Deciphering the Mechanism of Silver Catalysis of “Click” Chemistry in Water by Combining Experimental and MEDT Studies †

 ,
,  ,
,  ,
, 

Abstract
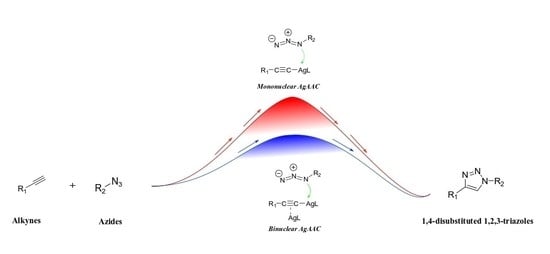
:
1. Introduction
2. Results and Discussion
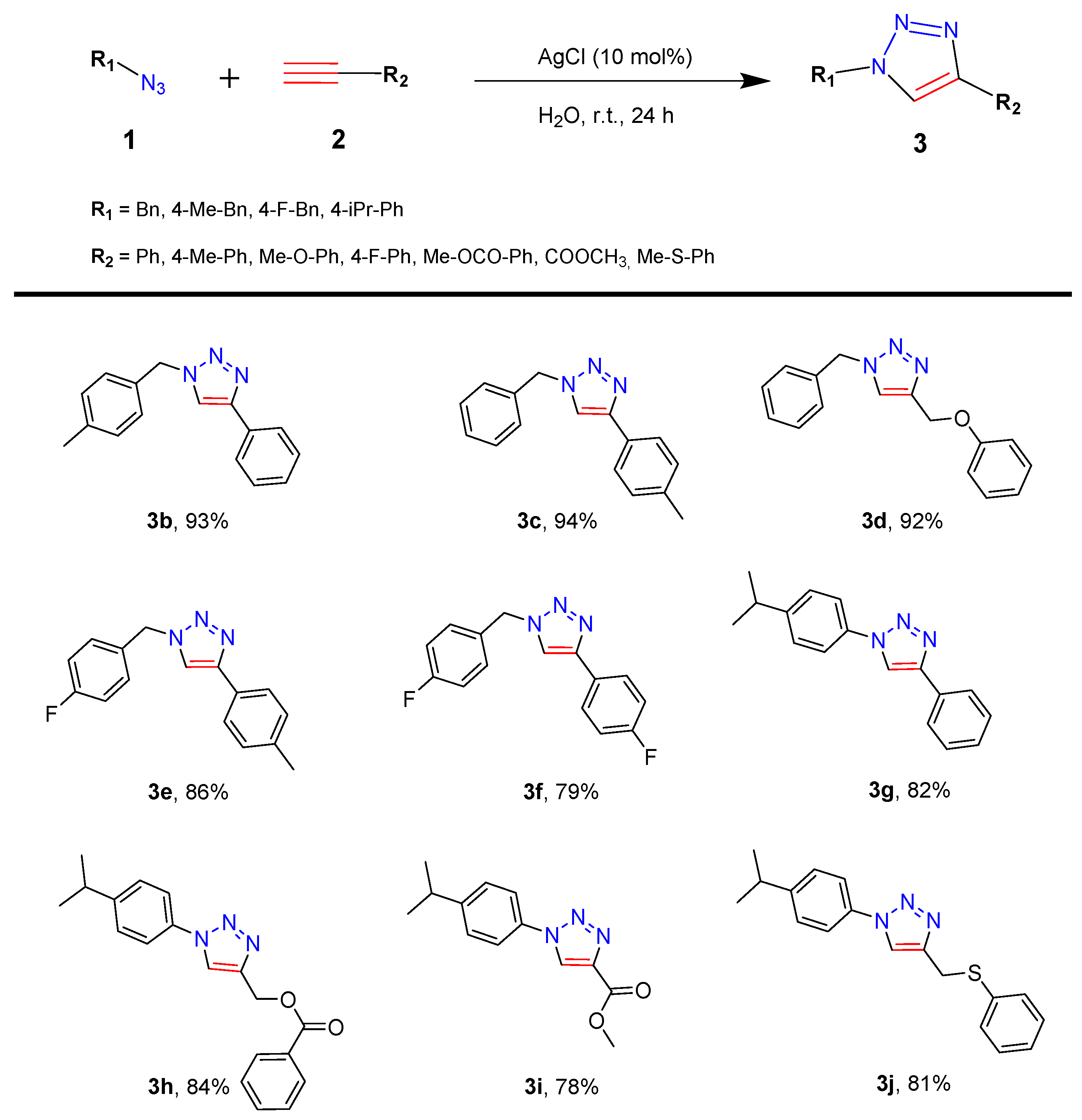
2.1. Setup and Scope of the Click AgAAC
2.2. MEDT Study
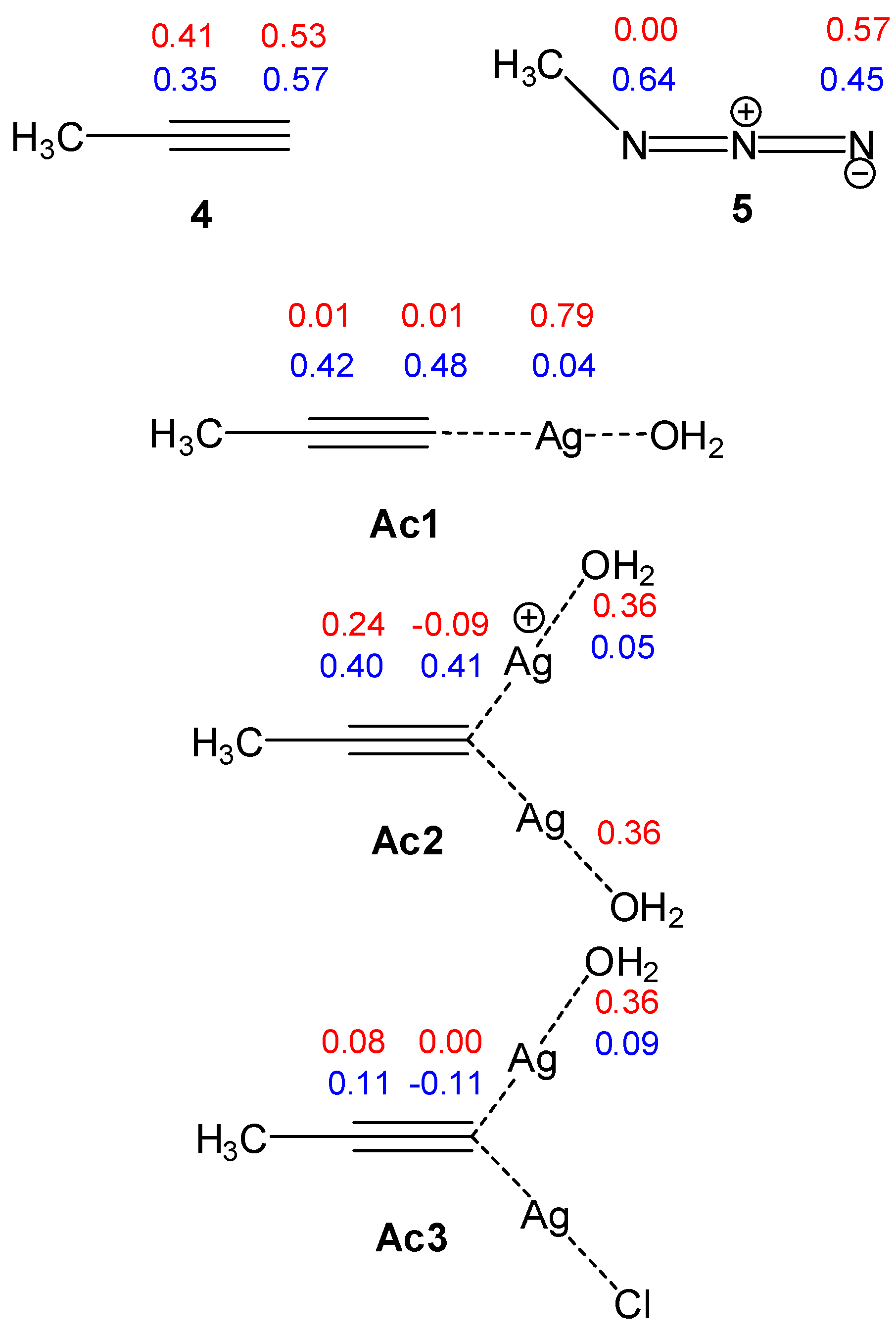
2.2.1. Analysis of the Global and Local CDFT Reactivity Indices
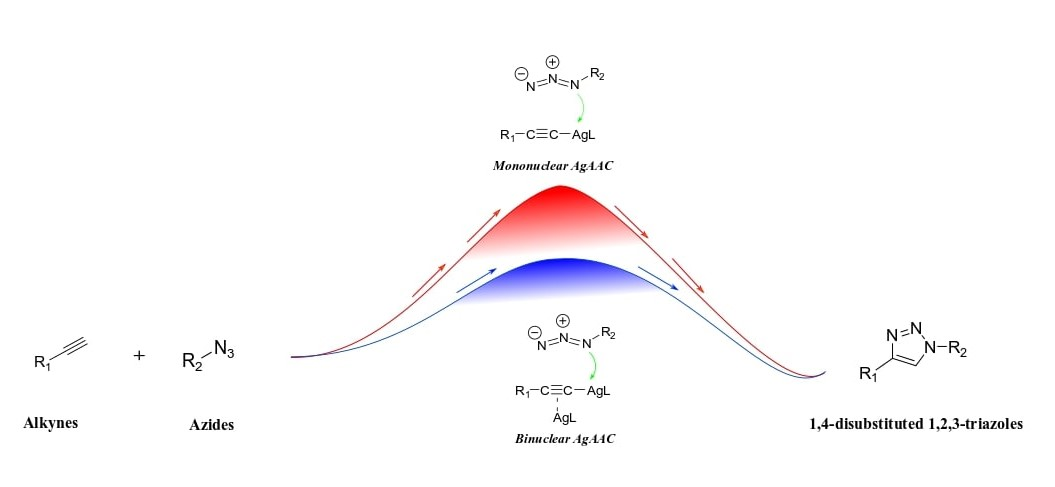
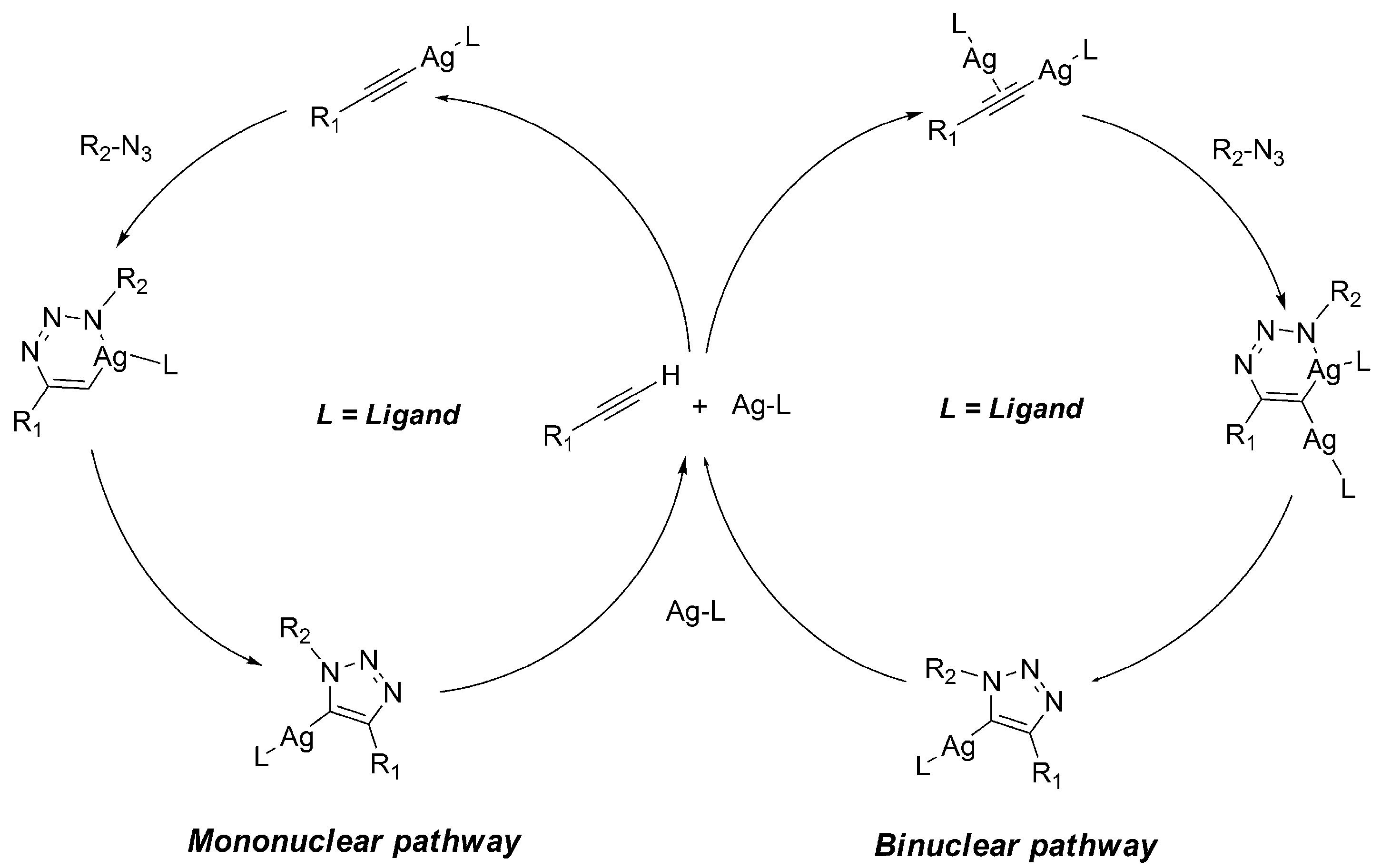
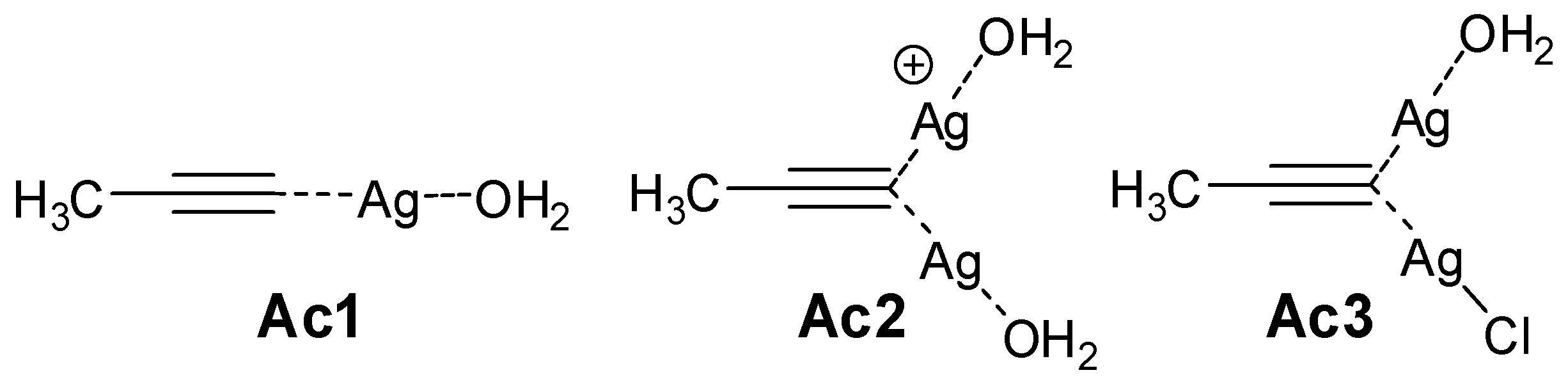
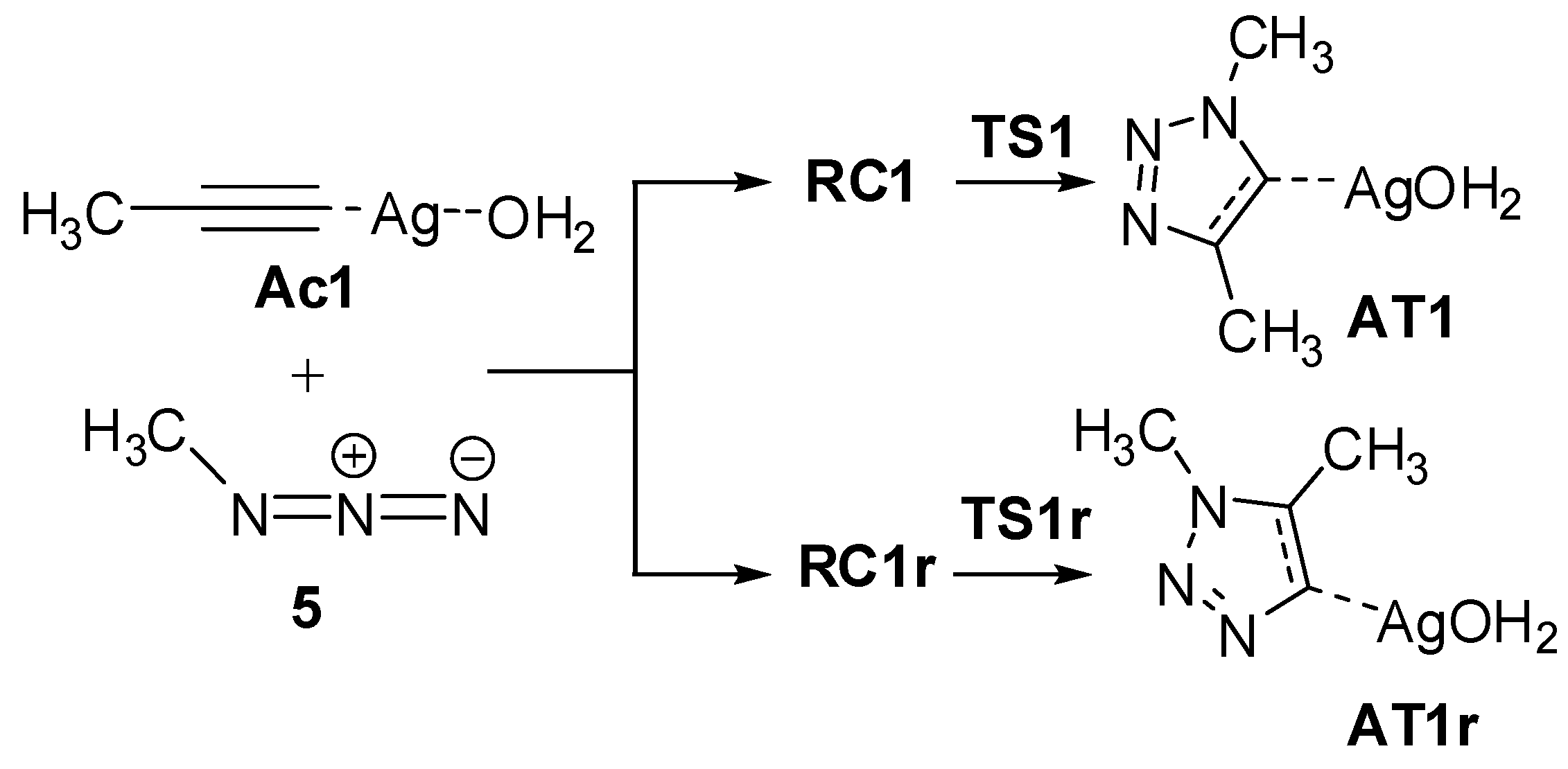
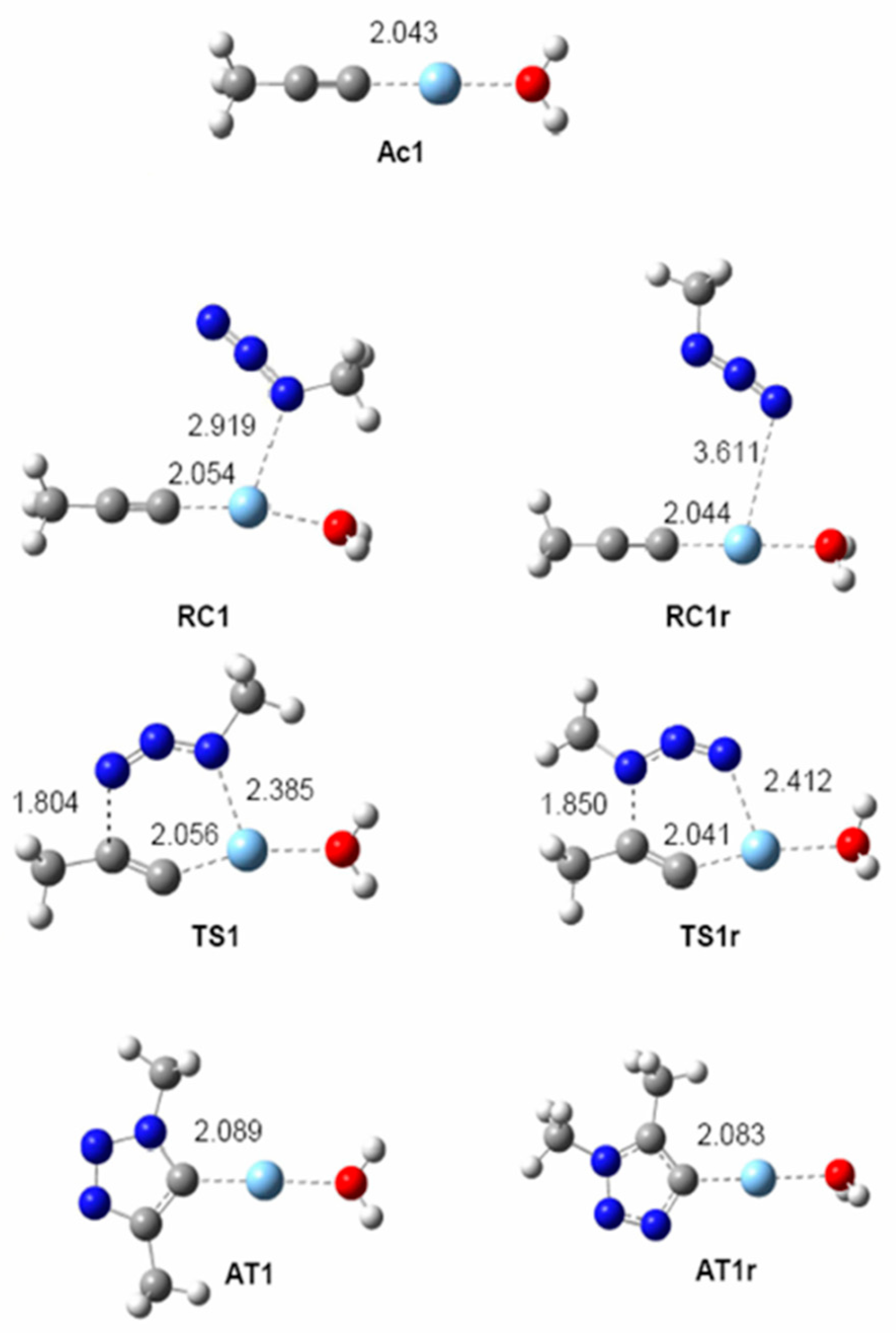
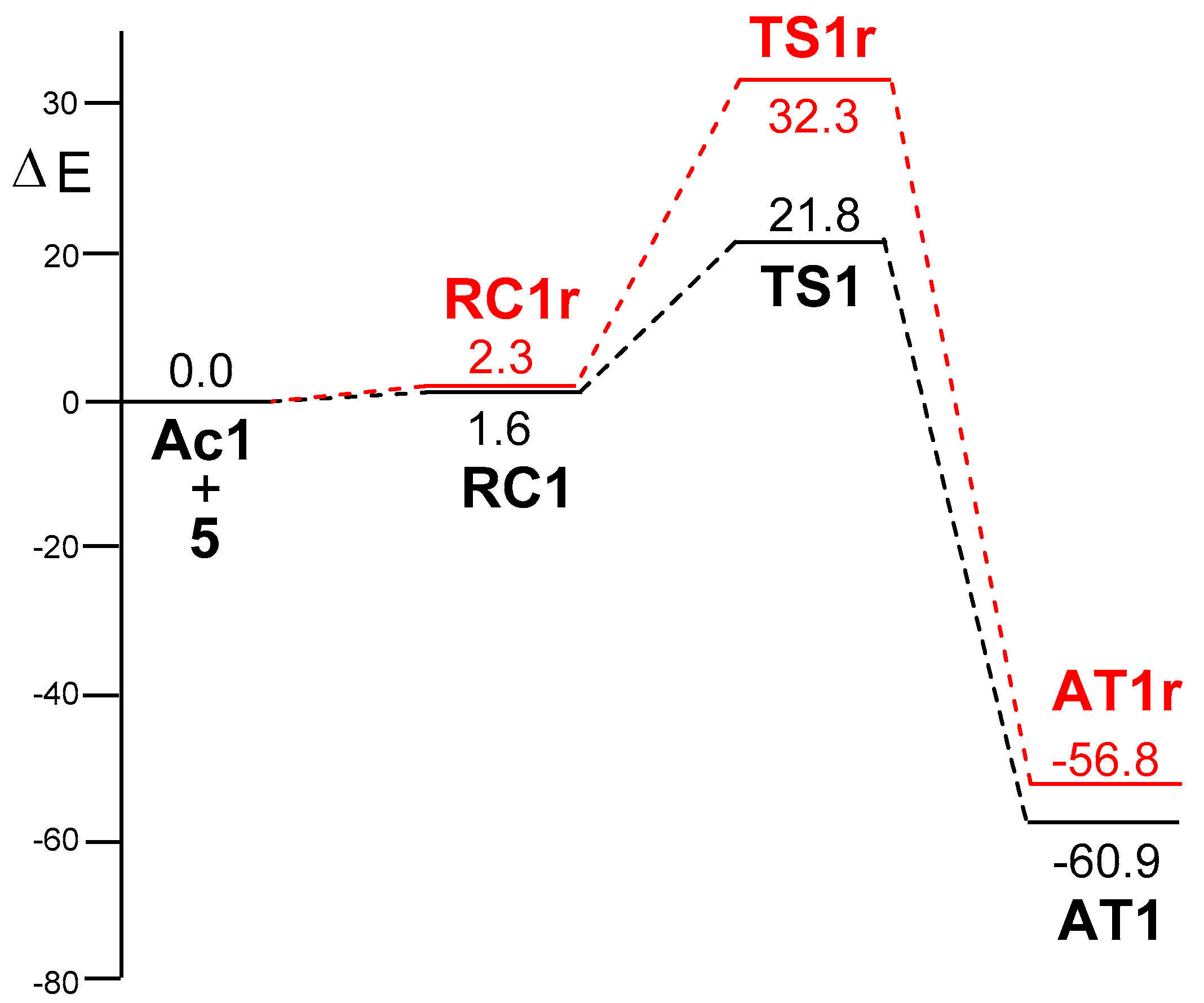
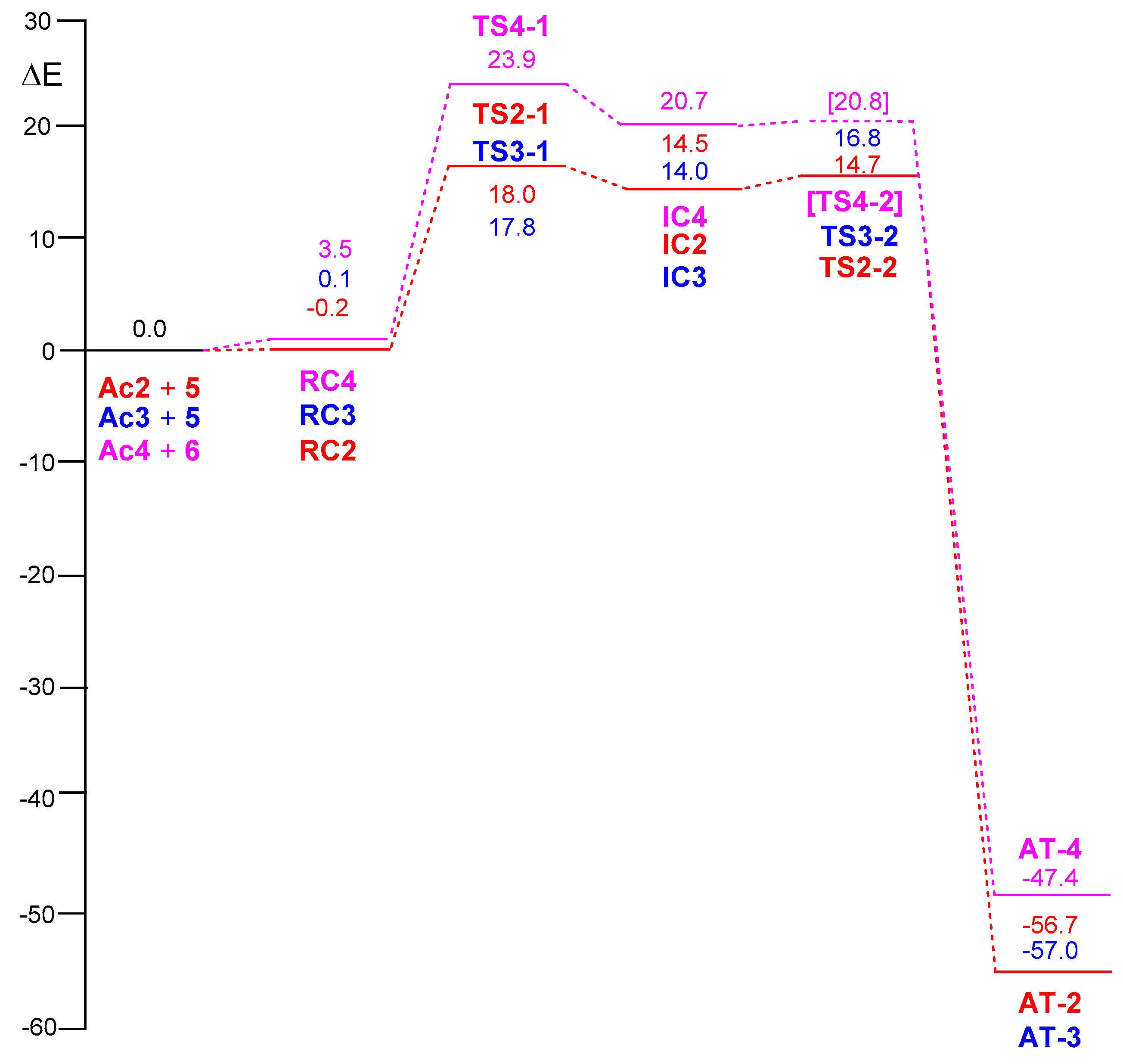
2.2.2. Mononuclear Mechanism
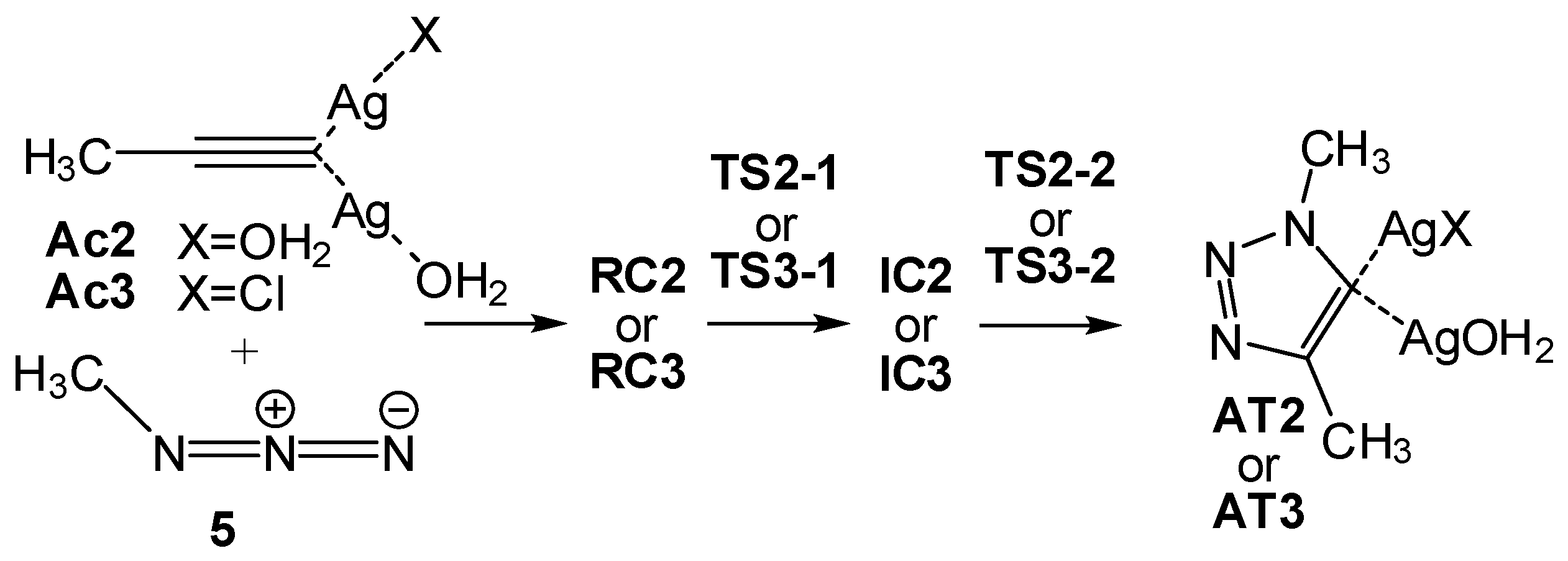
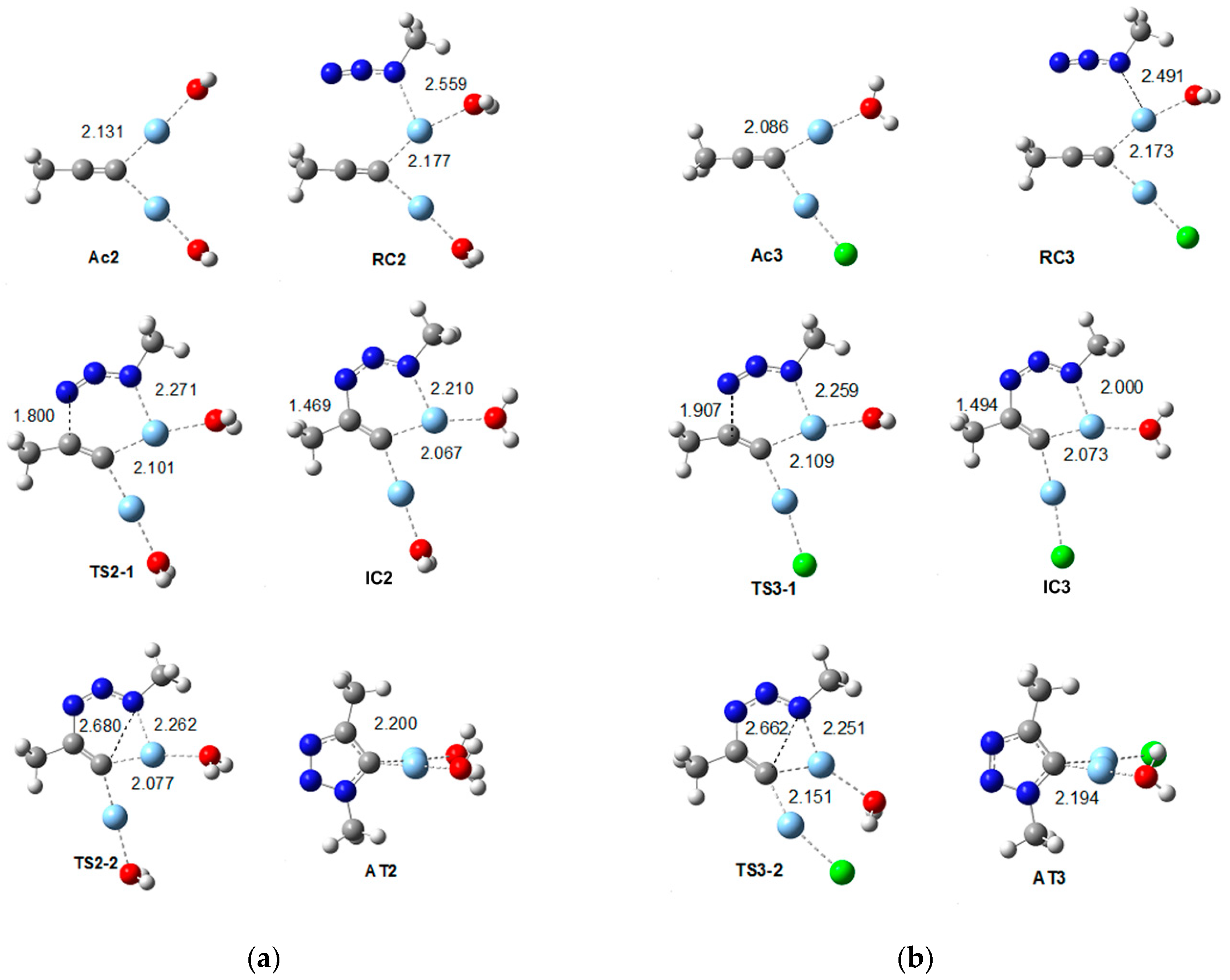
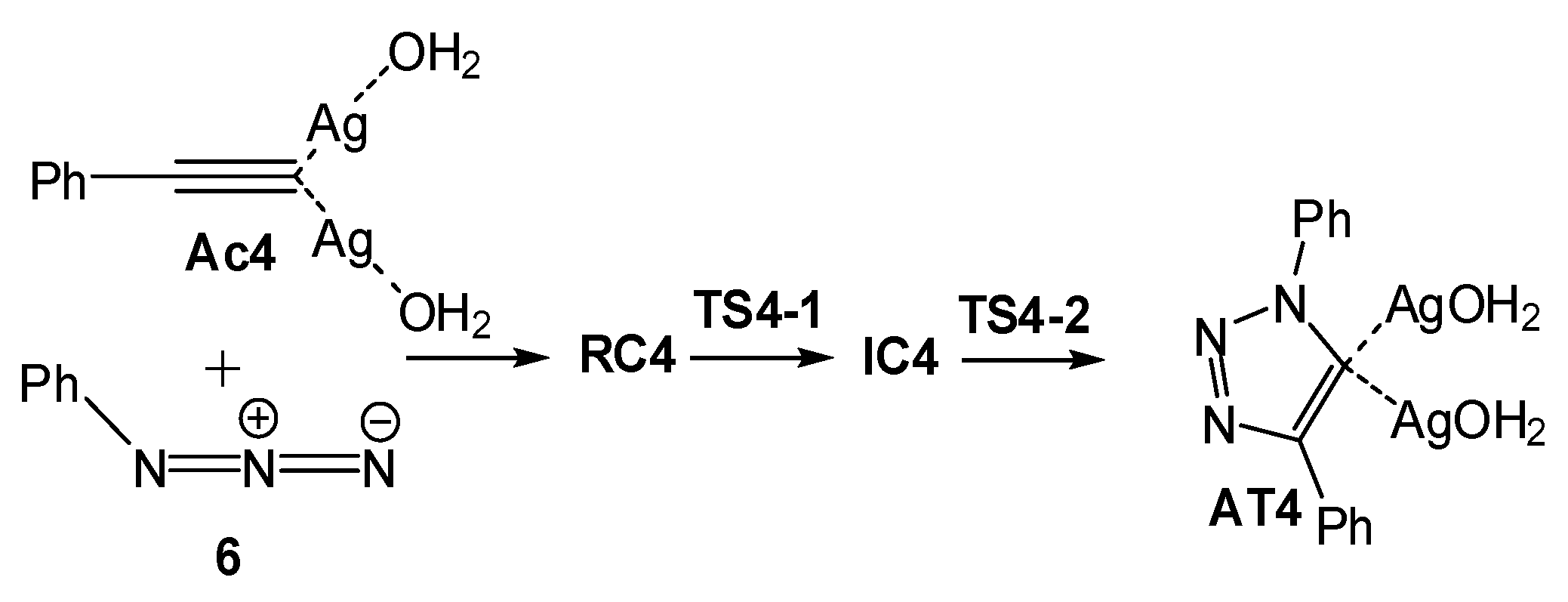
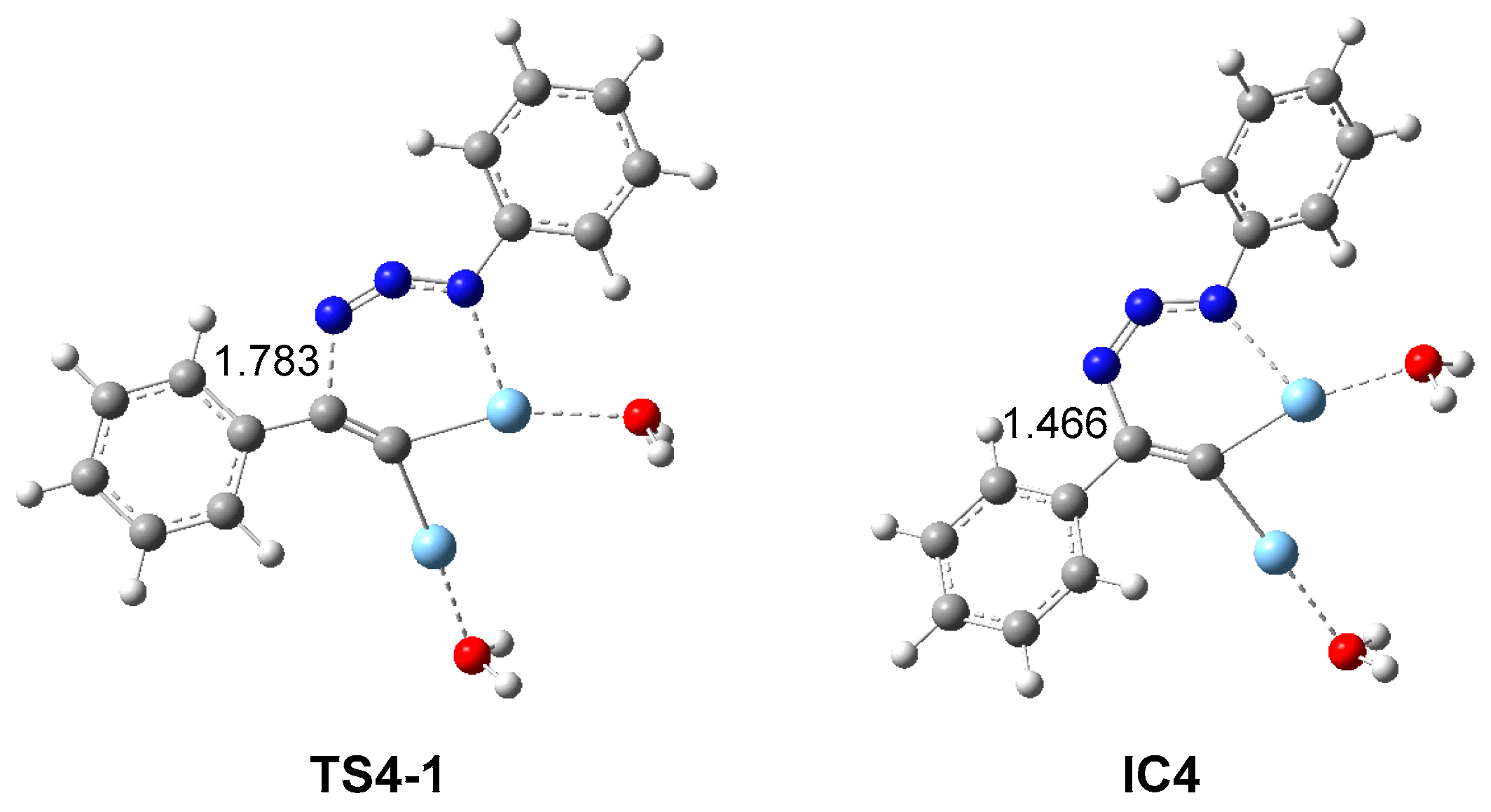
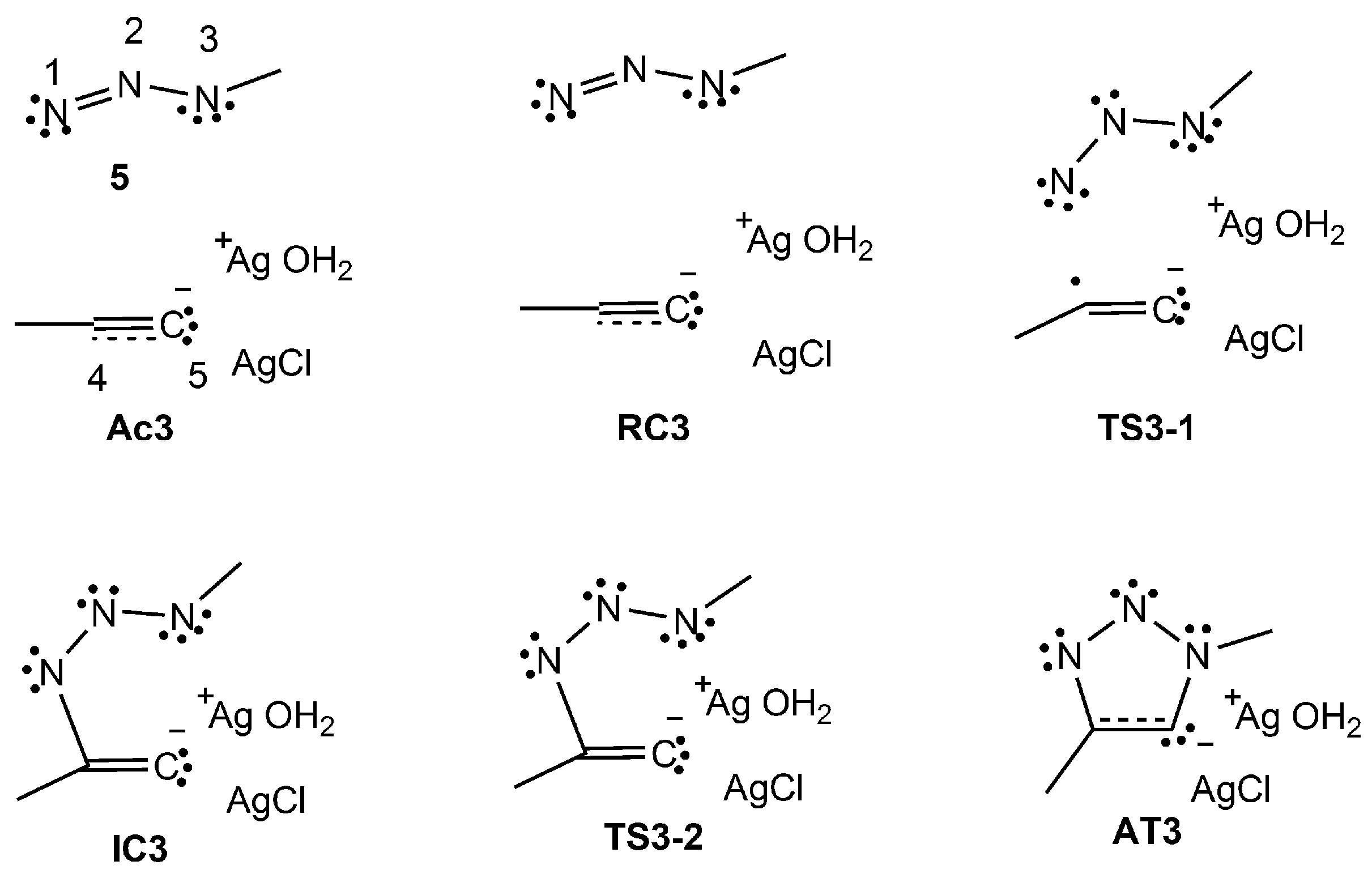
2.2.3. Dinuclear Mechanism
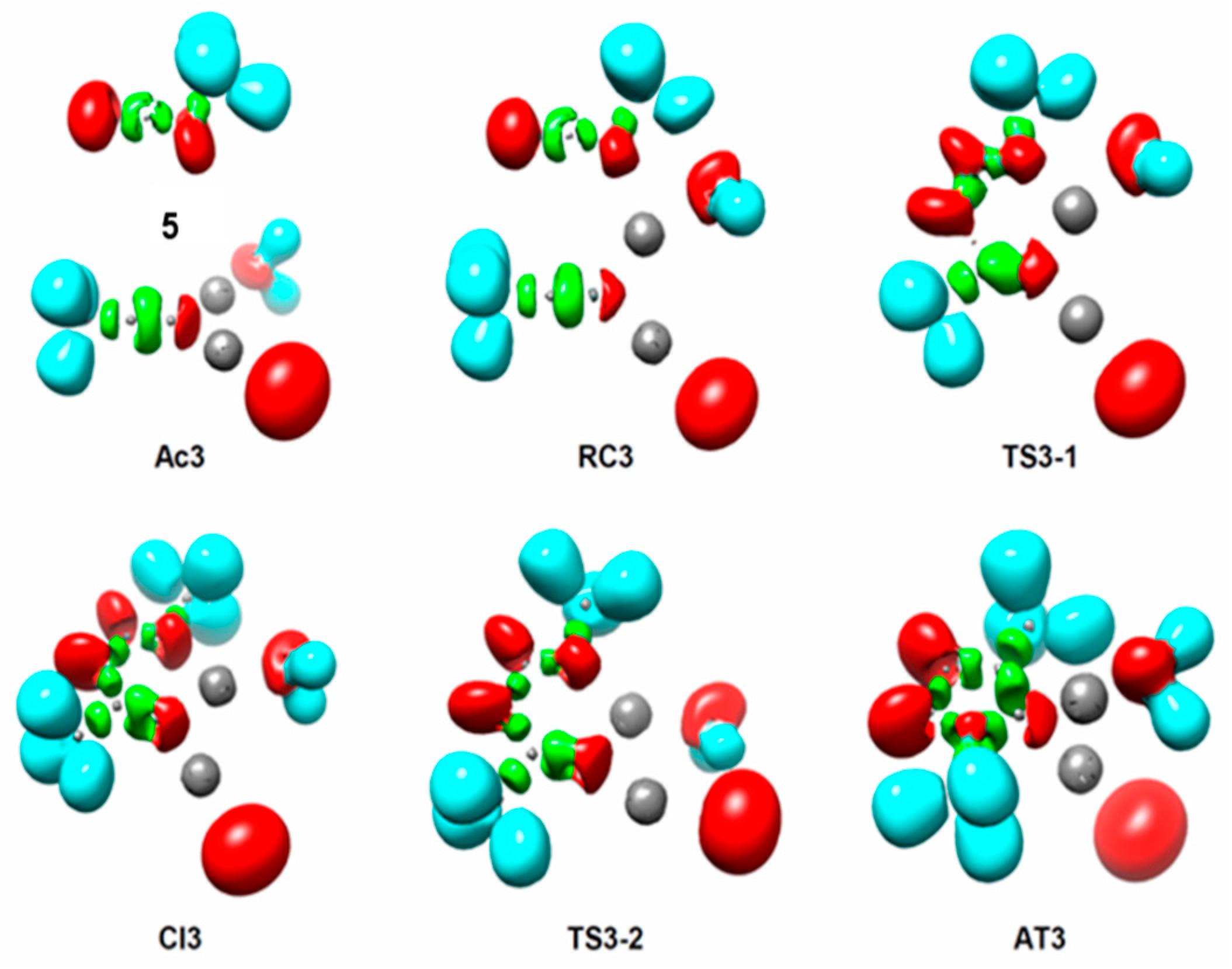
2.3. ELF Topological Analysis of the Stationary Points Involved in the AgAAC Dinucear Reaction of Ac3
3. Materials and Methods
3.1. Experimental Details
3.1.1. Reagents and Physical Measurements
3.1.2. Typical Procedure for the Ag-Catalyzed Cycloaddition of Alkyne with Azide
3.2. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Costa, M.S.; Boechat, N.; Rangel, É.A.; da Silva, F.D.C.; de Souza, A.M.T.; Rodrigues, C.R.; Castro, H.C.; Junior, I.N.; Lourenço, M.C.S.; Wardell, S.M.S.V.; et al. Synthesis, tuberculosis inhibitory activity, and SAR study of N-substituted-phenyl-1,2,3-triazole derivatives. Bioorg. Med. Chem. 2006, 14, 8644–8653. [Google Scholar] [CrossRef] [PubMed]
- Nandivada, H.; Jiang, X.; Lahann, J. Click Chemistry: Versatility and Control in the Hands of Materials Scientists. Adv. Mater. 2007, 19, 2197–2208. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Billington, R.A.; Canonico, P.L.; Sorba, G.; Genazzani, A.A. Click chemistry reactions in medicinal chemistry: Applications of the 1,3-dipolar cycloaddition between azides and alkynes. Med. Res. Rev. 2008, 28, 278–308. [Google Scholar] [CrossRef] [PubMed]
