Description of Transport Tunnel in Haloalkane Dehalogenase Variant LinB D147C+L177C from Sphingobium japonicum

 and
and 
Abstract
:1. Introduction
2. Results and Discussion
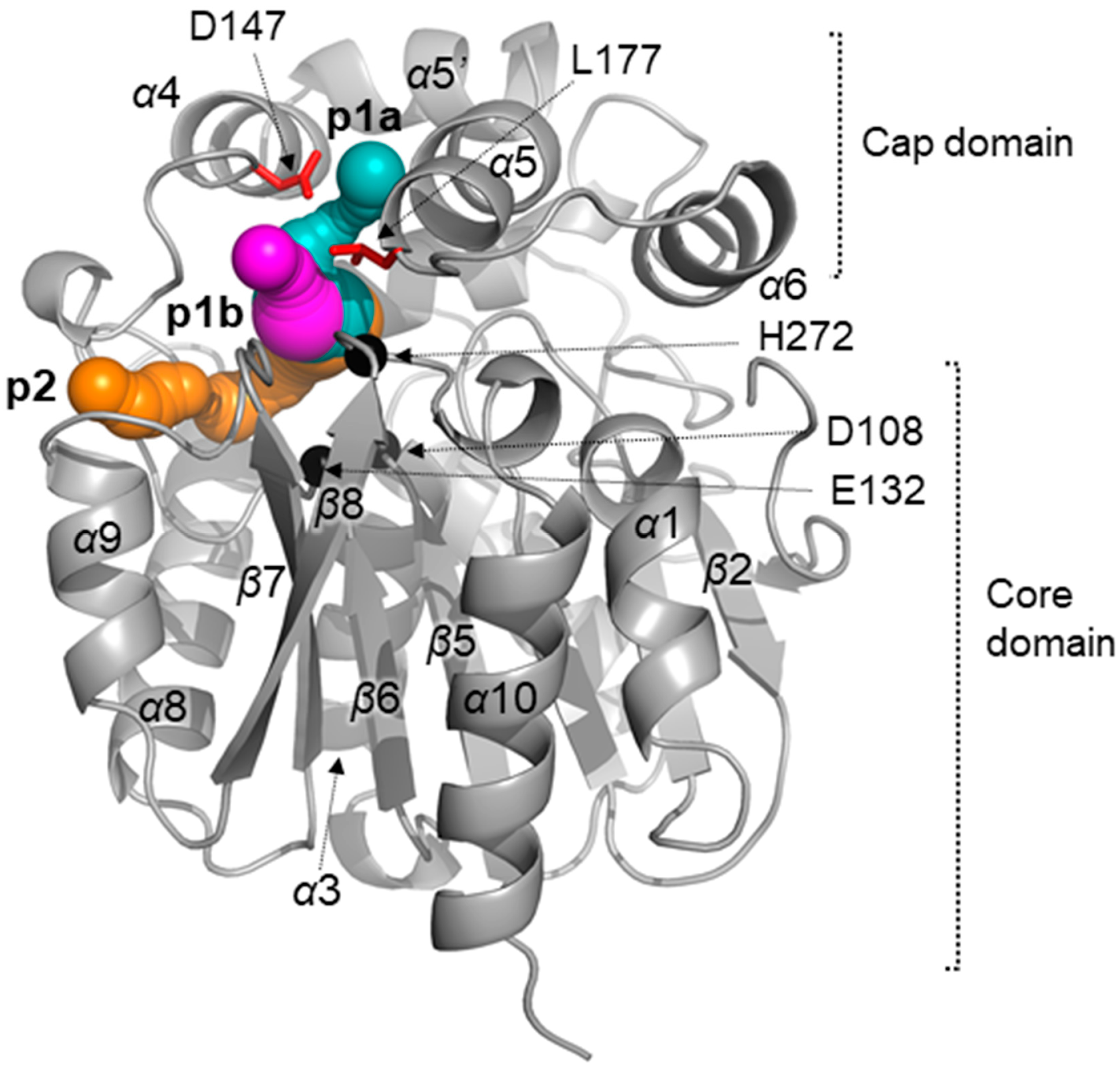
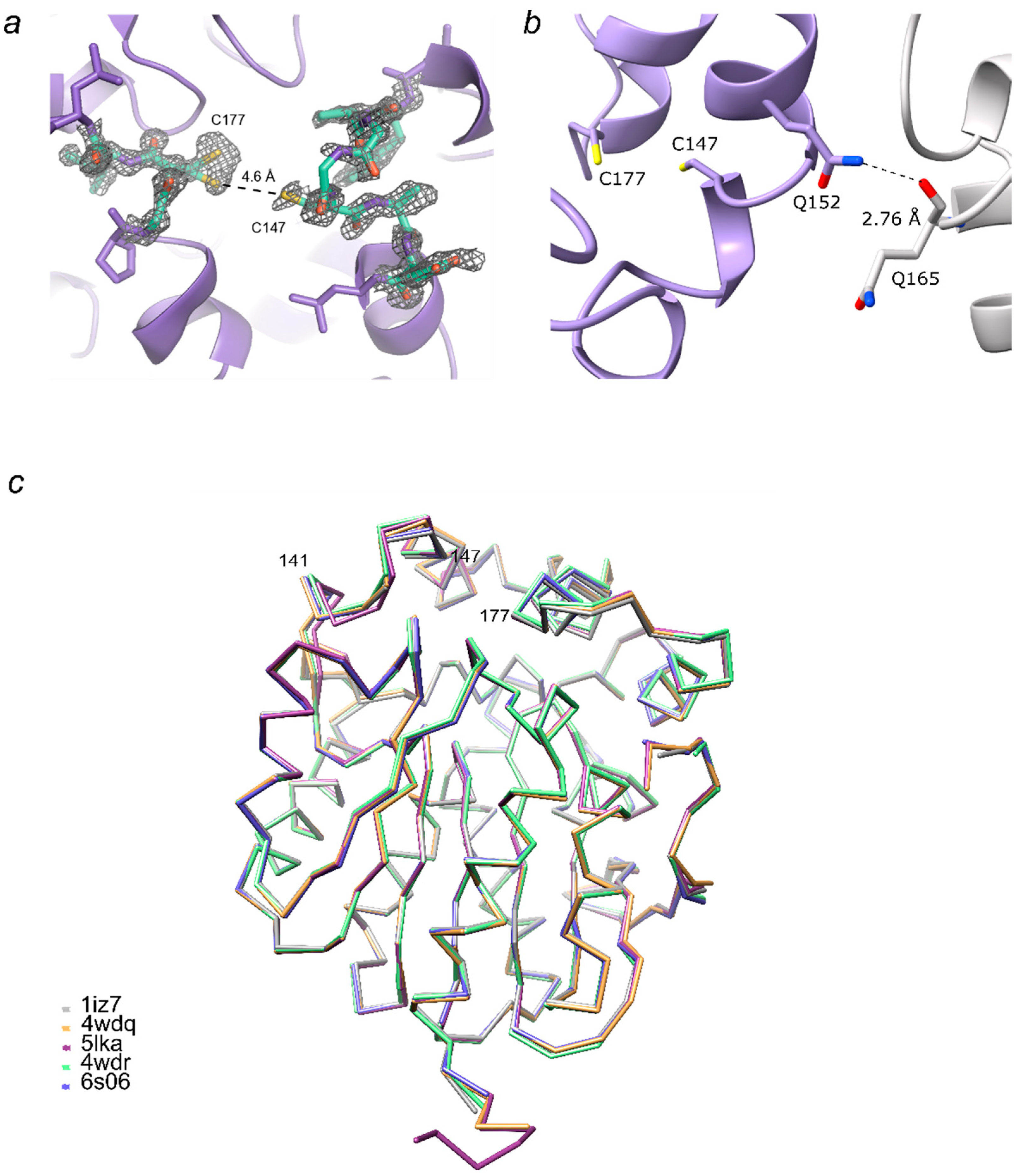
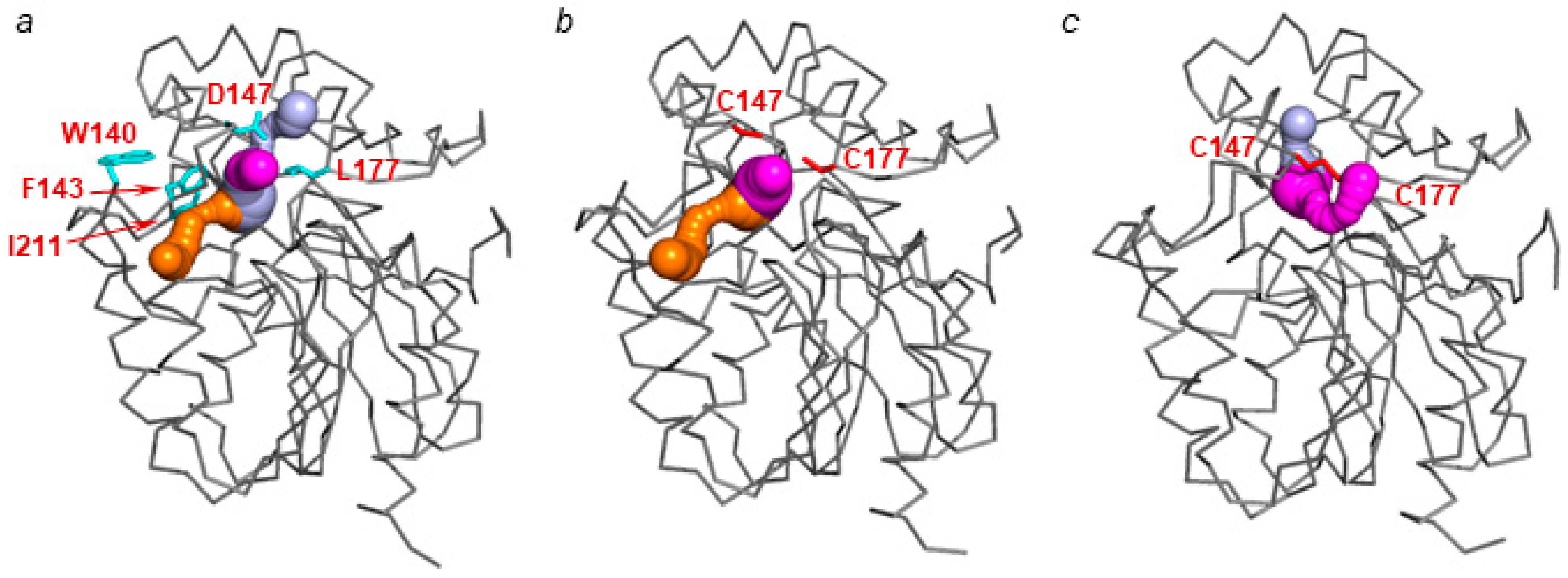
2.1. Crystal Structure of LinB D147C+L177C

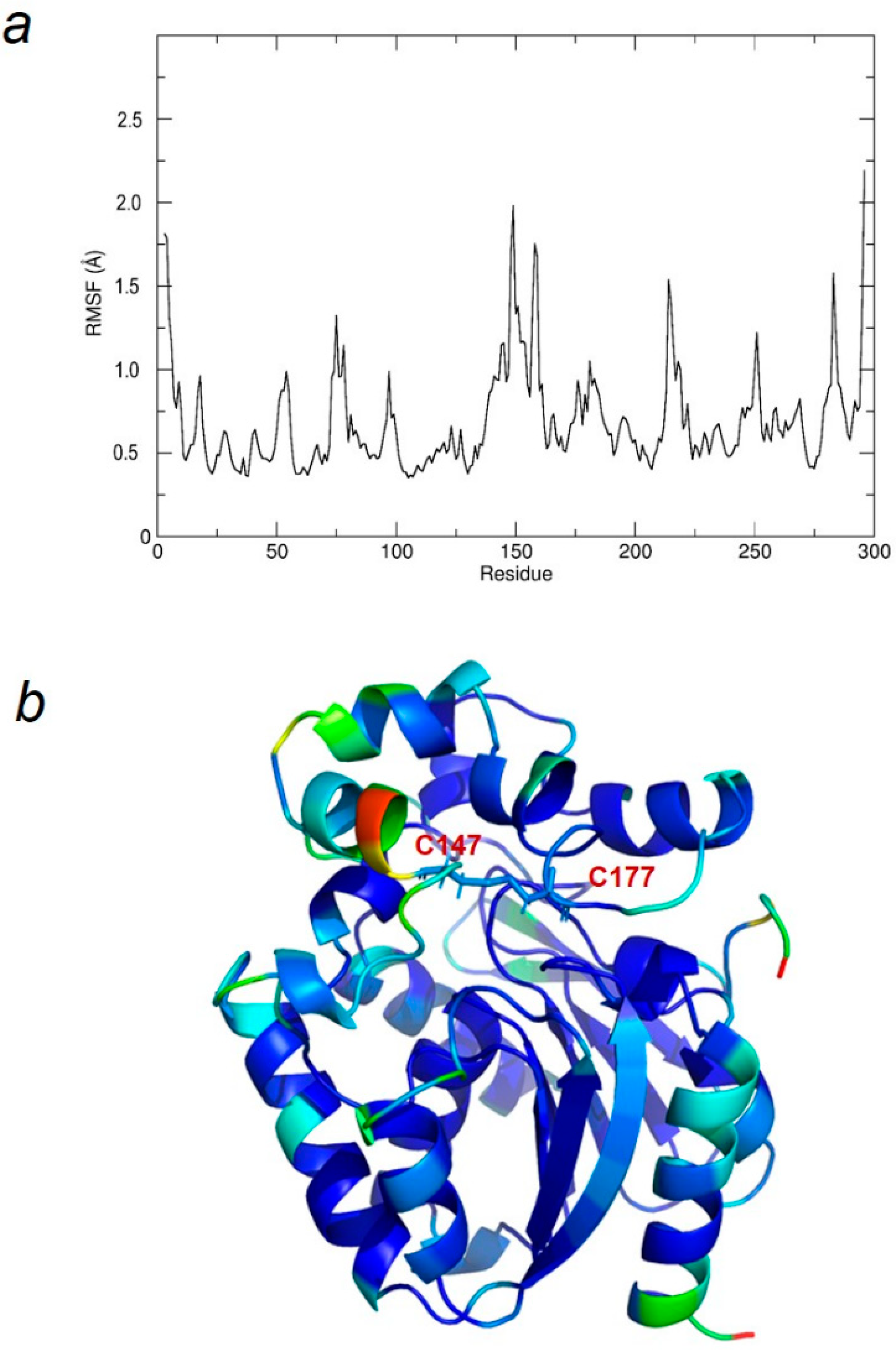
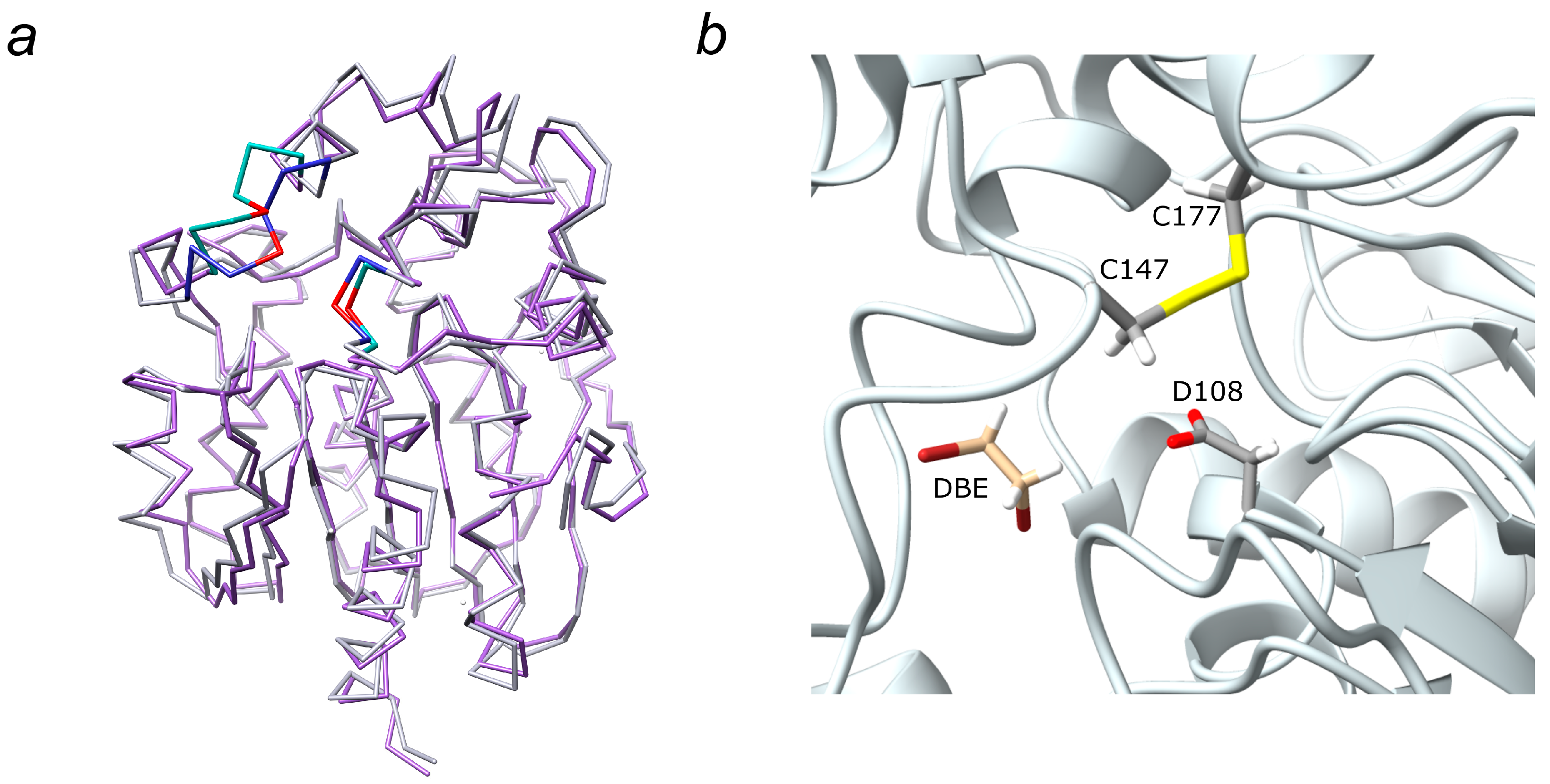
2.2. Molecular Dynamics Simulations and Molecular Docking Studies with LinB D147C+L177C
2.3. Analysis of access Tunnels in LinB Variants
3. Materials and Methods
3.1. Macromolecule Production
3.2. Crystallization
3.3. Data collection and Processing, Structure Solution and Refinement
3.4. Molecular Dynamics Simulations and Docking Studies
3.5. Analysis of Access Tunnel
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Janssen, D.B.; Dinkla, I.J.T.; Poelarends, G.J.; Terpstra, P. Bacterial degradation of xenobiotic compounds: Evolution and distribution of novel enzyme activities: Evolution and distribution of novel enzyme activities. Environ. Microbiol. 2005, 7, 1868–1882. [Google Scholar] [CrossRef] [Green Version]
- Hynkova, K.; Nagata, Y.; Takagi, M.; Damborsky, J. Identification of the catalytic triad in the haloalkane dehalogenase from Sphingomonas paucimobilis UT26. FEBS Lett. 1999, 446, 14747–14753. [Google Scholar] [CrossRef] [Green Version]
- Nagata, Y.; Miyauchi, K.; Damborsky, J.; Manova, K.; Ansorgova, A.; Takagi, M. Degradation of β-Hexachlorocyclohexane by Haloalkane Dehalogenase LinB from Sphingomonas paucimobilis UT26. Appl. Environ. Microbiol. 1997, 63, 3707–3710. [Google Scholar] [CrossRef] [Green Version]
