1. Introduction
Metal-organic frameworks (MOFs) are a class of crystalline porous materials where rigid bi- or multipodal organic linkers interact with metal ions or metal oxo clusters by Coulombic attraction and/or coordinative metal ligand bonds [
1,
2,
3]. The directionality of the coordinative bonds around the metal ions, typically octahedral, tetrahedral or square planar, combined with the rigidity and directionality of the organic molecules acting as a ligand, define a crystal structure that exhibits high porosity and large surface area [
4]. The dimensions of the pores depend on the geometry of the internal voids, the molecular dimension of the organic linkers, and the coordination geometry [
5].
Frequently, one of the coordination positions around the metal ions is not compromised with the organic linker and it is often occupied by exchangeable solvent molecules. There is also the possibility of structural defects consisting in metal nodes not totally coordinated. In these cases, the metal ion can interact by ligand exchange with substrates and reagents and in this way they can act as Lewis acid centers in catalytic reactions [
6,
7,
8,
9]. In fact, MOFs had been intensively studied as solid catalysts for a large variety of chemical transformations [
10,
11].
Transition metals, and in particular cobalt, which is the focus of the present review, are well-known oxidation catalysts for a variety of organic functional groups and organic molecules [
12]. Homogeneous cobalt-based catalysts can exhibit a high intrinsic activity due to easy accessibility of Co sites by substrate. However, these homogeneous systems may have serious limitations, particularly fast deactivation by oligomerization of the metal, difficult isolation of these catalysts from the reaction medium in identical state to the fresh catalyst, and complicate continuous flow operation. One way to circumvent these issues, while still maintaining high catalytic activity, is to graft metal ions into a porous solid possessing high surface area [
13,
14]. Among them, zeolites and mesoporous silicates were the preferred options to incorporate active transitions metals due to the high stability of these materials [
15,
16,
17]. While there have been successful cases of transition metals grafted on the framework of silicates and mesoporous silicates, they still have some drawbacks, particularly the low volumetric space and surface area. Besides, one of the unique features of MOFs compared to silicates is their high mass and volumetric loading of Co ions, making them more convenient from an engineering point of view, since it may diminish reactor costs that are related to the volume of the reactor [
2]. In addition, these transition metals are highly accessible depending on the pore dimensions and geometry of the MOF [
2]. Furthermore, although mesoporous oxides and MOFs can have accessible pores, MOFs have some additional features like tailoring surface area [
18], as a suitable host for high drug loading [
19], possibility to install the desired functional groups in the linker moiety [
20], entrapment of a wide range of guests (metal nanoparticles, polyoxometalates, tungstic acid) [
21], and the development of photoactive materials [
22], among others [
23].
MOFs have been extensively studied as heterogeneous catalysts for the oxidation of a series of benzylic hydrocarbons, cycloalkanes, alcohols, amines, sulfides, and other substrates both using oxidizing reagents and under aerobic conditions [
24,
25,
26]. The unsaturated metal sites that are not involved in the coordination with the organic linkers often play often the role of catalytic centers binding the oxidant and activating it, thus promoting the oxidation of various functional groups. On the other hand, homogeneous transition metal complexes installed over satellite positions of the organic linkers can also behave as Lewis acid sites to activate oxidants [
25,
26]. In addition, mixed-metal MOFs have also been employed as heterogeneous solids for the promotion of oxidation reactions [
27]. In another strategy, MOFs can be used as passive hosts for the encapsulation of metal nanoparticles, offering the advantage of controlling their size and environment. These encapsulated metal nanoparticles can then be active sites promoting oxidation reactions [
28,
29,
30].
Although MOFs have been employed in many ways as heterogeneous solid catalysts for the oxidation of a broad range of substrates, one of the important issues to be addressed is the stability of the MOF during the oxidation reaction. This issue is often addressed by comparing the structural integrity of the reused solid in successive cycles with that of the fresh solid by powder X-ray diffraction (XRD) and determining whether or not changes in the diffraction peaks have occurred. On the other hand, the metal content in the fresh solid catalyst is also compared with that of the reused solid and the analysis of the metal content in the MOF should always be complemented with the measurement of the metal content in the liquid phase by inductively coupled plasma (ICP) analysis. Further, elemental analysis and spectroscopic characterization of the MOF catalysts before and after catalytic reaction may not provide sufficient evidence on metal leaching. One of the issues in heterogeneous catalysis is the promotion of reaction by leached metals, even in trace amounts [
31,
32,
33,
34]. Hence, after determining the leached metal species from the solid by the above techniques, additional control experiments may be performed by purposely adding the leached metal content to the reaction mixture to precisely rule the contribution of these added metals. These leaching tests are especially important in the case of oxidation reactions, in which very minor concentrations of metals in solution can promote the formation of a large yield of products due to the operation of chain reaction mechanisms with long propagation steps. Surface and particle morphologies are often surveyed by measuring scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images before and after the oxidation reaction. The oxidation state of the metals inserted within the framework can be confirmed by analysis and deconvolution of the X-ray photoelectron spectra (XPS). The thermal stability of MOF before and after catalysis may be derived from thermogravimetry and differential thermal analysis.
Considering the available information on the use of MOFs as heterogeneous catalysts and focusing especially on oxidation reactions, cobalt compounds are known to exhibit a general catalytic activity in oxidation reactions [
35]. Therefore, the catalytic activity of Co–MOFs is presumed from the general activity of Co ions and complexes in homogeneous phase. Typical oxidizing reagents are hydroperoxides and organic hydroperoxides, but also other oxidizing species containing halogens, oxo acid compounds, and molecular oxygen, among others [
36].
The ability of cobalt compounds to activate molecular oxygen to give metal-peroxyl intermediates that subsequently afford the corresponding oxidation products is well established. Considering the intrinsic activity of homogeneous cobalt salts and complexes in promoting various oxidation reactions [
37,
38], an obvious development is the design of cobalt-based MOFs as oxidation solid catalysts in any of the above-commented active centers.
The objective of the present review is to illustrate the catalytic activity of cobalt MOFs for oxidation of organic compounds. The review is organized according to the organic substrates undergoing oxidations. Whenever possible, a comparison of the catalytic performance of the cobalt MOFs with those of homogeneous counterparts, other analogous MOFs of different metals, or even benchmark catalysts is provided. Due to the importance of cobalt MOFs and their related solids in electrocatalysis [
39,
40], a section is also focused on the recent developments of Co–MOFs as electrocatalysts for H
2O electrolysis in the oxygen evolution reaction. The last section of the review summarizes the current state of the art of cobalt MOFs as heterogeneous catalysts and provides our view of future developments in the field.
Table 1 provides a list of various Co–MOFs as heterogeneous catalysts for the oxidation of a wide range of functional groups, including water oxidation. The main purpose of this table is to compile reaction type and evidences for catalyst stability given in the original references.
2. Alcohol Oxidation
Alcohol oxidation to its corresponding carbonyl compounds is one of the challenging organic reactions due to the widespread applications of carbonyl compounds as precursors for the manufacturing of fine chemicals, drug molecules. Authors are encouraged to refer to a recent review on the use of MOFs for alcohol oxidation to carbonyl compounds [
23,
58]. This review is restricted exclusively to the use of Co–MOFs for alcohol oxidation. Selective oxidation of alcohols using MOFs as catalysts is one of the important tasks since the by-product, carboxylic acid, can easily deactivate catalytic sites through a strong coordination. Hence, the following papers are grouped to demonstrate the contribution of Co–MOFs in the oxidation of alcohols and special attention is given to the catalyst stability.