- Abu-Orabi, S.T.; Atfah, M.A.; Jibril, I.; Mari’i, F.M.; Ali, A.A.-S. Dipolar cycloaddition reactions of organic azides with some acetylenic compounds. J. Heterocycl. Chem. 1989, 26, 1461–1468. [Google Scholar] [CrossRef]
- Gouault, N.; Cupif, J.-F.; Sauleau, A.; David, M. γ-Methyl-substituted-γ-butyrolactones: Solid-phase synthesis employing a cyclisation–cleavage strategy. Tetrahedron Lett. 2000, 41, 7293–7297. [Google Scholar] [CrossRef]
- Lees, A.C.; Evrard, B.; Keyes, T.E.; Vos, J.G.; Kleverlaan, C.J.; Alebbi, M.; Bignozzi, C.A. Synthesis, Spectroscopy and Photophysical Properties of Ruthenium Triazole Complexes and Their Application as Dye-Molecules in Regenerative Solar Cells. Eur. J. Inorg. Chem. 1999, 1999, 2309–2317. [Google Scholar] [CrossRef]
- Lutz, J.-F. Nanotechnology for Life Science Research Group 1,3-dipolar cycloadditions of azides and alkynes: A universal ligation tool in polymer and materials science. Angew. Chem. Int. Ed. Engl. 2007, 46, 1018–1025. [Google Scholar] [CrossRef]
- Becer, C.R.; Hoogenboom, R.; Schubert, U.S. Click chemistry beyond metal-catalyzed cycloaddition. Angew. Chem. Int. Ed. Engl. 2009, 48, 4900–4908. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-Dipolar Cycloadditions. Past and Future. Angew. Chem. Int. Ed. Engl. 1963, 2, 565–598. [Google Scholar] [CrossRef]
- Huisgen, R. Kinetics and reaction mechanisms: Selected examples from the experience of forty years. Pure Appl. Chem. 1989, 61, 613–628. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Cu-Catalyzed Azide−Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Jalloh, A.S.; Wei, W.; Zhao, J.; Wu, P.; Chen, P.R. Biocompatible click chemistry enabled compartment-specific pH measurement inside E. coli. Nat. Commun. 