
The Roles of Precursor-Induced Metal–Support Interaction on the Selective Hydrogenation of Crotonaldehyde over Ir/TiO2 Catalysts

Abstract
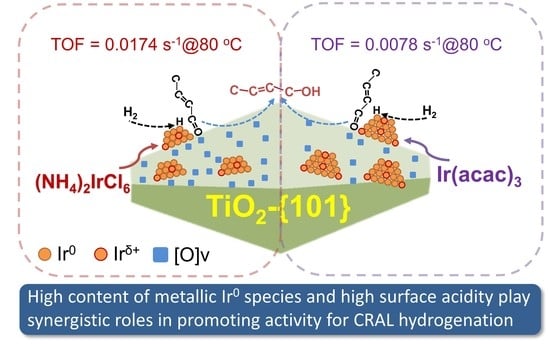
:
1. Introduction
2. Experimental
2.1. Synthesis of Anatase TiO2 Nanocrystals and Preparation of Ir/TiO2 Catalysts
2.2. Catalyst Characterizations
2.3. Catalytic Reaction
3. Results and Discussion
3.1. Characterizations of the Catalyst
3.2. Catalytic Behaviors of the Catalysts
3.3. In Situ FTIR Studies of CRAL Reaction over Ir/TiO2 Catalysts
3.4. Discussion on the Effect of Ir Precursors and Reaction Mechanisms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Claus, P. Selective Hydrogenation of α, β-Unsaturated Aldehydes and Other C=O and C=C Bonds Containing Compounds. Top. Catal. 1998, 5, 51–62. [Google Scholar] [CrossRef]
- Lan, X.; Wang, T. Highly Selective Catalysts for the Hydrogenation of Unsaturated Aldehydes: A Review. ACS Catal. 2020, 10, 2764–2790. [Google Scholar] [CrossRef]
- Mäki–Arvela, P.; Hájek, J.; Salmi, T.; Murzin, D.Y. Chemoselective Hydrogenation of Carbonyl Compounds over Heterogeneous Catalysts. Appl. Catal. A 2005, 292, 1–49. [Google Scholar] [CrossRef]
- Dandekar, A.; Vannice, M.A. Crotonaldehyde Hydrogenation on Pt/TiO2 and Ni/TiO2 SMSI Catalysts. J. Catal. 1999, 183, 344–354. [Google Scholar] [CrossRef]
- Delbecq, F.; Li, Y.; Loffreda, D. Metal–support Interaction Effects on Chemo–regioselectivity: Hydrogenation of Crotonaldehyde on Pt13/CeO2(1 1 1). J. Catal. 2016, 334, 68–78. [Google Scholar] [CrossRef]
- Yang, X.; Mueanngern, Y.; Baker, Q.A.; Baker, L.R. Crotonaldehyde Hydrogenation on Platinum–Titanium Oxide and Platinum–Cerium Oxide catalysts: Selective C=O Bond Hydrogen Requires Platinum Sites Beyond the Oxide–Metal Interface. Catal. Sci. Technol. 2016, 6, 6824–6835. [Google Scholar] [CrossRef]
- Gebauer-Henke, E.; Grams, J.; Szubiabiewicz, E.; Farbotko, J.; Touroude, R.; Rynkowske, J. Pt/Ga2O3 Catalysts of Selective Hydrogenation of Crotonaldehyde. J. Catal. 2007, 250, 195–208. [Google Scholar] [CrossRef]
- Abid, M.; Touroude, R. Pt/CeO2 Catalysts in Selective Hydrogenation of Crotonaldehyde: High Performance of Chlorine-free Catalysts. Catal. Lett. 2000, 69, 139–144. [Google Scholar] [CrossRef]
- Wang, D.; Ammari, F.; Touroude, R.; Su, D.S.; Schlögl, R. Promotion Effect in Pt–ZnO Catalysts for Selective Hydrogenation of Crotonaldehyde to Crotyl alcohol: A Structural Investigation. Catal. Today 2009, 147, 224–230. [Google Scholar] [CrossRef]
- Campo, B.; Petit, C.; Volpe, M.A. Hydrogenation of crotonaldehyde on different Au/CeO2 catalysts. J. Catal. 2008, 254, 71–78. [Google Scholar] [CrossRef]
- Ramos-Fernández, E.V.; Silvestre-Albero, J.; Sepúlveda-Escribano, A.; Rodríguez-Reinoso, F. Effect of the Metal Precursor on the Properties of Ru/ZnO Catalysts. Appl. Catal. A Gen. 2010, 374, 221–227. [Google Scholar] [CrossRef]
