
Chain Transfer to Solvent and Monomer in Early Transition Metal Catalyzed Olefin Polymerization: Mechanisms and Implications for Catalysis

 ,
,  ,
,  , ,
, ,
Abstract
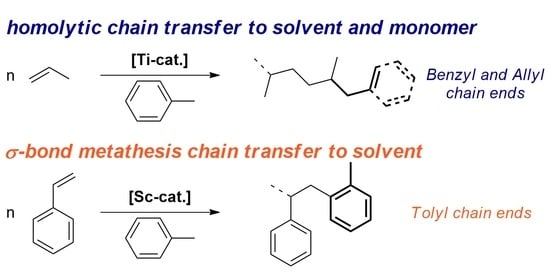
:
1. Introduction
2. CTS and CTM in Ti-Catalyzed Propene Polymerization
2.1. First Observation and General Considerations
2.2. Mechanistic Studies
3. CTS in Sc-Catalyzed Styrene Polymerization
3.1. Additive Triggered CTS
3.2. CTS by Highly Active β-Diketiminate Sc-Catalysts in Additive-Free Polymerization
4. Further Considerations on the Mechanisms of CTS and CTM, and Their Relevance in Catalysis
4.1. Connections with Catalyst Activity and Thermal Stability
4.2. Functionalized Polymers and Switchable Polymerization Catalysts
4.3. Connections with Small Molecule Activation and the Synthesis of Allyl Complexes
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Cossee, P. On the reaction mechanism of the ethylene polymerization with heterogeneous ziegler-natta catalysts. Tetrahedron Lett. 1960, 1, 12–16. [Google Scholar] [CrossRef]
- Arlman, E.J.; Cossee, P. Ziegler-Natta catalysis III. Stereospecific polymerization of propene with the catalyst system TiCl3 AlEt3. J. Catal. 1964, 3, 99–104. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R. Microstructure of polypropylene. Prog. Polym. Sci. 2001, 26, 443–533. [Google Scholar] [CrossRef]
- Resconi, L.; Cavallo, L.; Fait, A.; Piemontesi, F. Selectivity in Propene Polymerization with Metallocene Catalysts. Chem. Rev. 2000, 100, 1253–1346. [Google Scholar] [CrossRef]
- Resconi, L.; Camurati, I.; Sudmeijer, O. Chain transfer reactions in propylene polymerization with zirconocene catalysts. Top. Catal. 1999, 7, 145–163. [Google Scholar] [CrossRef]
- Bochmann, M. Cationic Group 4 metallocene complexes and their role in polymerisation catalysis: The chemistry of well defined Ziegler catalysts. J. Chem. Soc. Dalton Trans. 1996, 255–270. [Google Scholar] [CrossRef]
- Zaccaria, F.; Sian, L.; Zuccaccia, C.; Macchioni, A. Ion pairing in transition metal catalyzed olefin polymerization. In Advances in Organometallic Chemistry; Perez, P.J., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 73, pp. 1–78. [Google Scholar]
- Brintzinger, H.H.; Fischer, D.; Mülhaupt, R.; Rieger, B.; Waymouth, R.M. Stereospecific Olefin Polymerization with Chiral Metallocene Catalysts. Angew. Chem. Int. Ed. Engl. 1995, 34, 1143–1170. [Google Scholar] [CrossRef] [Green Version]
- Busico, V.; Cipullo, R.; Pellecchia, R.; Ronca, S.; Roviello, G.; Talarico, G. Design of stereoselective Ziegler-Natta propene polymerization catalysts. Proc. Natl. Acad. Sci. USA 2006, 103, 15321–15326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehm, C.; Vittoria, A.; Goryunov, G.P.; Kulyabin, P.S.; Budzelaar, P.H.M.; Voskoboynikov, A.Z.; Busico, V.; Uborsky, D.V.; Cipullo, R. Connection of Stereoselectivity, Regioselectivity, and Molecular Weight Capability in rac-R′2Si(2-Me-4-R-indenyl)2ZrCl2 Type Catalysts. Macromolecules 2018, 51, 8073–8083. [Google Scholar] [CrossRef]
- Uborsky, D.V.; Mladentsev, D.Y.; Guzeev, B.A.; Borisov, I.S.; Vittoria, A.; Ehm, C.; Cipullo, R.; Hendriksen, C.; Friederichs, N.; Busico, V.; et al. C1-Symmetric Si-bridged (2-indenyl)(1-indenyl) ansa-metallocenes as efficient ethene/1-hexene copolymerization catalysts. Dalton Trans. 2020, 49, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Ehm, C.; Vittoria, A.; Goryunov, P.G.; Izmer, V.V.; Kononovich, S.D.; Samsonov, V.O.; Di Girolamo, R.; Budzelaar, H.M.P.; Voskoboynikov, Z.A.; Busico, V.; et al. An Integrated High Throughput Experimentation/Predictive QSAR Modeling Approach to ansa-Zirconocene Catalysts for Isotactic Polypropylene. Polymers 2020, 12, 1005. [Google Scholar] [CrossRef]