2014, 5, 4981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, V.; Steinmetz, N.F.; Manchester, M.; Finn, M.G. Labeling Live Cells by Copper-Catalyzed Alkyne−Azide Click Chemistry. Bioconjugate Chem. 2010, 21, 1912–1916. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Chen, X.; Xue, P.; Sun, H.H.Y.; Williams, I.D.; Sharpless, K.B.; Fokin, V.V.; Jia, G. Ruthenium-Catalyzed Cycloaddition of Alkynes and Organic Azides. J. Am. Chem. Soc. 2005, 127, 15998–15999. [Google Scholar] [CrossRef]
- Liu, P.N.; Li, J.; Su, F.H.; Ju, K.D.; Zhang, L.; Shi, C.; Sung, H.H.Y.; Williams, I.D.; Fokin, V.V.; Lin, Z.; et al. Selective Formation of 1,4-Disubstituted Triazoles from Ruthenium-Catalyzed Cycloaddition of Terminal Alkynes and Organic Azides: Scope and Reaction Mechanism. Organometallics 2012, 31, 4904–4915. [Google Scholar] [CrossRef]
- Powers, A.R.; Ghiviriga, I.; Abboud, K.A.; Veige, A.S. Au-iClick mirrors the mechanism of copper catalyzed azide–alkyne cycloaddition (CuAAC). Dalton Trans. 2015, 44, 14747–14752. [Google Scholar] [CrossRef]
- Castillo, T.J.D.; Sarkar, S.; Abboud, K.A.; Veige, A.S. 1,3-Dipolar cycloaddition between a metal–azide (Ph3PAuN3) and a metal–acetylide (Ph3PAuCCPh): An inorganic version of a click reaction. Dalton Trans. 2011, 40, 8140–8144. [Google Scholar] [CrossRef]
- Meng, X.; Xu, X.; Gao, T.; Chen, B. Zn/C-Catalyzed Cycloaddition of Azides and Aryl Alkynes. Eur. J. Org. Chem. 2010, 2010, 5409–5414. [Google Scholar] [CrossRef]
- Smith, C.D.; Greaney, M.F. Zinc Mediated Azide–Alkyne Ligation to 1,5- and 1,4,5-Substituted 1,2,3-Triazoles. Org. Lett. 2013, 15, 4826–4829. [Google Scholar] [CrossRef] [PubMed]
- Rasolofonjatovo, E.; Theeramunkong, S.; Bouriaud, A.; Kolodych, S.; Chaumontet, M.; Taran, F. Iridium-Catalyzed Cycloaddition of Azides and 1-Bromoalkynes at Room Temperature. Org. Lett. 2013, 15, 4698–4701. [Google Scholar] [CrossRef] [PubMed]
