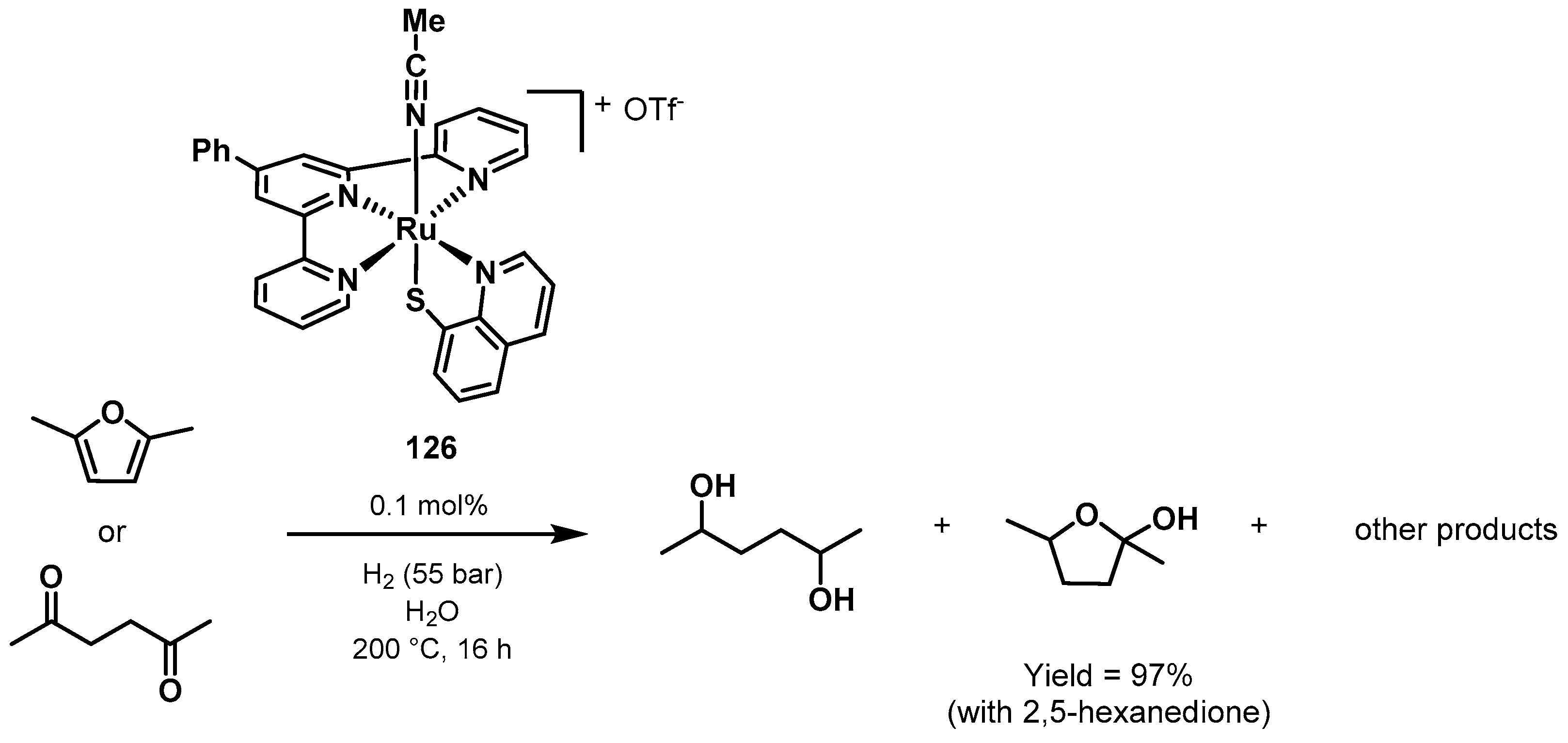{kind=link}
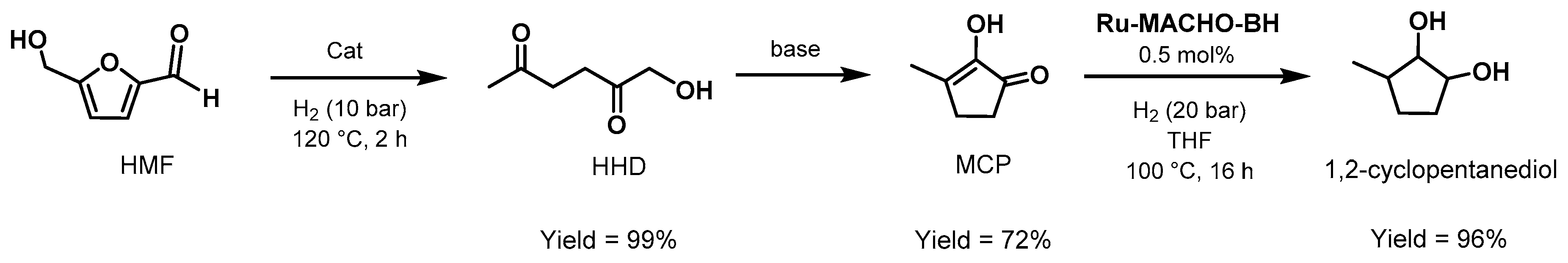{kind=link}
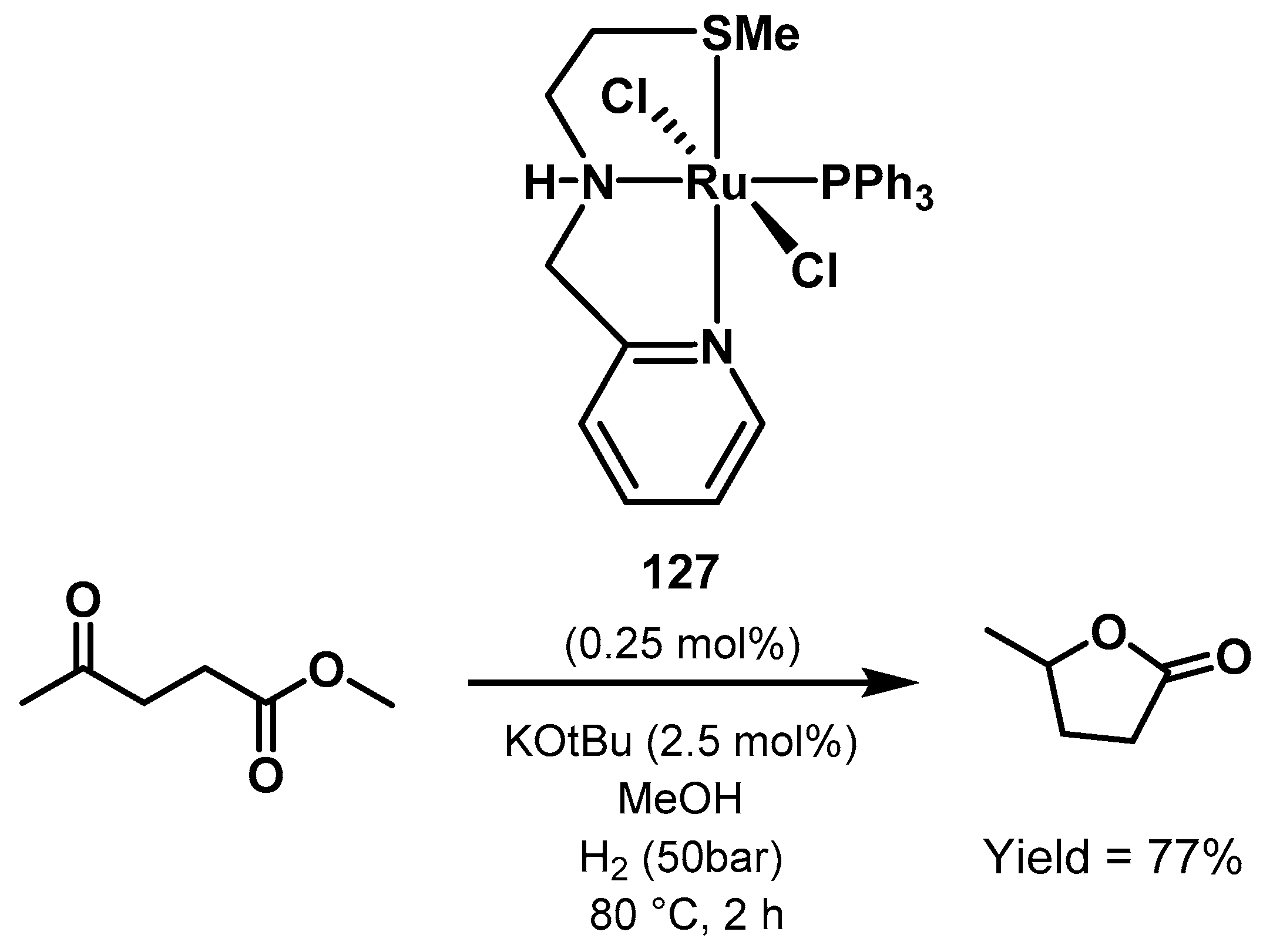{kind=link}
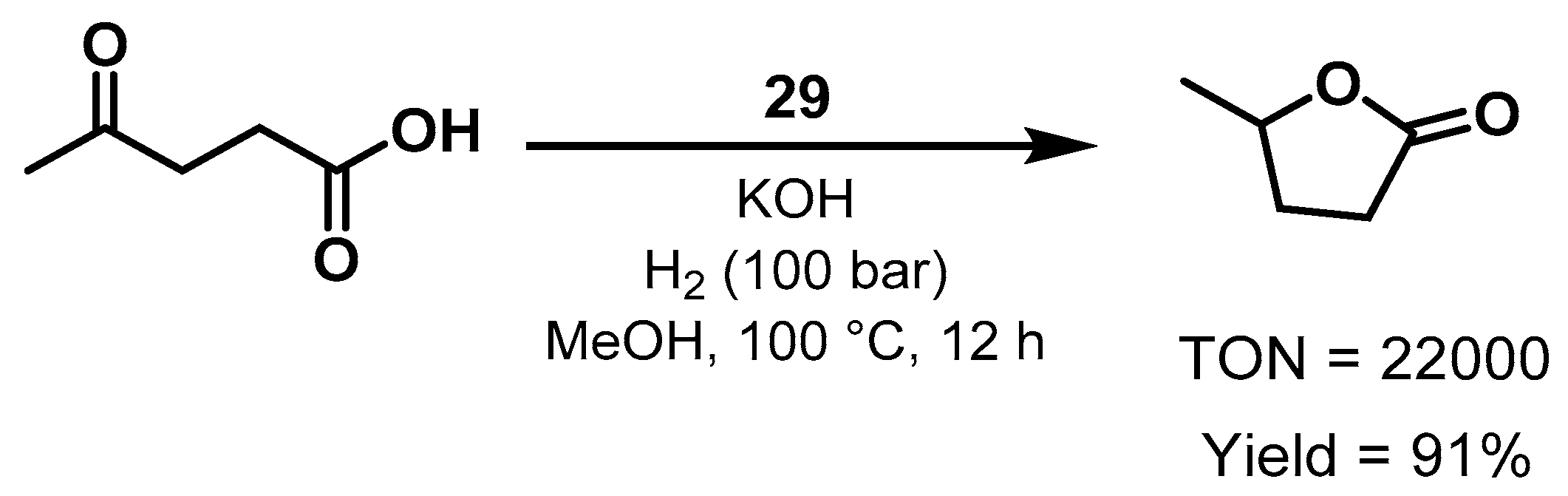{kind=link}