- Ehm, C.; Vittoria, A.; Goryunov, G.P.; Izmer, V.V.; Kononovich, D.S.; Samsonov, O.V.; Budzelaar, P.H.M.; Voskoboynikov, A.Z.; Busico, V.; Uborsky, D.V.; et al. On the limits of tuning comonomer affinity of ‘Spaleck-type’ ansa-zirconocenes in ethene/1-hexene copolymerization: A high-throughput experimentation/QSAR approach. Dalton Trans. 2020, 49, 10162–10172. [Google Scholar] [CrossRef] [PubMed]
- Möhring, P.C.; Coville, N.J. Group 4 metallocene polymerisation catalysts: Quantification of ring substituent steric effects. Coord. Chem. Rev. 2006, 250, 18–35. [Google Scholar] [CrossRef]
- Cruz, V.L.; Martinez, S.; Ramos, J.; Martinez-Salazar, J. 3D-QSAR as a Tool for Understanding and Improving Single-Site Polymerization Catalysts. A Review. Organometallics 2014, 33, 2944–2959. [Google Scholar] [CrossRef]
- Baier, M.C.; Zuideveld, M.A.; Mecking, S. Post-Metallocenes in the Industrial Production of Polyolefins. Angew. Chem. Int. Ed. 2014, 53, 9722–9744. [Google Scholar] [CrossRef]
- Klosin, J.; Fontaine, P.P.; Figueroa, R. Development of Group IV Molecular Catalysts for High Temperature Ethylene-α-Olefin Copolymerization Reactions. Acc. Chem. Res. 2015, 48, 2004–2016. [Google Scholar] [CrossRef] [PubMed]
- Fox, T.G.; Flory, P.J. Second-Order Transition Temperatures and Related Properties of Polystyrene. I. Influence of Molecular Weight. J. Appl. Phys. 1950, 21, 581–591. [Google Scholar] [CrossRef]
- Hintermeyer, J.; Herrmann, A.; Kahlau, R.; Goiceanu, C.; Rössler, E.A. Molecular Weight Dependence of Glassy Dynamics in Linear Polymers Revisited. Macromolecules 2008, 41, 9335–9344. [Google Scholar] [CrossRef]
- de Gennes, P.G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1981. [Google Scholar] [CrossRef]
- Rudin, A.; Burgin, D. Effects of molecular weight and chain ends on glass transition of polystyrene. Polymer 1975, 16, 291–297. [Google Scholar] [CrossRef]
- Tsui, O.K.C.; Zhang, H.F. Effects of Chain Ends and Chain Entanglement on the Glass Transition Temperature of Polymer Thin Films. Macromolecules 2001, 34, 9139–9142. [Google Scholar] [CrossRef]
- Stürzel, M.; Mihan, S.; Mülhaupt, R. From Multisite Polymerization Catalysis to Sustainable Materials and All-Polyolefin Composites. Chem. Rev. 2016, 116, 1398–1433. [Google Scholar] [CrossRef] [PubMed]
- Goring, P.D.; Morton, C.; Scott, P. End-functional polyolefins for block copolymer synthesis. Dalton Trans. 2019, 48, 3521–3530. [Google Scholar] [CrossRef]
- Kim, J.; Jung, H.Y.; Park, M.J. End-Group Chemistry and Junction Chemistry in Polymer Science: Past, Present, and Future. Macromolecules 2020. [Google Scholar] [CrossRef] [Green Version]
- Talarico, G.; Budzelaar, P.H.M. Variability of Chain Transfer to Monomer Step in Olefin Polymerization. Organometallics 2008, 27, 4098–4107. [Google Scholar] [CrossRef]
- Stehling, U.; Diebold, J.; Kirsten, R.; Roell, W.; Brintzinger, H.H.; Juengling, S.; Muelhaupt, R.; Langhauser, F. ansa-Zirconocene Polymerization Catalysts with Anelated Ring Ligands—Effects on Catalytic Activity and Polymer Chain Length. Organometallics 1994, 13, 964–970. [Google Scholar] [CrossRef]
- Margl, P.; Deng, L.; Ziegler, T. A Unified View of Ethylene Polymerization by d0 and d0fn Transition Metals. 3. Termination of the Growing Polymer Chain. J. Am. Chem. Soc. 1999, 121, 154–162. [Google Scholar] [CrossRef]