A new Co–MOF was reported by reacting cobalt acetate with 4,4′-[benzene-1,4-diylbis (methylylidenenitrilo)] dibenzoic acid (H
2bdda) under ultrasound irradiation in water medium [
41]. The catalytic activity of this solid was tested in the oxidation of primary and secondary alcohols as well as in Henry reaction. A wide range of primary benzylic alcohols consisting of electron donating and withdrawing groups was facilely oxidized to their respective aldehydes in 81–95% yields using tert-butyl hydroperoxide (TBHP) as oxidant at 65 °C. Similarly, 1-phenylethanol, diphenylcarbinol were oxidized to acetophenone, benzophenone in 82 and 75% yields, respectively, in the presence of Co–MOF under identical conditions. On the other hand, series of aldehydes were reacted with nitromethane to produce a wide array of nitro-aldol products ranging in their yields of 62–88% at 70 °C in water. The heterogeneity of the reaction was proven by the hot-filtration test. The catalyst stability was demonstrated by reusing the solid in five runs with no noticeable change in the yields in both reactions. The authors concluded that there were no significant changes in the FT-IR spectra between the fresh and the solid recovered after five runs. However, additional characterization data are essential to understand the structural and textural properties of Co–MOF during the oxidation reaction.
Recently, two isostructural MOFs with Co(II) and Zn(II) metal ions were synthesized by employing an amide appended organic ligand with three-dimensional structures (
Scheme 1). The catalytic activity of these MOFs was studied in the oxidation of benzyl alcohol and C–C bond forming reactions (Knoevenagel and Henry) under environmentally benign conditions [
42]. The catalytic performance of Co–MOF was remarkably higher in the oxidation of benzyl alcohol to benzaldehyde using microwave under solvent free conditions with TBHP as an oxidant compared to Zn–MOF. For instance, Co–MOF exhibited 89% yield of benzaldehyde with the reported turnover frequency (TOF) value of 148 h
−1 at 90 °C using 15 W microwave irradiation after 1.5 h in the presence of TBHP as an oxidant. In contrast, Zn–MOF afforded 27% yield with TOF value of 45 h
−1 under identical conditions. Interestingly, CoCl
2.6H
2O and Zn(NO
3)
2.6H
2O showed TOF values of 22 and 8 h
−1 under similar conditions. These catalytic data clearly indicate that the Co–MOF provides favourable conditions to achieve high yield of benzaldehyde compared to other heterogeneous and homogeneous catalysts under identical conditions. The superior activity of Co–MOF compared to Zn–MOFs is due to the redox behaviour of Co(II) sites. Further, the non-redox behaviour of Zn–MOF was proven by density-functional theory (DFT) studies for the decomposition of TBHP. In contrast, Zn–MOF was more active than Co–MOF in Henry and Knoevenagel reactions due to the stronger Lewis acid character of Zn(II) ions. The Henry reaction between benzaldehyde and nitroethane using Zn–MOF showed 99% yield in water medium at room temperature after 72 h. In contrary, the activity of Co–MOF provided 77% yield under identical conditions. The activity of homogeneous salts of CoCl
2.6H
2O and Zn(NO
3)
2.6H
2O was 10 and 23% yields under similar experimental conditions. Furthermore, the better catalytic data were obtained in water medium for Henry reaction using Zn–MOF while Knoevenagel condensation reaction is promoted by ultrasonication. The activity of Zn–MOF was mostly retained up to 4th cycle by providing similar yields, while it decreased in the 5th and 6th cycles (
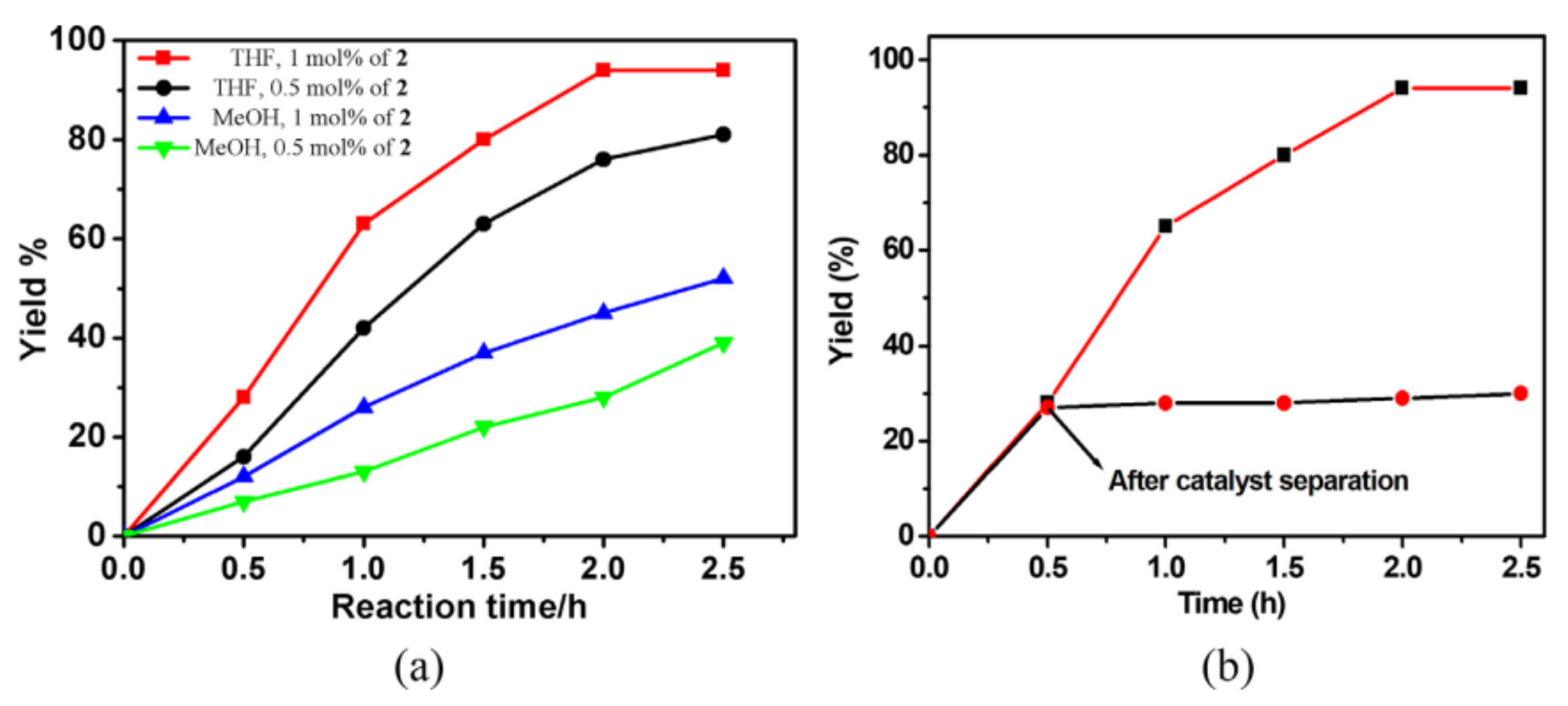
Figure 1). Among the various conditions optimized, Zn–MOFs exhibited better activity in the Knoevenagel condensation reaction between benzaldehyde and malononitrile in THF at room temperature under ultrasound waves after 2 h (
Figure 2a). The heterogeneity of the reaction was shown by the hot-filtration test, which indicates that the removal of the solid after 0.5 h at around 30% conversion significantly inhibits the formation of the product (
Figure 2b). The TOF values of Co–MOF and Zn–MOFs were 39 and 47 h
−1 under identical conditions. It is interesting to note that the number of precedents employing ultrasounds for catalytic reactions with MOFs is limited and this work nicely illustrates the greener protocol for these reactions using MOFs as heterogeneous catalysts. Some of the salient features of these catalysts are the use of mild reaction conditions like room temperature, alternative non-conventional energy sources, water as solvent medium, facile separation of catalyst and reusability.