{kind=link}
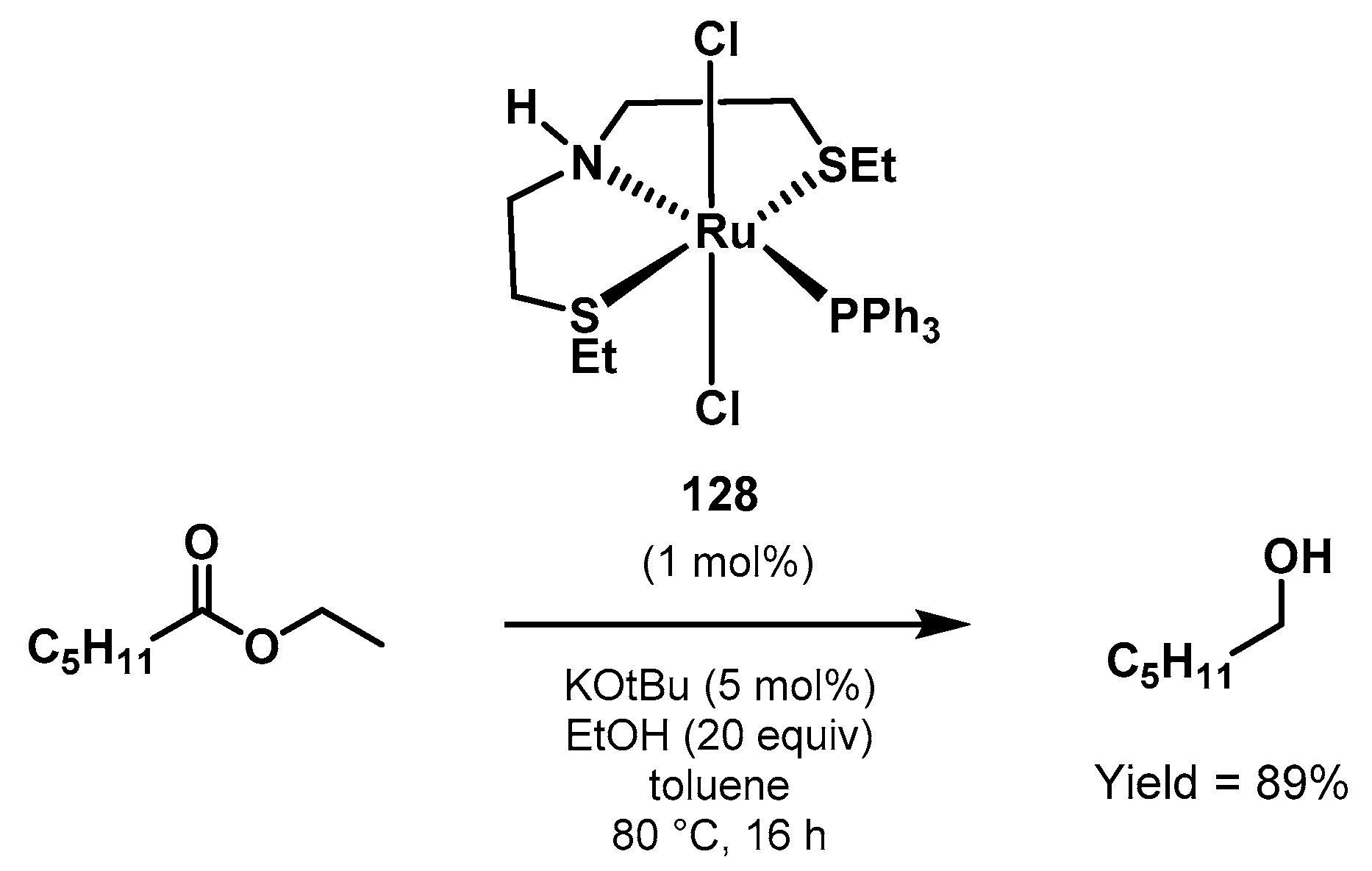{kind=link}
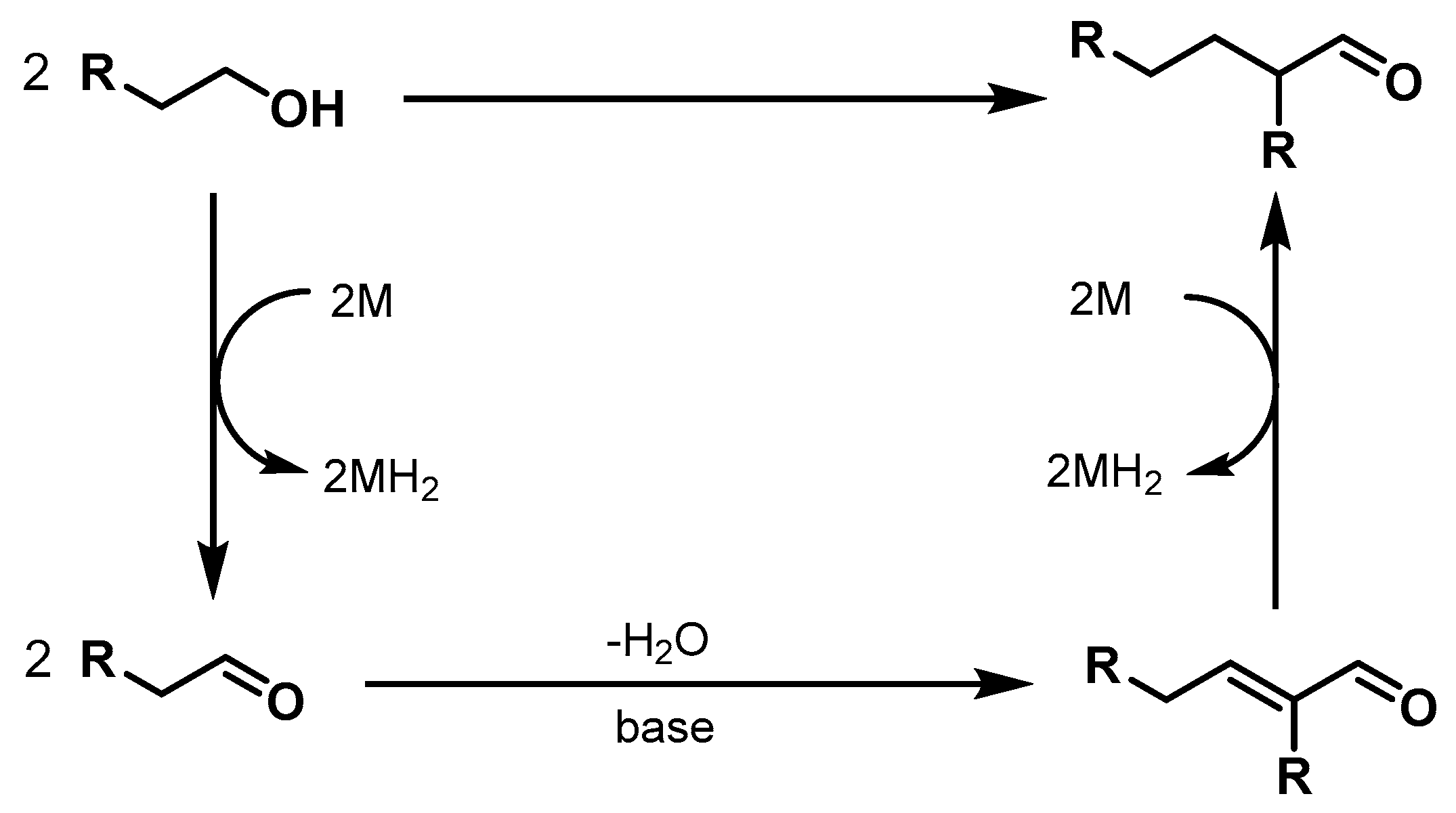{kind=link}
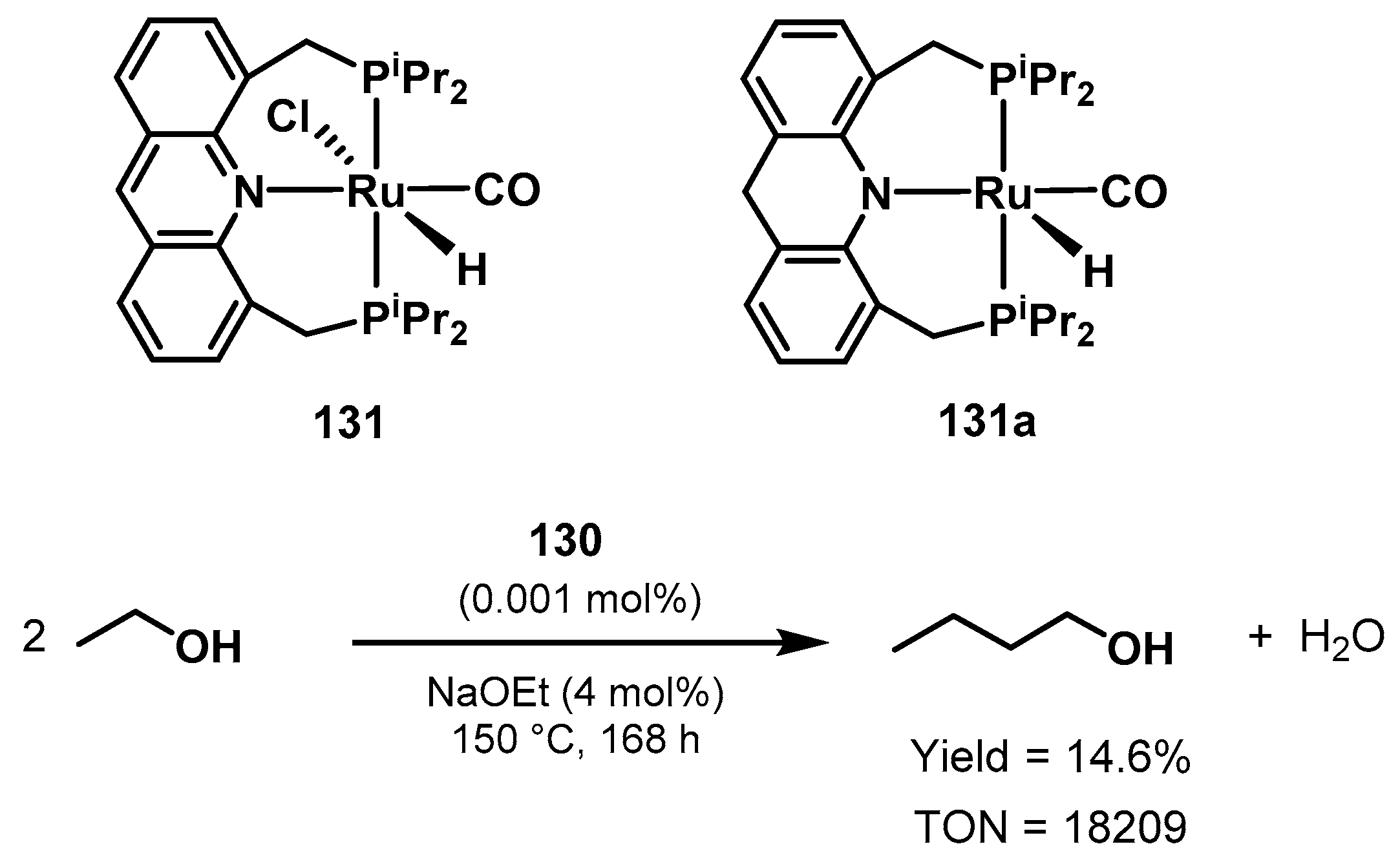{kind=link}
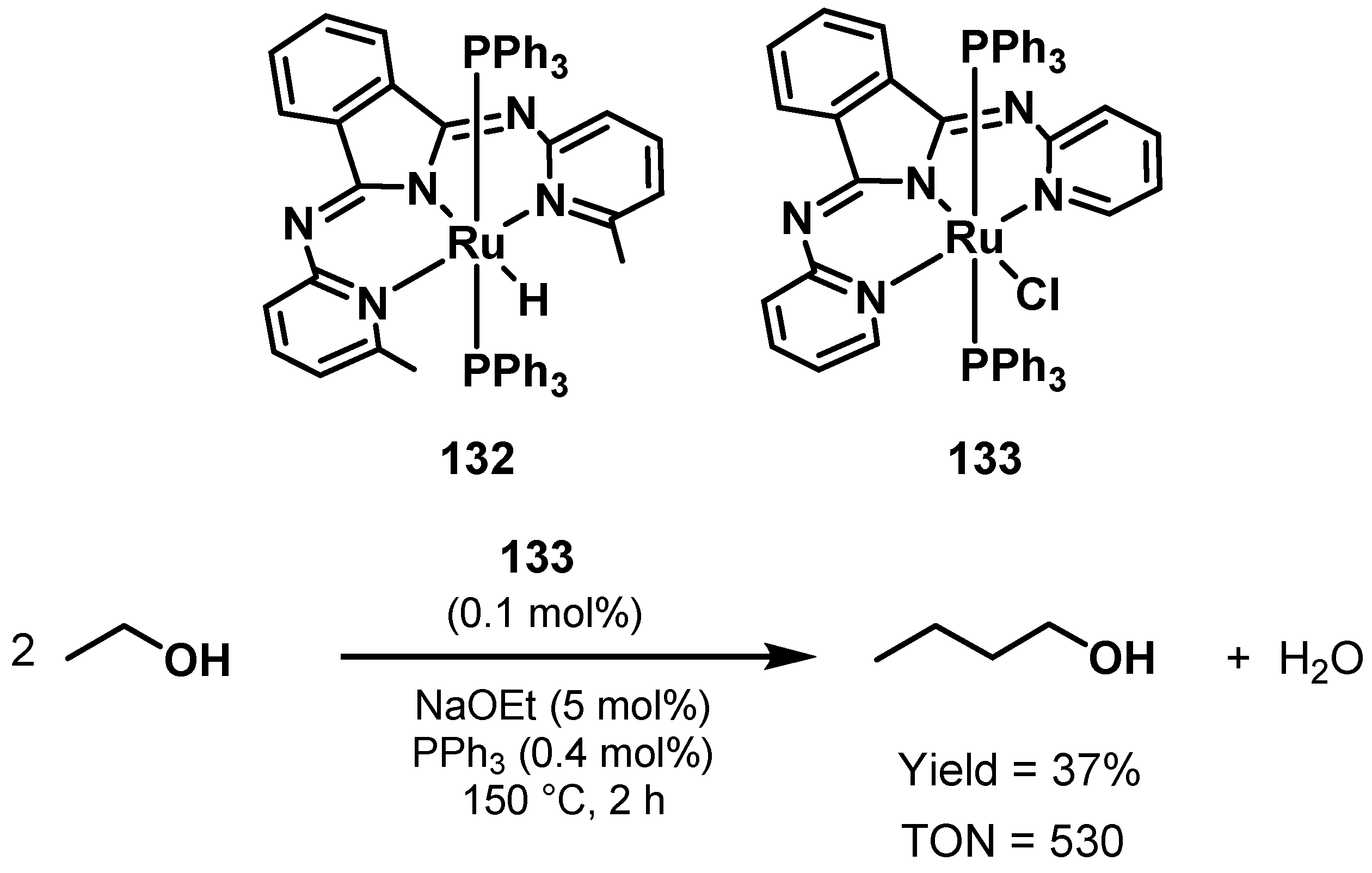{kind=link}