- Jeske, G.; Schock, L.E.; Swepston, P.N.; Schumann, H.; Marks, T.J. Highly reactive organolanthanides. Synthesis, chemistry, and structures of 4f hydrocarbyls and hydrides with chelating bis(polymethylcyclopentadienyl) ligands. J. Am. Chem. Soc. 1985, 107, 8103–8110. [Google Scholar] [CrossRef]
- Bunel, E.; Burger, B.J.; Bercaw, J.E. Carbon-carbon bond activation via. beta.-alkyl elimination. Reversible branching of 1,4-pentadienes catalyzed by scandocene hydride derivatives. J. Am. Chem. Soc. 1988, 110, 976–978. [Google Scholar] [CrossRef]
- Chien, J.C.W.; Wang, B.-P. Metallocene–methylaluminoxane catalysts for olefin polymerization. I. Trimethylaluminum as coactivator. J. Polym. Sci. Part A Polym. Chem. 1988, 26, 3089–3102. [Google Scholar] [CrossRef]
- Resconi, L.; Bossi, S.; Abis, L. Study on the role of methylalumoxane in homogeneous olefin polymerization. Macromolecules 1990, 23, 4489–4491. [Google Scholar] [CrossRef]
- Chadwick, J.C.; Van Kessel, G.M.M.; Sudmeijer, O. Regio- and stereospecificity in propene polymerization with MgCl2-supported Ziegler-Natta catalysts: Effects of hydrogen and the external donor. Macromol. Chem. Phys. 1995, 196, 1431–1437. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R.; Chadwick, J.C.; Modder, J.F.; Sudmeijer, O. Effects of Regiochemical and Stereochemical Errors on the Course of Isotactic Propene Polyinsertion Promoted by Homogeneous Ziegler-Natta Catalysts. Macromolecules 1994, 27, 7538–7543. [Google Scholar] [CrossRef]
- Ehm, C.; Cipullo, R.; Passaro, M.; Zaccaria, F.; Budzelaar, P.H.M.; Busico, V. Chain Transfer to Solvent in Propene Polymerization with Ti Cp-phosphinimide Catalysts: Evidence for Chain Termination via Ti–C Bond Homolysis. ACS Catal. 2016, 6, 7989–7993. [Google Scholar] [CrossRef]
- Ehm, C.; Budzelaar, P.H.M.; Busico, V. Metal–carbon bond strengths under polymerization conditions: 2,1-insertion as a catalyst stress test. J. Catal. 2017, 351, 146–152. [Google Scholar] [CrossRef]
- Zaccaria, F.; Ehm, C.; Budzelaar, P.H.M.; Busico, V.; Cipullo, R. Catalyst Mileage in Olefin Polymerization: The Peculiar Role of Toluene. Organometallics 2018, 37, 2872–2879. [Google Scholar] [CrossRef]
- Zaccaria, F.; Zuccaccia, C.; Cipullo, R.; Budzelaar, P.H.M.; Macchioni, A.; Busico, V.; Ehm, C. Toluene and α-Olefins as Radical Scavengers: Direct NMR Evidence for Homolytic Chain Transfer Mechanism Leading to Benzyl and “Dormant” Titanium Allyl Complexes. Organometallics 2018, 37, 4189–4194. [Google Scholar] [CrossRef]
- Liu, Z.; Yao, C.; Wu, C.; Zhao, Z.; Cui, D. Additive-Triggered Chain Transfer to a Solvent in Coordination Polymerization. Macromolecules 2020, 53, 1205–1211. [Google Scholar] [CrossRef]
- Lin, F.; Liu, Z.; Wang, M.; Liu, B.; Li, S.; Cui, D. Chain Transfer to Toluene in Styrene Coordination Polymerization. Angew. Chem. Int. Ed. 2020, 59, 4324–4328. [Google Scholar] [CrossRef]
- Magee, C.; Sugihara, Y.; Zetterlund, P.B.; Aldabbagh, F. Chain transfer to solvent in the radical polymerization of structurally diverse acrylamide monomers using straight-chain and branched alcohols as solvents. Polym. Chem. 2014, 5, 2259–2265. [Google Scholar] [CrossRef] [Green Version]
- Zetterlund, P.B.; Saka, Y.; McHale, R.; Nakamura, T.; Aldabbagh, F.; Okubo, M. Nitroxide-mediated radical polymerization of styrene: Experimental evidence of chain transfer to monomer. Polymer 2006, 47, 7900–7908. [Google Scholar] [CrossRef]
