Rationalizing the Reactivity of Mixed Allyl Rare-Earth Borohydride Complexes with DFT Studies



 and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
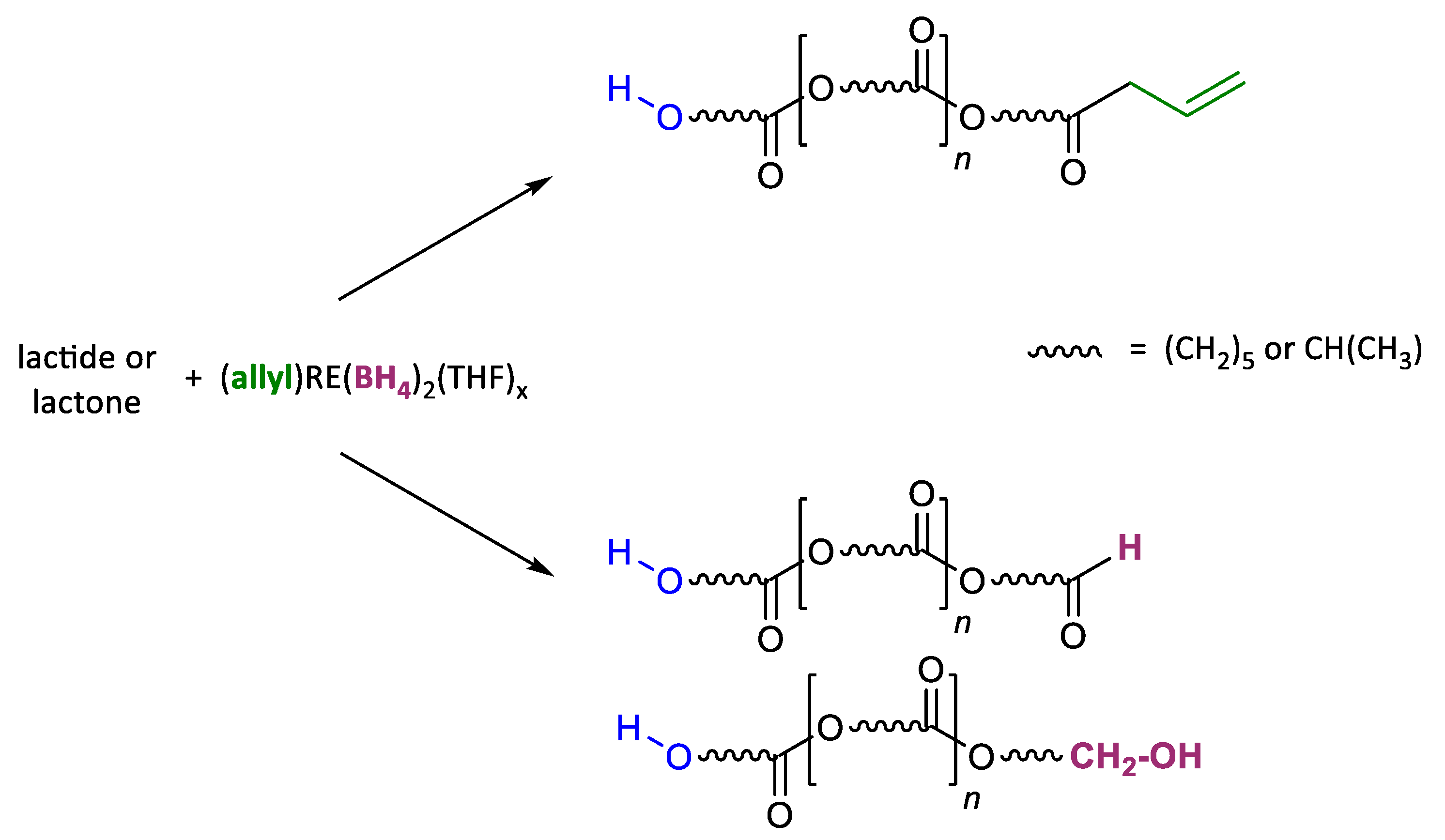{kind=link}
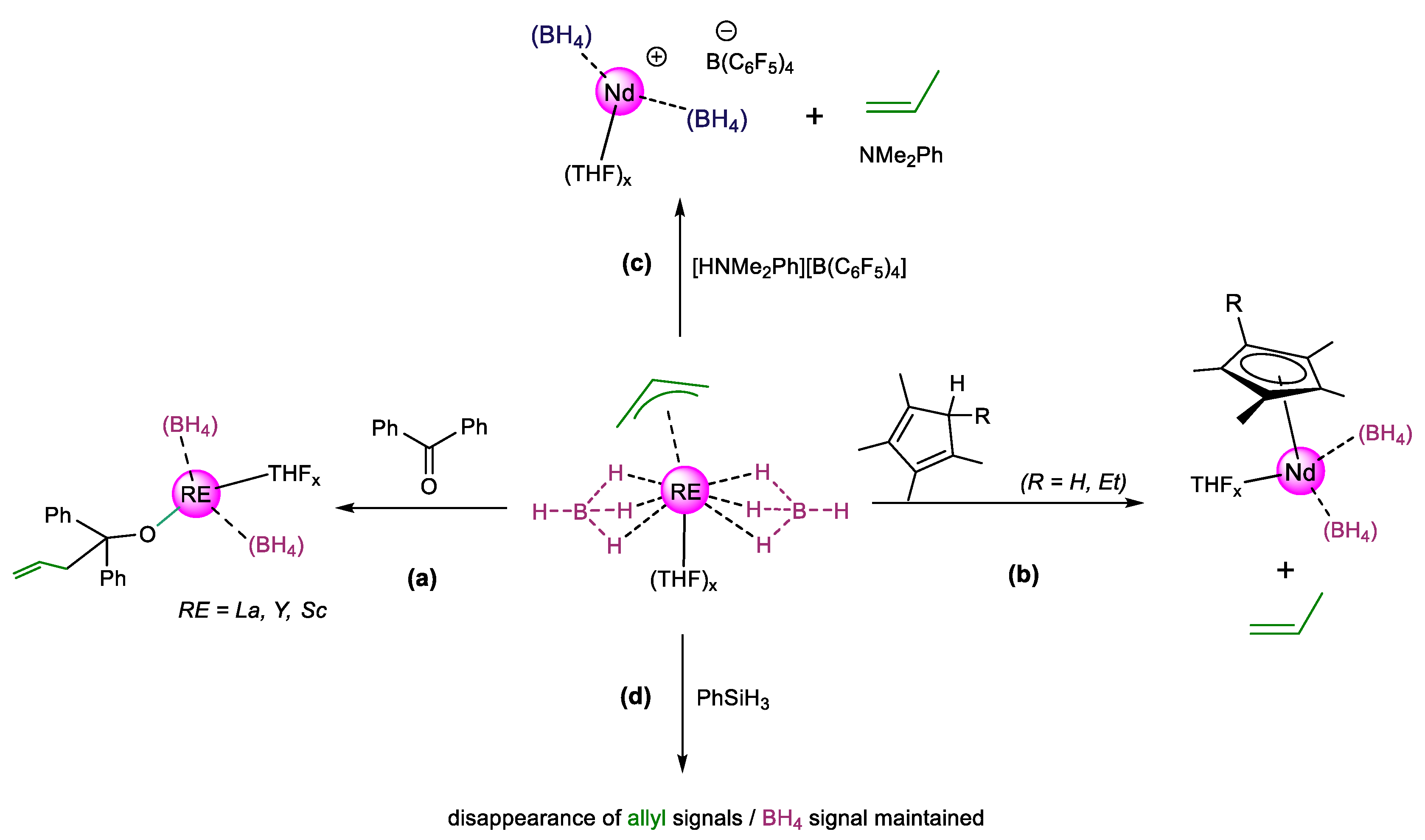{kind=link}
1. Introduction
2. Results and Discussion
2.1. 1H NMR Monitoring Experiments
2.2. DFT Studies
2.2.1. Allyl Initiation
2.2.2. Borohydride Initiation
2.2.3. Propagation Step
3. Materials and Methods
3.1. Experimental
3.2. NMR Monitoring Experiments
3.3. Methodological Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fadlallah, S.; Terrier, M.; Jones, C.; Roussel, P.; Bonnet, F.; Visseaux, M. Mixed Allyl–Borohydride Lanthanide Complexes: Synthesis of Ln(BH4)2(C3H5)(THF)3 (Ln = Nd, Sm), Characterization, and Reactivity toward Polymerization. Organometallics 2016, 35, 456–461. [Google Scholar] [CrossRef]
- Fadlallah, S.; Jothieswaran, J.; Capet, F.; Bonnet, F.; Visseaux, M. Mixed Allyl Rare-Earth Borohydride Complexes: Synthesis, Structure, and Application in (Co-)Polymerization Catalysis of Cyclic Esters. Chem. Eur. J. 2017, 23, 15644–15654. [Google Scholar] [CrossRef]
- Guillaume, S.M.; Maron, L.; Roesky, P.W. Catalytic Behavior of Rare-Earth Borohydride Complexes in Polymerization of Polar Monomers. In Handbook on the Physics and Chemistry of Rare Earths; Bünzli, J.-C.G., Pecharsky, V.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 244, pp. 1–86. [Google Scholar]
- Guillaume, S.M.; Schappacher, M.; Soum, A. Polymerization of ε-Caprolactone Initiated by Nd(BH4)3(THF)3: Synthesis of Hydroxytelechelic Poly(ε-caprolactone). Macromolecules 2003, 36, 54–60. [Google Scholar] [CrossRef]