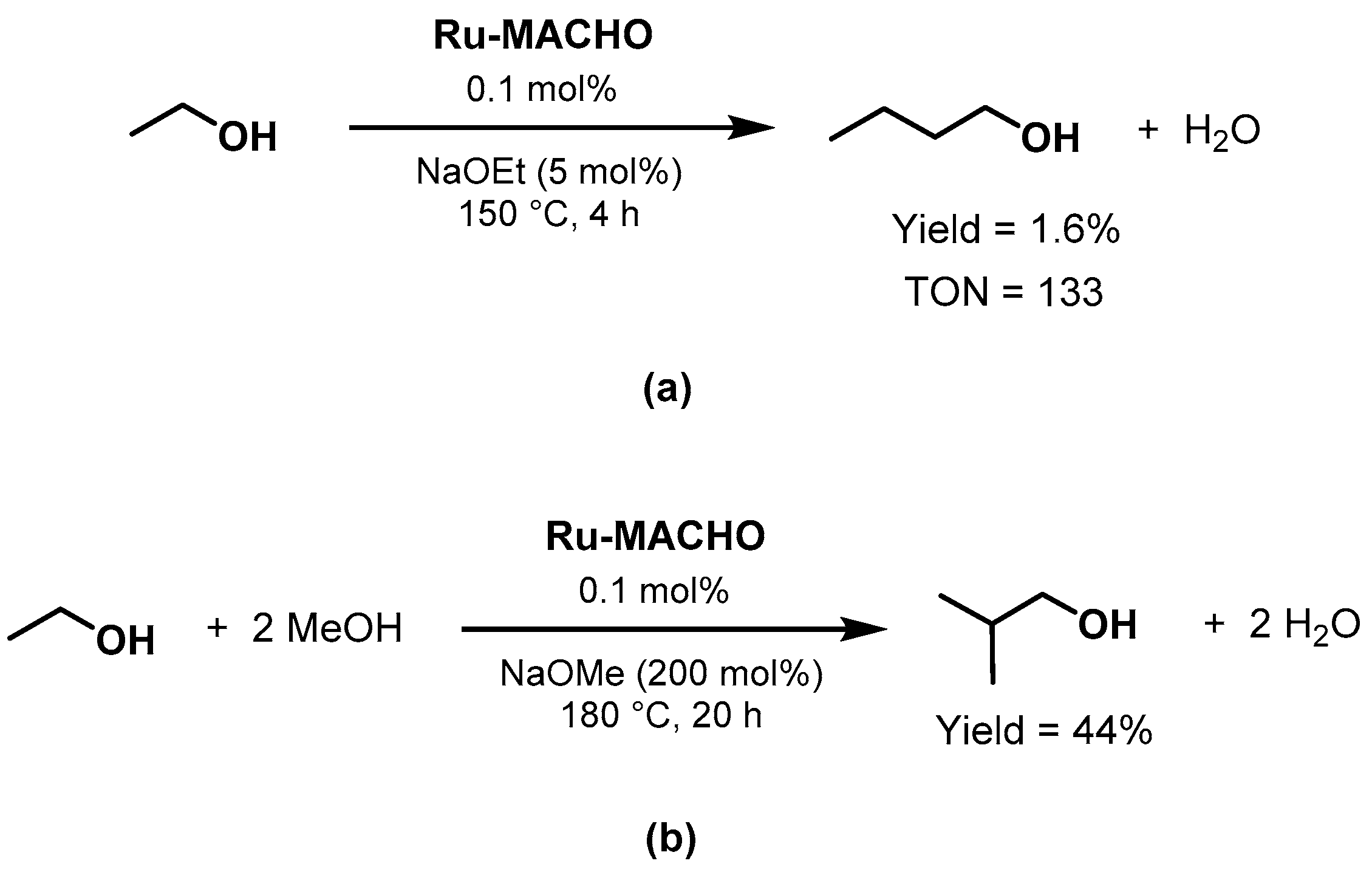{kind=link}
{kind=link}
| LOHC | H2 wt% |
|---|---|
| Methanol | 12.6 |
| Formic Acid | 4.4 |
| Ethanol | 12 |
| Formaldehyde | 6.6 |
| Glycerol | 9.6 |