- Peck, A.N.F.; Hutchinson, R.A. Secondary Reactions in the High-Temperature Free Radical Polymerization of Butyl Acrylate. Macromolecules 2004, 37, 5944–5951. [Google Scholar] [CrossRef]
- Moghadam, N.; Srinivasan, S.; Grady, M.C.; Rappe, A.M.; Soroush, M. Theoretical Study of Chain Transfer to Solvent Reactions of Alkyl Acrylates. J. Phys. Chem. A 2014, 118, 5474–5487. [Google Scholar] [CrossRef]
- Lessard, B.; Tervo, C.; Marić, M. High-Molecular-Weight Poly(tert-butyl acrylate) by Nitroxide-Mediated Polymerization: Effect of Chain Transfer to Solvent. Macromol. React. Eng. 2009, 3, 245–256. [Google Scholar] [CrossRef]
- Tan, S.; Li, J.; Zhang, Z. Study of Chain Transfer Reaction to Solvents in the Initiation Stage of Atom Transfer Radical Polymerization. Macromolecules 2011, 44, 7911–7916. [Google Scholar] [CrossRef]
- Kukulj, D.; Davis, T.P. Mechanism of catalytic chain transfer in the free-radical polymerisation of methyl methacrylate and styrene. Macromol. Chem. Phys. 1998, 199, 1697–1708. [Google Scholar] [CrossRef]
- Stephan, D.W.; Stewart, J.C.; Guérin, F.; Spence, R.E.V.H.; Xu, W.; Harrison, D.G. Phosphinimides as a Steric Equivalent to Cyclopentadienyl: An Approach to Ethylene Polymerization Catalyst Design. Organometallics 1999, 18, 1116–1118. [Google Scholar] [CrossRef]
- Rios, I.G.; Novarino, E.; van der Veer, S.; Hessen, B.; Bouwkamp, M.W. Amine Catalyzed Solvent C−H Bond Activation as Deactivation Route for Cationic Decamethylzirconocene Olefin Polymerization Catalysts. J. Am. Chem. Soc. 2009, 131, 16658–16659. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Dash, A.K.; Jordan, R.F. Structures and Reactivity of Zr(IV) Chlorobenzene Complexes. J. Am. Chem. Soc. 2004, 126, 15360–15361. [Google Scholar] [CrossRef]
- Ghiotto, F.; Pateraki, C.; Severn, J.R.; Friederichs, N.; Bochmann, M. Rapid evaluation of catalysts and MAO activators by kinetics: What controls polymer molecular weight and activity in metallocene/MAO catalysts? Dalton Trans. 2013, 42, 9040–9048. [Google Scholar] [CrossRef]
- Desert, X.; Carpentier, J.-F.; Kirillov, E. Quantification of active sites in single-site group 4 metal olefin polymerization catalysis. Coord. Chem. Rev. 2019, 386, 50–68. [Google Scholar] [CrossRef]
- van Doremaele, G.; van Duin, M.; Valla, M.; Berthoud, A. On the Development of Titanium κ1-Amidinate Complexes, Commercialized as Keltan ACE™ Technology, Enabling the Production of an Unprecedented Large Variety of EPDM Polymer Structures. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 2877–2891. [Google Scholar] [CrossRef] [Green Version]
- McKnight, A.L.; Waymouth, R.M. Group 4 ansa-Cyclopentadienyl-Amido Catalysts for Olefin Polymerization. Chem. Rev. 1998, 98, 2587–2598. [Google Scholar] [CrossRef]
- Ferreira, M.J.; Martins, A.M. Group 4 ketimide complexes: Synthesis, reactivity and catalytic applications. Coord. Chem. Rev. 2006, 250, 118–132. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R.; Cutillo, F.; Friederichs, N.; Ronca, S.; Wang, B. Improving the Performance of Methylalumoxane: A Facile and Efficient Method to Trap “Free” Trimethylaluminum. J. Am. Chem. Soc. 2003, 125, 12402–12403. [Google Scholar] [CrossRef] [PubMed]
- Zaccaria, F.; Zuccaccia, C.; Cipullo, R.; Budzelaar, P.H.M.; Macchioni, A.; Busico, V.; Ehm, C. BHT-Modified MAO: Cage Size Estimation, Chemical Counting of Strongly Acidic Al Sites, and Activation of a Ti-Phosphinimide Precatalyst. ACS Catal. 2019, 9, 2996–3010. [Google Scholar] [CrossRef]
