
Electrocatalysis by Heme Enzymes—Applications in Biosensing


Abstract
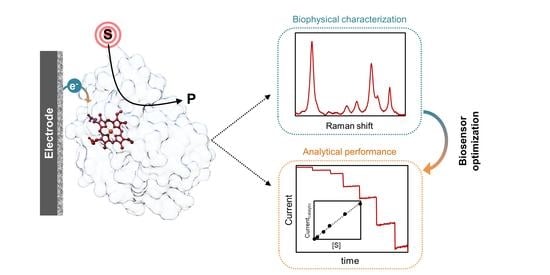
:
1. Introduction
2. Amperometric Enzyme-Based Biosensors
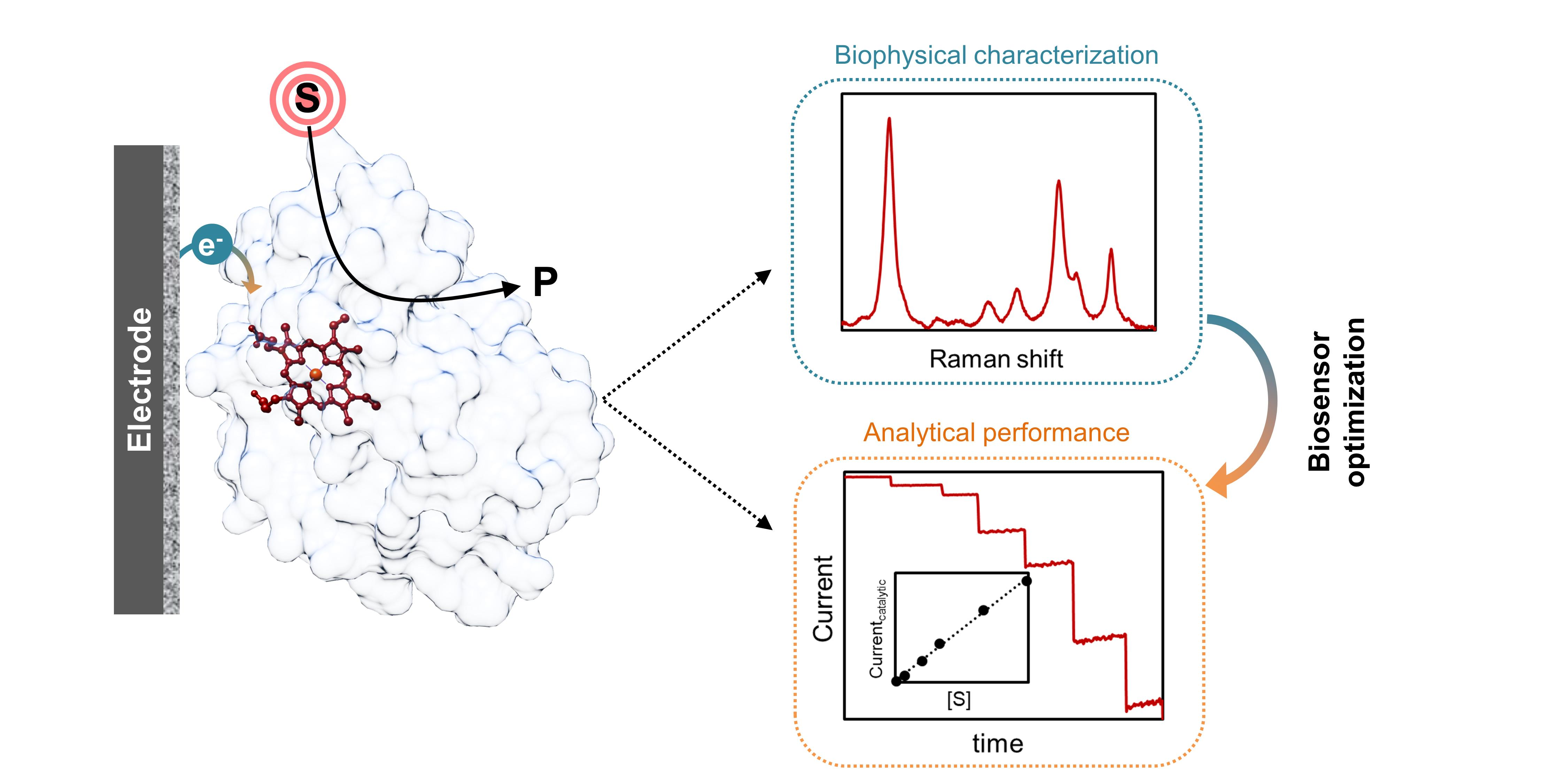
2.1. Electron Transfer in Enzyme Biosensors
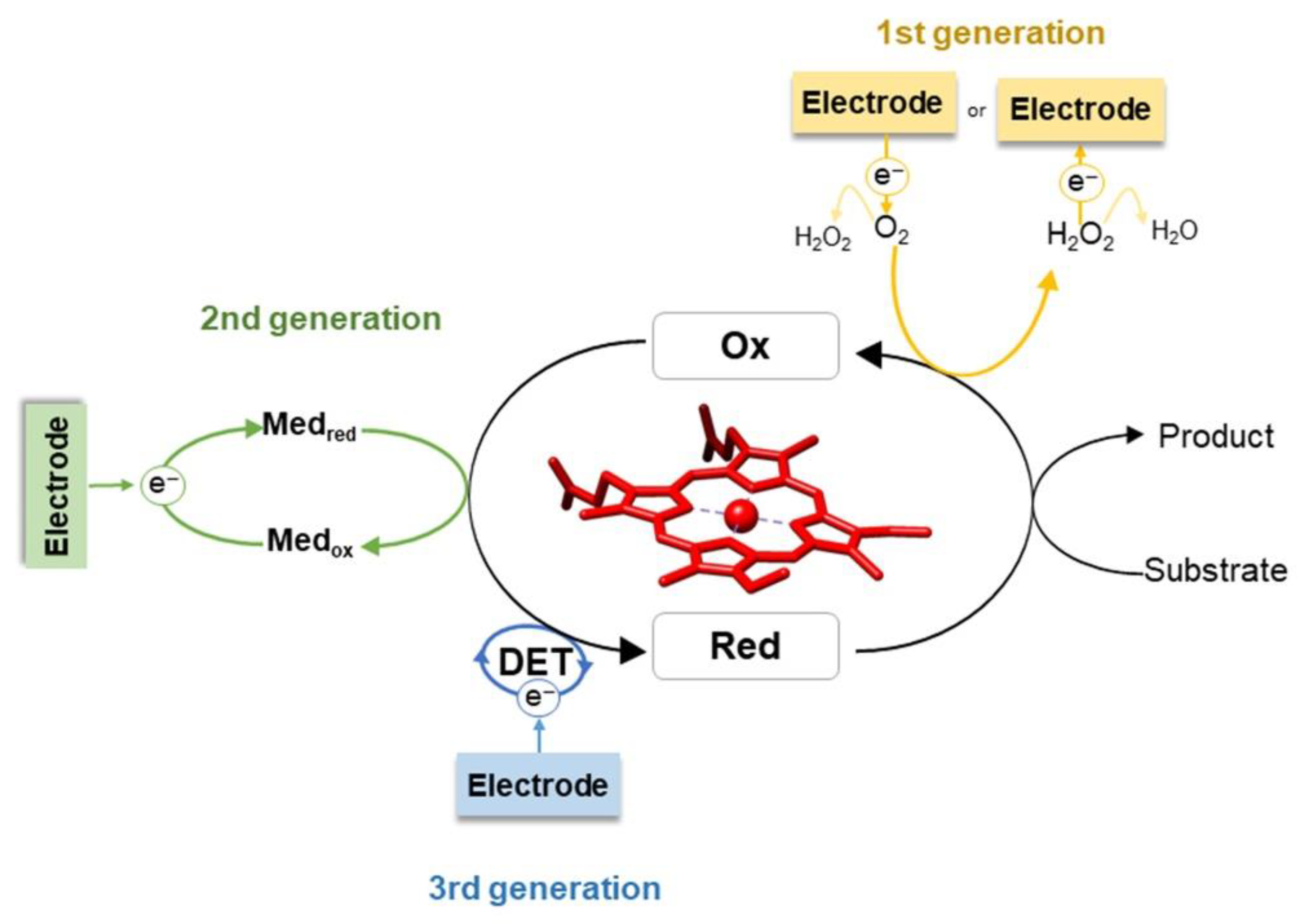
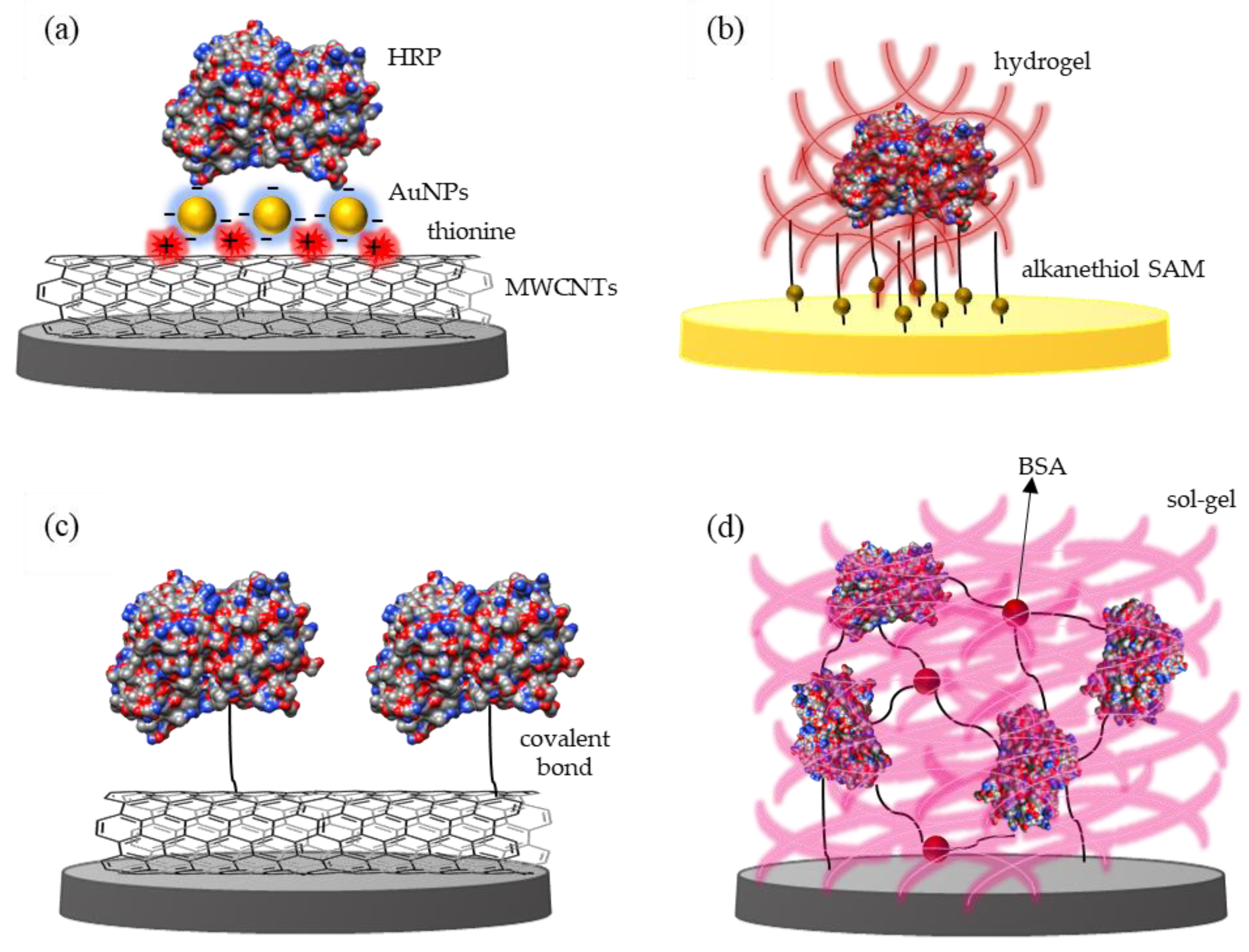
2.2. Enzyme Immobilization
2.3. Biophysical Characterization of Amperometric Enzyme Biosensors
2.3.1. Biosensor Performance
2.3.2. Structural Characterization of Immobilized Proteins
3. Biosensors Based on Heme Proteins and Enzymes
3.1. Peroxidases
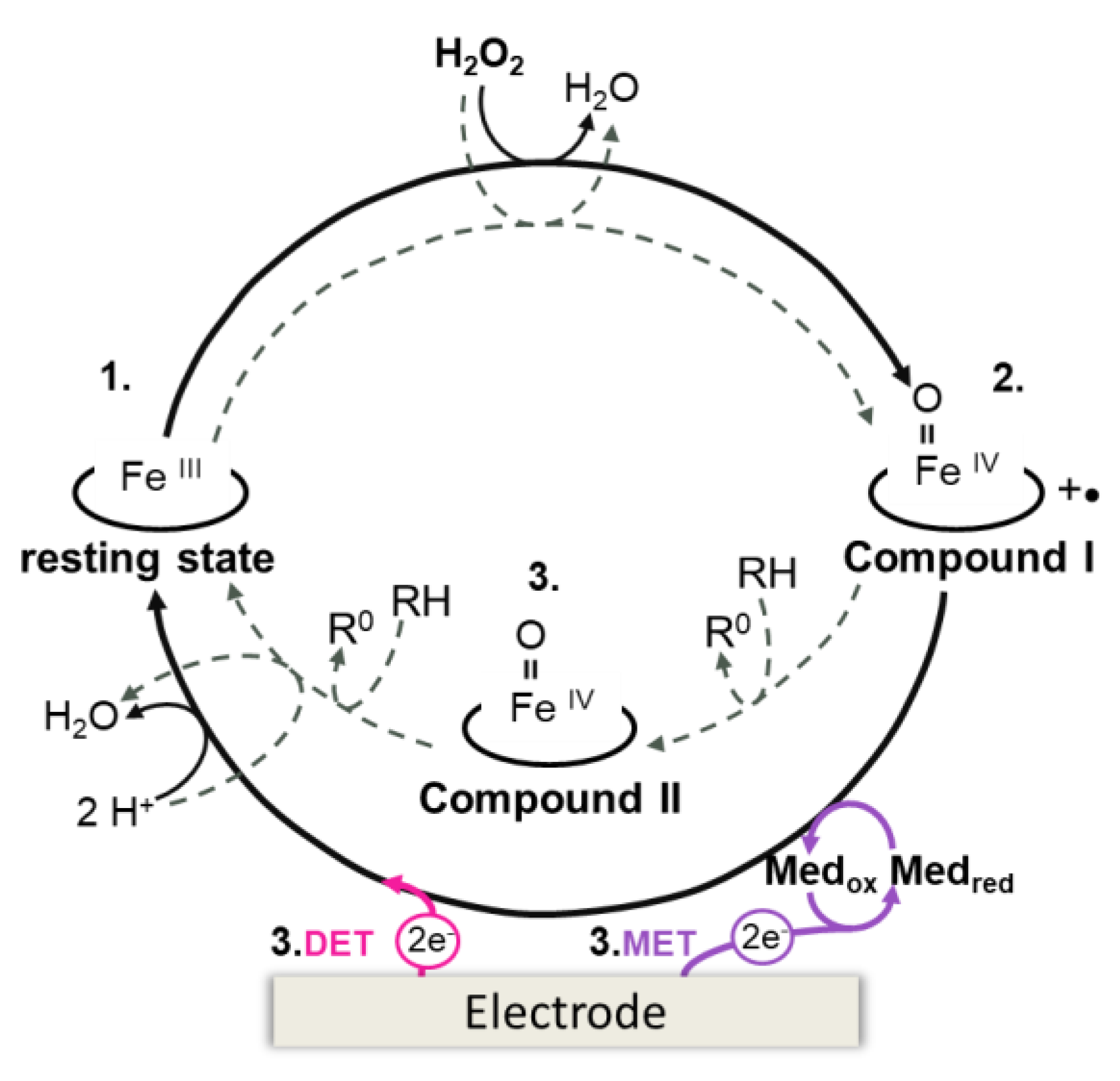
3.1.1. Detection Modes in Peroxidase-Based Biosensors
3.1.2. Plant Peroxidases
3.1.3. Dye-Decolorizing Peroxidases
3.1.4. Cytochrome c Peroxidases
3.2. Cytochromes P450
3.3. Catalases
3.4. Nitrite Reductases
3.5. Cytochrome c Oxidases
3.6. Cytochrome c and Microperoxidases
3.7. Globins
4. Outlook
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5c | penta-coordinated |
| 6c | hexa-coordinated |
| AFM | atomic force microscopy |
| AQS | anthraquinone disulfonate |
| BSA | bovine serum albumin |
| ccNiR | cytochrome c nitrite reductase |
| CcO | cytochrome c oxidase |
| CcP | cytochrome c peroxidase |
| CD | circular dichroism |
| cd1NiR | cytochrome cd1 nitrite reductase |
| CNT | carbon nanotube |
| CT | charge-transfer |
| CV | cyclic voltammetry |
| cyt c | cytochrome c |
| DDAB | didodecyldimethylammonium bromide |
| DET | direct electron transfer |
| DyP | dye-decolorizing peroxidase |
| EDC | 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide |
| ET | electron transfer |
| FTIR | Fourier-transform infrared |
| GC | glassy carbon |
| GOx | glucose oxidase |
| Hb | hemoglobin |
| HRP | horseradish peroxidase |
| IL | ionic liquid |
| ITO | indium tin oxide |
| LDH | layered double hydroxide |
| LOD | limit of detection |
| Mb | myoglobin |
| MET | mediated electron transfer |
| MP | microperoxidase |
| MWCNTs | multi-walled carbon nanotubes |
| NHS | N-hydroxysuccinimide |
| NiR | nitrite reductase |
| NPs | nanoparticles |
| P420 | cytochrome P420 |
| P450 | cytochrome P450 |
| PANI | polyaniline |
| PG | pyrolytic graphite |
| PPY | polypyrrole |
| ROS | reactive oxygen species |
| RR | resonance Raman |
| SAM | self-assembled monolayer |
| SEIRA | surface-enhanced infrared absorption |
| SEM | scanning electron microscopy |
| SERR | surface-enhanced resonance Raman |
| SPEs | screen-printed electrodes |
| SWCNTs | single-walled carbon nanotubes |
| TCA | trichloroacetic acid |
| TOP | tobacco peroxidase |
References
- Poulos, T.L. Heme Enzyme Structure and Function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [Green Version]
- Spiro, T.G.; Jarzecki, A.A. Heme-based sensors: Theoretical modeling of heme-ligand-protein interactions. Curr. Opin. Chem. Biol. 2001, 5, 715–723. [Google Scholar] [CrossRef]
- Turner, A.P.F. Biosensors: Sense and sensibility. Chem. Soc. Rev. 2013, 42, 3184–3196. [Google Scholar] [CrossRef] [Green Version]
- Labib, M.; Sargent, E.H.; Kelley, S.O. Electrochemical Methods for the Analysis of Clinically Relevant Biomolecules. Chem. Rev. 2016, 116, 9001–9090. [Google Scholar] [CrossRef]
- Monteiro, T.; Almeida, M.G. Electrochemical Enzyme Biosensors Revisited: Old Solutions for New Problems. Crit. Rev. Anal. Chem. 2019, 49, 44–66. [Google Scholar] [CrossRef]
- Sekretaryova, A.N.; Eriksson, M.; Turner, A.P. Bioelectrocatalytic systems for health applications. Biotechnol. Adv. 2016, 34, 177–197. [Google Scholar] [CrossRef]
- Ronkainen, N.J.; Halsall, H.B.; Heineman, W.R. Electrochemical biosensors. Chem. Soc. Rev. 2010, 39, 1747–1763. [Google Scholar] [CrossRef]
- Thevenot, D.R.; Toth, K.; Durst, R.A.; Wilson, G.S. Electrochemical Biosensors: Recommended Definitions and Classification. Biosens. Bioelectron. 2001, 16, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Elsebai, B.; Ghica, M.E.; Abbas, M.N.; Brett, C.M.A. Catalase Based Hydrogen Peroxide Biosensor for Mercury Determination by Inhibition Measurements. J. Hazard. Mater. 2017, 340, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Habermuller, K.; Mosbach, M.; Schuhmann, W. Electron-Transfer Mechanisms in Amperometric Biosensors. Fresenius J. Anal. Chem. 2000, 366, 560–568. [Google Scholar] [CrossRef]
- Gorton, L.; Lindgren, A.; Larsson, T.; Munteanu, F.D.; Ruzgas, T.; Gazaryan, I. Direct Electron Transfer between Heme-Containing Enzymes and Electrodes as Basis for Third Generation Biosensors. Anal. Chim. Acta 1999, 400, 91–108. [Google Scholar] [CrossRef]
- Clark, L.C., Jr.; Lyons, C. Electrode Systems for Continuous Monitoring In Cardiovascular Surgery. Ann. N. Y. Acad. Sci. 1962, 102, 29–45. [Google Scholar] [CrossRef]
- Wang, J. Electrochemical Glucose Biosensors. Chem. Rev. 2008, 108, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Heller, A.; Feldman, B. Electrochemical Glucose Sensors and Their Applications in Diabetes Management. Chem. Rev. 2008, 108, 2482–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, P.; Das, M.; Chinnadayyala, S.R.; Singha, I.M.; Goswami, P. Recent Advances on Developing 3rd Generation Enzyme Electrode for Biosensor Applications. Biosens. Bioelectron. 2016, 79, 386–397. [Google Scholar] [CrossRef]