- Prokop, Z.; Monincova, M.; Chaloupkova, R.; Klvana, M.; Nagata, Y.; Janssen, D.B.; Damborsky, J. Structure–Function Relationships and Engineering of Haloalkane Dehalogenases. J. Biol. Chem. 2003, 278, 45094–45100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marek, J.; Vevodova, J.; Kuta Smatanova, I.; Nagata, Y.; Svensson, L.A.; Newman, J.; Takagi, M.; Damborsky, J. Protein engineering of haloalkane dehalogenase LinB: Reconstruction of active site and modification of entrance tunnel. Biochemistry 2000, 39, 177–181. [Google Scholar]
- Chovancova, E.; Kosinski, J.; Bujnicki, M.J.; Damborsky, J. Phylogenetic Analysis of Haloalkane Dehalogenases. Proteins 2007, 67, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Bohac, M.; Nagata, Y.; Prokop, Z.; Prokop, M.; Monincova, M.; Koca, J.; Tsuda, M.; Damborsky, J. Halide-stabilizing residues of haloalkane dehalogenases studied by quantum mechanic calculations and site-directed mutagenesis. Biochemistry 2002, 41, 14272–14280. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Nariya, T.; Ohtomo, R.; Fukuda, M.; Yano, K.; Takagi, M. Cloning and sequencing of a dehalogenase gene encoding an enzyme with hydrolase activity involved in the degradation of gamma-hexachlorocyclohexane in Pseudomonas paucimobilis. J. Bacteriol. 1993, 175, 6403–6410. [Google Scholar] [CrossRef] [Green Version]
