Asymmetric Hydrogenation of 1-aryl substituted-3,4-Dihydroisoquinolines with Iridium Catalysts Bearing Different Phosphorus-Based Ligands


 ,
,  and
and
Abstract
:
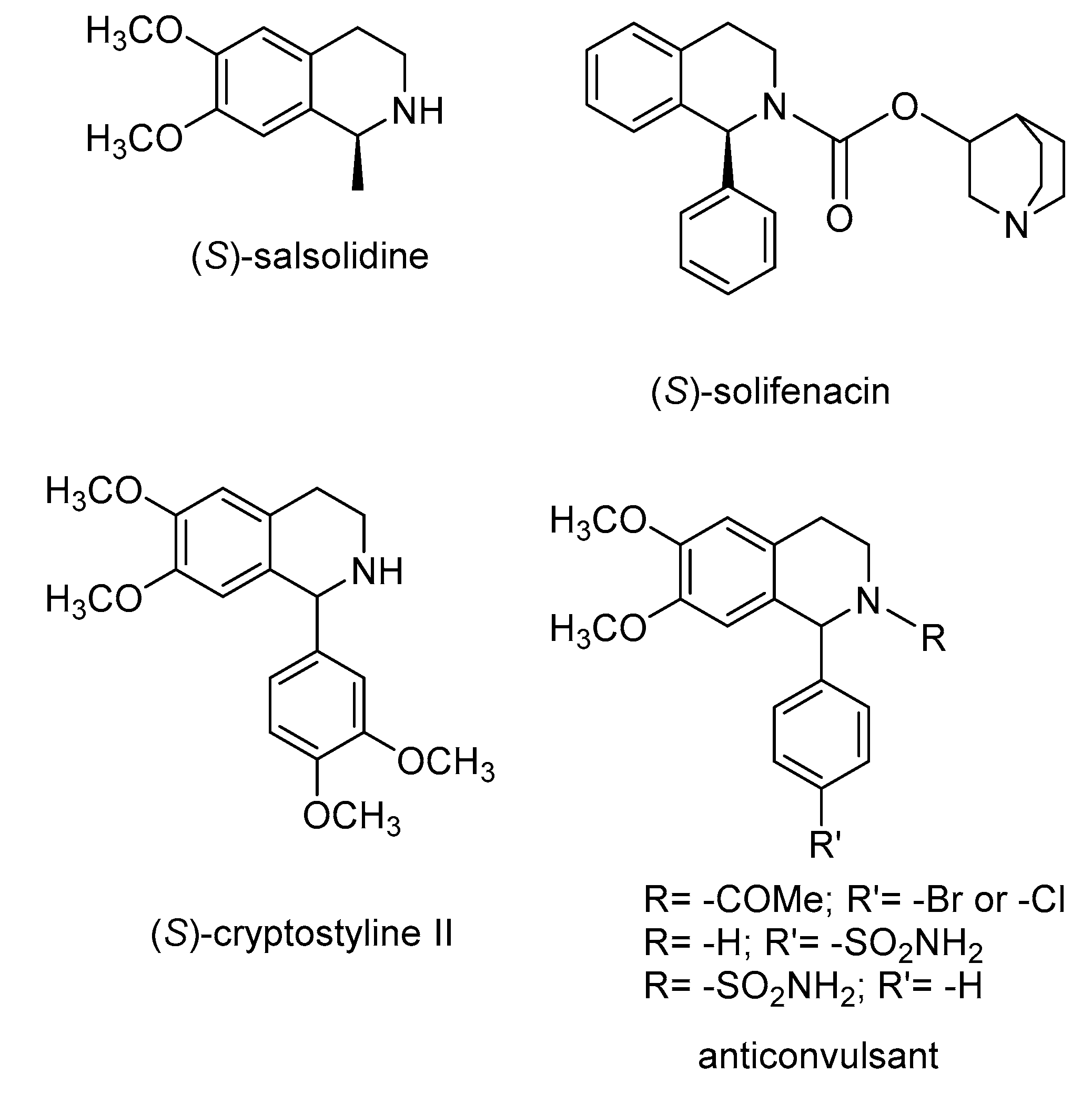
1. Introduction
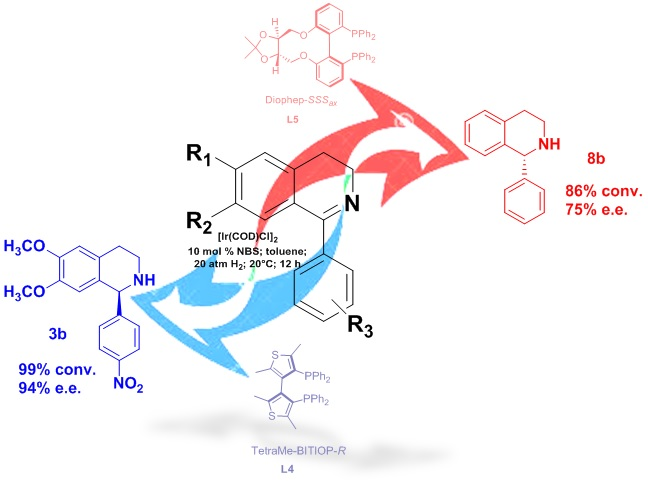
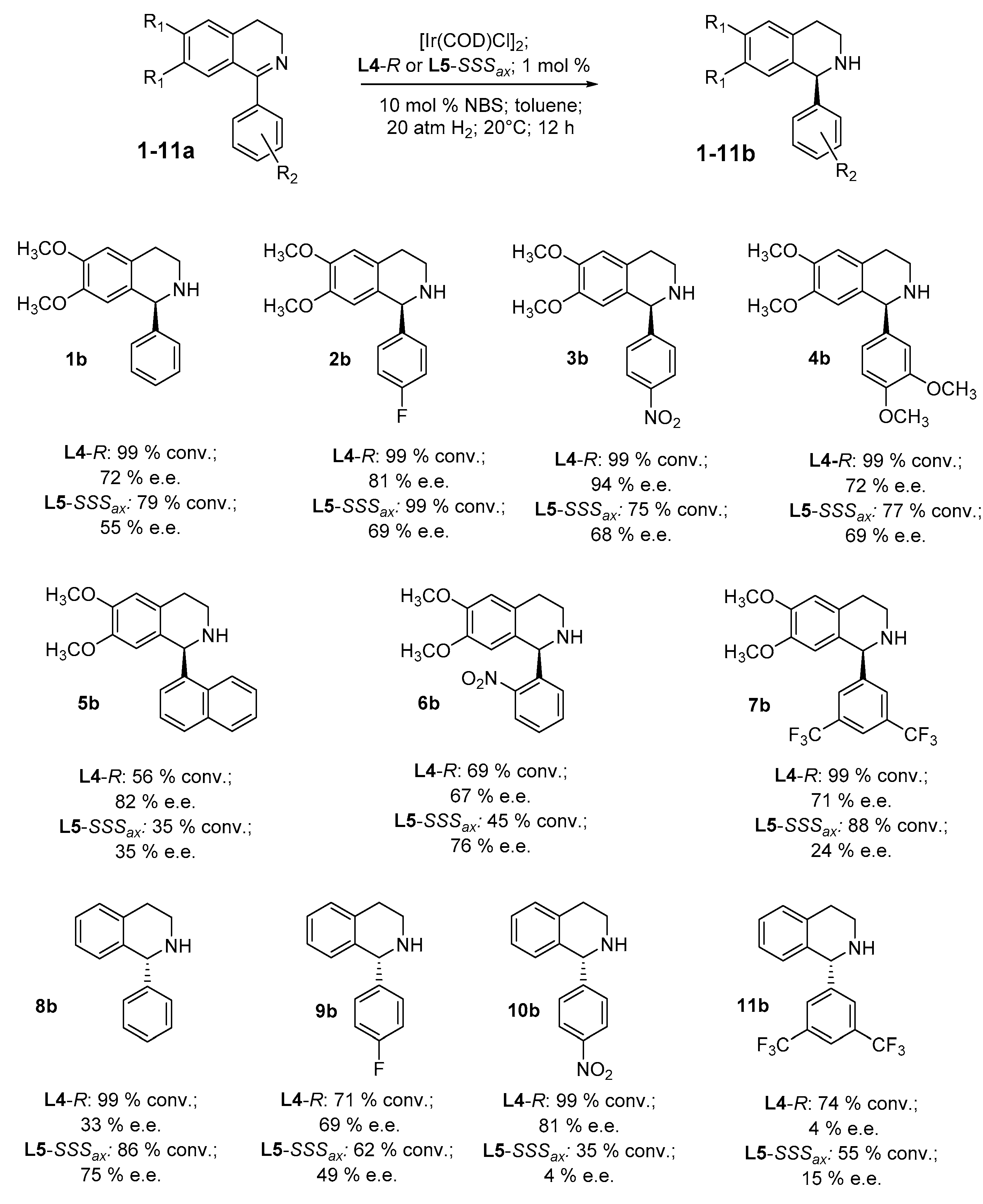
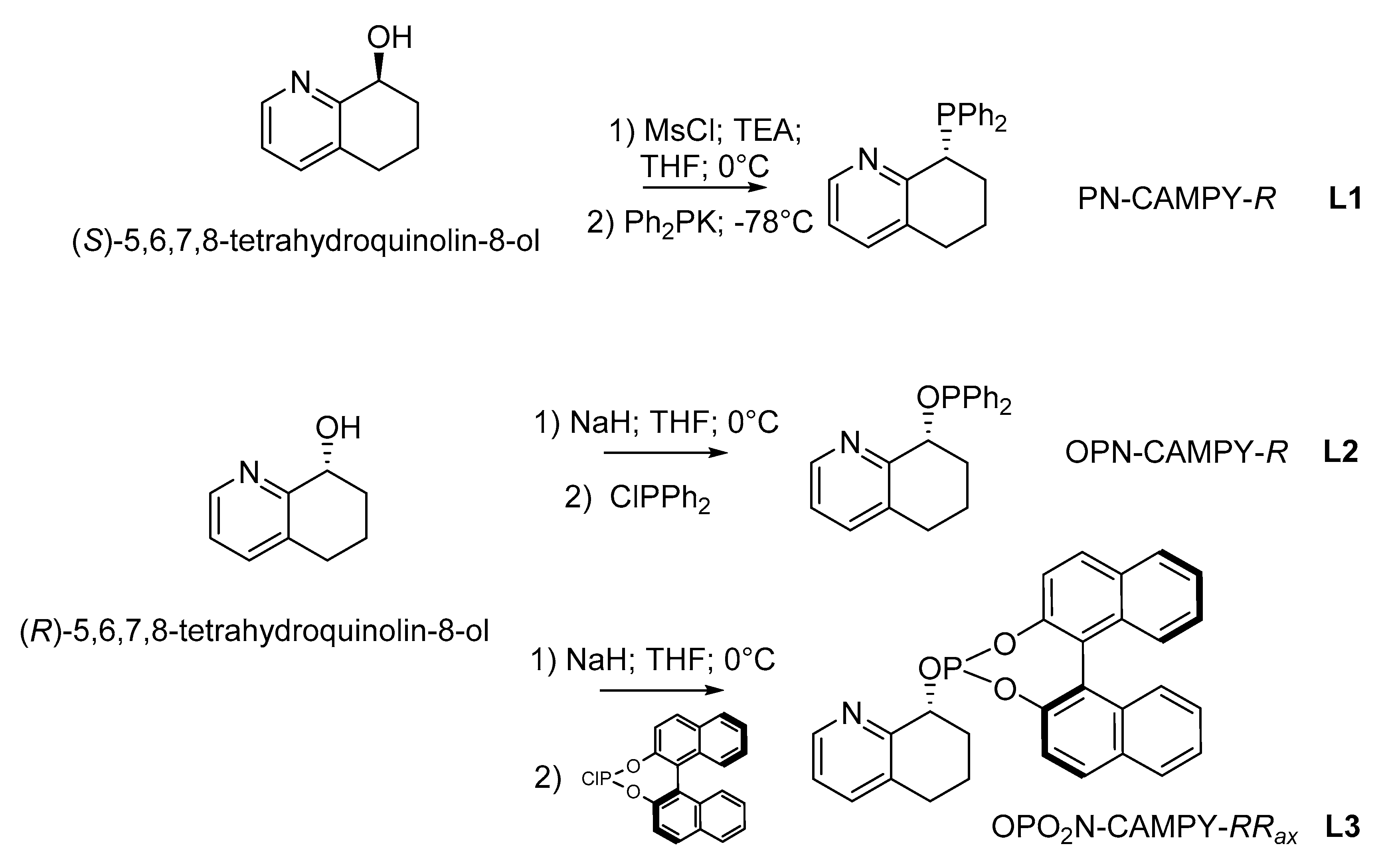
2. Results and Discussion
3. Experimental
3.1. General Procedure for Synthesis of Substrates
3.2. Synthesis of (R)-8-(Diphenylphosphanyl)-5,6,7,8-Tetrahydroquinoline, L1
3.3. Synthesis of (R)-8-((Diphenylphosphanyl)oxy)-5,6,7,8-Tetrahydroquinoline and (8R)-8-(((4R)-Dinaphtho[2,1-d:1′,2′-f][1,3,2]Dioxaphosphepin-4-yl)oxy)-5,6,7,8-Tetrahydroquinoline, L2 and L3
3.4. General Procedure for Asymmetric Hydrogenation
3.5. Analytical Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Scott, J.D.; Williams, R.M. Chemistry and Biology of the Tetrahydroisoquinoline Antitumor Antibiotics. Chem. Rev. 2002, 102, 1669–1730. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Saitoh, T.; Horiguchi, Y.; Utsunomiya, I.; Taguchi, K. Synthesis and Neurotoxicity of Tetrahydroisoquinoline Derivatives for Studying Parkinson’s Disease. Biol. Pharm. Bull. 2005, 28, 1355–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.R.; Safo, M.K.; Abraham, D.J. Identification of a series of tetrahydroisoquinoline derivatives as potential therapeutic agents for breast cancer. Bioorg. Med. Chem. Lett. 2007, 17, 2581–2589. [Google Scholar] [CrossRef] [PubMed]
- Quinto, T.; Schwizer, F.; Zimbron, J.M.; Morina, A.; Köhler, V.; Ward, T.R. Expanding the Chemical Diversity in Artificial Imine Reductases Based on the Biotin-Streptavidin Technology. ChemCatChem 2014, 6, 1010–1014. [Google Scholar] [CrossRef]
- Noyori, R.; Ohta, M.; Hsiao, Y.; Kitamura, M.; Ohta, T.; Takaya, H. Asymmetric synthesis of isoquinoline alkaloids by homogeneous catalysis. J. Am. Chem. Soc. 1986, 108, 7117–7119. [Google Scholar] [CrossRef]