- Reyes, P.; Aguirre, M.C.; Melián–Cabrera, I.; Granados, M.L.; Fierro, J.L.G. Interfacial Properties of an Ir/TiO2 System and Their Relevance in Crotonaldehyde Hydrogenation. J. Catal. 2002, 208, 229–237. [Google Scholar] [CrossRef]
- Chen, P.; Lu, J.-Q.; Xie, G.-Q.; Hu, G.-S.; Zhu, L.; Luo, L.-F.; Huang, W.-X.; Luo, M.-F. Effect of Reduction Temperature on Selective Hydrogenation of Crotonaldehyde over Ir/TiO2 Catalysts. Appl. Catal. A Gen. 2012, 433–434, 236–242. [Google Scholar] [CrossRef]
- Jia, A.P.; Zhang, Y.S.; Song, T.Y.; Zhang, Z.H.; Tang, C.; Hu, Y.M.; Zheng, W.B.; Luo, M.F.; Lu, J.Q.; Huang, W.X. Crystal-plane Effects of Anatase TiO2 on the Selective Hydrogenation of Crotonaldehyde over Ir/TiO2 Catalysts. J. Catal. 2021, 395, 10–22. [Google Scholar] [CrossRef]
- Gao, Y.X.; Wang, W.D.; Chang, S.J.; Huang, W.X. Morphology Effect of CeO2 Support in the Preparation, Metal–Support Interaction, and Catalytic Performance of Pt/CeO2 Catalysts. ChemCatChem 2013, 5, 3610–3620. [Google Scholar] [CrossRef]
- Ying, Z.; Doronkin, D.E.; Chen, M.; Wei, S.; Grunwaldt, J.D. Interplay of Pt and Crystal Facets of TiO2: CO Oxidation Activity and Operando XAS/DRIFTS Studies. ACS Catal. 2016, 6, 7799–7809. [Google Scholar] [CrossRef]
- Tamura, M.; Tokonami, K.; Nakagawa, Y.; Tomishige, K. Selective Hydrogenation of Crotonaldehyde to Crotyl Alcohol over Metal Oxide Modified Ir Catalysts and Mechanistic Insight. ACS Catal. 2016, 6, 3600–3609. [Google Scholar] [CrossRef]
- Yuan, J.F.; Luo, C.Q.; Yu, Q.; Jia, A.P.; Hu, G.S.; Lu, J.Q.; Luo, M.F. Great Improvement on the Selective Hydrogenation of Crotonaldehyde over CrOx- and FeOx-promoted Ir/SiO2 catalysts. Catal. Sci. Technol. 2016, 6, 4294–4305. [Google Scholar] [CrossRef]
- Xu, Y.M.; Zheng, W.B.; Hu, Y.M.; Tang, C.; Jia, A.P.; Lu, J.Q. The Effects of MoOx Decoration on the Selective Hydrogenation of Crotonaldehyde over MoOx-Promoted Ir/TUD-1 Catalysts. J. Catal. 2020, 381, 222–233. [Google Scholar] [CrossRef]
- Tian, S.; Gong, W.; Chen, W.; Lin, N.; Zhu, Y.; Feng, Q.; Xu, Q.; Fu, Q.; Chen, C.; Luo, J.; et al. Regulating the Catalytic Performance of Single-Atomic-Site Ir Catalyst for Biomass Conversion by Metal–Support Interactions. ACS Catal. 2019, 9, 5223–5230. [Google Scholar] [CrossRef]
- Lin, W.; Cheng, H.; Li, X.; Zhang, C.; Zhao, F.; Arai, M. Layered Double Hydroxide-like Mg3Al1−xFex Materials as Supports for Ir Catalysts: Promotional Effects of Fe Doping in Selective Hydrogenation of Cinnamaldehyde. Chin. J. Catal. 2018, 39, 988–996. [Google Scholar] [CrossRef]
- Tamura, M.; Tokonami, K.; Nakagawa, Y.; Tomishige, K. Effective NbOx-Modified Ir/SiO2 Catalyst for Selective Gas-Phase Hydrogenation of Crotonaldehyde to Crotyl Alcohol. ACS Sustain. Chem. Eng. 2017, 5, 3685–3697. [Google Scholar] [CrossRef]
- Yu, Q.; Bando, K.K.; Yuan, J.F.; Luo, C.Q.; Jia, A.P.; Hu, G.S.; Lu, J.Q.; Luo, M.F. Selective Hydrogenation of Crotonaldehyde over Ir–FeOx/SiO2 Catalysts: Enhancement of Reactivity and Stability by Ir–FeOx Interaction. J. Phys. Chem. C 2016, 120, 8663–8673. [Google Scholar] [CrossRef]