- Bonnet, F.; Cowley, A.R.; Mountford, P. Lanthanide Borohydride Complexes Supported by Diaminobis(phenoxide) Ligands for the Polymerization of ε-Caprolactone and l- and rac-Lactide. Inorg. Chem. 2005, 44, 9046–9055. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Okuda, S.; Yasuda, H.; Shiono, T. Synthesis of multiblock poly(l-lactide)-co-poly(ε-caprolactone) from hydroxy-telechelic prepolymers prepared by using neodymium tetrahydroborate. React. Funct. Polym. 2007, 67, 798–806. [Google Scholar] [CrossRef]
- Hu, C.; Louisy, E.; Fontaine, G.; Bonnet, F.J. Cyclic versus linear polylactide: Straightforward access using a single catalyst. Polym. Sci. Part A Polym. Chem. 2017, 55, 3175–3179. [Google Scholar] [CrossRef] [Green Version]
- Visseaux, M.; Bonnet, F. Borohydride complexes of rare earths, and their applications in various organic transformations. Coord. Chem. Rev. 2011, 255, 374–420. [Google Scholar] [CrossRef]
- Lyubov, D.M.; Tolpygin, A.O.; Trifonov, A.A. Rare-earth metal complexes as catalysts for ring-opening polymerization of cyclic esters. Coord. Chem. Rev. 2019, 392, 83–145. [Google Scholar] [CrossRef]
- Palard, I.; Soum, A.; Guillaume, S.M. Rare Earth Metal Tris(borohydride) Complexes as Initiators for ε-Caprolactone Polymerization: General Features and IR Investigations of the Process. Macromolecules 2005, 38, 6888–6894. [Google Scholar] [CrossRef]
- Barros, N.; Mountford, P.; Guillaume, S.M.; Maron, L. A DFT Study of the Mechanism of Polymerization of ε-Caprolactone Initiated by Organolanthanide Borohydride Complexes. Chem. Eur. J. 2008, 14, 5507–5518. [Google Scholar] [CrossRef]
- Jenter, J.; Roesky, P.W.; Ajellal, N.; Guillaume, S.M.; Susperregui, N.; Maron, L. Bis(phosphinimino)methanide Borohydride Complexes of the Rare-Earth Elements as Initiators for the Ring-Opening Polymerization of ε-Caprolactone: Combined Experimental and Computational Investigations. Chem. Eur. J. 2010, 16, 4629–4638. [Google Scholar] [CrossRef]