- Chaubey, A.; Malhotra, B.D. Mediated Biosensors. Biosens. Bioelectron. 2002, 17, 441–456. [Google Scholar] [CrossRef]
- Silveira, C.M.; Almeida, M.G. Small Electron-Transfer Proteins as Mediators in Enzymatic Electrochemical Biosensors. Anal. Bioanal. Chem. 2013, 405, 3619–3635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astier, Y.; Canters, G.W.; Davis, J.J.; Hill, H.A.O.; Verbeet, M.P.; Wijma, H.J. Sensing Nitrite through a Pseudoazurin–Nitrite Reductase Electron Transfer Relay. ChemPhysChem 2005, 6, 1114–1120. [Google Scholar] [CrossRef]
- Serra, A.S.; Jorge, S.R.; Silveira, C.M.; Moura, J.J.G.; Jubete, E.; Ochoteco, E.; Cabañero, G.; Grande, H.; Almeida, M.G. Cooperative Use of Cytochrome Cd1 Nitrite Reductase and Its Redox Partner Cytochrome C552 to Improve the Selectivity of Nitrite Biosensing. Anal. Chim. Acta 2011, 693, 41–46. [Google Scholar] [CrossRef]
- Da Silva, S.; Cosnier, S.; Almeida, M.G.; Moura, J.J.G. An Efficient Poly(Pyrrole–Viologen)-Nitrite Reductase Biosensor for the Mediated Detection of Nitrite. Electrochem. Commun. 2004, 6, 404–408. [Google Scholar] [CrossRef]
- Cosnier, S.; Da Silva, S.; Shan, D.; Gorgy, K. Electrochemical Nitrate Biosensor Based on Poly(Pyrrole–Viologen) Film–Nitrate Reductase–Clay Composite. Bioelectrochemistry 2008, 74, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Schuhmann, W. Electron-Transfer Pathways in Amperometric Biosensors. Ferrocene-Modified Enzymes Entrapped in Conducting-Polymer Layers. Biosens. Bioelectron. 1995, 10, 181–193. [Google Scholar] [CrossRef]
- Willner, I.; Willner, B.; Katz, E. Functional Biosensor Systems via Surface-Nanoengineering of Electronic Elements. Rev. Mol. Biotechnol. 2002, 82, 325–355. [Google Scholar] [CrossRef]
- Fruk, L.; Kuo, C.-H.; Torres, E.; Niemeyer, C.M. Apoenzyme Reconstitution as a Chemical Tool for Structural Enzymology and Biotechnology. Angew. Chem. Int. Ed. 2009, 48, 1550–1574. [Google Scholar] [CrossRef]
- Nöll, T.; Nöll, G. Strategies for “Wiring” Redox-Active Proteins to Electrodes and Applications in Biosensors, Biofuel Cells, and Nanotechnology. Curr. Soc. Rev. 2011, 40, 3564–3576. [Google Scholar] [CrossRef]
- Léger, C.; Bertrand, P. Direct Electrochemistry of Redox Enzymes as a Tool for Mechanistic Studies. Chem. Rev. 2008, 108, 2379–2438. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chakraborty, S.; Hosseinzadeh, P.; Yu, Y.; Tian, S.; Petrik, I.; Bhagi, A.; Lu, Y. Metalloproteins Containing Cytochrome, Iron–Sulfur, or Copper Redox Centers. Chem. Rev. 2014, 114, 4366–4469. [Google Scholar] [CrossRef]
- Borgmann, S.; Hartwich, G.; Schulte, A.; Schuhmann, W. Amperometric Enzyme Sensors based on Direct and Mediated Electron Transfer. In Perspectives in Bioanalysis; Paleček, E., Scheller, F., Wang, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 1, pp. 599–655. ISBN 1871-0069. [Google Scholar]
- Bollella, P.; Gorton, L. Enzyme Based Amperometric Biosensors. Curr. Opin. Electrochem. 2018, 10, 157–173. [Google Scholar] [CrossRef]
- Taurino, I.; Sanzò, G.; Antiochia, R.; Tortolini, C.; Mazzei, F.; Favero, G.; De Micheli, G.; Carrara, S. Recent Advances in Third Generation Biosensors Based on Au and Pt Nanostructured Electrodes. TrAC Trends Anal. Chem. 2016, 79, 151–159. [Google Scholar] [CrossRef]
- Redeker, E.S.; Ta, D.T.; Cortens, D.; Billen, B.; Guedens, W.; Adriaensens, P. Protein Engineering for Directed Immobilization. Bioconjug. Chem. 2013, 24, 1761–1777. [Google Scholar] [CrossRef] [PubMed]
- Bollella, P.; Katz, E. Chapter Ten—Bioelectrocatalysis at carbon nanotubes. In Methods Enzymology; Kumar, C.V., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 630, pp. 215–247. ISBN 0076-6879. [Google Scholar]
- Mazurenko, I.; Hitaishi, V.P.; Lojou, E. Recent Advances in Surface Chemistry of Electrodes to Promote Direct Enzymatic Bioelectrocatalysis. Curr. Opin. Electrochem. 2020, 19, 113–121. [Google Scholar] [CrossRef]
- Ju, H. Functional Nanomaterials and Nanoprobes for Amplified Biosensing. Appl. Mater. Today 2018, 10, 51–71. [Google Scholar] [CrossRef]
- Sassolas, A.; Blum, L.J.; Leca-Bouvier, B.D. Immobilization Strategies to Develop Enzymatic Biosensors. Biotechnol. Adv. 2012, 30, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Choudhary, S.; Chaudhari, R.; Jayant, R.D.; Joshi, A. 9-Enzyme-based biosensors. In Bioelectronics and Medical Devices; Pal, K., Kraatz, H.-B., Khasnobish, A., Bag, S., Banerjee, I., Kuruganti, U., Eds.; Woodhead Publishing: Sawston, UK, 2019; pp. 211–240. ISBN 978-0-08-102420-1. [Google Scholar]
- Wang, Z.; Li, M.; Su, P.; Zhang, Y.; Shen, Y.; Han, D.; Ivaska, A.; Niu, L. Direct Electron Transfer of Horseradish Peroxidase and Its Electrocatalysis Based on Carbon Nanotube/Thionine/Gold Composites. Electrochem. Commun. 2008, 10, 306–310. [Google Scholar] [CrossRef]
- Rosca, V.; Popescu, I.C. Kinetic Analysis of Horseradish Peroxidase “Wiring” in Redox Polyelectrolyte–Peroxidase Multilayer Assemblies. Electrochem. Commun. 2002, 4, 904–911. [Google Scholar] [CrossRef]
