Direct Decomposition of NO over Co-Mn-Al Mixed Oxides: Effect of Ce and/or K Promoters

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Chemical Analysis
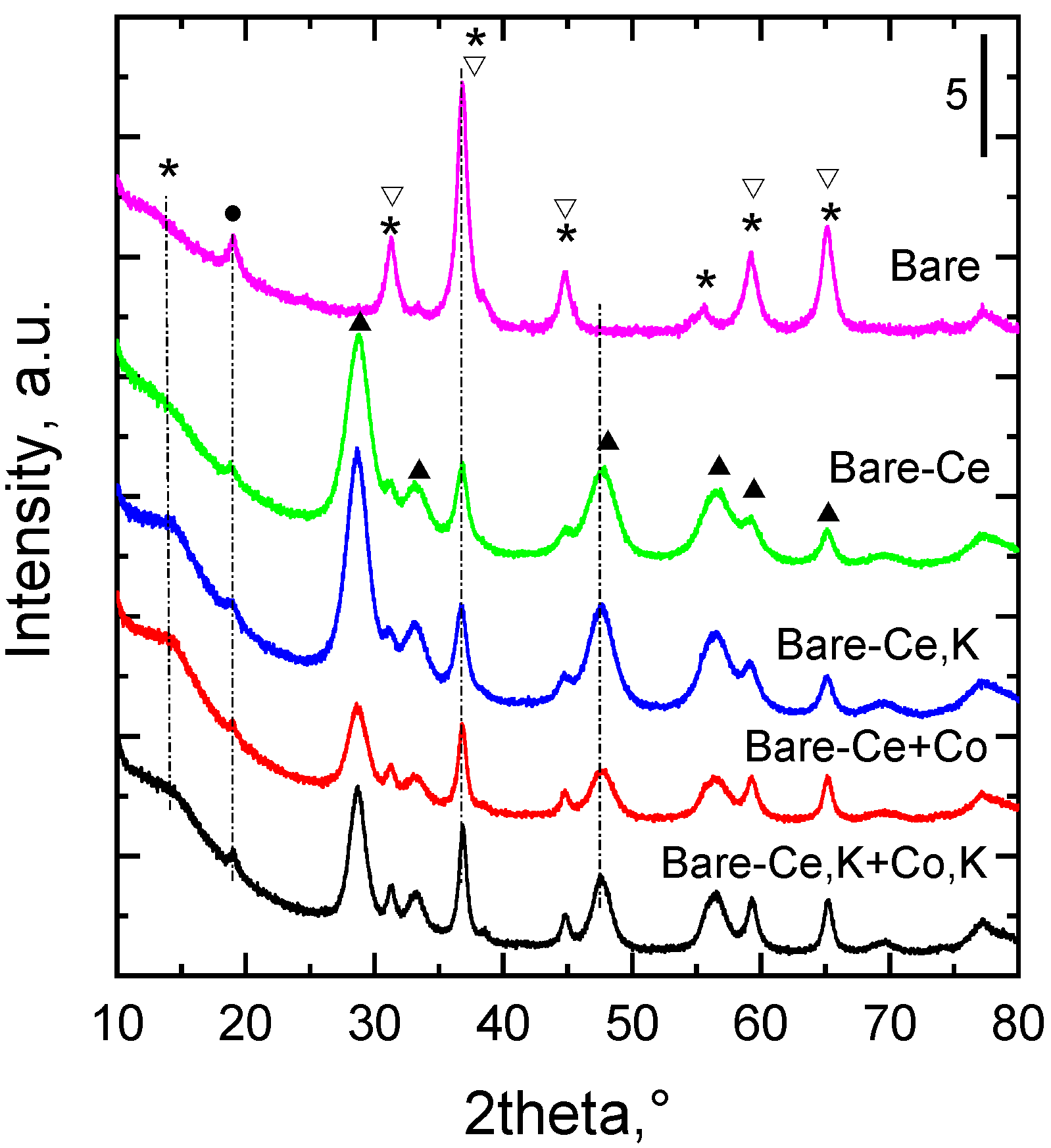
2.2. XRD
2.3. Surface Area and Pore Size Distribution
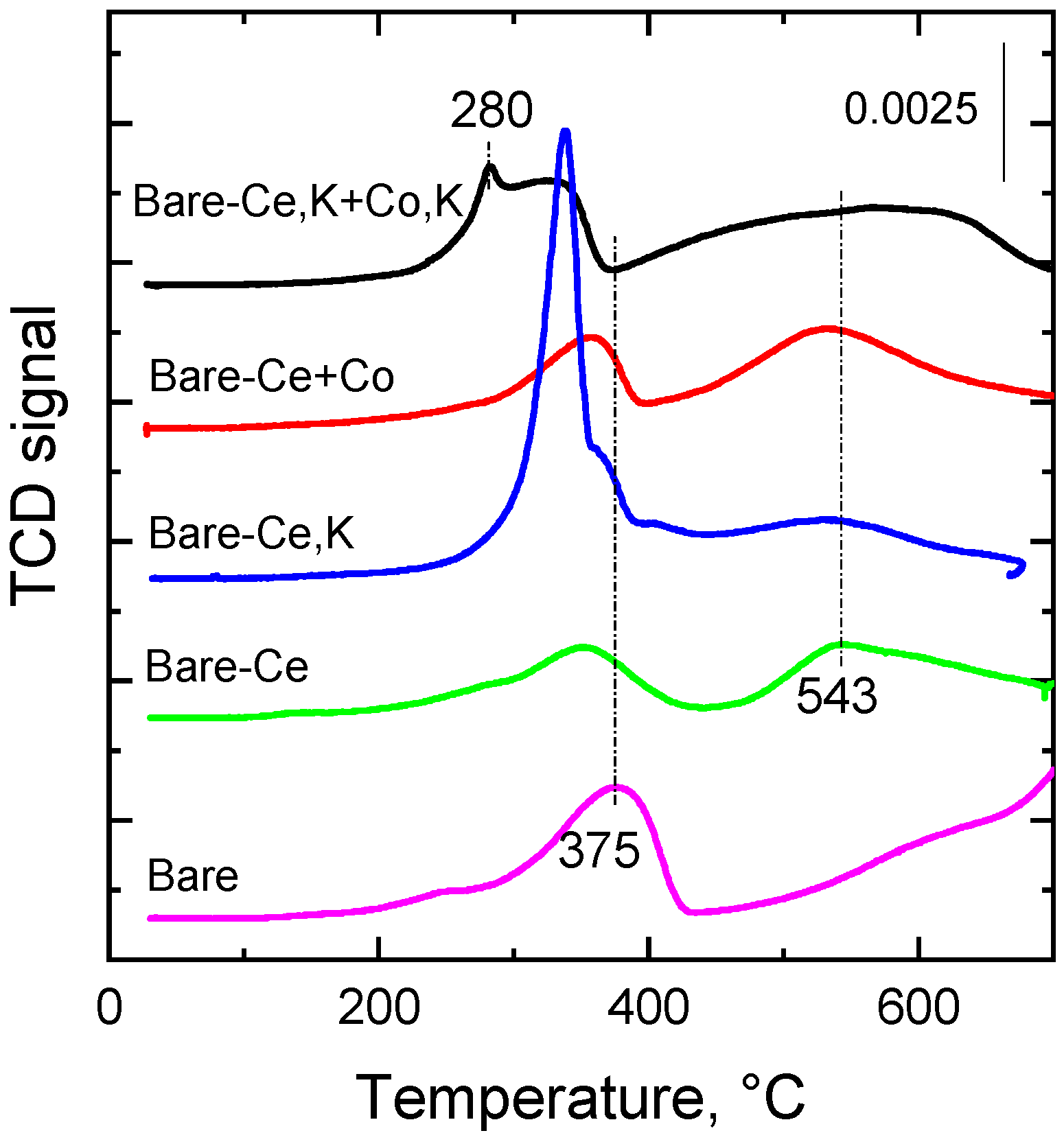