- Skvortsov, G.G.; Yakovenko, M.V.; Castro, P.M.; Fukin, G.K.; Cherkasov, A.V.; Carpentier, J.-F.; Trifonov, A.A. Lanthanide Borohydride Complexes of Bulky Guanidinate Ligands [(Me3Si)2NC(N-Cy)2]2Ln(μ-BH4)2Li(THF)2 (Ln = Nd, Sm, Yb): Synthesis, Structure and Catalytic Activity in Lactide Polymerization. Eur. J. Inorg. Chem. 2007, 3260–3267. [Google Scholar] [CrossRef]
- Dyer, H.E.; Huijser, S.; Susperregui, N.; Bonnet, F.; Schwarz, A.D.; Duchateau, R.; Maron, L.; Mountford, P. Ring-Opening Polymerization of rac-Lactide by Bis(phenolate)amine-Supported Samarium Borohydride Complexes: An Experimental and DFT Study. Organometallics 2010, 29, 3602–3621. [Google Scholar] [CrossRef]
- Bonnet, F.; Stoffelbach, F.; Fontaine, G.; Bourbigot, S. Continuous cyclo-polymerization of l-lactide by reactive extrusion using atoxic metal-based catalysts: Easy access to well-defined polylactide macrocycles. RSC Adv. 2015, 5, 31303–31310. [Google Scholar] [CrossRef]
- Woodman, T.J.; Schormann, M.; Hughes, D.L.; Bochmann, M. New Bulky Allyl Complexes of Lanthanide Metals: Role of Alkali-Metal Cations in Controlling Solid-State and Solution Assemblies in Precatalysts. Organometallics 2003, 22, 3028–3030. [Google Scholar] [CrossRef]
- Barbier-Baudry, D.; Bonnet, F.; Dormond, A.; Finot, E.; Visseaux, M. Diene/polar monomer copolymers, compatibilisers for polar/non-polar polymer blends. A controlled block copolymerization with a single-site component samarocene initiator. Macromol. Chem. Phys. 2002, 203, 1194–1200. [Google Scholar] [CrossRef]
- Woodman, T.J.; Schormann, M.; Hughes, D.L.; Bochmann, M. Sterically Hindered Lanthanide Allyl Complexes and Their Use as Single-Component Catalysts for the Polymerization of Methyl Methacrylate and ε-Caprolactone. Organometallics 2004, 23, 2972–2979. [Google Scholar] [CrossRef]
- Sánchez-Barba, L.F.; Hughes, D.L.; Humphrey, S.M.; Bochmann, M. Ligand Transfer Reactions of Mixed-Metal Lanthanide/Magnesium Allyl Complexes with β-Diketimines: Synthesis, Structures, and Ring-Opening Polymerization Catalysis. Organometallics 2006, 25, 1012–1020. [Google Scholar] [CrossRef]