One of the characteristic properties of MOFs is their relatively high surface area and pore volume that can facilely accommodate guests like metal nanoparticles and organocatalysts. In this way, the activity of the incorporated guest is enhanced by avoiding its deactivation or aggregation. Metal phthalocyanines are macrocyclic aromatic compounds showing chemical, thermal, and photoelectrical property [
59,
60]. Phthalocyanine compounds exhibit catalytic activity through the generation of intermediate products upon interaction with substrates through axial coordination [
61]. However, phthalocyanine undergoes aggregation in solution, thus showing a decrease in catalytic activity [
62]. One way to overcome this issue is to encapsulate phthalocyanine on a suitable host like MOFs. Hence, β-mCoPc/Cu-BDC was prepared by loading β-mCoPc over Cu-BDC MOF through impregnation strategy and the activity of this composite was studied in the oxidation of benzyl alcohol to benzaldehyde [
43] The catalytic data revealed that the activity of β-mCoPc and Cu-BDC was relatively lower than their composite, β-mCoPc/Cu-BDC by affording high stability and selectivity. The conversion of benzyl alcohol with β-mCoPc/Cu-BDC was 61% using H
2O
2 as oxidant at 80 °C after 4 h while the activity of β-mCoPc and Cu-BDC was 28.4 and 34.9%, respectively, under identical conditions. Furthermore, the activity of the composite was influenced by optimal loading of β-mCoPc. The superior activity of β-mCoPc/Cu-BDC was due to the effective interaction of O
2 and H
2O
2 with the active Cu(II) sites leading to the formation of active Cu(I) and HOO
· radical species. Later, the active Cu(I) complex reacted with H
2O
2 to afford β-mCoPc/Cu-BDC solid and HO
· radical. Sequentially, this highly reactive species was readily reacted with benzyl alcohol providing an unstable intermediate which upon dehydrogenation followed by rearrangement gives the desired benzaldehyde as the final oxidation product (
Figure 3). The composite catalyst was recycled for five times with no significant decrease in its activity. Further, powder XRD of the fresh and recycled solids showed no changes in their structural integrity, thus indicating the robust nature of the composite.
Wu and co-workers have demonstrated the catalytic efficiency and kinetics of microporous MOFs like Co
3(BTC)
2(H
2O)
12 (BTC: 1,3,5-benzenetricarboxylate) in the aerobic oxidation of hydroquinone to
p-benzoquinone [
44]. Structural analysis of this MOF indicated an extended one-dimensional channel possessing an elliptically shaped pore opening of 4 × 5 Å diameter. The kinetic investigations have shown the first-order rate as constant, which is increased in respect to increase in temperature, molar ratio of catalyst/hydroquinone, and pH of the medium.
3. Benzylic Hydrocarbon Oxidation
The pioneering work from Ishii group on the use of N-hydroxyphthalimide (NHPI) as a co-catalyst with Co-based catalysts under homogeneous conditions for the aerobic oxidation of hydrocarbons [
63] have prompted many researchers to employ NHPI and related analogous in combination with MOFs as catalysts for the aerobic oxidation of cycloalkanes [
64] and cycloalkenes [
65]. In this context, N,N′-dihydroxypyromellitimide (NDHPI) was incorporated within the pores of Co–MOF-74 to obtain Co–MOF-74@NDHPI composite and its activity was tested in the aerobic oxidation of toluene under solvent-free conditions [
45]. The employment of Co–MOF-74 or NDHPI as catalysts resulted in 4% and 2% toluene conversions, respectively. In contrast, Co–MOF-74@NDHPI composite showed significantly higher toluene conversion of 16% with 18, 48 and 34% of benzyl alcohol, benzaldehyde, and benzoic acid, respectively. Further, these results are relatively better with Cu-BTC@NHPI for the aerobic oxidation of toluene under identical conditions [
66]. On the other hand, the physical mixture of Co–MOF-74 and NDHPI with identical loading as in the case of composite showed 10% conversion of toluene with 29 and 71% of benzyl alcohol and benzaldehyde, respectively. The authors of this work believed that the superior activity of the composite is due to the operation of a synergistic effect by forming catalytically active species between NDHPI and coordinatively unsaturated sites in the activated Co–MOF-74. Although the structural integrity of the recovered Co–MOF-74@NDHPI composite remained identical with the fresh solid as evidenced by powder XRD and FT-IR, the activity data in successive cycles were not reported. These evidences are not sufficient to demonstrate the robust nature of the catalyst in oxidation reactions and thorough characterizations of the spent catalyst are required to ascertain its stability.
Selective aerobic oxidation of cumene to cumene hydroperoxide (CHP) is an important industrially relevant process since it is one of the intermediates for the production of phenol [
67,
68]. On the other hand, CHP is also employed in the Sumitomo process for the large scale production of propylene oxide [
69,
70]. In this aspect, Xamena and co-workers have reported the facile synthesis of mixed metal Co–Ni and Mn–Ni trimesate MOFs by fast aqueous synthesis and their catalytic activity was investigated in the selective aerobic oxidation of cumene to CHP (
Scheme 2) [
46]. The experimental catalytic results have shown that the isolation of Co
2+ (or Mn
2+) in an inert Ni-BTC framework is a facile strategy to attain more than 90% CHP selectivity. Further, it was shown that Co
2+ sites are responsible for the decomposition of CHP to PP and AP; thus, a significant decrease in the CHP selectivity is noticed upon increasing the cobalt content in the mixed-metal MOFs (
Figure 4). These catalytic data clearly indicate that the selectivity of CHP is influenced by a catalyst structure rather than the conversion of cumene. Among the various reaction conditions optimized to achieve the maximum CHP selectivity, mixed-metal Co
5Ni-BTC (Co:Ni ratio of 5:95) afforded 30% conversion of cumene with 91% selectivity to CHP at 90 °C after 7 h. This superior activity of this solid is due to the isolation of 73% of the total Co
2+ ions in the mixed-metal MOFs, and as a consequence, the desorption of CHP is a predominant process than to the CHP decomposition/over oxidation processes at the surface of the solid (
Figure 5). This work nicely illustrates how the site isolation of the active sites in the mixed-metal MOFs in controlling the selectivity of the desired products (
Figure 5). Furthermore, the mixed-metal solid, Co
5Ni-BTC, was found to retain its catalytic performance for at least five catalytic cycles with identical CHP selectivity (
Figure 6). Analysis of the reaction mixture by ICP-OES after the catalytic reaction showed no significant amounts of Co
2+, Ni
2+ or Mn
2+, thus proving the stability of this solid under the optimized reaction conditions.
4. Olefin Oxidation
A challenging industrially relevant chemical process is the selective oxidation of cyclohexene to its corresponding alcohol, ketone, and epoxides since these products are employed as chemical intermediates for the production of drugs, agrochemicals, and polymers [
71]. This is one of the challenging substrates for oxidation reaction since C=C double bond and allylic positions are prone to oxidization, leading to the formation of many products [
72]. Hence, one of the unique roles of MOFs in this reaction is to control the restriction of cyclohexene by pore confinement, thus tuning the reactivity.