- Feizabadi, M.; Soleymanpour, A.; Faridnouri, H.; Ajloo, D. Improving Stability of Biosensor Based on Covalent Immobilization of Horseradish Peroxidase by Gamma-Aminobutyric Acid and Application in Detection of H2O2. Int. J. Biol. Macromol. 2019, 136, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Satvekar, R.K.; Rohiwal, S.S.; Raut, A.V.; Karande, V.A.; Tiwale, B.M.; Pawar, S.H. A Silica-Dextran Nanocomposite as a Novel Matrix for Immobilization of Horseradish Peroxidase, and Its Application to Sensing Hydrogen Peroxide. Microchim. Acta 2014, 181, 71–77. [Google Scholar] [CrossRef]
- Datta, S.; Christena, L.R.; Rajaram, Y.R.S. Enzyme Immobilization: An Overview on Techniques and Support Materials. 3 Biotech 2013, 3, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arya, S.K.; Solanki, P.R.; Datta, M.; Malhotra, B.D. Recent Advances in Self-Assembled Monolayers Based Biomolecular Electronic Devices. Biosens. Bioelectron. 2009, 24, 2810–2817. [Google Scholar] [CrossRef]
- Samanta, D.; Sarkar, A. Immobilization of Bio-Macromolecules on Self-Assembled Monolayers: Methods and Sensor Applications. Curr. Soc. Rev. 2011, 40, 2567–2592. [Google Scholar] [CrossRef]
- Hitaishi, V.P.; Clement, R.; Bourassin, N.; Baaden, M.; De Poulpiquet, A.; Sacquin-Mora, S.; Ciaccafava, A.; Lojou, E. Controlling Redox Enzyme Orientation at Planar Electrodes. Catalysts 2018, 8, 192. [Google Scholar] [CrossRef] [Green Version]
- Liébana, S.; Drago, G.A. Bioconjugation and Stabilisation of Biomolecules in Biosensors. Essays Biochem. 2016, 60, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Yates, N.D.J.; Fascione, M.A.; Parkin, A. Methodologies for “Wiring” Redox Proteins/Enzymes to Electrode Surfaces. Chem. Eur. J. 2018, 24, 12164–12182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Guo, C.; Zhang, M.; Park, B.; Xu, B. High-Resolution Single-Molecule Recognition Imaging of the Molecular Details of Ricin–Aptamer Interaction. J. Phys. Chem. B 2012, 116, 5316–5322. [Google Scholar] [CrossRef] [PubMed]
- Asturias-Arribas, L.; Alonso-Lomillo, M.A.; Domínguez-Renedo, O.; Arcos-Martínez, M.J. Cytochrome P450 2D6 Based Electrochemical Sensor for the Determination of Codeine. Talanta 2014, 129, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, C.; Rajkumar, R.; Bhargava, K. (Eds.) Introduction to Biosensors. In Biosens. Bioelectronics; Elsevier: Amsterdam, The Netherlands, 2015; Chapter 1; pp. 1–68. ISBN 978-0-12-803100-1. [Google Scholar]
- Zhou, J.; Liao, C.; Zhang, L.; Wang, Q.; Tian, Y. Molecular Hydrogel-Stabilized Enzyme with Facilitated Electron Transfer for Determination of H 2 O 2 Released from Live Cells. Anal. Chem. 2014, 86, 4395–4401. [Google Scholar] [CrossRef]
- Battistuzzi, G.; Bellei, M.; Bortolotti, C.A.; Sola, M. Redox Properties of Heme Peroxidases. Arch. Biochem. Biophys. 2010, 500, 21–36. [Google Scholar] [CrossRef]
- Schneider, E.; Clark, D.S. Cytochrome P450 (CYP) Enzymes and the Development of CYP Biosensors. Biosens. Bioelectron. 2013, 39, 1–13. [Google Scholar] [CrossRef]
- McNaught, A.D.; Wilkinson, A. IUPAC Compendium of Chemical Terminology, 2nd ed.; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar]
- Ruzgas, T.; Csöregi, E.; Emnéus, J.; Gorton, L.; Marko-Varga, G. Peroxidase-Modified Electrodes: Fundamentals and Application. Anal. Chim. Acta 1996, 330, 123–138. [Google Scholar] [CrossRef]
- Gazaryan, I.G.; Gorton, L.; Ruzgas, T.; Csoregi, E.; Schuhmann, W.; Lagrimini, L.M.; Khushpul’yan, D.M.; Tishkov, V.I. Tobacco Peroxidase as a New Reagent for Amperometric Biosensors. J. Anal. Chem. 2005, 60, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Castilho, T.J.; Sotomayor, M.P.T.; Kubota, L.T. Amperometric Biosensor Based on Horseradish Peroxidase for Biogenic Amine Determinations in Biological Samples. J. Pharm. Biomed. Anal. 2005, 37, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Kermad, A.; Sam, S.; Ghellai, N.; Khaldi, K.; Gabouze, N. Horseradish Peroxidase-Modified Porous Silicon for Phenol Monitoring. MSEB 2013, 178, 1159–1164. [Google Scholar] [CrossRef]
- Rusling, J.F. Electrochemistry of Redox Enzymes. In Bioelectrochemistry; Bartlett, P.N., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2008; pp. 39–85. [Google Scholar]
- Zigah, D.; Lojou, E.; de Poulpiquet, A. Micro- and Nanoscopic Imaging of Enzymatic Electrodes: A Review. ChemElectroChem 2019, 6, 5524–5546. [Google Scholar] [CrossRef]
- Kornienko, N.; Ly, K.H.; Robinson, W.E.; Heidary, N.; Zhang, J.Z.; Reisner, E. Advancing Techniques for Investigating the Enzyme–Electrode Interface. Acc. Chem. Res. 2019, 52, 1439–1448. [Google Scholar] [CrossRef] [Green Version]
- López-Lorente, Á.I.; Kranz, C. Recent Advances in Biomolecular Vibrational Spectroelectrochemistry. Curr. Opin. Electrochem. 2017, 5, 106–113. [Google Scholar] [CrossRef]
- Sezer, M.; Millo, D.; Weidinger, I.M.; Zebger, I.; Hildebrandt, P. Analyzing the Catalytic Processes of Immobilized Redox Enzymes by Vibrational Spectroscopies. IUBMB Life 2012, 64, 455–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenfield, N.J. Using Circular Dichroism Spectra to Estimate Protein Secondary Structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef]