2.4. Reducibility of the Catalysts
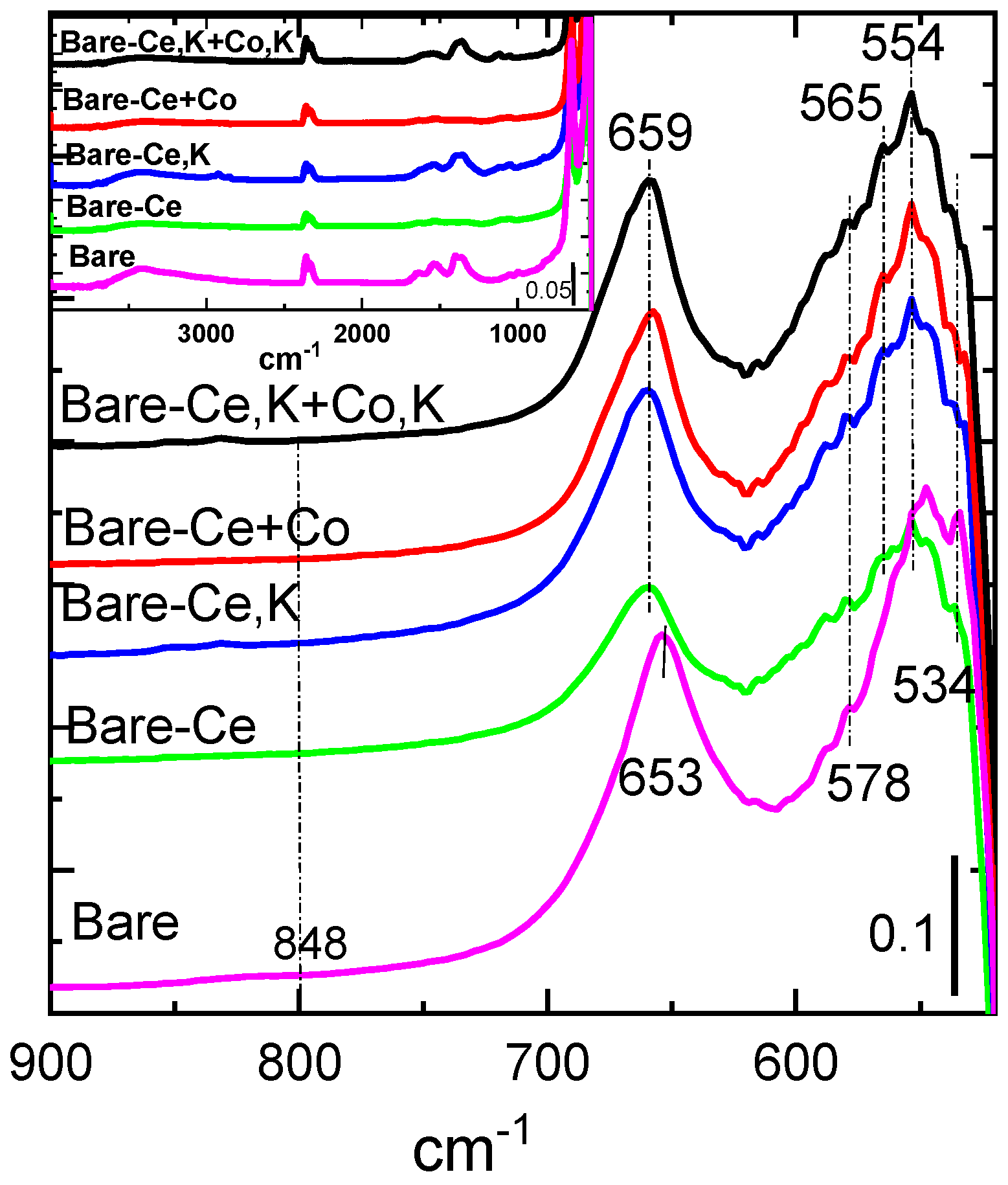
2.5. FTIR
2.6. Basicity
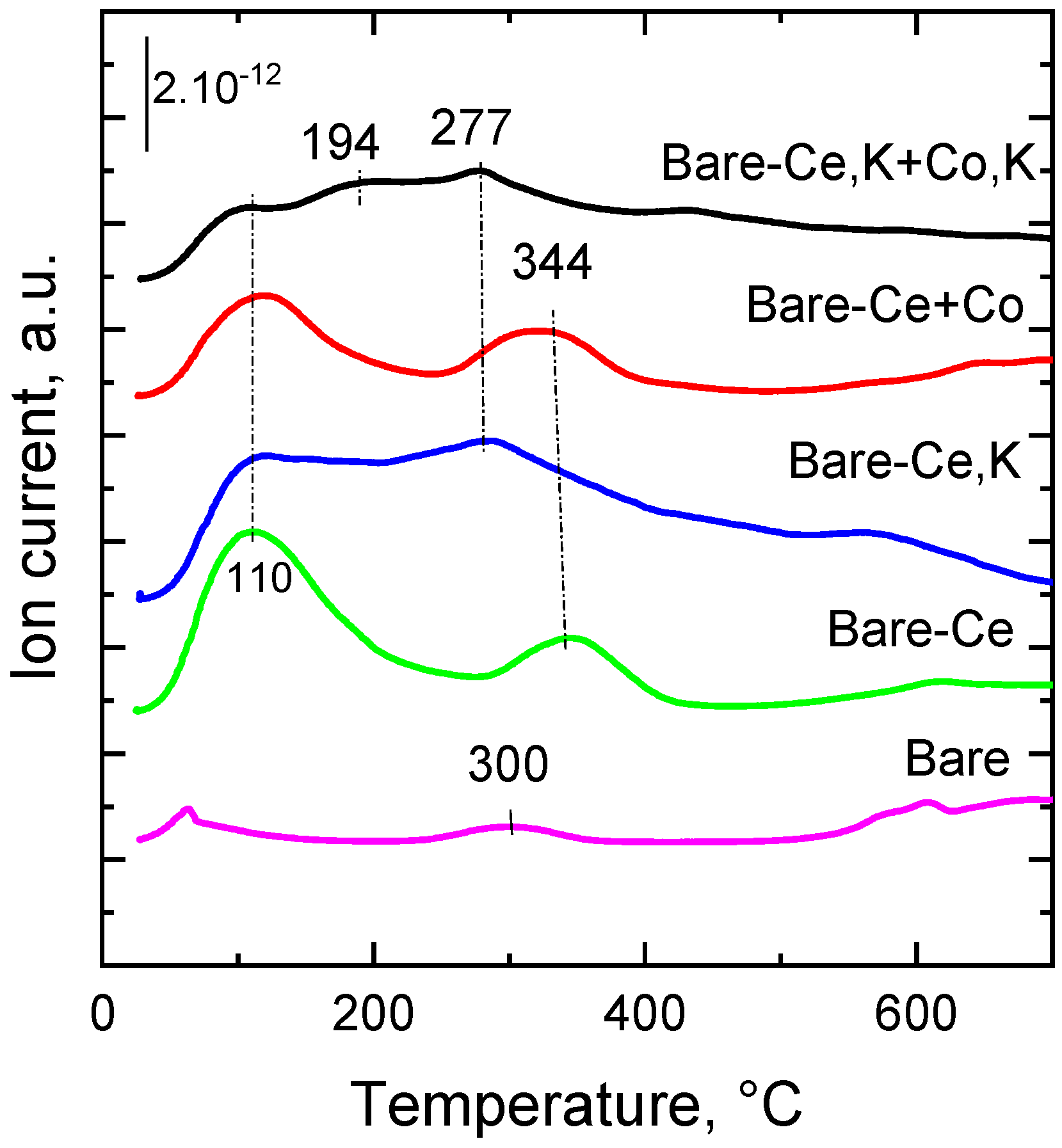
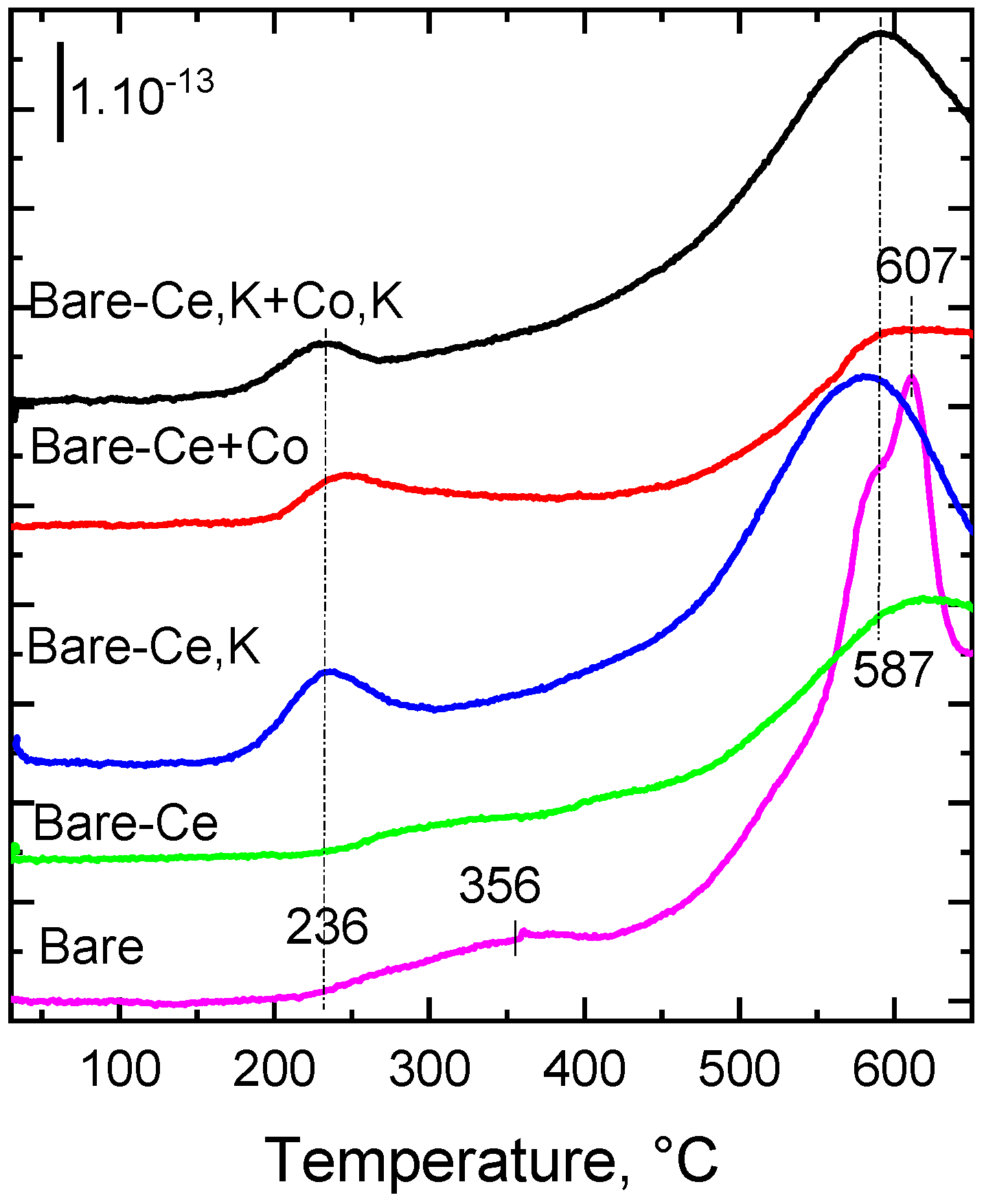
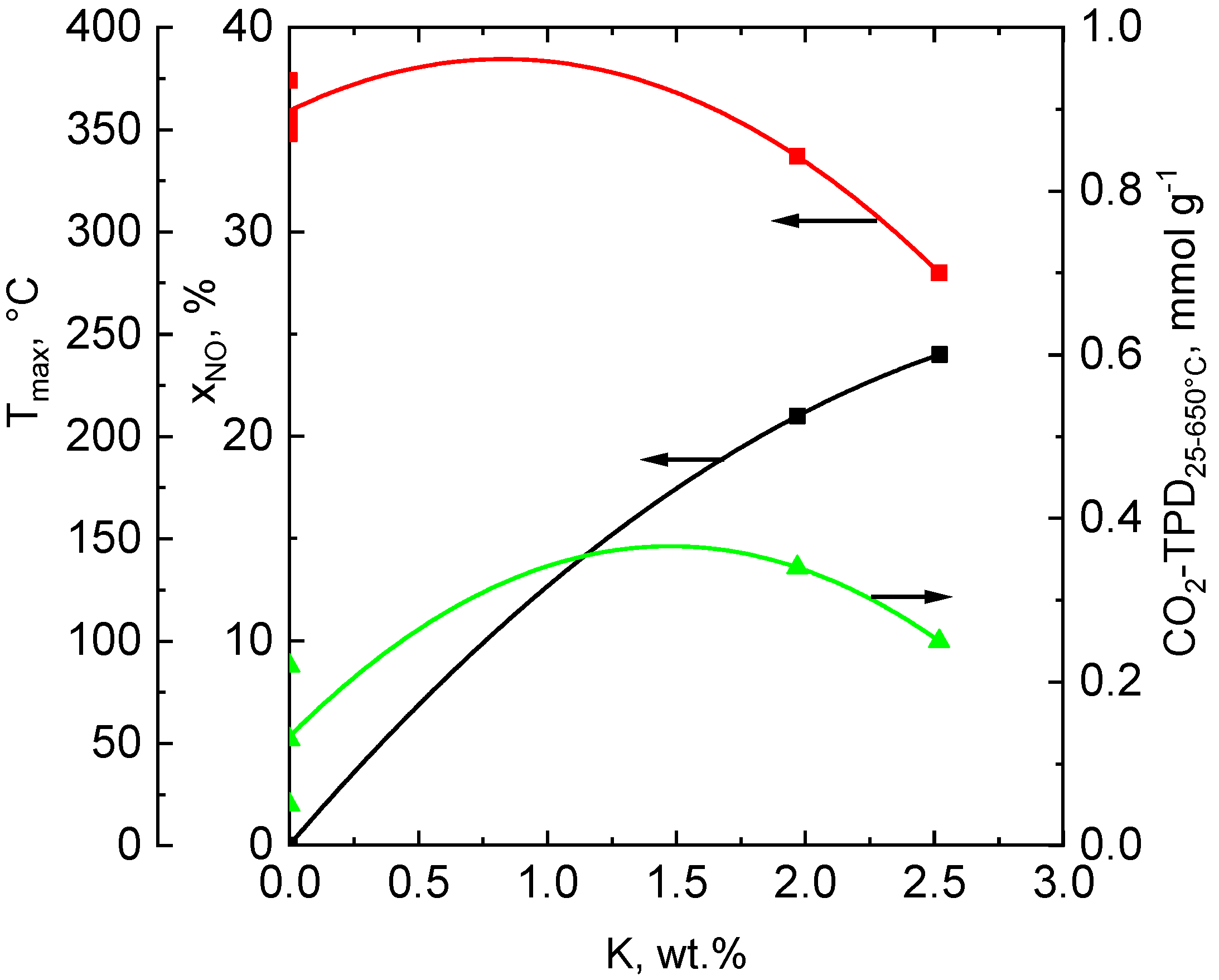
2.7. Desorption of O2
2.8. Surface Composition
2.9. Catalytic Activity
3. Discussion
4. Materials and Methods
4.1. Catalyst Preparation
4.2. Catalyst Characterization
4.3. Catalyst Testing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carslaw, D.; Beevers, S.; Westmoreland, E.; Williams, M.; Tate, J.; Murrells, T.; Stedman, J.; Li, Y.; Grice, S.; Kent, A.; et al. Trends in NO and NO2 Emissions and Ambient Measurements in the UK. Version 18th. Available online: http://ukair.defra.gov.uk/reports/cat05/1108251149_110718_AQ0724_Final_report.pdf (accessed on 31 July 2011).