In one of their seminal contributions, Dirk Volkmer and co-workers reported the synthesis of [Co
II4O(bdpb)
3] (MFU-1) MOF through the reaction of the ligand 1,4-bis[(3,5-dimethyl)-pyrazol-4-yl]benzene (H
2bdpb) with cobalt(II) salts under solvothermal conditions [
47]. The structure of MFU-1 mostly resembles MOF-5, which has a CaB
6-type framework topology. In addition, the MFU-1 network encloses octahedral {Co
4O(dmpz)
6} (3,5-dmpz: 3,5-dimethylpyrazolate) nodes that are reminiscent of the {Zn
4O(CO
2)
6} MOF-5 secondary building units. Phenylene rings constitute the edges of the cubic CaB
6 network. The framework of the MFU-1 solid has three-dimensional intersecting channels that encompass almost spherical voids with a diameter of 18.1 Å. UV-Vis diffuse reflectance spectra of MFU-1 showing a broad absorption band at 610 nm, which is due to the spin-allowed d-d transition of tetrahedral Co(II) ions within the framework. The catalytic performance of MFU-1 was evaluated in the oxidation of cyclohexene using TBHP as an oxidant. The experimental data clearly indicated that the oxidation of cyclohexene is very facile in the presence of MFU-1 (
Figure 7a), while poor conversion of cyclohexene (around 1% after 12 h) occurred in the absence of a solid catalyst. Under the optimized reaction conditions, the maximum cyclohexene conversion (27.5%) was observed at 70 °C and the oxidation products are tert-butyl-2-cyclohexenyl-1-peroxide, 2-cyclohexen-1-one and cyclohexene oxide. Furthermore,
Figure 7b shows the catalytic performance of MFU-1 in repeated cycles and the observed results suggest that there is a drop in the activity after the first cycle; however, no further loss in the activity is seen up to the fourth cycle. Powder XRD of the recovered solid showed no signs of decomposition. The decrease in the catalytic activity after the first cycle is believed to be due to pore clogging by polar reaction products and is evidenced by the decrease in BET surface area from 1485 to 1018 m
2/g for the fresh and after the first cycle solids, respectively. The hot filtration test proved the heterogeneity of the reaction and the analysis of the filtrate by atomic absorption spectroscopy showed the presence of low concentration of free Co(II) ions. Interestingly, the rate of the cyclohexene oxidation reaction with MFU-1 as solid catalyst was significantly dropped with Ph
3COOH compared to TBHP as an oxidant. Considering significant activities in the conversion of cyclohexene using these oxidants with MFU-1, the authors of this work concluded that catalytic cyclohexene oxidation occurs within the pores of the MFU-1 solid.
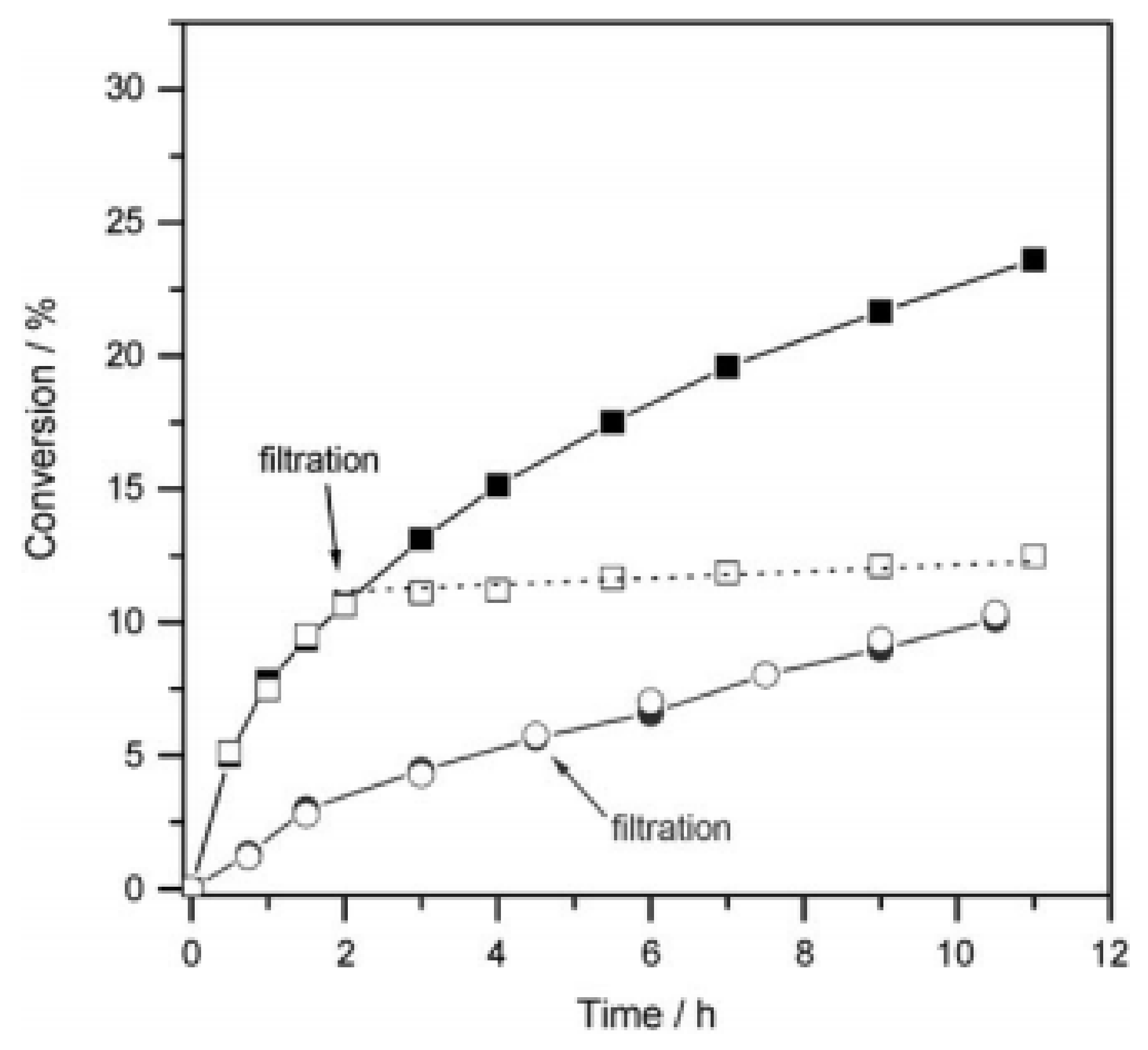
Later, the same group reported the synthesis of MFU-2, which is isostructural to MFU-1, consisting of redox-active Co
II sites linked to 1,4-bis[(3,5-dimethyl)pyrazol-4-yl] ligands. Although these solids are isostructural, they possess a significant difference in their textural properties. The pore aperture for MFU-1 and MFU-2 was 9 and 6.4 Å, respectively, while BET surface area was 1485 and 1477 m
2/g, for MFU-1 and MFU-2 solids, respectively. Furthermore, DFT studies have shown that MFU-1 activates molecular oxygen more facilely than MFU-2. The activity of these two solids was compared in the liquid phase oxidation of cyclohexene using TBHP as an oxidant [
48]. The catalytic data revealed that the activity of MFU-2 is almost similar to the results of MFU-1, while the oxidation of cyclohexene in the absence of solid was less than 1% after 12 h. The maximum cyclohexene conversion was 16% with MFU-2 after 22 h and the oxidation products were tert-butyl-2-cyclohexenyl-1-peroxide, 2-cyclohexen-1-one, and cyclohexene oxide (
Figure 8). This conversion was lower than the conversion achieved by using MFU-1 (27.5% cyclohexene conversion after 22 h) under similar experimental conditions. Interestingly, the product distributions in both cases were also identical, thus supporting the operation of similar mechanism with both solid catalysts. Although the catalytic activity and the product distributions were identical in these solids, MFU-2 exhibited marked changes compared to MFU-1 after the oxidation of cyclohexene with TBHP. In contrast to MFU-1, no colour change was observed with MFU-2 as a solid catalyst during the oxidation reaction. Although the crystal morphology of the recovered solid remains identical to the fresh solid, powder XRD of the recovered solid suggested an amorphous nature. This significant change in the textural properties of MFU-2 clearly reflected in the complete loss of porosity and the BET surface area decreased from 1675 to 3 m
2/g for the fresh and the recovered MFU-2, respectively. These results clearly indicate the decomposition of MFU-2 under these conditions, while no such changes were seen with MFU-1. Leaching experiments were performed with MFU-1 and MFU-2 solids to ascertain heterogeneity of the oxidation reaction and the observed results are shown in
Figure 9. The removal of MFU-1 from the reaction mixture showed no further conversion, indicating the absence of leached species in the reaction medium (
Figure 9). In contrary, the filtrate after removal of MFU-2 solid showed a slight decay in the catalytic activity and no significant differences with and without MFU-2 was observed. These results indicate that MFU-1 behaves as a heterogeneous solid while the activity of MFU-2 is mostly from the soluble metal complexes from the framework of MFU-2. This observation is in good agreement with the BET analysis.