- Schwizer, F.; Okamoto, Y.; Heinisch, T.; Gu, Y.; Pellizzoni, M.M.; Lebrun, V.; Reuter, R.; Köhler, V.; Lewis, J.C.; Ward, T.R. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. [Google Scholar] [CrossRef] [Green Version]
- Facchetti, G.; Rimoldi, I. 8-Amino-5,6,7,8-tetrahydroquinoline in iridium(iii) biotinylated Cp* complex as artificial imine reductase. New J. Chem. 2018, 42, 18773–18776. [Google Scholar] [CrossRef]
- Pyne, S.G.; Bloem, P.; Chapman, S.L.; Dixon, C.E.; Griffith, R. Chiral sulfur compounds. 9. Stereochemistry of the intermolecular and intramolecular conjugate additions of amines and anions to chiral (E)- and (Z)-vinyl sulfoxides. Total syntheses of (R)-(+)-carnegine and (+)- and (-)-sedamine. J. Org. Chem. 1990, 55, 1086–1093. [Google Scholar] [CrossRef]
- Ružič, M.; Pečavar, A.; Prudič, D.; Kralj, D.; Scriban, C.; Zanotti-Gerosa, A. The Development of an Asymmetric Hydrogenation Process for the Preparation of Solifenacin. Org. Process Res. Dev 2012, 16, 1293–1300. [Google Scholar] [CrossRef]
- Zhu, R.; Xu, Z.; Ding, W.; Liu, S.; Shi, X.; Lu, X. Efficient and Practical Syntheses of Enantiomerically Pure (S)-(−)-Norcryptostyline I, (S)-(−)-Norcryptostyline II, (R)-(+)-Salsolidine and (S)-(−)-Norlaudanosine via a Resolution-Racemization Method. Chin. J. Chem. 2014, 32, 1039–1048. [Google Scholar] [CrossRef]
- Zhurakulov, S.N.; Babkin, V.A.; Chernyak, E.I.; Morozov, S.V.; Grigor’ev, I.A.; Levkovich, M.G.; Vinogradova, V.I. Aminomethylation of 1-Aryl-6,7-Dimethoxy-1,2,3,4-Tetrahydroisoquinolines by Dihydroquercetin. Chem. Nat. Compd. 2015, 51, 57–61. [Google Scholar] [CrossRef]
- Guranova, N.I.; Varlamov, A.V.; Novikov, R.A.; Aksenov, A.V.; Borisova, T.N.; Sorokina, E.A.; Voskressensky, L.G. Reaction of benzyne with 1,2,3,4-tetrahydroisoquinolines as an access to 1H-3-benzazepines. Mendeleev Commun. 2018, 28, 22–24. [Google Scholar] [CrossRef]
- Chrzanowska, M.; Rozwadowska, M.D. Asymmetric Synthesis of Isoquinoline Alkaloids. Chem. Rev. 2004, 104, 3341–3370. [Google Scholar] [CrossRef] [PubMed]