- Chovancova, E.; Pavelka, A.; Benes, P.; Strnad, O.; Brezovsky, J.; Kozlikova, B.; Gora, A.; Sustr, V.; Klvana, M.; Medek, P.; et al. CAVER 3.0: A Tool for the Analysis of Transport Pathways in Dynamic Protein Structures. PLoS Comput. Biol. 2012, 8, e1002708. [Google Scholar] [CrossRef] [Green Version]
- Biedermannova, L.; Prokop, Z.; Gora, A.; Chovancova, E.; Kovacs, M.; Damborsky, J.; Wade, R.C. A Single Mutation in a Tunnel to the Active Site Changes the Mechanism and Kinetics of Product Release in Haloalkane Dehalogenase LinB. J. Biol. Chem. 2012, 287, 29062–29074. [Google Scholar] [CrossRef] [Green Version]
- Oakley, A.J.; Klvana, M.; Otyepka, M.; Nagata, Y.; Wilce, M.C.; Damborsky, J. Crystal structure of haloalkane dehalogenase LinB from Sphingomonas paucimobilis UT26 at 0.95 A resolution: Dynamics of catalytic residues. Biochemistry 2004, 43, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Brezovsky, J.; Babkova, P.; Degtjarik, O.; Fortova, A.; Gora, A.; Iermak, I.; Rezacova, P.; Dvorak, P.; Kuta Smatanova, I.; Prokop, Z.; et al. Engineering a de Novo Transport Tunnel. ACS Catal. 2016, 6, 7597–7610. [Google Scholar] [CrossRef]
- Pavlova, M.; Klvana, M.; Prokop, Z.; Chaloupkova, R.; Banas, P.; Otyepka, M.; Wade, R.C.; Tsuda, M.; Nagata, Y.; Damborsky, J. Redesigning dehalogenase access tunnels as a strategy for degrading an anthropogenic substrate. Nat. Chem. Biol. 2009, 5, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Prokop, Z.; Gora, A.; Brezovsky, J.; Chaloupkova, R.; Stepankova, V.; Damborsky, J. Engineering of protein tunnels: Keyhole-lock-key model for catalysis by the enzymes with buried active sites. Prot. Eng. Handb. 2012, 3, 421–464. [Google Scholar]