In another precedent, the chiral Co(II) MOF, [Co(L-RR)(H
2O)·H
2O]
∞ (L-RR: L-RR = (R,R)-thiazolidine-2,4-dicarboxylate) was synthesized, characterized, and its activity was tested in the oxidation of wide range of alkenes under anaerobic and aerobic conditions [
49]. The experimental catalytic data indicated that the activities and selectivities were different for each olefin and even in some cases a moderate enantiomeric excess (ee) was observed. The activity of [Co(L-RR)(H
2O)·H
2O]
∞ was examined in the liquid-phase cyclohexene oxidation using TBHP as an oxidant and observing 18.6% conversion of cyclohexene with complete selectivity to
tert-butyl-2-cyclohexenyl-1-peroxide after 24 h at 70 °C. Notably, a blank control experiment in the absence of catalyst showed ∼1.6% conversion of cyclohexene under similar conditions. The activity of [Co(L-RR)(H
2O)·H
2O]∞ in the oxidation of cyclohexene was lower compared to MFU-1 (27%, at 22 h) [
47], [Co
3(BTC)
2(HCOO)
4(DMF)]·H
2O (84% at 24 h) [
73], and MFU-3 (62% at 12 h) [
74] under identical conditions. On the other hand, the activity of [Co(L-RR)(H
2O)·H
2O]∞ was relatively higher compared to MFU-2 (16% at 22 h) [
48]. Interestingly, [Co(L-RR)(H
2O)·H
2O]∞ showed superior chemoselectivity towards tert-butyl-2-cyclohexenyl-1-peroxide while the selectivity to this product with MFU-3 was 83% and 66% with MFU-1 or MFU-2 under similar reaction conditions. Furthermore, the solid [Co
3(BTC)
2(HCOO)
4(DMF)]·H
2O exhibited 95% of cyclohexenone after 24 h, which is completely different to previous reports. In addition,
tert-butyl-2-cyclohexenyl-1-peroxide was the major oxidation product with cobalt-mediated oxidation of allylic position in cyclohexene using TBHP as oxidant [
75]. The catalyst, [Co(L-RR)(H
2O)·H
2O]
∞ retained its activity unchanged for three successive cycles; however, a slight decrease in the selectivity of the oxidation products was noticed from the second cycle. Interestingly, the highest conversion of 37% was achieved for the aerobic oxidation of cyclohexene after 24 h using Co–MOF but affording 2-cyclohexenone (49%) as the major product. These catalytic data under aerobic conditions are also complemented with DFT studies, clearly proving the formation of the η
1-superoxo nature of bound oxygen species and no evidence is seen for the interaction of the olefin with Co sites in the chiral environment.
Then, the activity of [Co(L-RR)(H
2O)·H
2O]
∞ was tested in the oxidation of cyclooctene (
Scheme 3) and reaching 28% after 24 h at 70 °C, but the selectivity of the oxidation products was lower than cyclohexene. The activity of this catalyst was lower than that of [Co
3(BTC)
2(HCOO)
4(DMF)]·H
2O (64%) [
73] under identical conditions. The analysis of the reaction mixture revealed that cyclooctene oxide and tert-butyl-2-cyclooctenyl-1-peroxide constitute ∼90% selectivity of the mixture, while around 10% is the selectivity to cyclooct-2-enone. Further, the catalyst showed lower selectivity of cyclooctene epoxide (41%) compared to [Co
3(BTC)
2(HCOO)
4(DMF)]·H
2O, for which 78% selectivity was reported [
73].
Furthemore, the oxidation of 1-octene (
Scheme 3) was performed with [Co(L-RR)(H
2O)·H
2O]
∞ and observing 19% conversion after 24 h at 70 °C with product selectivity to oct-1-en-3-ol (37%), (E/Z)-oct-2-en-1-ol (52%) and the 2-hexyloxirane (11%). In addition, 18% ee was observed for the formation of oct-1-en-3-ol with the present chiral Co–MOF solid, thus showing the ability of the enantiomerically pure cobalt coordination environment promoting chiral induction. One of the probable reasons for the moderate ee in the oxidation of 1-octene is the lack of direct interaction of 1-octene with the metal centre.
Co(II)-based MOF, {[Co
2(-btec)(2,2′-bipy)
2]H
2O}n (btec: 1,2,4,5-benzenetetracarboxylic acid; 2,2′-bipy: 2,2′-bipyridine) was prepared and its catalytic activity was studied in the aerobic oxidation of cyclohexene under solvent-free conditions [
50]. The structural X-ray analysis of {[Co
2(-btec)(2,2′-bipy)
2]H
2O}
n revealed that it crystallizes in the orthorhombic space group C
2221.
Figure 10a shows that the Co(II) centre provides a distorted octahedral geometry arrangement through coordinating by two nitrogen atoms of 2,2′-bpy linker, three oxygen atoms of two different carboxylate groups of btec, and an oxygen atom of a water molecule.
Figure 10b shows an infinite two-dimensional lamellar structure. The four carboxylate groups of btec exhibit two different types of coordination mode to Co atoms. Two carboxylate groups on the same side provide four oxygen atoms forming two bidentate chelating structures, and two carboxylate groups on the other side each provide an oxygen atom leading two monodentate bridging structures. The bridging mode of btec led to the formation of zig-zag chains, as shown in
Figure 10c. Furthermore, the coordinating water molecule acted as a bridge connecting two Co atoms, as shown in
Figure 10d. The crude Co–MOF that was filtered, washed with distilled water, and air-dried at room temperature was named Co–MOF-A. On the other hand, the crude solid that was crushed, washed with ethanol, and air-dried was named Co–MOF-B. Powder XRD analysis of Co–MOF-A and Co–MOF-B catalysts showed similar characteristic diffraction peaks, suggesting that the residual btec ligand does not affect the crystallinity. However, the diffraction peaks of Co–MOF-B are found to be more crystalline with higher intensity and are due to the removal of the residual btec by washing with ethanol. A blank control experiment was performed for the aerobic oxidation of cyclohexene in the absence of a catalyst, resulting in 2.7% conversion at 70 °C after 6 h. The conversion of cyclohexene was elevated to 12.3% using Co–MOF-A as solid under identical conditions and the conversion was further raised to 30.6% conversion using Co–MOF-B as a catalyst under similar conditions. The use of the Co–MOF-B catalyst provided relatively higher catalytic activity and the decomposition of cyclohexylhydroperoxide was facilitated better by decreasing its selectivity. The difference in the activities between Co–MOF-A and Co–MOF-B was due to the impurities of residual btec in Co–MOF-A. The catalyst stability of Co–MOF-B was examined by performing reusability experiments. The conversion of cyclohexene was notably decreased from 33.2% in the first run to 19.7% in the second run, but it remained identical in the subsequent 3–5 runs. Hot-filtration experiments proved the heterogeneity of the reaction. The decrease in the activity of Co–MOF-B after the first run was due to the adsorption of active sites by organic residues, as shown in SEM images and powder XRD (
Figure 11). The deactivated solid was treated with the scCO
2-expanded ethanol system and the recovered solid is subjected to reuse in the second run. Gratifyingly, the conversion of cyclohexene was 53.6%, which is higher than the activity shown by the fresh Co–MOF-B catalyst. These results clearly indicate that this regeneration process not only recovered the activity but also enhanced the activity of the catalyst.