- Haneda, M.; Hamada, H. Recent progress in catalytic NO decomposition. C. R. Chim. 2016, 19, 1254–1265. [Google Scholar] [CrossRef]
- Falsig, H.; Bligaard, T. Trends in catalytic NO decomposition over transition metal surfaces. Top. Catal. 2007, 45, 117–120. [Google Scholar] [CrossRef]
- Brown, W.A.; King, D.A. No Chemisorption and Reactions on Metal Surfaces: A New Perspective. J. Phys. Chem. B 2000, 104, 2578–2595. [Google Scholar] [CrossRef]
- Falsig, H.; Bligaard, T.; Christensen, C.H.; Nørskov, J.K. Direct No decomposition over stepped transition-metal surfaces. Pure Appl. Chem. 2007, 79, 1895–1903. [Google Scholar] [CrossRef]
- Wu, Z.; Xu, L.; Zhang, W.; Ma, Y.; Yuan, Q.; Jin, Y.; Yang, J.; Huang, W. Structure sensitivity of low-temperature NO decomposition on Au surfaces. J. Catal. 2013, 304, 112–122. [Google Scholar] [CrossRef]
- Obalová, L.; Fíla, V. Kinetic analysis of N2O decomposition over calcined hydrotalcites. Appl. Catal. B Environ. 2007, 70, 353. [Google Scholar] [CrossRef]
- Cheng, H.; Huang, Y.; Wang, A.; Li, L.; Wang, X.; Zhang, T. N2O decomposition over K-promoted Co-Al catalysts prepared from hydrotalcite-like precursors. Appl. Catal. B 2009, 89, 391–397. [Google Scholar] [CrossRef]
- Li, Q.; Meng, M.; Tsubaki, N.; Li, X.; Li, Z.; Xioe, Y.; Hu, T.; Zhang, J. Performance of K-promoted hydrotalcite-derived CoMgAlO catalysts used for soot combustion, NOx storage and simultaneous soot–NOx removal. Appl. Catal. B 2009, 91, 406–415. [Google Scholar] [CrossRef]
- Obalová, L.; Karásková, K.; Jirátová, K.; Kovanda, F. Effect of potassium in calcined Co–Mn–Al layered double hydroxide on the catalytic decomposition of N2O. Appl. Catal. B 2009, 90, 132–140. [Google Scholar] [CrossRef]
- Pacultová, K.; Draštíková, V.; Chromčáková, Ž.; Bílková, T.; Mamulová Kutláková, K.; Kotarba, A.; Obalová, L. On the stability of alkali metal promoters in Co mixed oxides during direct NO catalytic decomposition. J. Mol. Catal. A Chem. 2017, 428, 33–40. [Google Scholar] [CrossRef]
- Pacultová, K.; Bílková, T.; Klegová, A.; Karásková, K.; Fridrichová, D.; Jirátová, K.; Kiška, T.; Balabánová, J.; Koštejn, M.; Kotarba, A.; et al. Co-Mn-Al Mixed Oxides Promoted by K for Direct NO Decomposition: Effect of Preparation Parameters. Catalysts 2019, 9, 593. [Google Scholar] [CrossRef] [Green Version]
- Jirátová, K.; Pacultová, K.; Balabánová, J.; Karásková, K.; Klegová, A.; Bílková, T.; Jandová, V.; Koštejn, M.; Martaus, A.; Kotarba, A.; et al. Precipitated K-Promoted Co–Mn–Al Mixed Oxides for Direct NO Decomposition: Preparation and Properties. Catalysts 2019, 9, 592. [Google Scholar] [CrossRef] [Green Version]
- Figueredo, M.J.M.; Andana, T.; Bensaid, S.; Dosa, M.; Fino, D.; Russo, N.; Piumetti, M. Cerium–Copper–Manganese Oxides Synthesized via Solution Combustion Synthesis (SCS) for Total Oxidation of VOCs. Catal. Lett. 2020, 150, 1821–1840. [Google Scholar] [CrossRef]
- Lin, S.S.-Y.; Kim, D.H.; Ha, S.Y. Metallic phases of cobalt-based catalysts in ethanol steam reforming: The effect of cerium oxide. Appl. Catal. A Gen. 2009, 355, 69–77. [Google Scholar] [CrossRef]
- Katta, L.; Sudarsanam, P.; Thrimurthulu, G.B.; Reddy, M. Doped nanosized ceria solid solutions for low temperature soot oxidation: Zirconium versus lanthanum promoters. Appl. Catal. B 2010, 101, 101–108. [Google Scholar] [CrossRef]
- Liotta, L.F.; Ousmane, M.; Di Carlo, G.; Pantaleo, G.; Deganello, G.; Marci, G.; Retailleau, L.; Giroir-Fendler, A. Total oxidation of propene at low temperature over Co3O4–CeO2 mixed oxides: Role of surface oxygen vacancies and bulk oxygen mobility in the catalytic activit A. Appl. Catal. A Gen. 2008, 347, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Dong, F.; Suda, A.; Tanabe, T.; Nagai, Y.; Sobukawa, H.; Shinjoh, H.; Sugiura, M.; Descorme, C.; Duprez, D. Dynamic oxygen mobility and a new insight into the role of Zr atoms in three-way catalysts of Pt/CeO2–ZrO2. Catal. Today 2004, 93–95, 827–832. [Google Scholar] [CrossRef]
- Yu, S.-W.; Huang, H.-H.; Tang, C.-W.; Wang, C.-B. The effect of accessible oxygen over Co3O4-CeO2 catalysts on the steam reforming of ethanol. Int. J. Hydrog. Energy 2014, 39, 20700–20711. [Google Scholar] [CrossRef]