- Wu, H.; Wang, X.; Qiao, M.; Zhang, H.; Jin, X.; Fan, S. Enhancing Sensitivity of Hemoglobin-Based Electrochemical Biosensor by Using Protein Conformational Intermediate. Sens. Actuators B 2015, 221, 694–699. [Google Scholar] [CrossRef]
- Gao, J.; Liu, H.; Tong, C.; Pang, L.; Feng, Y.; Zuo, M.; Wei, Z.; Li, J. Hemoglobin-Mn3(PO4)2 Hybrid Nanoflower with Opulent Electroactive Centers for High-Performance Hydrogen Peroxide Electrochemical Biosensor. Sens. Actuators B 2020, 307, 127628. [Google Scholar] [CrossRef]
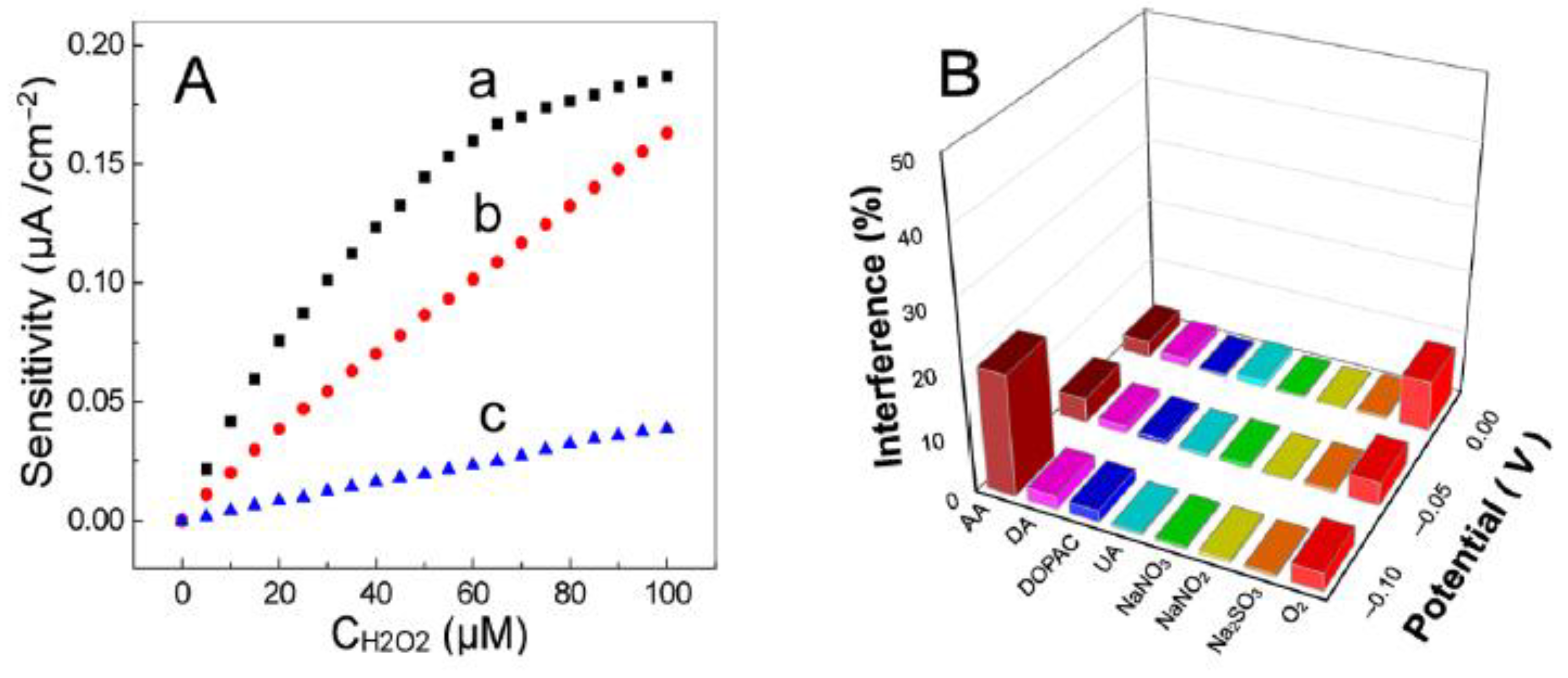
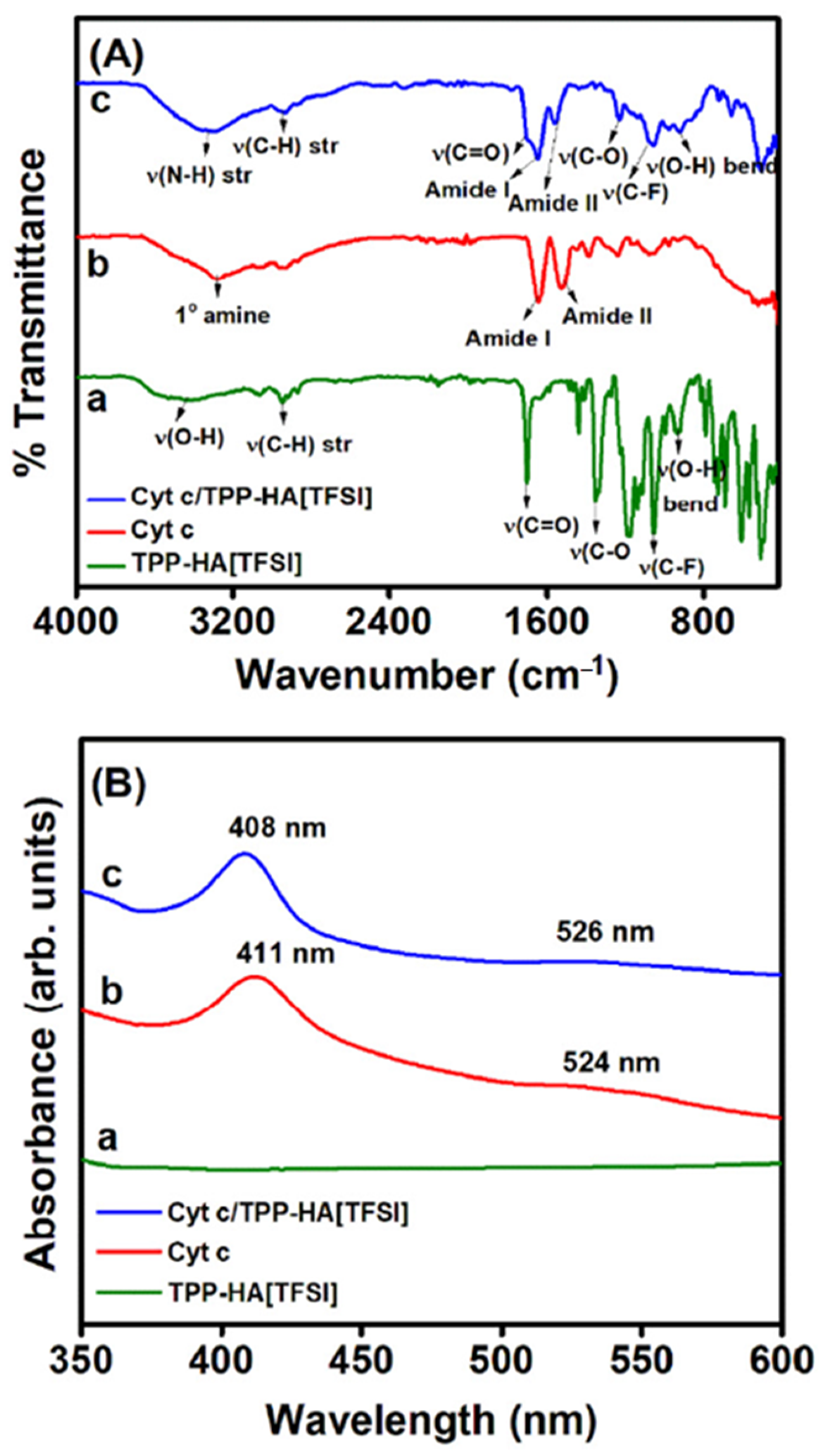
- Murphy, M.; Theyagarajan, K.; Ganesan, P.; Senthilkumar, S.; Thenmozhi, K. Electrochemical Biosensor for the Detection of Hydrogen Peroxide Using Cytochrome c Covalently Immobilized on Carboxyl Functionalized Ionic Liquid/Multiwalled Carbon Nanotube Hybrid. Appl. Surf. Sci. 2019, 492, 718–725. [Google Scholar] [CrossRef]
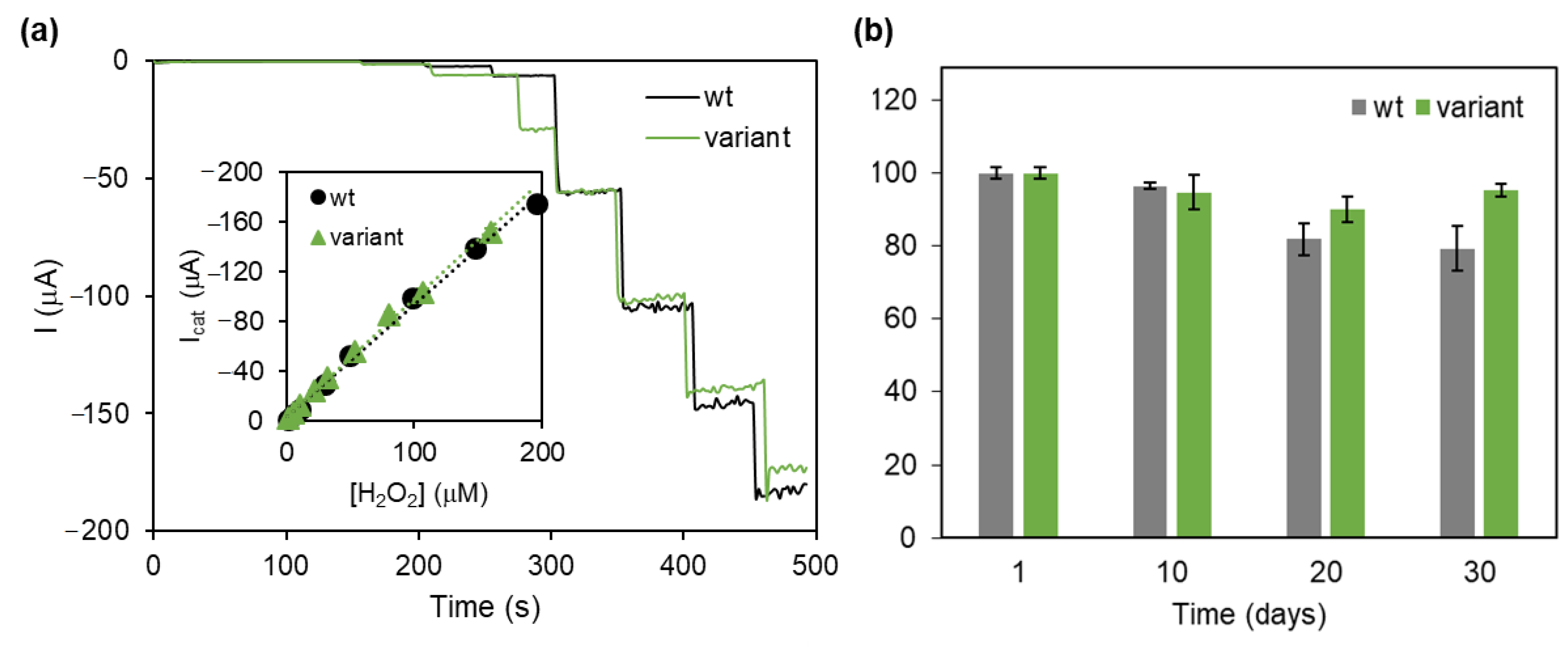
- Barbosa, C.; Silveira, C.M.; Silva, D.; Brissos, V.; Hildebrandt, P.; Martins, L.O.; Todorovic, S. Immobilized Dye-Decolorizing Peroxidase (DyP) and Directed Evolution Variants for Hydrogen Peroxide Biosensing. Biosens. Bioelectron. 2020, 153, 112055. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Peng, X.; Kong, J.; Deng, J. Facilitated Electron Transfer from an Electrode to Horseradish Peroxidase in a Biomembrane-like Surfactant Film. J. Electroanal. Chem. 2000, 480, 26–33. [Google Scholar] [CrossRef]
- Chen, X.; Xie, H.; Kong, J.; Deng, J. Characterization for Didodecyldimethylammonium Bromide Liquid Crystal Film Entrapping Catalase with Enhanced Direct Electron Transfer Rate. Biosens. Bioelectron. 2001, 16, 115–120. [Google Scholar] [CrossRef]
- Todorovic, S.; Jung, C.; Hildebrandt, P.; Murgida, D.H. Conformational Transitions and Redox Potential Shifts of Cytochrome P450 Induced by Immobilization. J. Biol. Inorg. Chem. 2006, 11, 119–127. [Google Scholar] [CrossRef]
- Liu, X.; Feng, H.; Zhao, R.; Wang, Y.; Liu, X. A Novel Approach to Construct a Horseradish Peroxidase|hydrophilic Ionic Liquids|Au Nanoparticles Dotted Titanate Nanotubes Biosensor for Amperometric Sensing of Hydrogen Peroxide. Biosens. Bioelectron. 2012, 31, 101–104. [Google Scholar] [CrossRef]
- Dong, J.; Wen, Y.; Miao, Y.; Xie, Z.; Zhang, Z.; Yang, H. A Nanoporous Zirconium Phytate Film for Immobilization of Redox Protein and the Direct Electrochemical Biosensor. Sens. Actuators B 2010, 150, 141–147. [Google Scholar] [CrossRef]
- Zhang, Y.; He, P.; Hu, N. Horseradish Peroxidase Immobilized in TiO2 Nanoparticle Films on Pyrolytic Graphite Electrodes: Direct Electrochemistry and Bioelectrocatalysis. Electrochim. Acta 2004, 49, 1981–1988. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, X.; Wen, Y.; Xing, Y.; Zhang, Z.; Yang, H. Direct Electrochemistry and Bioelectrocatalysis of Horseradish Peroxidase Based on Gold Nano-Seeds Dotted TiO2 Nanocomposite. Biosens. Bioelectron. 2010, 25, 2442–2446. [Google Scholar] [CrossRef] [PubMed]
- Siebert, F.; Hildebrandt, P. Heme Proteins. In Vibrational Spectroscopy in Life Science; Wiley-VCH Verlag GmbH & Co. KGaA: Hoboken, NJ, USA, 2008; pp. 227–282. ISBN 978-3-527-62134-7. [Google Scholar]
- Todorovic, S.; Verissimo, A.; Wisitruangsakul, N.; Zebger, I.; Hildebrandt, P.; Pereira, M.M.; Teixeira, M.; Murgida, D.H. SERR-Spectroelectrochemical Study of a Cbb3 Oxygen Reductase in a Biomimetic Construct. J. Phys. Chem. B 2008, 112, 16952–16959. [Google Scholar] [CrossRef]
- Murgida, D.H.; Hildebrandt, P. Heterogeneous Electron Transfer of Cytochrome c on Coated Silver Electrodes. Electric Field Effects on Structure and Redox Potential. J. Phys. Chem. B 2001, 105, 1578–1586. [Google Scholar] [CrossRef]
- Murgida, D.H.; Hildebrandt, P. Electron-Transfer Processes of Cytochrome c at Interfaces. New Insights by Surface-Enhanced Resonance Raman Spectroscopy. Acc. Chem. Res. 2004, 37, 854–861. [Google Scholar] [CrossRef]
- Moyo, M.; Okonkwo, J.O.; Agyei, N.M. A Novel Hydrogen Peroxide Biosensor Based on Adsorption of Horseradish Peroxidase onto a Nanobiomaterial Composite Modified Glassy Carbon Electrode. Electroanalysis 2013, 25, 1946–1954. [Google Scholar] [CrossRef]
- Tulli, F.; Gulotta, F.A.; Martino, D.M.; Zanini, V.I.P.; Borsarelli, C.D. Ultrasensitive Amperometric Biosensing of Polyphenols Using Horseradish Peroxidase Immobilized in a Laponite/Au/DNA-Bioinspired Polycation Nanocomposite. J. Electrochem. Soc. 2018, 165, B452–B457. [Google Scholar] [CrossRef]
- Zhu, Z.; Li, X.; Wang, Y.; Zeng, Y.; Sun, W.; Huang, X. Direct Electrochemistry and Electrocatalysis of Horseradish Peroxidase with Hyaluronic Acid–Ionic Liquid–Cadmium Sulfide Nanorod Composite Material. Anal. Chim. Acta 2010, 670, 51–56. [Google Scholar] [CrossRef]
- Song, H.; Ni, Y.; Kokot, S. Investigations of an Electrochemical Platform Based on the Layered MoS2–Graphene and Horseradish Peroxidase Nanocomposite for Direct Electrochemistry and Electrocatalysis. Biosens. Bioelectron. 2014, 56, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Cahuantzi-Muñoz, S.L.; González-Fuentes, M.A.; Ortiz-Frade, L.A.; Torres, E.; Ţălu, Ş.; Trejo, G.; Méndez-Albores, A. Electrochemical Biosensor for Sensitive Quantification of Glyphosate in Maize Kernels. Electroanalysis 2019, 31, 927–935. [Google Scholar] [CrossRef] [Green Version]