A series of zinc-based MOFs with the formula
3∞[Zn1−xMx(tdc)(bpy)] (M: Co
2+ and Fe
2+) (tdc: 2,5-thiophene dicarboxylate; bipy: 4,4’-bipyridine) consisting of Co
2+ and Fe
2+ ions were prepared and their activity was examined in the oxidation of cyclooctene using TBHP as an oxidant in toluene [
51]. The conversion of cyclooctene using
3∞[Zn(tdc)(bpy)] as catalyst was around 4%, which is similar to a blank experiment in the absence of any catalyst at 75 °C after 24 h. On the other hand, Fe(II)-substituted MOF
3∞[Zn
0.9Fe
0.1(tdc)(bpy)] was catalytically inactive under identical conditions. In contrast, Co
2+-based MOF
3∞[Co(tdc)(bpy)] was the active catalyst by affording 42% conversion of cyclooctene under identical conditions with the respective epoxide as the sole product. On the other hand, Cu-based Basolite C300 afforded 22% conversion of cyclooctene after 24 h. While Zn-based MOF was catalytically inactive, the incorporation of Co
2+ ions within the framework facilely promotes the oxidation, thus suggesting the formation of active Co sites in the solid through the dissociation of the longer Co–O bond. The heterogeneity of the reaction was shown by hot filtration experiment. Furthermore, many control experiments clearly established that the catalytic activity of the resulting solid is mainly influenced by the degree of Co
2+-substitution in the MOF solid.
Later, a series of isostructural, heteronuclear MOFs possessing Co
2+ and Zn
2+ ions within [Co
xZn
1−x(tdc)(bipy)] was reported as a heterogeneous catalyst for the epoxidation of cyclooctene with TBHP at 75 °C [
52]. Catalytic experiments have shown that the conversion of cyclooctene gradually increases in respect to Co-content in the MOF solid; thus, Zn-free MOF exhibited the highest conversion, while the Co-free MOF showed poor activity. For example, Zn(tdc)(bipy) solid showed 4% cyclooctene conversion at 75 °C after 24 h, while Co(tdc)(bipy) afforded the maximum conversion of 42% cyclooctene with 53% selectivity to its respective epoxide under identical conditions. Furthermore, Basolite C300 [Cu
3(BTC)
2] gave 22% conversion with 64% selectivity of epoxide under similar conditions. These results indicate that the conversion of cyclooctene is highly influenced by the nature of transition metals and their chemical environments in the MOF along with their Lewis/redox properties. Furthermore, the reusability test indicated that the activity of Co(tdc)(bipy) is retained up to the second run but the epoxide selectivity decreases to 42%. Characterization of the reused solid should have provided causes for the decrease of epoxide selectivity.
5. Sulphur Compounds Oxidation
A mustard gas, bis-(2-chloroethyl)sulfide, is a widely used chemical warfare reagent and possesses high toxicity and causes serious skin damages, respiratory problems, DNA damage, and leads to death with high dosage. Hence, one of the easiest ways to reduce its toxicity is the selective oxidation of mustard gas to its corresponding sulfoxide only, since over oxidation product, sulfone is more toxic [
76,
77,
78]. Hence, recently, two polyoxovanadate-based MOFs, [Co(bib)]{V
2O
6} (V-Co–MOF) and [Ni(en)(bib)]{V
2O
6}.2H
2O (V-Ni–MOF) (bib: 1,4-bis(1H-imidazoly-1-yl)benzene; en: ethylenediamine) were hydrothermally synthesized under mild reaction conditions [
53]. Single-crystal X-ray diffraction studies showed that the V sites in both solids exist as {VO
4} tetrahedral coordination geometries. The Co sites in V–Co–MOF exist as a four-coordinated distorted tetrahedron configuration while the Ni site in V–Ni–MOF has a six-coordinated octahedral geometry. The structural arrangements in both solids with these geometries offer coordinatively unsaturated metal sites that can be exploited as Lewis acid sites promoting oxidation reactions. Hence, the activity of these two solids was compared in the oxidation of 2-chloroethyl ethyl sulfide (CEES), a well-known mustard gas stimulant [
53]. One of the features of this oxidation reaction is to convert CEES to only 2-chloroethyl ethyl sulfoxide (CEESO), which is relatively non-toxic compared to its precursor. The catalytic data revealed that V–Co–MOF provides a complete conversion of CEES in 10 min at 25 °C while V–Ni–MOF converts only 47.5% of CEES under similar conditions (
Figure 12). A series of mechanistic studies with further experiments indicated that the superior activity of V–Co–MOF is attributed due to the synergetic effects through the involvement of two active sites. The interaction of H
2O
2 with V site in the MOF produces peroxovanadium with higher oxidation ability; meanwhile, the S atom in CEES coordinates with the four-coordinated Co(II) centre to produce 2-chloroethyl ethyl sulfonium cation (CEES