- McNulty, J.; Keskar, K.; Vemula, R. The First Well-Defined Silver(I)-Complex-Catalyzed Cycloaddition of Azides onto Terminal Alkynes at Room Temperature. Chem. A Eur. J. 2011, 17, 14727–14730. [Google Scholar] [CrossRef] [PubMed]
- McNulty, J.; Keskar, K. Discovery of a Robust and Efficient Homogeneous Silver(I) Catalyst for the Cycloaddition of Azides onto Terminal Alkynes. Eur. J. Org. Chem. 2012, 2012, 5462–5470. [Google Scholar] [CrossRef]
- Ortega-Arizmendi, A.I.; Aldeco-Pérez, E.; Cuevas-Yañez, E. Alkyne-Azide Cycloaddition Catalyzed by Silver Chloride and “Abnormal” Silver N-Heterocyclic Carbene Complex. Sci. World J. 2013. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, A.M.; Ponti, A.; Molteni, G. Silver(I) oxide nanoparticles as a catalyst in the azide–alkyne cycloaddition. Tetrahedron Lett. 2015, 56, 5727–5730. [Google Scholar] [CrossRef]
- Ali, A.A.; Chetia, M.; Saikia, B.; Saikia, P.J.; Sarma, D. AgN(CN)2/DIPEA/H2O-EG: A highly efficient catalytic system for synthesis of 1,4-disubstituted-1,2,3 triazoles at room temperature. Tetrahedron Lett. 2015, 56, 5892–5895. [Google Scholar] [CrossRef]
- Worrell, B.T.; Malik, J.A.; Fokin, V.V. Direct evidence of a dinuclear copper intermediate in Cu(I)-catalyzed azide-alkyne cycloadditions. Science 2013, 340, 457–460. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Romero, E.A.; Melaimi, M.; Bertrand, G. The Janus Face of the X Ligand in the Copper-Catalyzed Azide–Alkyne Cycloaddition. J. Am. Chem. Soc. 2015, 137, 15696–15698. [Google Scholar] [CrossRef]
- Boren, B.C.; Narayan, S.; Rasmussen, L.K.; Zhang, L.; Zhao, H.; Lin, Z.; Jia, G.; Fokin, V.V. Ruthenium-Catalyzed Azide−Alkyne Cycloaddition: Scope and Mechanism. J. Am. Chem. Soc. 2008, 130, 8923–8930. [Google Scholar] [CrossRef]
- Boz, E.; Tüzün, N.Ş. Reaction mechanism of ruthenium-catalyzed azide–alkyne cycloaddition reaction: A DFT study. J. Organomet. Chem. 2013, 724, 167–176. [Google Scholar] [CrossRef]
- Boz, E.; Tüzün, N.Ş. Ag-catalyzed azide alkyne cycloaddition: A DFT approach. Dalton Trans. 2016, 45, 5752–5764. [Google Scholar] [CrossRef] [PubMed]