- Mai, Z.; Zhao, X.; Dai, Z.; Zou, X. Contributions of Components in Guanidine Hydrochloride to Hemoglobin Unfolding Investigated by Protein Film Electrochemistry. J. Phys. Chem. B 2010, 114, 7090–7097. [Google Scholar] [CrossRef]
- Li, X.; Zheng, W.; Zhang, L.; Yu, P.; Lin, Y.; Su, L.; Mao, L. Effective Electrochemical Method for Investigation of Hemoglobin Unfolding Based on the Redox Property of Heme Groups at Glassy Carbon Electrodes. Anal. Chem. 2009, 81, 8557–8563. [Google Scholar] [CrossRef]
- Smulevich, G.; Feis, A.; Howes, B.D.; Ivancich, A. Structure-Function Relationships among Heme Peroxidases: New Insights from Electronic Absorption, Resonance Raman and Multifrequency Electron Paramagnetic Resonance Spectroscopies. In Handbook of Porphyrin Science; World Scientific Publishing Co.: Singapore, 2010; pp. 367–453. ISBN 978-981-4307-19-2. [Google Scholar]
- Torres, E.; Ayala, M. Biocatalysis Based on Heme Peroxidases; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Poulos, T.L. Thirty Years of Heme Peroxidase Structural Biology. Arch. Biochem. Biophys. 2010, 500, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nóbrega, C.S.; Pauleta, S.R. Reduction of hydrogen peroxide in gram-negative bacteria—bacterial peroxidases. In Advances in Microbial Physiology; Poole, R.K., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 74, pp. 415–464. ISBN 0065-2911. [Google Scholar]
- Mossanha, R.; Erdmann, C.A.; Santos, C.S.; Wohnrath, K.; Fujiwara, S.T.; Pessoa, C.A. Construction of a Biosensor Based on SAM of Thiolactic Acid on Gold Nanoparticles Stabilized by Silsesquioxane Polyelectrolyte for Cathecol Determination. Sens. Actuators B 2017, 252, 747–756. [Google Scholar] [CrossRef]
- Ferapontova, E.E. Direct Peroxidase Bioelectrocatalysis on a Variety of Electrode Materials. Electroanalysis 2004, 16, 1101–1112. [Google Scholar] [CrossRef]
- Narvaez, A.; Suarez, G.; Popescu, I.C.; Katakis, I.; Dominguez, E. Reagentless Biosensors Based on Self-Deposited Redox Polyelectrolyte-Oxidoreductases Architectures. Biosens. Bioelectron. 2000, 15, 43–52. [Google Scholar] [CrossRef]
- Okawa, Y.; Yokoyama, N.; Sakai, Y.; Shiba, F. Direct Electron Transfer Biosensor for Hydrogen Peroxide Carrying Nanocomplex Composed of Horseradish Peroxidase and Au-Nanoparticle—Characterization and Application to Bienzyme Systems. Anal. Chem. Res. 2015, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
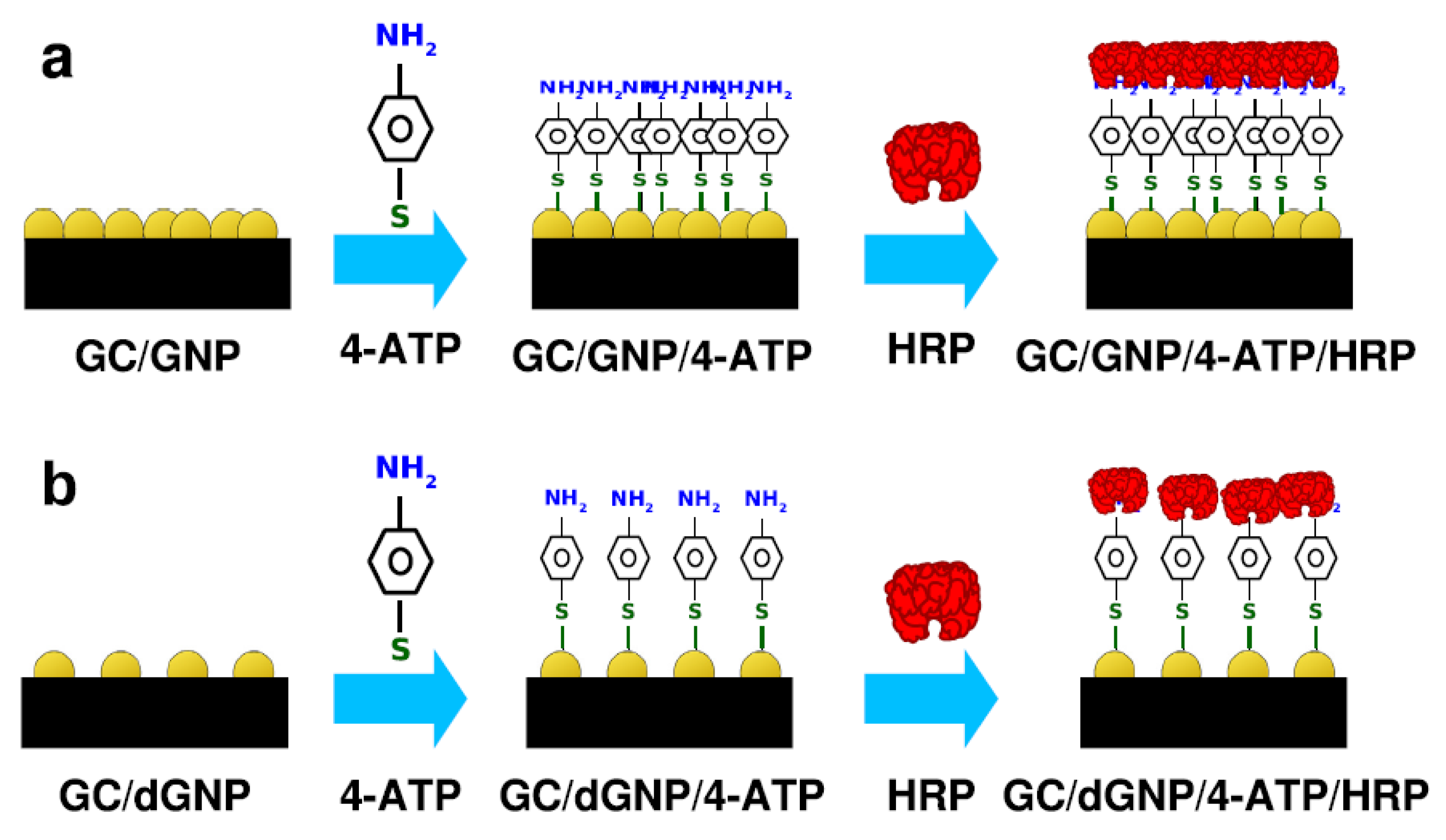
- Huerta-Miranda, G.A.; Arrocha-Arcos, A.A.; Miranda-Hernandez, M. Gold Nanoparticles/4-Aminothiophenol Interfaces for Direct Electron Transfer of Horseradish Peroxidase: Enzymatic Orientation and Modulation of Sensitivity towards Hydrogen Peroxide Detection. Bioelectrochemistry 2018, 122, 77–83. [Google Scholar] [CrossRef]
- Bollella, P.; Medici, L.; Tessema, M.; Poloznikov, A.A.; Hushpulian, D.M.; Tishkov, V.I.; Andreu, R.; Leech, D.; Megersa, N.; Marcaccio, M.; et al. Highly Sensitive, Stable and Selective Hydrogen Peroxide Amperometric Biosensors Based on Peroxidases from Different Sources Wired by Os-Polymer: A Comparative Study. Solid State Ion. 2018, 314, 178–186. [Google Scholar] [CrossRef]
- Olloqui-Sariego, J.L.; Zakharova, G.S.; Poloznikov, A.A.; Calvente, J.J.; Hushpulian, D.M.; Gorton, L.; Andreu, R. Fenton-like Inactivation of Tobacco Peroxidase Electrocatalysis at Negative Potentials. ACS Catal. 2016, 6, 7452–7457. [Google Scholar] [CrossRef]
- Rusling, J.F. Enzyme Bioelectrochemistry in Cast Biomembrane-Like Films. Acc. Chem. Res. 1998, 31, 363–369. [Google Scholar] [CrossRef]
- Zhang, Z.; Chouchane, S.; Magliozzo, R.S.; Rusling, J.F. Direct Voltammetry and Catalysis with Mycobacterium Tuberculosis Catalase−Peroxidase, Peroxidases, and Catalase in Lipid Films. Anal. Chem. 2002, 74, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Guo, K.; Lv, J.; Gao, Y.; Duan, C.; Deng, L.; Zhu, Z. A Novel Nitrite Biosensor Based on the Direct Electrochemistry of Horseradish Peroxidase Immobilized on Porous Co3O4 Nanosheets and Reduced Graphene Oxide Composite Modified Electrode. Sens. Actuators B 2017, 238, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Niu, Y.; Liu, J.; Chen, W.; Yin, C.; Weng, W.; Li, X.; Wang, X.; Li, G.; Sun, W. A Direct Electron Transfer Biosensor Based on a Horseradish Peroxidase and Gold Nanotriangle Modified Electrode and Electrocatalysis. Anal. Methods 2018, 10, 5297–5304. [Google Scholar] [CrossRef]
- Sun, W.; Guo, Y.; Li, T.; Ju, X.; Lou, J.; Ruan, C. Electrochemistry of Horseradish Peroxidase Entrapped in Graphene and DsDNA Composite Modified Carbon Ionic Liquid Electrode. Electrochim. Acta 2012, 75, 381–386. [Google Scholar] [CrossRef]
- Şenel, M.; Çevik, E.; Abasıyanık, M.F. Amperometric Hydrogen Peroxide Biosensor Based on Covalent Immobilization of Horseradish Peroxidase on Ferrocene Containing Polymeric Mediator. Sens. Actuators B 2010, 145, 444–450. [Google Scholar] [CrossRef]
- Tatsuma, T.; Okawa, Y.; Watanabe, T. Enzyme Monolayer- and Bilayer-Modified Tin Oxide Electrodes for the Determination of Hydrogen Peroxide and Glucose. Anal. Chem. 1989, 61, 2352–2355. [Google Scholar] [CrossRef]