- Zhou, G.; Somasundaram, T.; Blanc, E.; Parthasarathy, G.; Ellington, W.R.; Chapman, M.S. Transition state structure of arginine kinase: Implications for catalysis of bimolecular reactions. Proc. Natl. Acad. Sci. USA 1998, 95, 8449–8454. [Google Scholar] [CrossRef] [Green Version]
- Liebgott, P.-P.; Leroux, F.; Burlat, B.; Dementin, S.; Baffert, C.; Lautier, T.; Fourmond, V.; Ceccaldi, P.; Cavazza, C.; Meynial-Salles, I.; et al. Relating diffusion along the substrate tunnel and oxygen sensitivity in hydrogenase. Nat. Chem. Biol. 2010, 6, 63–70. [Google Scholar] [CrossRef]
- Chaloupkova, R.; Sykorova, J.; Prokop, Z.; Jesenska, A.; Monincova, M.; Pavlova, M.; Tsuda, M.; Nagata, Y.; Damborsky, J. Modification of activity and specificity of haloalkane dehalogenase from Sphingomonas paucimobilis UT26 by engineering of its entrance tunnel. J. Biol. Chem. 2003, 278, 52622–52628. [Google Scholar] [CrossRef] [Green Version]
- Minincova, M.; Prokop, Z.; Vevodova, J.; Nagata, Y.; Damborsky, J. Weak Activity of Haloalkane Dehalogenase Linb with 1,2,3-Trichloropropane Revealed by X-Ray Crystallography and Microcalorimetry. Appl. Environ. Microbiol. 2007, 73, 2005–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eldar, A.; Rozenberg, H.; Diskin-Posner, Y.; Rohs, R.; Shakked, Z. Structural studies of p53 inactivation by DNA-contact mutations and its rescue by suppressor mutations via alternative protein-DNA interactions. Nucleic Acids Res. 2013, 41, 8748–8759. [Google Scholar] [CrossRef] [Green Version]