- Ben El Ayouchia, H.; Bahsis, L.; Anane, H.; Domingo, L.R.; Stiriba, S.-E. Understanding the mechanism and regioselectivity of the copper(I) catalyzed [3 + 2] cycloaddition reaction between azide and alkyne: A systematic DFT study. RSC Adv. 2018, 8, 7670–7678. [Google Scholar] [CrossRef] [Green Version]
- Domingo, L.R. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef] [PubMed]
- Banerji, B.; Chandrasekhar, K.; Killi, S.K.; Pramanik, S.K.; Uttam, P.; Sen, S.; Maiti, N.C. Silver-catalysed azide–alkyne cycloaddition (AgAAC): Assessing the mechanism by density functional theory calculations. Open Sci. 2016, 3, 160090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultana, J.; Khupse, N.D.; Chakrabarti, S.; Chattopadhyay, P.; Sarma, D. Ag2CO3-catalyzed cycloaddition of organic azides onto terminal alkynes: A green and sustainable protocol accelerated by aqueous micelles of CPyCl. Tetrahedron Lett. 2019, 60, 1117–1121. [Google Scholar] [CrossRef]
- Baxter, C.W.; Higgs, T.C.; Bailey, P.J.; Parsons, S.; McLachlan, F.; McPartlin, M.; Tasker, P.A. Copper(I) Alkynyl Clusters, [Cux+y(hfac)x(C≡CR)y], with Cu10–Cu12 Cores. Chem. A Eur. J. 2006, 12, 6166–6174. [Google Scholar] [CrossRef]
- Özen, C.; Tüzün, N.Ş. The mechanism of copper-catalyzed azide–alkyne cycloaddition reaction: A quantum mechanical investigation. J. Mol. Graph. Model. 2012, 34, 101–107. [Google Scholar] [CrossRef]
- Calvo-Losada, S.; Pino-González, M.S.; Quirante, J.J. Rationalizing the Catalytic Activity of Copper in the Cycloaddition of Azide and Alkynes (CuAAC) with the Topology of ∇2ρ(r) and ∇∇2ρ(r). J. Phys. Chem. B 2015, 119, 1243–1258. [Google Scholar] [CrossRef]
- Cantillo, D.; Ávalos, M.; Babiano, R.; Cintas, P.; Jiménez, J.L.; Palacios, J.C. Assessing the whole range of CuAAC mechanisms by DFT calculations—on the intermediacy of copper acetylides. Org. Biomol. Chem. 2011, 9, 2952–2958. [Google Scholar] [CrossRef]