- Zaccaria, F.; Zuccaccia, C.; Cipullo, R.; Budzelaar, P.H.M.; Macchioni, A.; Busico, V.; Ehm, C. On the nature of the Lewis acidic sites in ‘TMA-free’ phenol-modified Methylaluminoxane. Eur. J. Inorg. Chem. 2020, 2020, 1088–1095. [Google Scholar] [CrossRef]
- Busico, V.; Cipullo, R.; Romanelli, V.; Ronca, S.; Togrou, M. Reactivity of Secondary Metal−Alkyls in Catalytic Propene Polymerization: How Dormant Are “Dormant Chains”? J. Am. Chem. Soc. 2005, 127, 1608–1609. [Google Scholar] [CrossRef]
- Landis, C.R.; Sillars, D.R.; Batterton, J.M. Reactivity of Secondary Metallocene Alkyls and the Question of Dormant Sites in Catalytic Alkene Polymerization. J. Am. Chem. Soc. 2004, 126, 8890–8891. [Google Scholar] [CrossRef] [PubMed]
- Flisak, Z.; Ziegler, T. “Dormant” secondary metal-alkyl complexes are not omnipresent. Proc. Natl. Acad. Sci. USA 2006, 103, 15338–15342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, F.; Cannon, R.D.; Bochmann, M. Zirconocene-Catalyzed Propene Polymerization: A Quenched-Flow Kinetic Study. J. Am. Chem. Soc. 2003, 125, 7641–7653. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.E.; Baxter, S.M.; Bulls, A.R.; Burger, B.J.; Nolan, M.C.; Santarsiero, B.D.; Schaefer, W.P.; Bercaw, J.E. σ-Bond metathesis for carbon-hydrogen bonds of hydrocarbons and Sc-R (R = H, alkyl, aryl) bonds of permethylscandocene derivatives. Evidence for noninvolvement of the. pi. system in electrophilic activation of aromatic and vinylic C-H bonds. J. Am. Chem. Soc. 1987, 109, 203–219. [Google Scholar] [CrossRef]
- Butschke, B.; Schwarz, H. Thermal C−H Bond Activation of Benzene, Toluene, and Methane with Cationic [M(X)(bipy)]+ (M = Ni, Pd, Pt; X = CH3, Cl; bipy = 2,2′-bipyridine): A Mechanistic Study. Organometallics 2011, 30, 1588–1598. [Google Scholar] [CrossRef]
- Henrici-olivé, G.; Olivé, S. The Active Species in Homogeneous Ziegler-Natta Catalysts for the Polymerization of Ethylene. Angew. Chem. Int. Ed. Engl. 1967, 6, 790–798. [Google Scholar] [CrossRef]
- Machat, M.R.; Jandl, C.; Rieger, B. Titanocenes in Olefin Polymerization: Sustainable Catalyst System or an Extinct Species? Organometallics 2017, 36, 1408–1418. [Google Scholar] [CrossRef]
- Machat, M.R.; Fischer, A.; Schmitz, D.; Vöst, M.; Drees, M.; Jandl, C.; Pöthig, A.; Casati, N.P.M.; Scherer, W.; Rieger, B. Behind the Scenes of Group 4 Metallocene Catalysis: Examination of the Metal–Carbon Bond. Organometallics 2018, 37, 2690–2705. [Google Scholar] [CrossRef]
- Flisak, Z. Thermodynamics of Titanium and Vanadium Reduction in Non-Aqueous Environment Calculated at Various Levels of Theory. J. Phys. Chem. A 2012, 116, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Simoes, J.A.M.; Beauchamp, J.L. Transition metal-hydrogen and metal-carbon bond strengths: The keys to catalysis. Chem. Rev. 1990, 90, 629–688. [Google Scholar] [CrossRef]
- Reints, W.; Pratt, D.A.; Korth, H.-G.; Mulder, P. O−O Bond Dissociation Enthalpy in Di(trifluoromethyl) Peroxide (CF3OOCF3) as Determined by Very Low Pressure Pyrolysis. Density Functional Theory Computations on O−O and O−H Bonds in (Fluorinated) Derivatives. J. Phys. Chem. A 2000, 104, 10713–10720. [Google Scholar] [CrossRef]
- Fong, A.; Peters, B.; Scott, S.L. One-Electron-Redox Activation of the Reduced Phillips Polymerization Catalyst, via Alkylchromium(IV) Homolysis: A Computational Assessment. ACS Catal. 2016, 6, 6073–6085. [Google Scholar] [CrossRef]