- Xue, L.; Zhang, C.; He, H.; Teraoka, Y. Promotion effect of residual K on the decomposition of N2O over cobalt–cerium mixed oxide catalyst. Catal. Today 2007, 126, 449–455. [Google Scholar] [CrossRef]
- Xue, L.; He, H.; Liu, C.; Zhang, C.; Zhang, B. Promotion Effects and Mechanism of Alkali Metals and Alkaline Earth Metals on Cobalt-Cerium Composite Oxide Catalysts for N2O Decomposition. Environ. Sci. Technol. 2009, 43, 890–895. [Google Scholar] [CrossRef]
- Argyle, M.D. Supported, Alkali-Promoted Cobalt Oxide Catalysts for NOx Removal from Coal Combustion Flue Gases; University of Wyoming: Laramie, WY, USA, 2005. [Google Scholar]
- Stoyanova, D.; Georgieva, P.; Avramova, I.; Aleksieva, K.; Marinova, D.; Mehandjiev, D. Nitric oxide (NO) decomposition on catalysts, containing oxides of lanthanum and cerium, supported on γ-alumina. J. Rear Earths 2019, 37, 151–159. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, Y.; Li, N.; Feng, Y. Catalytic N2O decomposition over CeMeOy/γ-Al2O3 (Me = Mn, Cu, Zn) catalysts prepared by impregnation method. Asia Pac. J. Chem. Eng. 2018, 13, e2233. [Google Scholar] [CrossRef]
- Hong, W.-J.; Iwamoto, S.; Inoue, M. Direct NO decomposition over a Ce-Mn mixed oxide modified with alkali and alkali earth species and CO2-TPD behavior of the catalyst. Catal. Today 2011, 164, 489–494. [Google Scholar] [CrossRef]
- Hong, W.-J.; Ueda, M.; Iwamoto, S.; Hosokawa, S.; Wada, K.; Inoue, M. Synthesis of Highly Effective CeOx-MnOy-BaO Catalysts for direct NO decomposition. Catal. Lett. 2012, 142, 32–41. [Google Scholar] [CrossRef]
- Klug, H.P.; Alexander, L.E. X-ray Diffraction Procedures for Polycrystalline and Amorphous Materials; Wiley: New York, NY, USA, 1954. [Google Scholar]
- Schneider, P.; Hudec, P.; Šolcová, O. Pore-volume and surface area in microporous–mesoporous solids. Micropor. Mesopor. Mater. 2008, 115, 491–496. [Google Scholar] [CrossRef]
- Gregg, S.J.; Sing, K.S.W. Adsorption Surface Area and Porosity; Academic Press: New York, NY, USA, 1982. [Google Scholar]
- Leofanti, G.; Padovan, M.; Tozzola, G.; Venturelli, B. Surface area and pore structure of catalysts. Catal. Today 1998, 41, 207–219. [Google Scholar] [CrossRef]
- Schneider, P. Adsorption isotherms of microporous-mesoporous solids revisited. Appl. Catal. A 1995, 157–165. [Google Scholar] [CrossRef]
- Tang, C.-W.; Leu, T.-Y.; Yu, W.-Y.; Wang, C.-B.; Chien, S.-H. Low temperature oxidation of carbon monoxide: The influence of water and oxygen on the reactivity of a Co3O4 powder surface. Appl. Catal. B 2004, 48, 267–274. [Google Scholar] [CrossRef]
- Chromčáková, Ž.; Obalová, L.; Kovanda, F.; Michalik, S.; Kuśtrowski, P.; Jirátová, K. Effect of precursor synthesis on catalytic activity of Co3O4 in N2O decomposition. Catal. Today 2015, 257, 18–25. [Google Scholar] [CrossRef]
- Venkataswamy, P.; Jampaiah, D.; Rao, K.N.; Reddy, B.M. Nanostructured Ce0.7Mn0.3O2−δ and Ce0.7Fe0.3O2−δ solid solutions for diesel soot oxidation. Appl. Catal. A Gen. 2011, 488, 1–10. [Google Scholar] [CrossRef]
- Guan, F.; Ma, Y.; Yang, W.; Kang, J.; Deng, H.; Qi, H. Preparation of CeO2 nanoparticles and its application to Ion-selective Electrodes Based on Acetylcellulose. Rare Metals 2001, 20, 217–220. [Google Scholar]
- Ho, C.; Yu, J.C.; Kwang, T.; Mark, A.C.; Lai, S. Morphology-Controllable Synthesis of Mesoporous CeO2 Nano- and Microstructures. Chem. Mater. 2006, 17, 4514–4522. [Google Scholar] [CrossRef]
- Nyquist, R.A.; Nagel, R.O. Infrared Spectra of Inorganic Compounds; Academic Press: New York, NY, USA, 1971. [Google Scholar]
- Bentley, F.F.; Smithson, L.D.; Rozek, A.L. Infrared Spectra and Characteristic Frequencies 700–300 cm−1: A Collection of Spectra, Interpretation, and Bibliography; Interscience: New York, NY, USA, 1986. [Google Scholar]
- Jirátová, K.; Balabánová, J.; Kovanda, F.; Klegová, A.; Obalová, L.; Fajgar, R. Cobalt Oxides Supported Over Ceria–Zirconia Coated Cordierite Monoliths as Catalysts for Deep Oxidation of Ethanol and N2O Decomposition. Catal. Lett. 2017, 147, 1379–1391. [Google Scholar] [CrossRef]
- Habibi, M.H.; Bagheri, P. Spinel cobalt manganese oxide nano-composites grown hydrothermally on nanosheets for enhanced photocatalytic mineralization of Acid Black 1 textile dye: XRD, FTIR, FESEM, EDS and TOC studies. J. Iran Chem. Soc. 2017, 14, 1643–1649. [Google Scholar] [CrossRef]