- Özkılıç, Y.; Tüzün, N.Ş. A DFT Study on the Binuclear CuAAC Reaction: Mechanism in Light of New Experiments. Organometallics 2016, 35, 2589–2599. [Google Scholar] [CrossRef]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide–alkyne cycloaddition (CuAAC) and beyond: New reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Straub, B.F. µ-Acetylide and µ-alkenylidene ligands in “click” triazole syntheses. Chem. Commun. 2007, 0, 3868–3870. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, I.P.; Andemarian, F.; Khairallah, G.N.; Yap, S.W.; Quach, T.; Tsegay, S.; Williams, C.M.; O’Hair, R.A.J.; Donnelly, P.S.; Williams, S.J. Copper(I)-catalyzed cycloaddition of silver acetylides and azides: Incorporation of volatile acetylenes into the triazole core. Org. Biomol. Chem. 2011, 9, 6082–6088. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Aurell, M.J.; Pérez, P. A DFT analysis of the participation of zwitterionic TACs in polar [3+2] cycloaddition reactions. Tetrahedron 2014, 70, 4519–4525. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [Green Version]
- Domingo, L.R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef] [Green Version]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Jaramillo, P.; Domingo, L.R.; Chamorro, E.; Pérez, P. A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J. Mol. Struct. THEOCHEM 2008, 865, 68–72. [Google Scholar] [CrossRef]
- Silvi, B. The synaptic order: A key concept to understand multicenter bonding. J. Mol. Struct. 2002, 614, 3–10. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M. A Molecular Electron Density Theory Study of the Reactivity of Azomethine Imine in [3+2] Cycloaddition Reactions. Molecules 2017, 22, 750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Roy, L.E.; Hay, P.J.; Martin, R.L. Revised Basis Sets for the LANL Effective Core Potentials. J. Chem. Theory Comput. 2008, 4, 1029–1031. [Google Scholar] [CrossRef]