- Wang, Y.; Hasebe, Y. Carbon-Felt-Based Bioelectrocatalytic Flow-Detectors: Optimization of the Adsorption Conditions of Horseradish Peroxidase and Thionine onto Carbon-Felt for Highly Sensitive Amperometric Determination of H2O2. Anal. Sci. 2011, 27, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindgren, A.; Tanaka, M.; Ruzgas, T.; Gorton, L.; Gazaryan, I.; Ishimori, K.; Morishima, I. Direct Electron Transfer Catalysed by Recombinant Forms of Horseradish Peroxidase: Insight into the Mechanism. Electrochem. Commun. 1999, 1, 171–175. [Google Scholar] [CrossRef]
- Ruzgas, T.; Emnéus, J.; Gorton, L.; Marko-Varga, G. The Development of a Peroxidase Biosensor for Monitoring Phenol and Related Aromatic Compounds. Anal. Chim. Acta 1995, 311, 245–253. [Google Scholar] [CrossRef]
- Pandey, V.P.; Awasthi, M.; Singh, S.; Tiwari, S.; Dwivedi, U.N. A Comprehensive Review on Function and Application of Plant Peroxidases. Biochem. Anal. Biochem. 2017, 6, 308. [Google Scholar] [CrossRef]
- Krainer, F.W.; Glieder, A. An Updated View on Horseradish Peroxidases: Recombinant Production and Biotechnological Applications. Appl. Microbiol. Biotechnol. 2015, 99, 1611–1625. [Google Scholar] [CrossRef] [Green Version]
- Castillo, J.; Ferapontova, E.; Hushpulian, D.; Tasca, F.; Tishkov, V.; Chubar, T.; Gazaryan, I.; Gorton, L. Direct Electrochemistry and Bioelectrocatalysis of H2O2 Reduction of Recombinant Tobacco Peroxidase on Graphite. Effect of Peroxidase Single-Point Mutation on Ca2+-Modulated Catalytic Activity. J. Electroanal. Chem. 2006, 588, 112–121. [Google Scholar] [CrossRef]
- Gaspar, S.; Popescu, I.C.; Gazaryan, I.G.; Gerardo Bautista, A.; Sakharov, I.Y.; Mattiasson, B.; Csöregi, E. Biosensors Based on Novel Plant Peroxidases: A Comparative Study. Electrochim. Acta 2000, 46, 255–264. [Google Scholar] [CrossRef]
- Centeno, D.A.; Solano, X.H.; Castillo, J.J. A New Peroxidase from Leaves of Guinea Grass (Panicum Maximum): A Potential Biocatalyst to Build Amperometric Biosensors. Bioelectrochemistry 2017, 116, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Ferapontova, E.E.; Grigorenko, V.G.; Egorov, A.M.; Börchers, T.; Ruzgas, T.; Gorton, L. Mediatorless Biosensor for H2O2 Based on Recombinant Forms of Horseradish Peroxidase Directly Adsorbed on Polycrystalline Gold. Biosens. Bioelectron. 2001, 16, 147–157. [Google Scholar] [CrossRef]
- Ferapontova, E.; Gorton, L. Effect of PH on Direct Electron Transfer in the System Gold Electrode–Recombinant Horseradish Peroxidase. Bioelectrochemistry 2002, 55, 83–87. [Google Scholar] [CrossRef]
- Andreu, R.; Ferapontova, E.E.; Gorton, L.; Calvente, J.J. Direct Electron Transfer Kinetics in Horseradish Peroxidase Electrocatalysis. J. Phys. Chem. B 2007, 111, 469–477. [Google Scholar] [CrossRef]
- Sun, Y.-X.; Zhang, J.-T.; Huang, S.-W.; Wang, S.-F. Hydrogen Peroxide Biosensor Based on the Bioelectrocatalysis of Horseradish Peroxidase Incorporated in a New Hydrogel Film. Sens. Actuators B 2007, 124, 494–500. [Google Scholar] [CrossRef]
- Guo, C.; Song, Y.; Wei, H.; Li, P.; Wang, L.; Sun, L.; Sun, Y.; Li, Z. Room Temperature Ionic Liquid Doped DNA Network Immobilized Horseradish Peroxidase Biosensor for Amperometric Determination of Hydrogen Peroxide. Anal. Bioanal. Chem. 2007, 389, 527–532. [Google Scholar] [CrossRef]
- Tian, J.; Wang, J.; Li, Y.; Huang, M.; Lu, J. Electrochemically Driven Omeprazole Metabolism via Cytochrome P450 Assembled on the Nanocomposites of Ceria Nanoparticles and Graphene. J. Electrochem. Soc. 2017, 164, H470–H476. [Google Scholar] [CrossRef]
- Lu, J.; Cui, D.; Li, H.; Zhang, Y.; Liu, S. Cytochrome P450 Bienzymes Assembled on Au/Chitosan/Reduced Graphene Oxide Nanosheets for Electrochemically-Driven Drug Cascade Metabolism. Electrochim. Acta 2015, 165, 36–44. [Google Scholar] [CrossRef]
- Salimi, A.; Sharifi, E.; Noorbakhsh, A.; Soltanian, S. Direct Electrochemistry and Electrocatalytic Activity of Catalase Immobilized onto Electrodeposited Nano-Scale Islands of Nickel Oxide. Biophys. Chem. 2007, 125, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Saadati, S.; Salimi, A.; Hallaj, R.; Rostami, A. Layer by Layer Assembly of Catalase and Amine-Terminated Ionic Liquid onto Titanium Nitride Nanoparticles Modified Glassy Carbon Electrode: Study of Direct Voltammetry and Bioelectrocatalytic Activity. Anal. Chim. Acta 2012, 753, 32–41. [Google Scholar] [CrossRef]
- Rahimi, P.; Rafiee-Pour, H.-A.; Ghourchian, H.; Norouzi, P.; Ganjali, M.R. Ionic-Liquid/NH2-MWCNTs as a Highly Sensitive Nano-Composite for Catalase Direct Electrochemistry. Biosens. Bioelectron. 2010, 25, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Vilian, A.T.E.; Chen, S.-M.; Lou, B.-S. A Simple Strategy for the Immobilization of Catalase on Multi-Walled Carbon Nanotube/Poly (l-Lysine) Biocomposite for the Detection of H2O2 and Iodate. Biosens. Bioelectron. 2014, 61, 639–647. [Google Scholar] [CrossRef]
- Chen, H.; Mousty, C.; Cosnier, S.; Silveira, C.; Moura, J.J.G.; Almeida, M.G. Highly Sensitive Nitrite Biosensor Based on the Electrical Wiring of Nitrite Reductase by [ZnCr-AQS] LDH. Electrochem. Commun. 2007, 9, 2240–2245. [Google Scholar] [CrossRef]