- Okai, M.; Ohtsuka, J.; Imai, L.F.; Mase, T.; Moriuchi, R.; Tsuda, M.; Nagata, K.; Nagata, Y.; Tanokura, M. Crystal structure and site-directed mutagenesis analyses of haloalkane dehalogenase linB from sphingobium sp. Strain MI1205. J. Bacteriol. 2013, 195, 2642–2651. [Google Scholar] [CrossRef] [Green Version]
- Prudnikova, T.; Mozga, T.; Rezacova, P.; Chaloupkova, R.; Sato, Y.; Nagata, Y.; Brynda, J.; Kuty, M.; Damborsky, J.; Kuta Smatanova, I. Crystallization and Preliminary X-ray Analysis of a Novel Haloalkane Dehalogenase DbeA from Bradyrhizobium elkani USDA94. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Degtjarik, O.; Chaloupkova, R.; Rezacova, P.; Kuty, M.; Damborsky, J.; Kuta Smatanova, I. Differences in crystallization of two LinB variants from Sphingobium japonicum UT26. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2013, 69, 284–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 13, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. PyMol. The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. 2002. Available online: http://www.pymol.org (accessed on 14 September 2020).
- Streltsov, V.A.; Prokop, Z.; Damborsky, J.; Nagata, Y.; Oakley, A.; Wilce, M.C.J. Haloalkane dehalogenase LinB from Sphingomonas paucimobilis UT26: X-ray crystallographic studies of dehalogenation of brominated substrates. Biochemistry 2003, 42, 10104–10112. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Detection of Protein Assemblies in Crystals. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2256–2268. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Negri, A.; Marco, E.; Damborsky, J.; Gago, F. Stepwise Dissection and Visualization of the Catalytic Mechanism of Haloalkane Dehalogenase LinB using Molecular Dynamics Simulations and Computer Graphics. J. Mol. Graph. Model. 2007, 26, 643–651. [Google Scholar] [CrossRef]
- Kokkonen, P.; Bednar, D.; Pinto, G.; Prokop, Z.; Damborsky, J. Engineering Enzyme Access Tunnels. Biotechnol. Adv. 2019, 37, 107386. [Google Scholar] [CrossRef]
- Petrek, M.; Otyepka, M.; Banas, P.; Kosinova, P.; Koca, J.; Damborsky, J. CAVER: A New Tool to Explore Routes from Protein Clefts, Pockets and Cavities. BMC Bioinform. 2006, 7, 316. [Google Scholar] [CrossRef] [Green Version]
- Gabaldinho, J.; Beteva, A.; Guijarro, M.; Rey-Bakaikoa, S.D.; Bowler, M.W.; Brockhauser, S.; Flot, D.; Gordon, D.F.; Hall, D.R.; Lavault, B.; et al. MxCuBE: A synchrotron beamline control environment customized for macromolecular crystallography experiments. J. Synchrotron Radiat. 2010, 17, 700–707. [Google Scholar] [CrossRef]
- Battye, T.G.G.; Kontogiannis, L.; Jonson, O.; Powell, H.R.; Leslie, A.G.W. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, A.G. The integration of macromolecular diffraction data. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krug, M.; Weiss, M.S.; Heinemann, U.; Mueller, U. XDSAPP2.0. J. Appl. Crystallogr. 2010, 45, 568–572. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. MOLREP: An Automated Program for Molecular Replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Sheldrick, G.M.; Schneider, T.R. SHELXL: High-resolution refinement. Meth. Enzymol. 