- Ning, X.; Xu, Y.M.; Wu, A.Q.; Tang, C.; Jia, A.P.; Luo, M.F.; Lu, J.Q. Kinetic Study of Selective Hydrogenation of Crotonaldehyde over Fe-promoted Ir/BN Catalysts. Appl. Surf. Sci. 2019, 463, 463–473. [Google Scholar] [CrossRef]
- Reyes, P.; Rojas, H.; Fierro, J.L.G. Effect of Fe/Ir Ratio on the Surface and Catalytic Properties in Citral Hydrogenation on Fe–Ir/TiO2 Catalysts. J. Mol. Catal. A Chem. 2003, 203, 203–211. [Google Scholar] [CrossRef]
- Rojas, H.A.; Martínez, J.J.; Díaz, G.; Gómez–Cortés, A. The Effect of Metal Composition on the Performance of Ir–Au/TiO2 Catalysts for Citral Hydrogenation. Appl. Catal. A Gen. 2015, 503, 196–202. [Google Scholar] [CrossRef]
- Liu, L.; Gu, X.; Ji, Z.; Zou, W.; Tang, C.; Gao, F.; Dong, L. Anion-Assisted Synthesis of TiO2 Nanocrystals with Tunable Crystal Forms and Crystal Facets and Their Photocatalytic Redox Activities in Organic Reactions. J. Phys. Chem. C 2013, 117, 18578–18587. [Google Scholar] [CrossRef]
- Chen, S.; Abdel-Mageed, A.M.; Li, D.; Bansmann, J.; Cisneros, S.; Biskupek, J.; Huang, W.; Behm, R.J. Morphology-Engineered Highly Active and Stable Ru/TiO2 Catalysts for Selective CO Methanation. Angew. Chem. Int. Ed. 2019, 58, 10732–10736. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Z.; Wang, W.; Huang, W. Engineering Highly Active TiO2 Photocatalysts via the Surface-Phase Junction Strategy Employing a Titanate Nanotube Precursor. J. Catal. 2014, 310, 16–23. [Google Scholar] [CrossRef]
- Tanaka, K.; Watters, K.L.; Howe, R.F. Characterization of Supported Iridium Catalysts Prepared from Ir4(CO)12. J. Catal. 1982, 75, 23–38. [Google Scholar] [CrossRef]
- Lòpez-De Jesús, Y.M.; Vicente, A.; Lafaye, G.; Marécot, P.; Williams, C.T. Synthesis and Characterization of Dendrimer-Derived Supported Iridium Catalysts. J. Phys. Chem. C 2008, 112, 13837–13845. [Google Scholar] [CrossRef]
- Toolenar, F.J.C.M.; Bastein, A.G.T.M.; Ponec, V. The Effect of Particle Size in the Adsorption of Carbon Monoxide on Iridium: An Infrared Investigation. J. Catal. 1983, 82, 35–44. [Google Scholar] [CrossRef]
- Okumura, M.; Coronado, J.M.; Soria, J.; Haruta, M.; Conesa, J.C. EPR Study of CO and O2 Interaction with Supported Au Catalysts. J. Catal. 2001, 203, 168–174. [Google Scholar] [CrossRef]
- Li, Y.; Xu, B.; Fan, Y.; Feng, N.; Qiu, A.; Miao, J.; He, J.; Yang, H.; Chen, Y. The Effect of Titania Polymorph on the Strong Metal-Support Interaction of Pd/TiO2 Catalysts and Their Application in the Liquid Phase Selective Hydrogenation of Long Chain Alkadienes. J. Mol. Catal. A Chem. 2004, 216, 107–114. [Google Scholar] [CrossRef]
- Conesa, J.C.; Soria, J. Reversible Ti3+ Formation by Hydrogen Adsorption on M/ TiO2 Catalysts. J. Phys. Chem. 1982, 86, 1392–1395. [Google Scholar] [CrossRef]