- Susperregui, N.; Kramer, M.U.; Okuda, J.; Maron, L. Theoretical Study on the Ring-Opening Polymerization of ε-Caprolactone by [YMeX(THF)5]+ with X = BH4, NMe2. Organometallics 2011, 30, 1326–1333. [Google Scholar] [CrossRef]
- Kramer, M.; Robert, D.; Arndt, S.; Zeimentz, P.M.; Spaniol, T.P.; Yahia, A.; Maron, L.; Eisenstein, O.; Okuda, J. Cationic Methyl Complexes of the Rare-Earth Metals: An Experimental and Computational Study on Synthesis, Structure, and Reactivity. Inorg. Chem. 2008, 9265–9278. [Google Scholar] [CrossRef]
- Robert, D.; Abinet, E.; Spaniol, T.P.; Okuda, J. Cationic Allyl Complexes of the Rare-Earth Metals: Synthesis, Structural Characterization, and 1,3-Butadiene Polymerization Catalysis. Chem. Eur. J. 2009, 15, 11937–11947. [Google Scholar] [CrossRef]
- Visseaux, M.; Baudry, D.; Dormond, A.; Qian, C. Co-ordinatively unsaturated neodymium hydrides, stability in solution. J. Organomet. Chem. 1999, 574, 213–218. [Google Scholar] [CrossRef]
- Bonnet, F.; Visseaux, M.; Barbier-Baudry, D.; Hafid, A.; Vigier, E.; Kubicki, M.M. Organometallic Early Lanthanide Clusters: Syntheses and X-ray Structures of New Monocyclopentadienyl Complexes. Inorg. Chem. 2004, 43, 3682–3690. [Google Scholar] [CrossRef]
- Robert, D.; Kondracka, M.; Okuda, J. Cationic rare-earth metal bis(tetrahydridoborato) complexes: Direct synthesis, structure and ring-opening polymerization activity toward cyclic esters. Dalton Trans. 2008, 2667–2669. [Google Scholar] [CrossRef]
- Visseaux, M.; Mainil, M.; Terrier, M.; Mortreux, A.; Roussel, P.; Mathivet, T.; Destarac, M. Cationic borohydrido–neodymium complex: Synthesis, characterization and its application as an efficient pre-catalyst for isoprene polymerization. Dalton Trans. 2008, 34, 4558–4561. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Erratum: Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1993, 48, 4978. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Wang, Y. Erratum: Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1998, 57, 14999–15033. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P. Unified Theory of Exchange and Correlation beyond the Local Densityapproximation; Akademie Verlag: Berlin, Germany, 1991; Volume 11. [Google Scholar]
- Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the rare earth elements. J. Chem. Phys. 1989, 90, 1730–1734. [Google Scholar] [CrossRef]
- Cao, X.; Dolg, M. Segmented contraction scheme for small-core lanthanide pseudopotential basis sets. J. Mol. Struct. 2002, 581, 139–147. [Google Scholar] [CrossRef]
- Pople, P.C.; Hariharan, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near-Hartree–Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fadlallah, S.; Jothieswaran, J.; Del Rosal, I.; Maron, L.; Bonnet, F.; Visseaux, M. Rationalizing the Reactivity of Mixed Allyl Rare-Earth Borohydride Complexes with DFT Studies. Catalysts 2020, 10, 820. https://doi.org/10.3390/catal10080820
Fadlallah S, Jothieswaran J, Del Rosal I, Maron L, Bonnet F, Visseaux M. Rationalizing the Reactivity of Mixed Allyl Rare-Earth Borohydride Complexes with DFT Studies. Catalysts. 2020; 10(8):820. https://doi.org/10.3390/catal10080820
Chicago/Turabian StyleFadlallah, Sami, Jashvini Jothieswaran, Iker Del Rosal, Laurent Maron, Fanny Bonnet, and Marc Visseaux. 2020. "Rationalizing the Reactivity of Mixed Allyl Rare-Earth Borohydride Complexes with DFT Studies" Catalysts 10, no. 8: 820. https://doi.org/10.3390/catal10080820
APA StyleFadlallah, S., Jothieswaran, J., Del Rosal, I., Maron, L., Bonnet, F., & Visseaux, M. (2020). Rationalizing the Reactivity of Mixed Allyl Rare-Earth Borohydride Complexes with DFT Studies. Catalysts, 10(8), 820. https://doi.org/10.3390/catal10080820