- Falivene, L.; Credendino, R.; Poater, A.; Petta, A.; Serra, L.; Oliva, R.; Scarano, V.; Cavallo, L. SambVca 2. A Web Tool for Analyzing Catalytic Pockets with Topographic Steric Maps. Organometallics 2016, 35, 2286–2293. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.R. Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Rayton, FL, USA, 2007. [Google Scholar]
- Babushkin, D.E.; Panchenko, V.N.; Brintzinger, H.-H. Zirconium Allyl Complexes as Participants in Zirconocene-Catalyzed α-Olefin Polymerizations. Angew. Chem. Int. Ed. 2014, 53, 9645–9649. [Google Scholar] [CrossRef] [PubMed]
- Landis, C.R.; Christianson, M.D. Metallocene-catalyzed alkene polymerization and the observation of Zr-allyls. Proc. Natl. Acad. Sci. USA 2006, 103, 15349–15354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauriol, F.; Wong, E.; Leung, A.M.H.; Donaghue, I.E.; Baird, M.C.; Wondimagegn, T.; Ziegler, T. Structures and Properties of Nonchelated, d0 Alkyl Alkene Complexes of the Type [Cp2ZrMe(alkene)]+: Elusive Intermediates during Ziegler-Natta Polymerizations of Alkenes. Angew. Chem. Int. Ed. 2009, 48, 3342–3345. [Google Scholar] [CrossRef] [PubMed]
- Moscardi, G.; Resconi, L.; Cavallo, L. Propene Polymerization with the Isospecific, Highly Regioselective rac-Me2C(3-t-Bu-1-Ind)2ZrCl2/MAO Catalyst. 2. Combined DFT/MM Analysis of Chain Propagation and Chain Release Reactions. Organometallics 2001, 20, 1918–1931. [Google Scholar] [CrossRef]
- Vatamanu, M. Observation of zirconium allyl species formed during zirconocene-catalyzed propene polymerization and mechanistic insights. J. Catal. 2015, 323, 112–120. [Google Scholar] [CrossRef]
- Liu, B.; Wang, L.; Wu, C.; Cui, D. Sequence-controlled ethylene/styrene copolymerization catalyzed by scandium complexes. Polym. Chem. 2019, 10, 235–243. [Google Scholar] [CrossRef]
- Dong, S.; Ou, Y.; Zhang, G. Preparations and Applications of the Brominated Polystyrenes with Low Molecular Weight. J. Funct. Polym. 1997, 10, 87–89. [Google Scholar]
- Liu, D.T.; Yao, C.G.; Wang, R.; Wang, M.Y.; Wang, Z.C.; Wu, C.J.; Lin, F.; Li, S.H.; Wan, X.H.; Cui, D.M. Highly Isoselective Coordination Polymerization of ortho-Methoxystyrene with β-Diketiminato Rare-Earth-Metal Precursors. Angew. Chem. Int. Ed. 2015, 54, 5205–5209. [Google Scholar] [CrossRef]
- Lenton, T.N.; Bercaw, J.E.; Panchenko, V.N.; Zakharov, V.A.; Babushkin, D.E.; Soshnikov, I.E.; Talsi, E.P.; Brintzinger, H.H. Formation of Trivalent Zirconocene Complexes from ansa-Zirconocene-Based Olefin-Polymerization Precatalysts: An EPR- and NMR-Spectroscopic Study. J. Am. Chem. Soc. 2013, 135, 10710–10719. [Google Scholar] [CrossRef]
- Yamamoto, A.; Nishiura, M.; Yang, Y.; Hou, Z. Cationic Scandium Anisyl Species in Styrene Polymerization Using Anisole and N,N-Dimethyl-o-toluidine as Chain-Transfer Agents. Organometallics 2017, 36, 4635–4642. [Google Scholar] [CrossRef]
- Teator, A.J.; Lastovickova, D.N.; Bielawski, C.W. Switchable Polymerization Catalysts. Chem. Rev. 2016, 116, 1969–1992. [Google Scholar] [CrossRef]
- Grassi, A.; Zambelli, A.; Laschi, F. Reductive Decomposition of Cationic Half-Titanocene(IV) Complexes, Precursors of the Active Species in Syndiospecific Styrene Polymerization. Organometallics 1996, 15, 480–482. [Google Scholar] [CrossRef]
- Nomura, K.; Izawa, I.; Yi, J.; Nakatani, N.; Aoki, H.; Harakawa, H.; Ina, T.; Mitsudome, T.; Tomotsu, N.; Yamazoe, S. Solution XAS Analysis for Exploring Active Species in Syndiospecific Styrene Polymerization and 1-Hexene Polymerization Using Half-Titanocene–MAO Catalysts: Significant Changes in the Oxidation State in the Presence of Styrene. Organometallics 2019. [Google Scholar] [CrossRef]