- Bentley, K.W. β-Phenylethylamines and the isoquinoline alkaloids. Nat. Prod. Rep. 2006, 23, 444–463. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Huang, N.; Jiang, Z.-Y.; Zhang, Q.; Zheng, Y.T.; Chen, J.J.; Zhang, X.M.; Ma, Y.B. 1-Aryl-tetrahydroisoquinoline analogs as active anti-HIV agents in vitro. Bioorg. Med. Chem. Lett. 2008, 18, 2475–2478. [Google Scholar] [CrossRef]
- Bruno, E.; Buemi, M.R.; De Luca, L.; Ferro, S.; Monforte, A.M.; Supuran, C.T.; Vullo, D.; De Sarro, G.; Russo, E.; Gitto, R. In Vivo Evaluation of Selective Carbonic Anhydrase Inhibitors as Potential Anticonvulsant Agents. ChemMedChem 2016, 11, 1812–1818. [Google Scholar] [CrossRef]
- Gitto, R.; Ferro, S.; Agnello, S.; De Luca, L.; De Sarro, G.; Russo, E.; Vullo, D.; Supuran, C.T.; Chimirri, A. Synthesis and evaluation of pharmacological profile of 1-aryl-6,7-dimethoxy-3,4-dihydroisoquinoline-2(1H)-sulfonamides. Bioorg. Med. Chem. 2009, 17, 3659–3664. [Google Scholar] [CrossRef]
- Singh, K.N.; Singh, P.; Kaur, M.; Sharma, E. Intermolecular Nucleophilic Addition of N-Diaminophosphinoyl-Protected α-Carbanions Derived from Secondary Amines to Arynes: Synthesis of 1-Aryl-1,2,3,4-tetrahydroisoquinolines. ChemistrySelect 2017, 2, 2213–2218. [Google Scholar] [CrossRef]
- Pesnot, T.; Gershater, M.C.; Ward, J.M.; Hailes, H.C. Phosphate mediated biomimetic synthesis of tetrahydroisoquinoline alkaloids. Chem. Commun. 2011, 47, 3242–3244. [Google Scholar] [CrossRef]
- Vanden Eynden, M.J.; Kunchithapatham, K.; Stambuli, J.P. Calcium-Promoted Pictet-Spengler Reactions of Ketones and Aldehydes. J. Org. Chem. 2010, 75, 8542–8549. [Google Scholar] [CrossRef]
- Vanden Eynden, M.J.; Stambuli, J.P. Calcium-Catalyzed Pictet−Spengler Reactions. Org. Lett. 2008, 10, 5289–5291. [Google Scholar] [CrossRef] [PubMed]
- Doi, S.; Shirai, N.; Sato, Y. Abnormal products in the Bischler–Napieralski isoquinoline synthesis. J. Chem. Soc. Perkin Trans. 1 1997, 15, 2217–2221. [Google Scholar] [CrossRef]