- Huang, J.; He, S.; Goodsell, J.L.; Mulcahy, J.R.; Guo, W.; Angerhofer, A.; Wei, W.D. Manipulating Atomic Structures at the Au/TiO2 Interface for O2 Activation. J. Am. Chem. Soc. 2020, 142, 6456–6460. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Hu, G.S.; Lu, J.Q.; Xie, G.Q.; Chen, P.; Luo, M.F. Effects of Ir Content on Selective Hydrogenation of Crotonaldehyde over Ir/ZrO2 Catalysts. Catal. Commun. 2012, 21, 5–8. [Google Scholar] [CrossRef]
- Ammari, F.; Lamotte, J.; Touroude, R. An Emergent Catalytic Material: Pt/ZnO Catalyst for Selective Hydrogenation of Crotonaldehyde. J. Catal. 2004, 221, 32–42. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | SBET /m2 g−1 | Ir Content a /wt.%a | dVA /nm | DIr b/ % | Surface Cl/Ti Molar Ratio c | |
|---|---|---|---|---|---|---|
| Calcined | Pre-Reduced | |||||
| Ir/TiO2-A | 92 | 4.93 | 1.7 | 64.7 | n.d. | n.d. |
| Ir/TiO2-Cl | 89 | 4.90 | 1.5 | 73.3 | 0.056 | 0.005 |
| Ir/TiO2-N | 90 | 4.86 | 1.3 | 84.6 | 0.061 | 0.006 |
| TiO2 | 98 | - | - | - | - | - |
| Sample a | Reduction Temperature/°C | Reaction Time/min | Reaction Rate b /μmol gIr−1 s−1 | TOF c/×10−3 | Selectivity /% | Reference | ||
|---|---|---|---|---|---|---|---|---|
| Crotyl Alcohol | Butanal | Others d | ||||||
| Ir/TiO2-A | 300 | 180 | 11.5 | 2.6 | 70.1 | 29.9 | 0 | This work |
| Ir/TiO2-Cl | 300 | 180 | 21.7 | 4.4 | 71.3 | 27.0 | 1.7 | This work |
| Ir/TiO2-N | 300 | 180 | 24.7 | 4.1 | 69.5 | 28.8 | 1.7 | This work |
| Ir/TiO2 | 300 | 500 | 16.2 | 3.8 | 74.6 | 14.6 | 10.8 | [13] |
| Ir/ZrO2 | 500 | 180 | 8.7 | 7.6 | 82.2 | 8.1 | 9.7 | [37] |
| 3Ir0.1Fe/SiO2 | 300 | 600 | 23.8 | 18.0 | 90.8 | 3.7 | 5.5 | [23] |
| 3Ir-0.05(Cr-Fe)/SiO2 | 300 | 600 | 27.1 | 16.9 | 85.9 | 8.3 | 5.8 | [18] |
| 3Ir-0.05Fe/BN | 300 | 180 | 16.4 | 6.7 | 84.4 | 9.0 | 6.6 | [24] |
| Ir-NbOx/SiO2 (Nb/Ir = 0.5) | 500 | 120 | 48.4 | 210 | 92.8 | 3.7 | 3.0 | [22] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, A.; Peng, H.; Zhang, Y.; Song, T.; Ye, Y.; Luo, M.; Lu, J.; Huang, W. The Roles of Precursor-Induced Metal–Support Interaction on the Selective Hydrogenation of Crotonaldehyde over Ir/TiO2 Catalysts. Catalysts 2021, 11, 1216. https://doi.org/10.3390/catal11101216
Jia A, Peng H, Zhang Y, Song T, Ye Y, Luo M, Lu J, Huang W. The Roles of Precursor-Induced Metal–Support Interaction on the Selective Hydrogenation of Crotonaldehyde over Ir/TiO2 Catalysts. Catalysts. 2021; 11(10):1216. https://doi.org/10.3390/catal11101216
Chicago/Turabian StyleJia, Aiping, Hantao Peng, Yunshang Zhang, Tongyang Song, Yanwen Ye, Mengfei Luo, Jiqing Lu, and Weixin Huang. 2021. "The Roles of Precursor-Induced Metal–Support Interaction on the Selective Hydrogenation of Crotonaldehyde over Ir/TiO2 Catalysts" Catalysts 11, no. 10: 1216. https://doi.org/10.3390/catal11101216
APA StyleJia, A., Peng, H., Zhang, Y., Song, T., Ye, Y., Luo, M., Lu, J., & Huang, W. (2021). The Roles of Precursor-Induced Metal–Support Interaction on the Selective Hydrogenation of Crotonaldehyde over Ir/TiO2 Catalysts. Catalysts, 11(10), 1216. https://doi.org/10.3390/catal11101216