- Silveira, C.M.; Gomes, S.P.; Araújo, A.N.; Montenegro, M.C.B.S.M.; Todorovic, S.; Viana, A.S.; Silva, R.J.C.; Moura, J.J.G.; Almeida, M.G. An Efficient Non-Mediated Amperometric Biosensor for Nitrite Determination. Biosens. Bioelectron. 2010, 25, 2026–2032. [Google Scholar] [CrossRef]
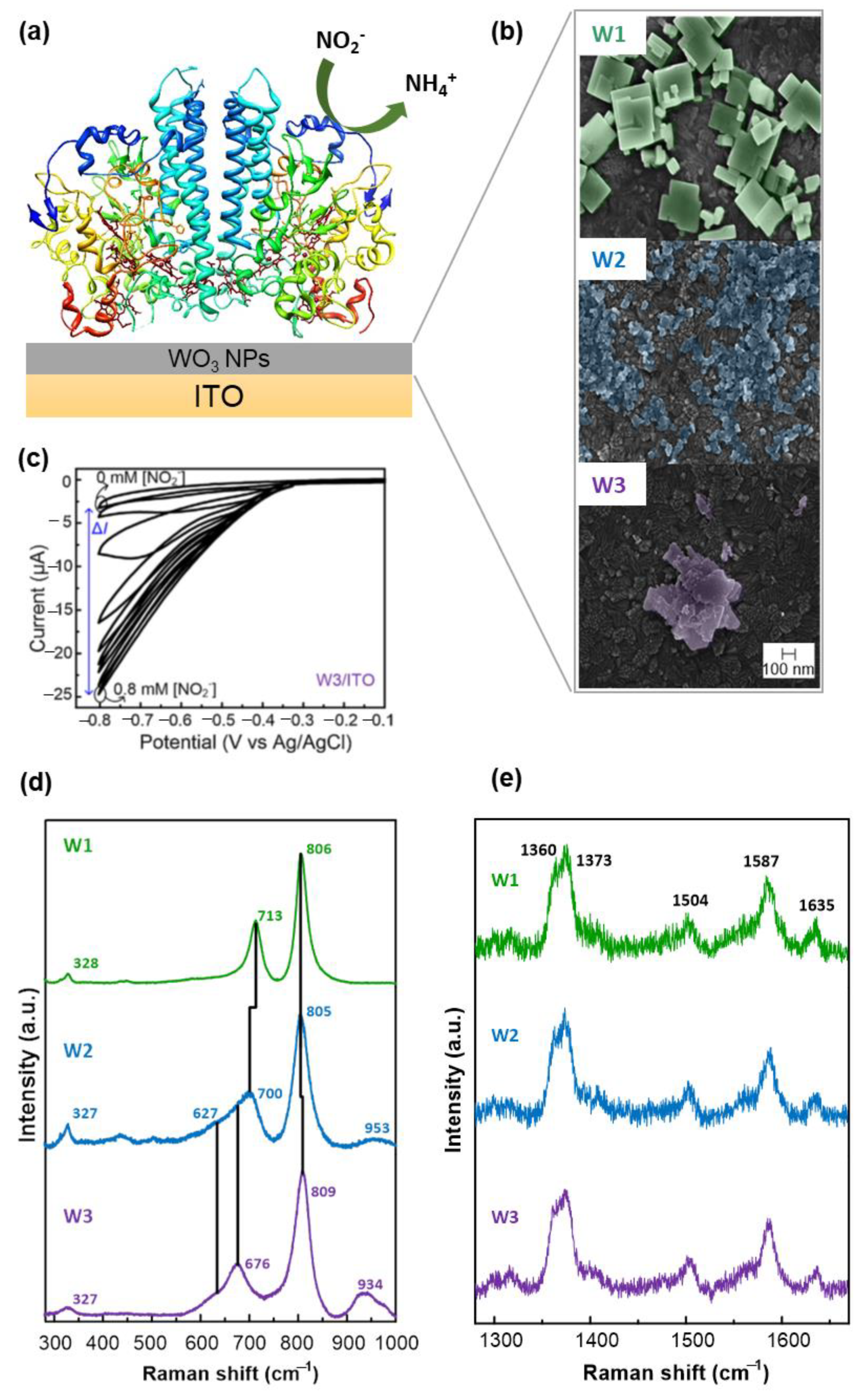
- Santos, L.; Silveira, C.M.; Elangovan, E.; Neto, J.P.; Nunes, D.; Pereira, L.; Martins, R.; Viegas, J.; Moura, J.J.G.; Todorovic, S.; et al. Synthesis of WO3 Nanoparticles for Biosensing Applications. Sens. Actuators B 2016, 223, 186–194. [Google Scholar] [CrossRef]
- Ashe, D.; Alleyne, T.; Iwuoha, E. Serum Cytochrome c Detection Using a Cytochrome c Oxidase Biosensor. Biotechnol. Appl. Biochem. 2007, 46, 185. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.L.; Lianyong, S.; Hawkridge, F.M.; Ward, K.R.; Rhoten, M.C. Immobilization of Cytochrome c Oxidase into Electrode-Supported Lipid Bilayer Membranes for in Vitro Cytochrome c Sensing. IEEE Sens. J. 2006, 6, 420–427. [Google Scholar] [CrossRef]
- Eguílaz, M.; Agüí, L.; Yáñez-Sedeño, P.; Pingarrón, J.M. A Biosensor Based on Cytochrome c Immobilization on a Poly-3-Methylthiophene/Multi-Walled Carbon Nanotubes Hybrid-Modified Electrode. Application to the Electrochemical Determination of Nitrite. J. Electroanal. Chem. 2010, 644, 30–35. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhi, J.; Zou, Y.; Zhang, W.; Lee, S.-T. Direct Electrochemistry and Electrocatalytic Activity of Cytochrome c Covalently Immobilized on a Boron-Doped Nanocrystalline Diamond Electrode. Anal. Chem. 2008, 80, 4141–4146. [Google Scholar] [CrossRef] [PubMed]
- Pashai, E.; Najafpour Darzi, G.; Jahanshahi, M.; Yazdian, F.; Rahimnejad, M. An Electrochemical Nitric Oxide Biosensor Based on Immobilized Cytochrome c on a Chitosan-Gold Nanocomposite Modified Gold Electrode. Int. J. Biol. Macromol. 2018, 108, 250–258. [Google Scholar] [CrossRef]
- Neumann, B.; Kielb, P.; Rustam, L.; Fischer, A.; Weidinger, I.M.; Wollenberger, U. Bioelectrocatalytic Reduction of Hydrogen Peroxide by Microperoxidase-11 Immobilized on Mesoporous Antimony-Doped Tin Oxide. ChemElectroChem 2017, 4, 913–919. [Google Scholar] [CrossRef]
- Gong, C.; Shen, Y.; Chen, J.; Song, Y.; Chen, S.; Song, Y.; Wang, L. Microperoxidase-11@PCN-333 (Al)/Three-Dimensional Macroporous Carbon Electrode for Sensing Hydrogen Peroxide. Sens. Actuators B 2017, 239, 890–897. [Google Scholar] [CrossRef]
- Abdelwahab, A.A.; Koh, W.C.A.; Noh, H.-B.; Shim, Y.-B. A Selective Nitric Oxide Nanocomposite Biosensor Based on Direct Electron Transfer of Microperoxidase: Removal of Interferences by Co-Immobilized Enzymes. Biosens. Bioelectron. 2010, 26, 1080–1086. [Google Scholar] [CrossRef]
- Liu, H.; Duan, C.; Su, X.; Dong, X.; Huang, Z.; Shen, W.; Zhu, Z. A Hemoglobin Encapsulated Titania Nanosheet Modified Reduced Graphene Oxide Nanocomposite as a Mediator-Free Biosensor. Sens. Actuators B 2014, 203, 303–310. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Q.; Wang, L.; Gao, F.; Wang, W.; Hu, Z. Imidazoline Derivative Templated Synthesis of Broccoli-like Bi2S3 and Its Electrocatalysis towards the Direct Electrochemistry of Hemoglobin. Biosens. Bioelectron. 2015, 66, 216–223. [Google Scholar] [CrossRef]
- Mazzochette, Z.; Newton, E.; Mugweru, A. Electrochemical Catalysis of Artemisinin on Hemoglobin Functionalized Carbon Nanofibers. Anal. Methods 2017, 9, 2997–3002. [Google Scholar] [CrossRef]
- Vilian, A.T.E.; Veeramani, V.; Chen, S.-M.; Madhu, R.; Kwak, C.H.; Huh, Y.S.; Han, Y.-K. Immobilization of Myoglobin on Au Nanoparticle-Decorated Carbon Nanotube/Polytyramine Composite as a Mediator-Free H2O2 and Nitrite Biosensor. Sci. Rep. 2015, 5, 18390. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Xie, H.; Niu, Y.; Liu, J.; Huang, Y.; Li, B.; Li, G.; Sun, W. Electrochemical Myoglobin Biosensor Based on Magnesium Metal-Organic Frameworks and Gold Nanoparticles Composite Modified Electrode. Int. J. Electrochem. Sci. 2019, 2405–2413. [Google Scholar] [CrossRef]
- Ozoner, S.K.; Yilmaz, F.; Celik, A.; Keskinler, B.; Erhan, E. A Novel Poly(Glycine Methacrylate-Co-3-Thienylmethyl Methacrylate)-Polypyrrole-Carbon Nanotube-Horseradish Peroxidase Composite Film Electrode for the Detection of Phenolic Compounds. Curr. Appl. Phys. 2011, 11, 402–408. [Google Scholar] [CrossRef]
- Cao, S.; Yuan, R.; Chai, Y.; Zhang, L.; Li, X.; Gao, F. A Mediator-Free Amperometric Hydrogen Peroxide Biosensor Based on HRP Immobilized on a Nano-Au/Poly 2,6-Pyridinediamine-Coated Electrode. Bioprocess Biosyst. Eng. 2007, 30, 71–78. [Google Scholar] [CrossRef]
- Ahirwal, G.K.; Mitra, C.K. Direct Electrochemistry of Horseradish Peroxidase-Gold Nanoparticles Conjugate. Sensors 2009, 9, 881–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, K.; Terse-Thakoor, T.; Mulchandani, A. Electrochemical Properties of Seamless Three-Dimensional Carbon Nanotubes-Grown Graphene Modified with Horseradish Peroxidase. Bioelectrochemistry 2016, 111, 57–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.-X.; Bao, W.-J.; Wang, J.; Lu, Q.-Q.; Xia, X.-H. Immobilization and Catalytic Activity of Horseradish Peroxidase on Molybdenum Disulfide Nanosheets Modified Electrode. Electrochem. Commun. 2013, 35, 146–148. [Google Scholar] [CrossRef]