- Bouyahyi, M.; Turki, Y.; Tanwar, A.; Jasinska-Walc, L.; Duchateau, R. Randomly Functionalized Polyethylenes: In Quest of Avoiding Catalyst Deactivation. ACS Catal. 2019, 9, 7779–7790. [Google Scholar] [CrossRef]
- Poli, R. 3.11—Organometallic-Mediated Radical Polymerization. In Polymer Science: A Comprehensive Reference; Matyjaszewski, K., Möller, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 351–375. [Google Scholar] [CrossRef]
- Allan, L.E.N.; Perry, M.R.; Shaver, M.P. Organometallic mediated radical polymerization. Prog. Polym. Sci. 2012, 37, 127–156. [Google Scholar] [CrossRef] [Green Version]
- Asandei, A.D.; Moran, I.W. TiCp2Cl-Catalyzed Living Radical Polymerization of Styrene Initiated by Oxirane Radical Ring Opening. J. Am. Chem. Soc. 2004, 126, 15932–15933. [Google Scholar] [CrossRef]
- Asandei, A.D.; Moran, I.W. The ligand effect in Ti-mediated living radical styrene polymerizations initiated by epoxide radical ring opening. 2. Scorpionate and half-sandwich LTiCl3 complexes. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 6039–6047. [Google Scholar] [CrossRef]
- Asandei, A.D.; Saha, G. Cp2TiCl-Catalyzed Epoxide Radical Ring Opening: A New Initiating Methodology for Graft Copolymer Synthesis. Macromolecules 2006, 39, 8999–9009. [Google Scholar] [CrossRef]
- Stadler, S.M.; Göttker-Schnetmann, I.; Mecking, S. Incorporation of Radicals during Ni(II)-Catalyzed Ethylene Insertion Polymerization. ACS Catal. 2019, 9, 2760–2767. [Google Scholar] [CrossRef]
- Stadler, S.M.; Göttker-Schnetmann, I.; Fuchs, A.S.; Fischer, S.R.R.; Mecking, S. Catalytic Chain Transfer Polymerization to Functional Reactive End Groups for Controlled Free Radical Growth. Macromolecules 2020, 53, 2362–2368. [Google Scholar] [CrossRef]
- Ölscher, F.; Göttker-Schnetmann, I.; Monteil, V.; Mecking, S. Role of Radical Species in Salicylaldiminato Ni(II) Mediated Polymer Chain Growth: A Case Study for the Migratory Insertion Polymerization of Ethylene in the Presence of Methyl Methacrylate. J. Am. Chem. Soc. 2015, 137, 14819–14828. [Google Scholar] [CrossRef] [PubMed]
- Keyes, A.; Basbug Alhan, H.E.; Ordonez, E.; Ha, U.; Beezer, D.B.; Dau, H.; Liu, Y.-S.; Tsogtgerel, E.; Jones, G.R.; Harth, E. Olefins and Vinyl Polar Monomers: Bridging the Gap for Next Generation Materials. Angew. Chem. Int. Ed. 2019, 58, 12370–12391. [Google Scholar] [CrossRef] [PubMed]
- Dau, H.; Keyes, A.; Basbug Alhan, H.E.; Ordonez, E.; Tsogtgerel, E.; Gies, A.P.; Auyeung, E.; Zhou, Z.; Maity, A.; Das, A.; et al. Dual Polymerization Pathway for Polyolefin-Polar Block Copolymer Synthesis via MILRad: Mechanism and Scope. J. Am. Chem. Soc. 2020, 142, 21469–21483. [Google Scholar] [CrossRef] [PubMed]
- Manßen, M.; Schafer, L.L. Titanium catalysis for the synthesis of fine chemicals—Development and trends. Chem. Soc. Rev. 2020, 49, 6947–6994. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Piers, W.E.; Gao, Y.; Parvez, M. Isolation and Characterization of a Monomeric Cationic Titanium Hydride. J. Am. Chem. Soc. 2004, 126, 5668–5669. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Piers, W.E.; Parvez, M. Competitive ArC−H and ArC−X (X = Cl, Br) Activation in Halobenzenes at Cationic Titanium Centers. J. Am. Chem. Soc. 2006, 128, 3303–3312. [Google Scholar] [CrossRef]