- Tholkappiyan, R.; Naveen, A.N.; Sumithra, S.; Vishista, K. Investigation on spinel MnCo2O4 electrode material prepared via controlled and uncontrolled synthesis route for supercapacitor application. J. Mater. Sci. 2015, 50, 5833–5843. [Google Scholar] [CrossRef]
- Rojas, R.M.; Vila, E.; García, O.; de Vidales, J.L.M. Thermal behaviour and reactivity of manganese cobaltites MnxCo3-xO4 (0.0 ≤ x ≤ 1.0) obtained at low temperature. J. Mater. Chem. 1994, 4, 1635–1639. [Google Scholar] [CrossRef]
- Babitha, K.K.; Sreedevi, A.; Priyanka, K.P.; Sabu, B.; Varghese, T. Structural characterization and optical studies of CeO2 nanoparticles synthesized by chemical precipitation. Indian J. Pure Appl. Phys. 2015, 53, 596–603. [Google Scholar]
- Imanaka, N.; Masui, T. Advances in direct NOx decomposition catalysts. Appl. Catal. A Gen. 2012, 431–432, 1–8. [Google Scholar] [CrossRef]
- Smoláková, L.; Frolich, K.; Troppová, I.; Kutálek, P.; Kroft, E.; Cˇapek, L. Determination of basic sites in Mg–Al mixed oxides by combination of TPD-CO2 and CO2 adsorption calorimetry. J. Therm. Anal. Calorim. 2017, 127, 1921–1929. [Google Scholar] [CrossRef]
- Shibata, K.; Kiyoura, T.; Kitagawa, J.; Sumiyoshi, T.; Tanabe, K. Acidic properties of Binary Metal Oxides. Bull. Chem. Soc. Jpn. 1973, 46, 2985–2988. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Ozaki, A. Acid-base properties and catalytic activity of solid surfaces. J. Catal. 1967, 8, 1–7. [Google Scholar] [CrossRef]
- Russo, N.; Finno, D.; Saracco, G.; Specchia, V. N2O catalytic decomposition over various spinel-type oxides. Catal. Today 2007, 119, 228–232. [Google Scholar] [CrossRef]
- Karaskova, K.; Obalova, L.; Jiratova, K.; Kovanda, F. Effect of promoters in Co–Mn–Al mixed oxide catalyst on N2O decomposition. Chem. Eng. J. 2010, 160, 480–487. [Google Scholar] [CrossRef]
- Karaskova, K.; Obalova, L.; Kovanda, F. N2O catalytic decomposition and temperature programmed desorption tests on alkali metals promoted Co–Mn–Al mixed oxide. Catal. Today 2011, 176, 208–211. [Google Scholar] [CrossRef]
- Scofield, J.H. Hartree-Slater subshell photoionization cross-sections at 1254 and 1487 eV. J. Electron Spectrosc. Relat. Phenom. 1976, 8, 129–137. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Kim, S.C.; Shim, W.G. Catalytic combustion of VOCs over a series of manganese oxide catalysts. Appl. Catal. B Environ. 2010, 98, 180–185. [Google Scholar] [CrossRef]
- Nefedov, V.I.; Salyn, Y.V.; Leonhardt, G.; Scheibe, R. A comparison of different spectrometers and charge corrections used in X-ray photoelectron spectroscopy. J. Electron Spectrosc. Relat. Phenom. 1977, 10, 121–124. [Google Scholar] [CrossRef]
- Available online: https://xpssimplified.com/elements/cerium.php (accessed on 30 June 2020).
- Obalová, L.; Karásková, K.; Wach, A.; Kustrowski, P.; Michalik, S.; Jirátová, K. Alkali metals as the promoters in Co-Mn-Al mixed oxide. Appl. Catal. A Chem. 2013, 462–463, 227–235. [Google Scholar]
- Haneda, M.; Kintaichi, Y.; Bion, N.; Hamada, H. Alkali metal-doped cobalt oxide catalysts for NO decomposition. Appl. Catal. B 2003, 46, 473–482. [Google Scholar] [CrossRef]
- Abu-Zied, B.M.; Asiri, A.M. The role of alkali promoters in enhancing the direct N2O decomposition reactivity over NiO catalysts. Chin. J. Catal. 2015, 36, 1837–1845. [Google Scholar] [CrossRef]
- Asano, K.; Ohnishi, C.; Iwamoto, S.; Shioya, Y.; Inoue, M. Potassium-doped Co3O4 catalyst for direct decomposition of N2O. Appl. Catal. B 2008, 78, 242–249. [Google Scholar] [CrossRef]
- Grzybek, G.; Wojcik, S.; Legutko, P.; Grybos, J.; Indyka, P.; Leszczynski, B.; Kotarba, A.; Sojka, Z. Thermal stability and repartition of potassium promoter between the support and active phase in the K-Co2.6Zn0.4O4 vertical bar α-Al2O3 catalyst for N2O decomposition: Crucial role of activation temperature on catalytic performance. Appl. Catal. B Environ. 2017, 205, 597–604. [Google Scholar] [CrossRef]
- Park, P.W.; Kil, J.K.; Kung, H.H.; Kung, M.C. NO decomposition over sodium-promoted cobalt oxide. Catal. Today 1998, 42, 51–60. [Google Scholar] [CrossRef]
- Pacultová, K.; Klegová, A.; Karásková, K.; Fridrichová, D.; Bílková, T.; Obalová, L. Oxygen effect in NO direct decomposition over K/Co-Mg-Mn-Al mixed oxide catalyst (in preparation).