1997, 277, 319–343. [Google Scholar]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. Mol. Model. Annu. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Trott, O.; Olson, J.A. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, 1202–1213. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | LinB WT | LinB D147C+L177C | LinBMD D147C+L177C | |
|---|---|---|---|---|
| PDB ID | 2bfn | 6s06 | n/a | |
| Bottleneck radius, Å | p1a | 1.1 | - a | 1.2 |
| p1b | 1.6 | 1.8 | 1.0 | |
| p2 | 1.0 | 1.1 | - a | |
| Tunnel length, Å | p1a | 13.4 | - a | 8.4 |
| p1b | 8.6 | 6.7 | 17.5 | |
| p2 | 13.8 | 15.0 | - a |
| Diffraction source | BL14.1, BESSY II |
| Wavelength (Å) | 0.9184 |
| Temperature (K) | 100 |
| Detector | PILATUS 6M |
| Crystal-detector distance (mm) | 210.545 |
| Rotation range per image (°) | 0.1 |
| Total rotation range (°) | 200 |
| Exposure time per image (s) | 0.5 |
| Space group | P212121 |
| a, b, c (Å) | 44.72, 68.71, 80.61 |
| α, β, γ (°) | 90.0 90.0 90.0 |
| Mosaicity (°) | 0.142 |
| Resolution range (Å) | 50.00–1.15 (1.22–1.15) |
| Total No. of reflections | 550,609 |
| No. of unique reflections | 88,614 |
| Completeness (%) | 99.l (98.4) |
| CC1/2 | 99.9 (86.8) |
| 〈I/σ(I)〉 | 14.93 (2.60) |
| Rr.i.m. | 6.9 (67.0) |
| Overall B factor from Wilson plot (Å2) | 18.18 |
| Refinement program | SHELXL |
| No. of reflections, working set | 84,016 |
| No. of reflections, test set | 4431 |
| Final Rcryst | 15.29 |
| Final Rfree | 20.41 |
| R.m.s. deviations | |
| Bonds (Å) | 0.0128 |
| Angles (°) | 0.0205 a |