- Paul, A.; Adili, A.; Seidel, D. Redox-Annulations of Cyclic Amines with Electron-Deficient o-Tolualdehydes. Org. Lett. 2019, 21, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Vedejs, E.; Trapencieris, P.; Suna, E. Substituted Isoquinolines by Noyori Transfer Hydrogenation: Enantioselective Synthesis of Chiral Diamines Containing an Aniline Subunit. J. Org. Chem. 1999, 64, 6724–6729. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Zhu, Y.; Hu, X.; Wei, Z.; Yao, L.; Zhou, G.; Wang, P.; Jiang, R.; Zhang, S. Josiphos-Type Binaphane Ligands for Iridium-Catalyzed Enantioselective Hydrogenation of 1-Aryl-Substituted Dihydroisoquinolines. Org. Lett. 2019, 21, 8641–8645. [Google Scholar] [CrossRef]
- Václavíková, B.; Budinská, A.; Václavík, J.; Matoušek, V.; Kuzma, M.; Červený, L. Asymmetric Transfer Hydrogenation of 1-Aryl-3,4-Dihydroisoquinolines Using a Cp*Ir(TsDPEN) Complex. Eur. J. Org. Chem. 2017, 2017, 5131–5134. [Google Scholar] [CrossRef]
- Lu, S.M.; Wang, Y.Q.; Han, X.W.; Zhou, Y.G. Asymmetric hydrogenation of quinolines and isoquinolines activated by chloroformates. Angew Chem. Int. Ed. Engl. 2006, 118, 2318–2321. [Google Scholar] [CrossRef]
- Chang, M.; Li, W.; Zhang, X. A Highly Efficient and Enantioselective Access to Tetrahydroisoquinoline Alkaloids: Asymmetric Hydrogenation with an Iridium Catalyst. Angew. Chem. Int. Ed. 2011, 50, 10679–10681. [Google Scholar] [CrossRef]
- Shi, L.; Ye, Z.S.; Cao, L.L.; Guo, R.N.; Hu, Y.; Zhou, Y.G. Enantioselective Iridium-Catalyzed Hydrogenation of 3,4-Disubstituted Isoquinolines. Angew. Chem. Int. Ed. 2012, 51, 8286–8289. [Google Scholar] [CrossRef]
- Wen, J.; Tan, R.; Liu, S.; Zhao, Q.; Zhang, X. Strong Bronsted acid promoted asymmetric hydrogenation of isoquinolines and quinolines catalyzed by a Rh-thiourea chiral phosphine complex via anion binding. Chem. Sci. 2016, 7, 3047–3051. [Google Scholar] [CrossRef] [Green Version]