| Sugar Alcohols | 8.9–9.3 |
| N-ethylcarbazole (NEC) | 5.8 |
| Dibenzyltoluene (DBT) | 6.2 |
| 1,2-BN-cyclohexane | 7.1 |
| BuPy | 3.14 |
| MePHI | 5.76 |
 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piccirilli, L.; Lobo Justo Pinheiro, D.; Nielsen, M. Recent Progress with Pincer Transition Metal Catalysts for Sustainability. Catalysts 2020, 10, 773. https://doi.org/10.3390/catal10070773
Piccirilli L, Lobo Justo Pinheiro D, Nielsen M. Recent Progress with Pincer Transition Metal Catalysts for Sustainability. Catalysts. 2020; 10(7):773. https://doi.org/10.3390/catal10070773
Chicago/Turabian StylePiccirilli, Luca, Danielle Lobo Justo Pinheiro, and Martin Nielsen. 2020. "Recent Progress with Pincer Transition Metal Catalysts for Sustainability" Catalysts 10, no. 7: 773. https://doi.org/10.3390/catal10070773
APA StylePiccirilli, L., Lobo Justo Pinheiro, D., & Nielsen, M. (2020). Recent Progress with Pincer Transition Metal Catalysts for Sustainability. Catalysts, 10(7), 773. https://doi.org/10.3390/catal10070773