- Bagno, A.; Bonchio, M. Effective core potential DFT calculations of nuclear shielding as a tool for the prediction and assignment of the tungsten chemical shift in mono- and polynuclear complexes. Chem. Phys. Lett. 2000, 317, 123–128. [Google Scholar] [CrossRef]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P. The nucleophilicity N index in organic chemistry. Org. Biomol. Chem. 2011, 9, 7168–7175. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
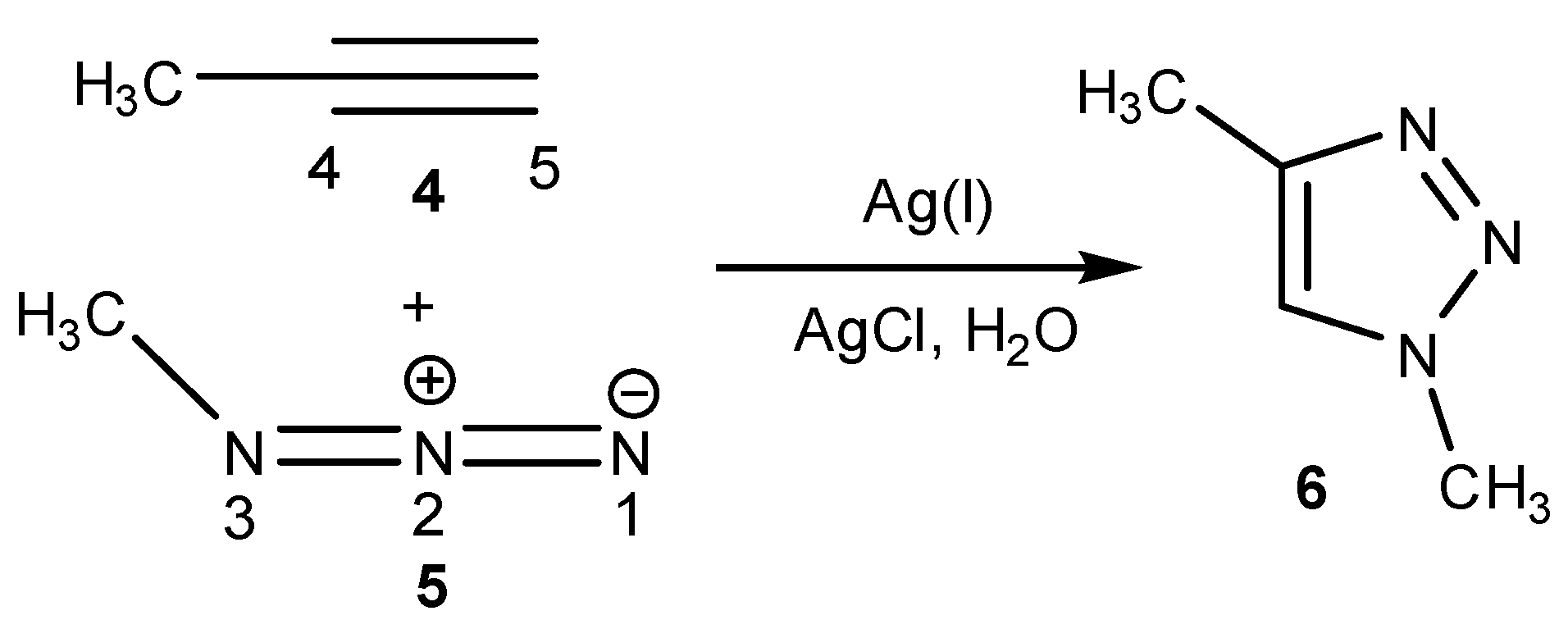{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
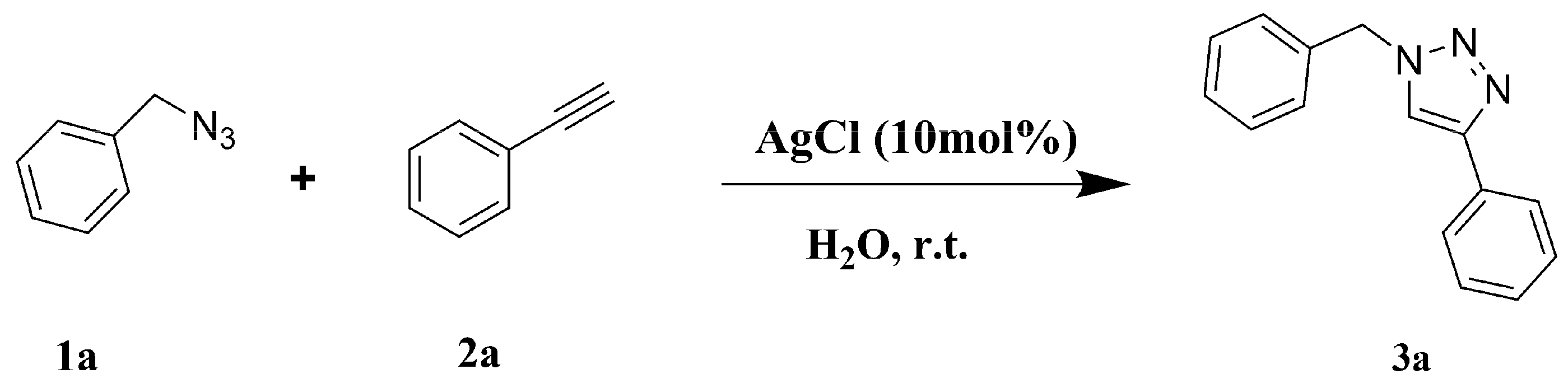{kind=link}
{kind=link}
| Entry | Ag-Cat. | Conditions | Yield (%) | Ref. |
|---|---|---|---|---|
| 1 | AgCl (10 mol%) | H2O/r.t./24 h | 92 | This work |
| 2 | AgCl (5 mol%) | H2O/acetone/r.t./24 h | 64 | [25] |
| 3 | AgCl (20 mol%) | THF/60 °C/TEA/4 h | 87 | [35] |
| 4 | AgN(CN)2 (10 mol%) | H2O/EG/DIPEA/2 h | 98 | [27] |
| 5 | AgCl (0.1 mol%)/L1 | THF/r.t./15 h | 77 | [25] |
| 6 | Ag(OOCCH3) (20 mol%)/L2 | Toluene/r.t./Caprylic acid/48 h | 98 | [23] |
| 7 | Ag(OOCCH3) (2 mol%)/L3 | Toluene/90 °C/Caprylic acid/24 h | 98 | [24] |
| 8 | Ag2CO3 (10 mol%) | H2O/CPyCl/r.t./2 h | 98 | [36] |
| Species | µ | η | ω | N |
|---|---|---|---|---|
| Propyne (4) | −2.69 | 8.72 | 0.41 | 2.06 |
| Methyl azide (5) | −3.85 | 6.19 | 1.20 | 2.17 |
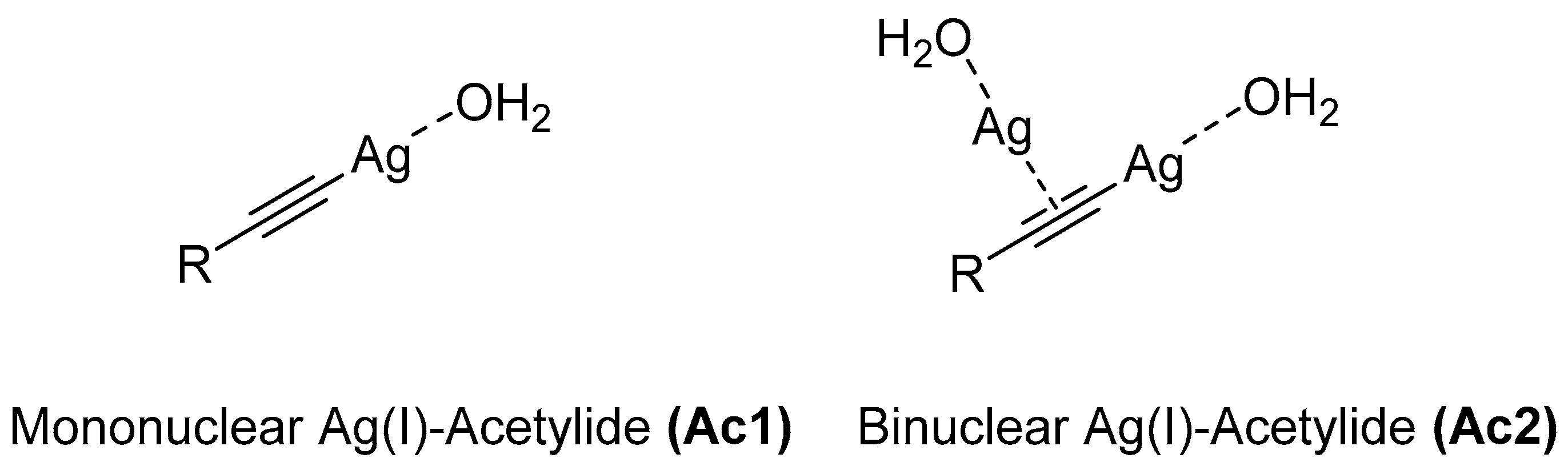
| Mononuclear Ag(I)-acetylide (Ac1) | −3.49 | 4.04 | 1.51 | 3.61 |
| Dinuclear Ag(I)-acetylide (Ac2) | −7.70 | 5.17 | 5.73 | −1.16 |
| Dinuclear Ag(I)-AgCl-acetylide (Ac3) | −4.09 | 3.86 | 2.16 | 3.10 |
| Bond Length | 5 | Ac3 | RC3 | TS3-1 | IC3 | TS3-2 | AT3 |
|---|---|---|---|---|---|---|---|
| d(N1-C4) | - | - | 4.246 | 1.808 | 1.467 | 1.445 | 1.362 |
| d(N3-Ag) | - | - | 2.555 | 2.277 | 2.219 | 2.251 | 3.108 |
| d(Ag-C5) | - | 2.134 | 2.178 | 2.101 | 2.074 | 2.157 | 2.194 |
| d(N3-C5) | - | - | 4.041 | 3.053 | 2.793 | 2.662 | 1.381 |
| GEDT | - | - | −0.05 | 0.17 | 0.38 | 0.42 | 0.31 |