- Schwenk, R.; Togni, A. P-Trifluoromethyl ligands derived from Josiphos in the Ir-catalysed hydrogenation of 3,4-dihydroisoquinoline hydrochlorides. Dalton Trans. 2015, 44, 19566–19575. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Wang, J.; Chen, M.; Shi, L.; Zhou, Y. Dual Stereocontrol for Enantioselective Hydrogenation of Dihydroisoquinolines Induced by Tuning the Amount of N-Bromosuccinimide. Chin. J. Chem. 2018, 36, 139–142. [Google Scholar] [CrossRef]
- Chen, M.; Ji, Y.; Shi, L.; Sun, L.; Zhou, Y. A Method Catalyzed by Iridium and Used for Bidirectional Enantioselective Synthesis of Chiral Tetrahydroisoquinoline. CN107540606 A, 2018. [Google Scholar]
- Berhal, F.; Wu, Z.; Zhang, Z.; Ayad, T.; Ratovelomanana-Vidal, V. Enantioselective Synthesis of 1-Aryl-tetrahydroisoquinolines through Iridium Catalyzed Asymmetric Hydrogenation. Org. Lett. 2012, 14, 3308–3311. [Google Scholar] [CrossRef]
- Perez, M.; Wu, Z.; Scalone, M.; Ayad, T.; Ratovelomanana-Vidal, V. Enantioselective Synthesis of 1-Aryl-Substituted Tetrahydroisoquinolines Through Ru-Catalyzed Asymmetric Transfer Hydrogenation. Eur. J. Org. Chem. 2015, 2015, 6503–6514. [Google Scholar] [CrossRef]
- Fusè, M.; Rimoldi, I.; Cesarotti, E.; Rampino, S.; Barone, V. On the relation between carbonyl stretching frequencies and the donor power of chelating diphosphines in nickel dicarbonyl complexes. Phys. Chem. Chem. Phys. 2017, 19, 9028–9038. [Google Scholar] [CrossRef] [Green Version]
- Fusè, M.; Rimoldi, I.; Facchetti, G.; Rampino, S.; Barone, V. Exploiting coordination geometry to selectively predict the σ-donor and π-acceptor abilities of ligands: A back-and-forth journey between electronic properties and spectroscopy. Chem. Commun. 2018, 54, 2397–2400. [Google Scholar] [CrossRef]
- Rimoldi, I.; Facchetti, G.; Cesarotti, E.; Pellizzoni, M.; Fuse, M.; Zerla, D. Enantioselective transfer hydrogenation of aryl ketones: Synthesis and 2D-NMR characterization of new 8-amino-5,6,7,8-tetrahydroquinoline Ru(II)-complexes. Curr. Org. Chem. 2012, 16, 2982–2988. [Google Scholar] [CrossRef]
- Gandolfi, R.; Facchetti, G.; Christodoulou, M.S.; Fusè, M.; Meneghetti, F.; Rimoldi, I. Cascade Reaction by Chemo and Biocatalytic Approaches to Obtain Chiral Hydroxy Ketones and anti 1,3–Diols. ChemistryOpen 2018, 7, 393–400. [Google Scholar] [CrossRef]