+) through electron transfer reaction. Later, this species undergoes a nucleophilic attack to provide CEESO as the final oxidized product.
Figure 13 shows the proposed reaction mechanism. Leaching test confirmed the heterogeneity of the process. Reusability experiment showed no decay in the conversion and selectivity even after five cycles, indicating high stability of V–Co–MOF under the present experimental conditions. Powder XRD patterns and the FT-IR spectra of the recovered solid coincided well with the fresh catalyst showing the maintenance of structural integrity.
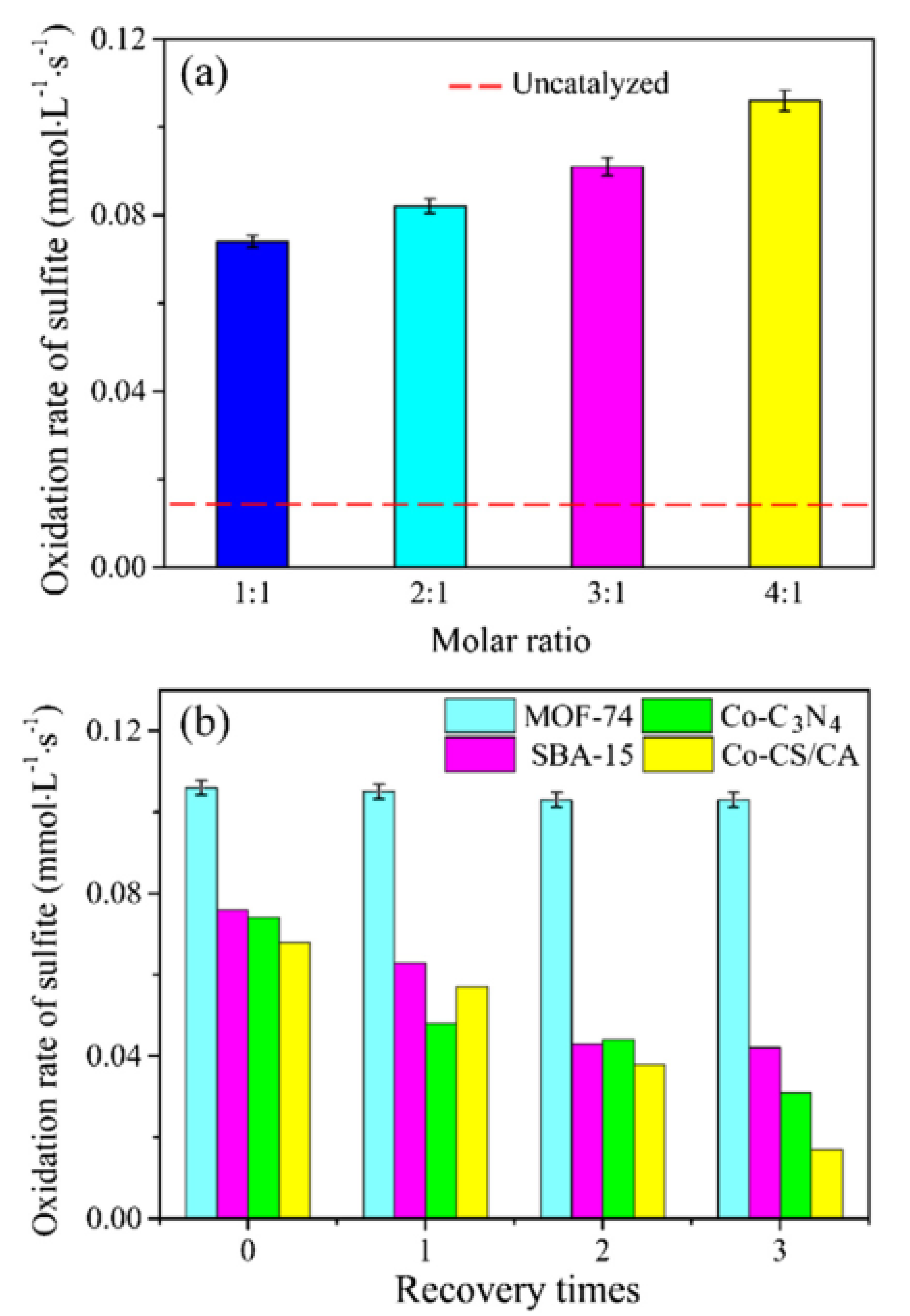
In another precedent, robust Co-based MOF catalysts with different Co
2+/organic ligand ratios of 1:1, 2:1, 3:1, and 4:1 were prepared leading to the formation of Co–MOF-74(1), Co–MOF-74(2), Co–MOF-74(3), and Co–MOF-74(4), respectively. The activity of these solids was tested in the oxidation of magnesium sulfite [
54]. Among the various catalysts tested, Co–MOF-74(4) exhibited the highest activity with the catalytic rate of 0.106 mmol L
−1s
−1 (
Figure 14a), which is 10 times higher than the control experiment in the absence of catalyst. The activity of Co–MOF-74(4) was compared with a series of supported catalysts like Co–SBA-15, Co–C
3N
4, and Co–CS/CA, and the observed results indicate that the former catalyst exhibited superior activity than the later catalysts. On the other hand, the activity of Co–MOF-74(4) was retained in three successive cycles for the oxidation of magnesium sulfite, while the other catalysts showed a significant decrease around 50% relative to their initial rate during three recycles [
79,
80,
81]. The inferior activity of these catalysts is due to the presence of agglomerated Co species prepared by the impregnation method, thus preventing the uniform distribution of Co sites, which potentially retard the reaction rate. In contrary, the oxidation rate of Co–MOF-74(4) was maintained very high after three cycles with no obvious attenuation in catalytic activity in the oxidation of magnesium sulfite. This enhanced activity shown by Co–MOF-74(4) is due to the distinct structural arrangements obtained by the hydrothermal method where the Co sites are uniformly distributed and embedded with organic linkers. Furthermore, the hollow structure of MOF-74 provides easy accessibility to active Co sites by sulfite, thus promoting the oxidation at a higher reaction rate. In addition, the higher activity of Co–MOF-74 is also due to the lack of agglomerated Co species in its structural arrangements. Furthermore, DFT calculations have shown the existence of unsaturated cobalt site on the open framework of Co–MOF-74(4), thus providing more opportunities to sulfite ions to react efficiently and enhance the reaction rate.
6. Water Oxidation
Electrocatalysis for the conversion of H
2O into H
2 and O
2 is an attractive strategy in the production of chemical fuels [
82,
83]. Although many catalytic systems have been developed for this reaction, the mechanistic aspects are still unclear and the reaction is hampered by sluggish kinetics. Among the various catalysts reported for this reaction, the most efficient catalysts are Ir and Ru-based catalysts, posing practical difficulties due to their high cost [
84,
85].
In this regard, a bioinspired cobalt-citrate (UTSA-16) MOF was synthesized by solvothermal route and its activity was demonstrated in an electrocatalytic oxygen evolution reaction (OER) [
55]. This MOF shows an open framework structure constructed from tetranuclear cobalt citrate clusters as octahedral linkers and tetrahedral Co
II atoms as trigonal nodes. The oxygen atoms in citrate chelate with octahedral cobalt atoms leading to the formation of a Co
4O
4 cubane arrangement (
Figure 15), which is characterized as a core unit for the promotion of OER catalysis. The electrocatalytic activity of UTSA-16 exhibited small onset potential (1.6 V), large anodic current density, and long-term durability in alkaline medium, which is superior to other MOF-based electrocatalysts and the standard Co
3O
4 counterpart.
Figure 16 provides the Tafel plots of the UTSA-16 and some reference catalysts derived from corresponding linear sweep voltammetry (LSV) curves. The UTSA-16 has the Tafel slope of 77 mV dec
−1, which is slightly larger than the benchmark catalyst RuO
2 (62 mVdec
−1) but much smaller than commercial catalysts like Pt/C (182 mVdec
−1) and the Co
3O
4 counterpart (91 mVdec
−1). These catalytic data clearly suggest the favourable kinetics offered by UTSA-16 for electrochemical water oxidation. This enhanced activity of UTSA-16 is due to the synergistic cooperation effect of an open porous structure and the high-valent cobalt formed in-situ, and the existence of Co
4O
4 cubane in UTSA-16.
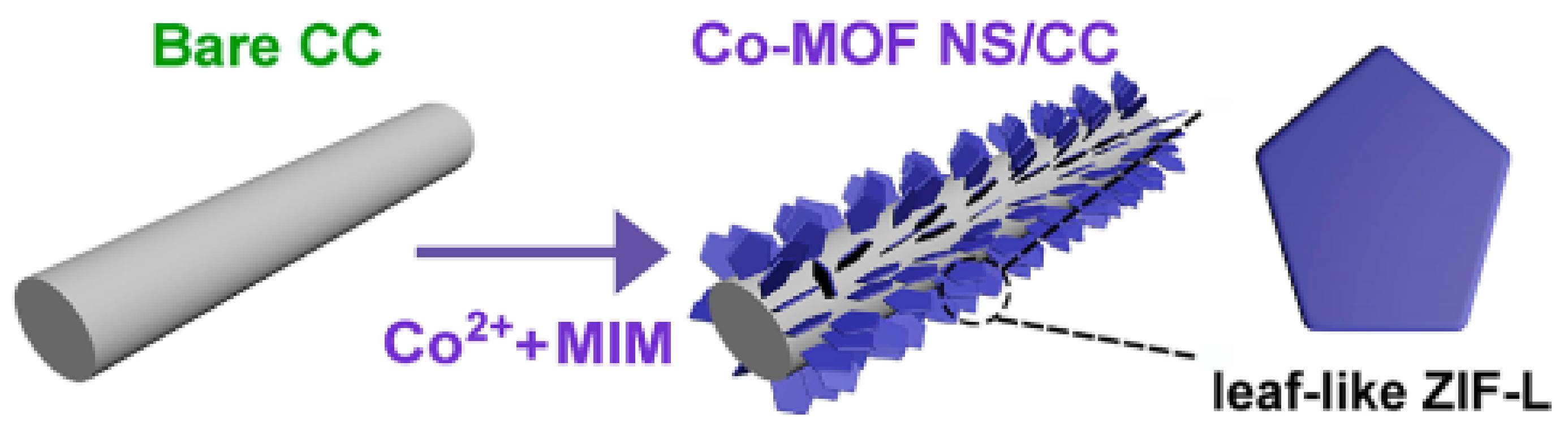