| V(N1,N2) | 1.74 | - | 1.79 | 2.49 | 2.17 | 2.12 | 1.87 |
| V′(N1,N2) | 2.26 | - | 1.79 | - | - | - | - |
| V(N2) | - | - | - | 2.29 | 2.65 | 2.70 | 3.24 |
| V(N2,N3) | 2.50 | - | 2.43 | 1.76 | 1.75 | 1.80 | 1.66 |
| V(C4,C5) | - | 2.48 | 2.41 | 2.10 | 1.75 | 1.74 | 2.93 |
| V′(C4,C5) | - | 2.32 | 2.41 | 2.03 | 1.83 | 1.71 | - |
| V(N1) | 3.94 | - | 3.89 | 4.05 | 3.36 | 3.35 | 3.07 |
| V(N3) | 3.53 | - | 3.59 | 1.81 | 1.70 | 3.50 | 0.77 |
| V′(N3) | - | - | - | 1.82 | 1.85 | - | 0.75 |
| V(C4) | - | - | - | 0.26 | - | - | - |
| V(C5) | - | 2.91 | 2.91 | 3.16 | 3.24 | 3.19 | 2.68 |
| V(N1,C4) | - | - | - | - | 1.64 | 1.75 | 2.29 |
| V(N3,C5) | - | - | - | - | - | - | 2.08 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben El Ayouchia, H.; Bahsis, L.; Fichtali, I.; Domingo, L.R.; Ríos-Gutiérrez, M.; Julve, M.; Stiriba, S.-E. Deciphering the Mechanism of Silver Catalysis of “Click” Chemistry in Water by Combining Experimental and MEDT Studies. Catalysts 2020, 10, 956. https://doi.org/10.3390/catal10090956
Ben El Ayouchia H, Bahsis L, Fichtali I, Domingo LR, Ríos-Gutiérrez M, Julve M, Stiriba S-E. Deciphering the Mechanism of Silver Catalysis of “Click” Chemistry in Water by Combining Experimental and MEDT Studies. Catalysts. 2020; 10(9):956. https://doi.org/10.3390/catal10090956
Chicago/Turabian StyleBen El Ayouchia, Hicham, Lahoucine Bahsis, Ismail Fichtali, Luis R. Domingo, Mar Ríos-Gutiérrez, Miguel Julve, and Salah-Eddine Stiriba. 2020. "Deciphering the Mechanism of Silver Catalysis of “Click” Chemistry in Water by Combining Experimental and MEDT Studies" Catalysts 10, no. 9: 956. https://doi.org/10.3390/catal10090956
APA StyleBen El Ayouchia, H., Bahsis, L., Fichtali, I., Domingo, L. R., Ríos-Gutiérrez, M., Julve, M., & Stiriba, S.-E. (2020). Deciphering the Mechanism of Silver Catalysis of “Click” Chemistry in Water by Combining Experimental and MEDT Studies. Catalysts, 10(9), 956. https://doi.org/10.3390/catal10090956