- Facchetti, G.; Ferri, N.; Lupo, M.G.; Giorgio, L.; Rimoldi, I. Monofunctional PtII Complexes Based on 8-Aminoquinoline: Synthesis and Pharmacological Characterization. Eur. J. Inorg. Chem. 2019, 2019, 3389–3395. [Google Scholar] [CrossRef]
- Kaiser, S.; Smidt, S.P.; Pfaltz, A. Iridium Catalysts with Bicyclic Pyridine–Phosphinite Ligands: Asymmetric Hydrogenation of Olefins and Furan Derivatives. Angew. Chem. Int. Ed. 2006, 45, 5194–5197. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.B.; Yu, C.B.; Zhou, Y.G. Synthesis of tunable phosphinite–pyridine ligands and their applications in asymmetric hydrogenation. Tetrahedron Lett. 2006, 47, 4733–4736. [Google Scholar] [CrossRef]
- Uenishi, J.; Hamada, M. Synthesis of Enantiomerically Pure 8-Substituted 5,6,7,8-Tetrahydroquinolines. Synthesis 2002, 2002, 0625–0630. [Google Scholar] [CrossRef]
- Benincori, T.; Brenna, E.; Sannicolò, F.; Trimarco, L.; Antognazza, P.; Cesarotti, E.; Demartin, F.; Pilati, T. New Class of Chiral Diphosphine Ligands for Highly Efficient Transition Metal-Catalyzed Stereoselective Reactions: The Bis(diphenylphosphino) Five-membered Biheteroaryls. J. Org. Chem. 1996, 61, 6244–6251. [Google Scholar] [CrossRef]
- Facchetti, G.; Cesarotti, E.; Pellizzoni, M.; Zerla, D.; Rimoldi, I. “In situ” Activation of Racemic RuII Complexes: Separation of trans and cis Species and Their Application in Asymmetric Reduction. Eur. J. Inorg. Chem. 2012, 2012, 4365–4370. [Google Scholar] [CrossRef]
- Cesarotti, E.; Abbiati, G.; Rossi, E.; Spalluto, P.; Rimoldi, I. DIOPHEP, a chiral diastereoisomeric bisphosphine ligand: Synthesis and applications in asymmetric hydrogenations. Tetrahedron 2008, 19, 1654–1659. [Google Scholar] [CrossRef]
- Cesarotti, E.; Araneo, S.; Rimoldi, I.; Tassi, S. Aminophosphonite-phosphite and aminophosphonite-phosphinite ligands with mixed chirality: Preparation and catalytic applications in asymmetric hydrogenation and hydroformylation. J. Mol. Catal. A 2003, 204-205, 211–220. [Google Scholar] [CrossRef]