| Average B factors (Å2) | 15.00 |
| No. of protein atoms | 2272 |
| No. of chloride ions | 1 |
| No. of magnesium ions | 3 |
| No. of water molecules | 233 |
| Clashscore, all atom (%) | 98 |
| Ramachandran plot | |
| Most favoured (%) | 97 |
| Allowed (%) | 3 |
| PDB ID | 6s06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iermak, I.; Degtjarik, O.; Havlickova, P.; Kuty, M.; Chaloupkova, R.; Damborsky, J.; Prudnikova, T.; Kuta Smatanova, I. Description of Transport Tunnel in Haloalkane Dehalogenase Variant LinB D147C+L177C from Sphingobium japonicum. Catalysts 2021, 11, 5. https://doi.org/10.3390/catal11010005
Iermak I, Degtjarik O, Havlickova P, Kuty M, Chaloupkova R, Damborsky J, Prudnikova T, Kuta Smatanova I. Description of Transport Tunnel in Haloalkane Dehalogenase Variant LinB D147C+L177C from Sphingobium japonicum. Catalysts. 2021; 11(1):5. https://doi.org/10.3390/catal11010005
Chicago/Turabian StyleIermak, Iuliia, Oksana Degtjarik, Petra Havlickova, Michal Kuty, Radka Chaloupkova, Jiri Damborsky, Tatyana Prudnikova, and Ivana Kuta Smatanova. 2021. "Description of Transport Tunnel in Haloalkane Dehalogenase Variant LinB D147C+L177C from Sphingobium japonicum" Catalysts 11, no. 1: 5. https://doi.org/10.3390/catal11010005
APA StyleIermak, I., Degtjarik, O., Havlickova, P., Kuty, M., Chaloupkova, R., Damborsky, J., Prudnikova, T., & Kuta Smatanova, I. (2021). Description of Transport Tunnel in Haloalkane Dehalogenase Variant LinB D147C+L177C from Sphingobium japonicum. Catalysts, 11(1), 5. https://doi.org/10.3390/catal11010005