In another report, a leaf-like ZIF-L nanosheet (NS) array assembled on the carbon cloth (Co–MOF NS/CC) was prepared by a facile liquid-phase deposition under ambient condition (
Figure 17) and its catalytic activity was examined as a versatile electrocatalysts in water oxidation [
56]. The OER activity of Co–MOF NS/CC was compared to analogous catalysts like CoMOF NS powder/CC, bare CC, and RuO
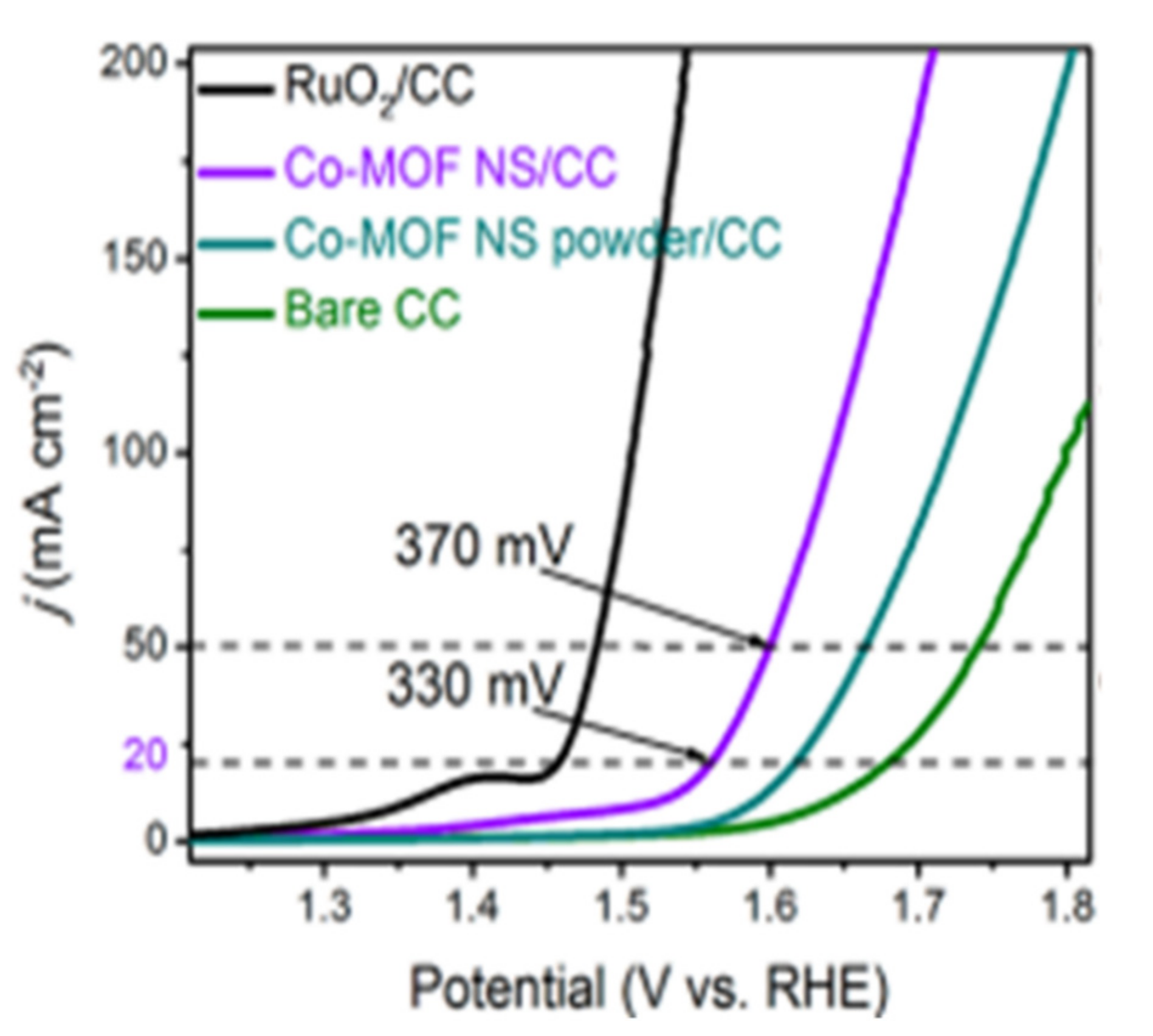
2/CC using LSV in a three electrode configuration using the electrodes as the working electrode, graphite plate as the counter electrode, and saturated calomel electrode as the reference electrode. Co–MOF NS/CC showed a higher current density and more negative OER onset potential (∼1.52 V vs. RHE) compared to Co–MOF NS powder/CC (∼1.58 V vs. RHE) and bare CC (∼1.63 V vs. RHE) (
Figure 18). However, there is still a gap between RuO
2/CC and Co–MOF NS/CC. Interestingly, the electrocatalytic activity of Co–MOF NS/CC showed much superior activity than the calcined product of Co
3O
4 NS/CC. This is due to the fact that the calcination treatment sacrifices Co–MOF intrinsic molecular metal active sites [
59,
86]. Furthermore, the Co–MOF NS/CC electrode showed small overpotential of 330 and 370 mV to achieve current densities of 20 and 50 mA cm
−2 with a relatively low Tafel slope of 106.6 mVdec
−1. This activity is higher than the Co–MOF NS powder/CC, MOF-based catalysts and transition metal based oxides, hydroxides, chalcogenides, and phosphides. The small overpotential and low Tafel slope achieved with Co–MOF NS/CC clearly suggests high activity and fast reaction kinetics for OER [
87,
88]. The stability of the Co–MOF NS/CC was performed in OER using multistep chronopotentiometric curve. The Co–MOF NS/CC exhibited impressive OER durability with the polarization curve remaining almost identical even after 5000 s electrolysis.
Very recently, a series of two-dimensional semiconductive CoNi mixed-metal MOFs (Co
xNi
3−x(HAB)
2) were prepared using hexaaminobenzene (HAB) as an organic linker (
Figure 19) [
57]. The structural characterizations of Co
xNi
3−x(HAB)
2 MOFs revealed the existence of multiple metal oxidation states (Co
0/Co
2+/Co
3+ and Ni
2+/Ni
3+) and graphene-like nanostructures embedded with high density of CoNi alloy nanoparticles. The electrocatalytic OER performance of Co
xNi
3−x(HAB)
2 MOFs was compared with their analogous catalysts like Co
3(HAB)
2 MOF, Ni
3(HAB)
2 MOF and commercial RuO
2 in 1.0 M KOH by LSV at a scan rate of 10 mVs
−1 [
57]. The experimental results have shown that the OER performances of the series of Co
xNi
3-x(HAB)
2 MOFs are superior to RuO
2 catalyst. In particular, the activity of Co
xNi
3−x(HAB)
2 MOF-1 and Co
xNi
3−x(HAB)
2 MOF-2 show identical η
0 of 1.35 V. These results indicate the involvement of a synergistic effect between Co and Ni ions that can effectively reduce the onset potential in OER. However, among all the samples examined, the use of Co
xNi
3−x(HAB)
2 MOF-2 solid showed the lowest η
10 of 1.40 V, suggesting its superior catalytic performance in OER. Interestingly, the electrocatalytic OER activity of Co
xNi
3-x(HAB)
2 MOF-2 was better compared to other reported OER catalysts like NiCo–POM/Ni (1.58 V) [
89], Co–Mo-S/CC (1.53 V) [
90], Ni-Mo (1.53 V) [
91], and Co
4Mo
2@NC (1.56 V) [
92]. Furthermore, the Tafel slope of Co
xNi
3-x(HAB)
2 MOF-2 solid was relatively smaller to the value of 26 mVdec
−1 compared to its analogous solids like Co
xNi
3-x(HAB)
2 MOF-1 (28 mVdec
−1), Co
xNi
3-x(HAB)
2 MOF-3 (92 mVdec
−1), and RuO
2 (52 mV dec
−1), thus clearly suggesting the faster OER kinetics of Co
xNi
3-x(HAB)
2 MOF-2 solid. The superior activity of this solid is due to the high electrical conductivity caused by the strong π-d conjunction, which can facilitate facile charge transport during electrolysis, thus showing lower overpotentials for OER. Further, the two-dimensional nature of this MOF with ultrathin nanosheets offers a large electrochemical surface area for the facile diffusion of reactants and the partial electron transfer from Ni
2+ to Co
3+ sites greatly increases the OER performance of mixed-metal MOFs. Additionally, chronoamperometry measurement was carried out at a constant potential of 1.40 V vs. RHE to attain the current density of 10 mA cm
−2 and examine the long-term stability of the Co
xNi
3-x(HAB)
2 MOF-2 catalyst. The experimental results clearly indicated that the current density decreases by 3.6% with its initial value after 48 h, while the current density remarkably decreased by 22.8% after 48 h with the commercial RuO
2 catalyst (
Figure 20). In addition, the LSV curve of the Co
xNi
3-x(HAB)
2 MOF-2 catalyst showed a negligible change even after 5000 cycles, thus indicating the superior stability of this solid (
Figure 20).

 ,
, 


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}