- Facchetti, G.; Bucci, R.; Fusè, M.; Rimoldi, I. Asymmetric Hydrogenation vs Transfer Hydrogenation in the Reduction of Cyclic Imines. ChemistrySelect 2018, 3, 8797–8800. [Google Scholar] [CrossRef]
- Zhang, D.; Zhou, X.; Liu, R.; Li, M.; Li, X.; Jiang, R.; Nie, H.; Zhang, S. Josiphos-type binaphane ligands for the asymmetric Ir-catalyzed hydrogenation of acyclic aromatic N-aryl imines. Catal. Commun. 2020, 136, 105906. [Google Scholar] [CrossRef]
- Min, L.; Yang, W.; Weng, Y.; Zheng, W.; Wang, X.; Hu, Y. A Method for Bischler–Napieralski-Type Synthesis of 3,4-Dihydroisoquinolines. Org. Lett. 2019, 21, 2574–2577. [Google Scholar] [CrossRef]
- Zhang, R.; Qin, Y.; Zhang, L.; Luo, S. Mechanistic Studies on Bioinspired Aerobic C–H Oxidation of Amines with an ortho-Quinone Catalyst. J. Org. Chem. 2019, 84, 2542–2555. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yi, H.; Lei, A. Electrochemical Acceptorless Dehydrogenation of N-Heterocycles Utilizing TEMPO as Organo-Electrocatalyst. ACS Catal. 2018, 8, 1192–1196. [Google Scholar] [CrossRef]
- Zheng, B.; Trieu, T.H.; Li, F.L.; Zhu, X.L.; He, Y.G.; Fan, Q.Q.; Shi, X.X. Copper-Catalyzed Benign and Efficient Oxidation of Tetrahydroisoquinolines and Dihydroisoquinolines Using Air as a Clean Oxidant. ACS Omega 2018, 3, 8243–8252. [Google Scholar] [CrossRef] [PubMed]
- Lucena-Serrano, C.; Lucena-Serrano, A.; Rivera, A.; López-Romero, J.M.; Valpuesta, M.; Díaz, A. Synthesis and dopaminergic activity of a series of new 1-aryl tetrahydroisoquinolines and 2-substituted 1-aryl-3-tetrahydrobenzazepines. Bioorg. Chem. 2018, 80, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.W.; Ji, Y.; Wang, J.; Chen, Q.A.; Shi, L.; Zhou, Y.G. Asymmetric Hydrogenation of Isoquinolines and Pyridines Using Hydrogen Halide Generated in Situ as Activator. Org. Lett. 2017, 19, 4988–4991. [Google Scholar] [CrossRef] [PubMed]