- Sato, F.; Urabe, H.; Okamoto, S. Synthesis of Organotitanium Complexes from Alkenes and Alkynes and Their Synthetic Applications. Chem. Rev. 2000, 100, 2835–2886. [Google Scholar] [CrossRef]
- Carpenetti, D.W. Models of intermediates in metallocene-catalyzed alkene polymerizations: Observation of a d0 cationic titanium–alkyl-alkene complex and decomposition by β-allyl elimination. Inorg. Chem. Commun. 2003, 6, 1287–1290. [Google Scholar] [CrossRef]
- Sauriol, F.; Sonnenberg, J.F.; Chadder, S.J.; Dunlop-Brière, A.F.; Baird, M.C.; Budzelaar, P.H.M. Remarkable Reactions and Intermediates in Titanocene(IV) Chemistry: Migratory Insertion Reactions of 2,2-Disubstituted-1-alkenes, Intramolecular 1,5-σ Bond Metathesis via ε-Agostic Interactions, and a Rare Example of a β-Agostic Alkyltitanocene Complex. J. Am. Chem. Soc. 2010, 132, 13357–13370. [Google Scholar] [CrossRef]
- Li, F.; Lin, S.; Chen, Y.; Shi, C.; Yan, H.; Li, C.; Wu, C.; Lin, L.; Duan, C.; Shi, L. Photocatalytic Generation of π-allyltitanium Complexes via Radical Intermediates. Angew. Chem. Int. Ed. 2020. [Google Scholar] [CrossRef]
- van der Heijden, H.; Hessen, B.; Orpen, A.G. A Zwitterionic Zirconocene Alkyl Complex as a Single-Component α-Olefin Dimerization Catalyst. J. Am. Chem. Soc. 1998, 120, 1112–1113. [Google Scholar] [CrossRef] [Green Version]
- Horton, A.D. Direct Observation of β-Methyl Elimination in Cationic Neopentyl Complexes: Ligand Effects on the Reversible Elimination of Isobutene. Organometallics 1996, 15, 2675–2677. [Google Scholar] [CrossRef]
- Vatamanu, M. Synthesis, Structures, and Dynamic Features of d0 Zirconocene–Allyl Complexes. Organometallics 2014, 33, 3683–3694. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Cat. | Mnb | Mwb | Ð | %SAT c | %UNSAT c | %Bn c | |
|---|---|---|---|---|---|---|---|---|
| Total | nBu | |||||||
| 1 | 1′-Ph | 3.3 | 6.4 | 2.0 | 160 | 79 | 19 | 21 |
| 2 | 1′-Cy | 1.6 | 2.7 | 1.7 | 137 | 30 | 54 | 9 |
| 3 d | 1′-tBu | 1.4 | 1.6 | 1.2 | 127 | 23 | 77 | n.d. e |
| 4 | 1-Ph | 6.4 | 13 | 2.0 | 179 | 40 | 16 | 5 |
| 5 | 1-Cy | 4.1 | 7.8 | 1.9 | 168 | 58 | 19 | 14 |
| 6 | 1-tBu | 2.3 | 3.9 | 1.8 | 165 | 69 | 26 | 9 |
| 7 | 2 | 5.9 | 13 | 2.2 | 131 | 69 | 19 | 50 |
| 8 | 3 | 4.1 | 7.4 | 1.8 | 148 | 49 | 48 | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaccaria, F.; Budzelaar, P.H.M.; Zuccaccia, C.; Cipullo, R.; Macchioni, A.; Busico, V.; Ehm, C. Chain Transfer to Solvent and Monomer in Early Transition Metal Catalyzed Olefin Polymerization: Mechanisms and Implications for Catalysis. Catalysts 2021, 11, 215. https://doi.org/10.3390/catal11020215
Zaccaria F, Budzelaar PHM, Zuccaccia C, Cipullo R, Macchioni A, Busico V, Ehm C. Chain Transfer to Solvent and Monomer in Early Transition Metal Catalyzed Olefin Polymerization: Mechanisms and Implications for Catalysis. Catalysts. 2021; 11(2):215. https://doi.org/10.3390/catal11020215
Chicago/Turabian StyleZaccaria, Francesco, Peter H. M. Budzelaar, Cristiano Zuccaccia, Roberta Cipullo, Alceo Macchioni, Vincenzo Busico, and Christian Ehm. 2021. "Chain Transfer to Solvent and Monomer in Early Transition Metal Catalyzed Olefin Polymerization: Mechanisms and Implications for Catalysis" Catalysts 11, no. 2: 215. https://doi.org/10.3390/catal11020215
APA StyleZaccaria, F., Budzelaar, P. H. M., Zuccaccia, C., Cipullo, R., Macchioni, A., Busico, V., & Ehm, C. (2021). Chain Transfer to Solvent and Monomer in Early Transition Metal Catalyzed Olefin Polymerization: Mechanisms and Implications for Catalysis. Catalysts, 11(2), 215. https://doi.org/10.3390/catal11020215