- Sanderson, R.T. Chemical Bonds and Bond Energy; Academic Press: New York, NY, USA, 1971. [Google Scholar]
- Peck, T.C.; Roberts, C.A.; Reddy, G.K. Contrasting Effects of Potassium Addition on M3O4 (M = Co, Fe, and Mn) Oxides during Direct NO Decomposition Catalysis. Catalysts 2020, 10, 561. [Google Scholar] [CrossRef]
- Zasada, F.; Stelmachowski, P.; Maniak, G.; Paul, J.-F.; Kotarba, A.; Sojka, Z. Potassium promotion of cobalt spinel catalyst for N2O decomposition—Accounted by work function measurements and DFT modelling. Catal. Lett. 2009, 127, 126–131. [Google Scholar]
- Deboer, J.H.; Lippens, B.C.; Linsen, B.G.; Broekhof, J.C.; Vandenhe, A.; Osinga, T.J. The t-curve of multimolecular N2-adsorption. J. Colloid Interface Sci. 1966, 21, 405–414. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Lecloux, A.; Pirard, J.P. The importance of standard isotherms in the analysis of adsorption isotherms for determining the porous texture of solids. J. Colloid Interface Sci. 1979, 70, 265–281. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Co_Mn_Al_Ce_K wt.% | Co:Mn:Al:Ce:K Molar Ratio | D (Co3O4) nm | D (CeO2) nm |
|---|---|---|---|---|
| Bare a | 52.0_11.0_3.7_0_0 | 4:1.0:1.0:0:0 | 10.21 | - |
| Bare-Ce b | 24.6_4.7_2.7_40.5_0 | 4:0.8:1.0:2.8:0 | 10.48 | 4.84 |
| Bare-Ce,K c | 25.0_4.8_2.7_41.0_2.0 | 4:0.8:1.0:2.8:0.5 | 10.28 | 5.07 |
| Bare-Ce+Co d | 36.3_3.7_2.2_32.4_0 | 4:0.6:0.8:2.2:0 | 13.13 | 6.10 |
| Bare-Ce,K+Co,K e | 35.6_3.6_2.1_30.7_2.5 | 4:0.6:0.7:2.1:0.6 | 15.17 | 6.85 |
| Sample | SBET m2 g−1 | Smeso m2 g−1 | Vtot mm3liq g−1 | Vmicro mm3liq g−1 |
|---|---|---|---|---|
| Bare | 85 | 84 | 454 | 2.0 |
| Bare-Ce | 118 | 118 | 604 | 0.7 |
| Bare-Ce,K | 93 | 92 | 456 | 0 |
| Bare-Ce+Co | 81 | 81 | 285 | 1.0 |
| Bare-Ce,K+Co,K | 59 | 49 | 204 | 5.0 |
| Sample | H2 a mmol g−1 | H2-TPR b mmol g−1 | Tmaxc °C | CO2-TPD b mmol g−1 | CO2-TPD d mmol g−1 | XNO (650 °C) % |
|---|---|---|---|---|---|---|
| Bare | 11.78 | 5.3 | 374 | 0.05 | 0.03 | 2 |
| Bare-Ce | 11.35 | 4.3 | 348 | 0.22 | 0.07 | 0 |
| Bare-Ce+K | 11.53 | 7.3 | 390 | 0.34 | 0.19 | 21 |
| Bare-Ce+Co | 12.84 | 5.6 | 356 | 0.13 | 0.06 | 0 |
| Bare-Ce.K+Co.K | 12.45 | 6.2 | 280 | 0.25 | 0.15 | 24 |
| Sample | C 1s | O 1s | Co 2p | Mn 2p | Al 2p | Ce 3d | K 2p | Na 1s | N 1s |
|---|---|---|---|---|---|---|---|---|---|
| Bare | 16.2 | 56.1 | 13.8 | 4.1 | 9.2 | 0.0 | 0.0 | 0.6 | 0.0 |
| Bare-Ce | 30.8 | 48.4 | 7.4 | 1.7 | 4.8 | 6.4 | 0.0 | 0.5 | 0.0 |
| Bare-Ce,K | 27.0 | 50.9 | 7.5 | 1.7 | 4.9 | 5.8 | 2.3 | 0.0 | 0.0 |
| Bare-Ce+Co | 25.0 | 51.4 | 9.0 | 2.1 | 4.5 | 7.6 | 0.0 | 0.4 | 0.0 |
| Bare-Ce,K+Co,K | 22.9 | 50.9 | 9.9 | 2.0 | 3.7 | 5.1 | 4.4 | 0.4 | 0.7 |
| Sample | O 1s (529.3 eV) | O 1s (531.6 eV) |
|---|---|---|
| Bare | 33.9 | 66.1 |
| Bare-Ce | 49.3 | 50.6 |
| Bare-Ce,K | 46.4 | 53.6 |
| Bare-Ce+Co | 55.8 | 44.2 |
| Bare-Ce,K+Co,K | 55.9 | 44.1 |
| Sample | Co | Mn | Al | Ce | K |
|---|---|---|---|---|---|
| Bare | 1.2 | 1.5 | 3.4 | ||
| Bare-Ce | 1.1 | 1.0 | 2.1 | 0.8 | |
| Bare-Ce,K | 1.1 | 1.0 | 2.1 | 0.7 | 4.6 |
| Bare-Ce+Co | 0.8 | 1.5 | 2.2 | 1.3 | |
| Bare-Ce,K+Co,K | 1.0 | 1.6 | 2.0 | 0.9 | 5.2 |
| Reaction Medium | Conversion of NO (700 °C), % |
|---|---|
| 1000 ppm NO/N2 | 30.0 |
| +2 mol.% O2 | 4.0 |
| After O2 removal | 29.0 |
| +2 mol.% CO2 | 10.4 |
| After CO2 removal | 29.0 |
| +2 mol.% O2 + 2 mol.% H2O | 5.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jirátová, K.; Pacultová, K.; Karásková, K.; Balabánová, J.; Koštejn, M.; Obalová, L. Direct Decomposition of NO over Co-Mn-Al Mixed Oxides: Effect of Ce and/or K Promoters. Catalysts 2020, 10, 808. https://doi.org/10.3390/catal10070808
Jirátová K, Pacultová K, Karásková K, Balabánová J, Koštejn M, Obalová L. Direct Decomposition of NO over Co-Mn-Al Mixed Oxides: Effect of Ce and/or K Promoters. Catalysts. 2020; 10(7):808. https://doi.org/10.3390/catal10070808
Chicago/Turabian StyleJirátová, Květa, Kateřina Pacultová, Kateřina Karásková, Jana Balabánová, Martin Koštejn, and Lucie Obalová. 2020. "Direct Decomposition of NO over Co-Mn-Al Mixed Oxides: Effect of Ce and/or K Promoters" Catalysts 10, no. 7: 808. https://doi.org/10.3390/catal10070808
APA StyleJirátová, K., Pacultová, K., Karásková, K., Balabánová, J., Koštejn, M., & Obalová, L. (2020). Direct Decomposition of NO over Co-Mn-Al Mixed Oxides: Effect of Ce and/or K Promoters. Catalysts, 10(7), 808. https://doi.org/10.3390/catal10070808