- Hati, S.; Sen, S. Cerium Chloride Catalyzed, 2-Iodoxybenzoic Acid Mediated Oxidative Dehydrogenation of Multiple Heterocycles at Room Temperature. Eur. J. Org. Chem. 2017, 2017, 1277–1280. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Ligand | Additive | Conversion %[b] | e.e. (S %) [b] |
|---|---|---|---|---|
| 1 | L1-R | - | - | - |
| 2 | L1-R | NBS | 50 | 20 |
| 3 | L2-R | - | - | - |
| 4 | L2-R | NBS | 27 | 32 |
| 5 | L3-RRax | - | - | - |
| 6 | L3-RRax | NBS | 41 | 39 |
| 7 | L4-R | DCDMH [c] | 83 | 73 |
| 8 | L4-R | - | 85 | 50 |
| 9 | L4-R | NBS | >99 | 72 |
| 10 | L4-R | FeCl3 | 85 | 72 |
| 11 | L4-R[a] | NBS | 90 | 71 |
| 12 | L5-SSSax | - | 10 | 38 |
| 13 | L5-SSSax | NBS | 69 | 55 |
| 14 | L6-RRax | - | 21 | 27 |
| 15 | L6-RRax | DCDMH | 5 | 25 |
| 16 | L6-RRax | NBS | 50 | 25 |
| 17 | L7-SRaxRax | - | - | - |
| 18 | L7-SRaxRax | NBS | 25 | 25 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Facchetti, G.; Christodoulou, M.S.; Binda, E.; Fusè, M.; Rimoldi, I. Asymmetric Hydrogenation of 1-aryl substituted-3,4-Dihydroisoquinolines with Iridium Catalysts Bearing Different Phosphorus-Based Ligands. Catalysts 2020, 10, 914. https://doi.org/10.3390/catal10080914
Facchetti G, Christodoulou MS, Binda E, Fusè M, Rimoldi I. Asymmetric Hydrogenation of 1-aryl substituted-3,4-Dihydroisoquinolines with Iridium Catalysts Bearing Different Phosphorus-Based Ligands. Catalysts. 2020; 10(8):914. https://doi.org/10.3390/catal10080914
Chicago/Turabian StyleFacchetti, Giorgio, Michael S. Christodoulou, Eleonora Binda, Marco Fusè, and Isabella Rimoldi. 2020. "Asymmetric Hydrogenation of 1-aryl substituted-3,4-Dihydroisoquinolines with Iridium Catalysts Bearing Different Phosphorus-Based Ligands" Catalysts 10, no. 8: 914. https://doi.org/10.3390/catal10080914
APA StyleFacchetti, G., Christodoulou, M. S., Binda, E., Fusè, M., & Rimoldi, I. (2020). Asymmetric Hydrogenation of 1-aryl substituted-3,4-Dihydroisoquinolines with Iridium Catalysts Bearing Different Phosphorus-Based Ligands. Catalysts, 10(8), 914. https://doi.org/10.3390/catal10080914