- Sugano, Y. DyP-Type Peroxidases Comprise a Novel Heme Peroxidase Family. Cell. Mol. Life Sci. 2009, 66, 1387–1403. [Google Scholar] [CrossRef] [PubMed]
- Colpa, D.I.; Fraaije, M.W.; van Bloois, E. DyP-Type Peroxidases: A Promising and Versatile Class of Enzymes. J. Ind. Microbiol. Biotechnol. 2014, 41, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Eltis, L.D. The Multihued Palette of Dye-Decolorizing Peroxidases. Arch. Biochem. Biophys. 2015, 574, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Sugano, Y. A Structural and Functional Perspective of DyP-Type Peroxidase Family. Arch. Biochem. Biophys. 2015, 574, 49–55. [Google Scholar] [CrossRef]
- Mendes, S.; Robalo, M.P.; Martins, L.O. Bacterial Enzymes and Multi-enzymatic Systems for Cleaning-up Dyes from the Environment. In Microbial Degradation of Synthetic Dyes in Wastewaters; Singh, S.N., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 27–55. ISBN 978-3-319-10941-1. [Google Scholar]
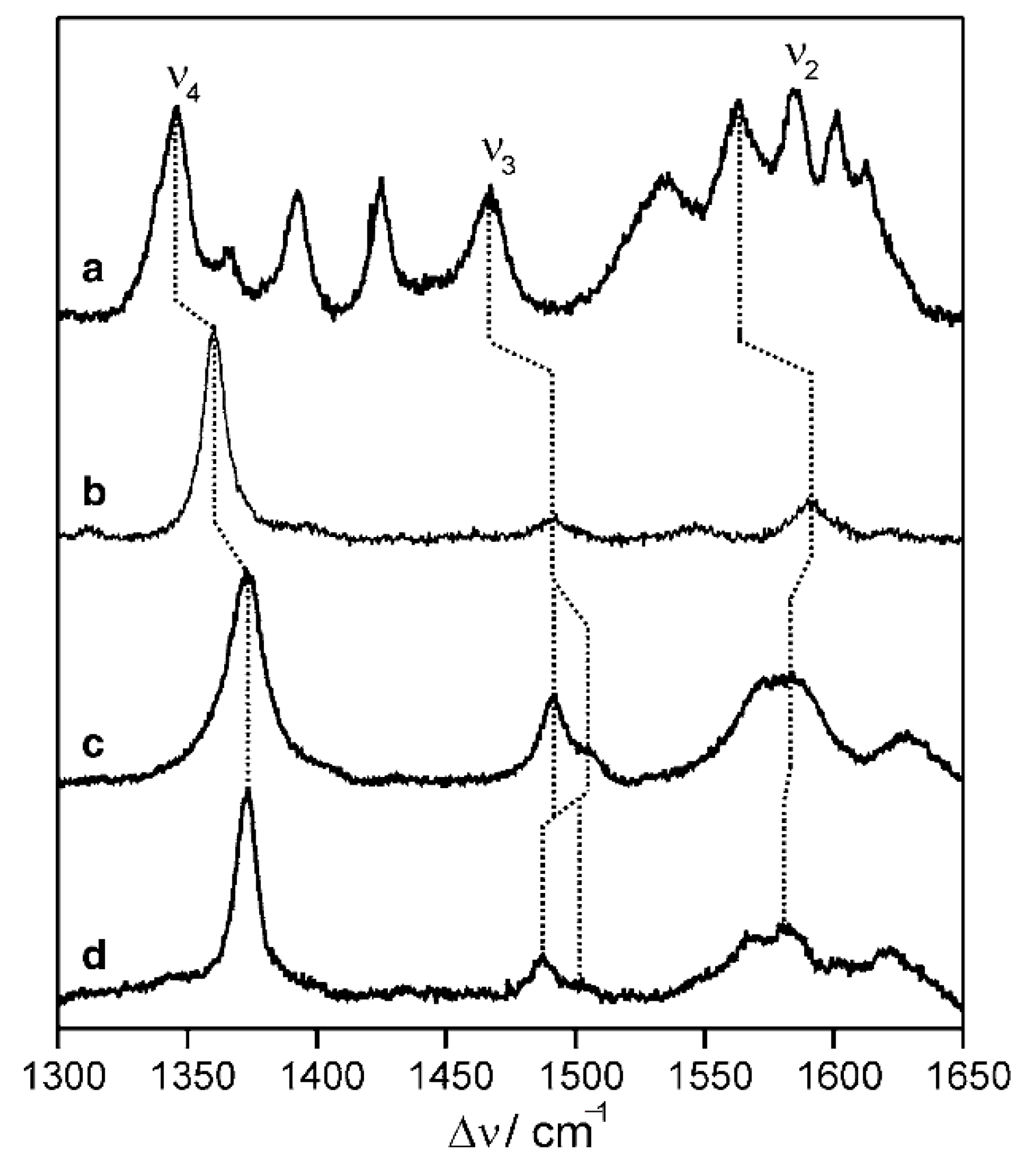
- Silveira, C.M.; Moe, E.; Fraaije, M.; Martins, L.O.; Todorovic, S. Resonance Raman View of the Active Site Architecture in Bacterial DyP-Type Peroxidases. RSC Adv. 2020, 10, 11095–11104. [Google Scholar] [CrossRef]
- Sezer, M.; Genebra, T.; Mendes, S.; Martins, L.O.; Todorovic, S. A DyP-Type Peroxidase at a Bio-Compatible Interface: Structural and Mechanistic Insights. Soft Matter 2012, 8, 10314–10321. [Google Scholar] [CrossRef]
- Brissos, V.; Tavares, D.; Sousa, A.C.; Robalo, M.P.; Martins, L.O. Engineering a Bacterial DyP-Type Peroxidase for Enhanced Oxidation of Lignin-Related Phenolics at Alkaline PH. ACS Catal. 2017, 7, 3454–3465. [Google Scholar] [CrossRef]
- Atack, J.M.; Kelly, D.J. Structure, Mechanism and Physiological Roles of Bacterial Cytochrome c Peroxidases. In Advances in Microbial Physiology; Poole, R.K., Ed.; Academic Press: Cambridge, MA, USA, 2006; Volume 52, pp. 73–106. ISBN 0065-2911. [Google Scholar]
- Ellis, K.E.; Seidel, J.; Einsle, O.; Elliott, S.J. Geobacter Sulfurreducens Cytochrome c Peroxidases: Electrochemical Classification of Catalytic Mechanisms. Biochemistry 2011, 50, 4513–4520. [Google Scholar] [CrossRef] [Green Version]
- Becker, C.F.; Watmough, N.J.; Elliott, S.J. Electrochemical Evidence for Multiple Peroxidatic Heme States of the Diheme Cytochrome c Peroxidase of Pseudomonas Aeruginosa. Biochemistry 2009, 48, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wael, K.; Buschop, H.; Heering, H.A.; De Smet, L.; Van Beeumen, J.; Devreese, B.; Adriaens, A. Electrochemical Determination of Hydrogen Peroxide Using Rhodobacter Capsulatus Cytochrome c Peroxidase at a Gold Electrode. Microchim. Acta 2008, 162, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Sousa, P.M.P.; Pauleta, S.R.; Simões Gonçalves, M.L.; Pettigrew, G.W.; Moura, I.; Correia dos Santos, M.M.; Moura, J.J.G. Mediated Catalysis of Paracoccus Pantotrophus Cytochrome c Peroxidase by P. Pantotrophus Pseudoazurin: Kinetics of Intermolecular Electron Transfer. JBIC J. Biol. Inorg. Chem. 2007, 12, 691–698. [Google Scholar] [CrossRef] [Green Version]
- Sousa, P.M.P.; Pauleta, S.R.; Rodrigues, D.; Simões Gonçalves, M.L.; Pettigrew, G.W.; Moura, I.; Moura, J.J.G.; Correia dos Santos, M.M. Benefits of Membrane Electrodes in the Electrochemistry of Metalloproteins: Mediated Catalysis of Paracoccus Pantotrophus Cytochrome c Peroxidase by Horse Cytochrome c: A Case Study. JBIC J. Biol. Inorg. Chem. 2008, 13, 779–787. [Google Scholar] [CrossRef] [PubMed]
- De Wael, K.; Bashir, Q.; Van Vlierberghe, S.; Dubruel, P.; Heering, H.A.; Adriaens, A. Electrochemical Determination of Hydrogen Peroxide with Cytochrome c Peroxidase and Horse Heart Cytochrome c Entrapped in a Gelatin Hydrogel. Bioelectrochemistry 2012, 83, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, R. Cytochrome P-450. In Encyclopedia of Biological Chemistry; Elsevier: Amsterdam, The Netherlands, 2013; pp. 607–612. ISBN 978-0-12-378631-9. [Google Scholar]
- Guengerich, F.P. Mechanisms of Cytochrome P450-Catalyzed Oxidations. ACS Catal. 2018, 8, 10964–10976. [Google Scholar] [CrossRef] [PubMed]
- Hannemann, F.; Bichet, A.; Ewen, K.M.; Bernhardt, R. Cytochrome P450 Systems—Biological Variations of Electron Transport Chains. Biochim. Biophys. Acta Gen. Subj. 2007, 1770, 330–344. [Google Scholar] [CrossRef]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and Chemistry of Cytochrome P450. Chem. Rev. 2005, 105, 2253–2278. [Google Scholar] [CrossRef]
- Reipa, V.; Mayhew, M.P.; Holden, M.J.; Vilker, V.L. Redox Control of the P450cam Catalytic Cycle: Effects of Y96F Active Site Mutation and Binding of a Non-Natural Substrate. Chem. Comm. 2002, 318–319. [Google Scholar] [CrossRef]
- Daff, S.N.; Chapman, S.K.; Holt, R.A.; Govindaraj, S.; Poulos, T.L.; Munro, A.W. Redox Control of the Catalytic Cycle of Flavocytochrome P-450 BM3. Biochemistry 1997, 36, 13816–13823. [Google Scholar] [CrossRef]
- Martinis, S.A.; Blanke, S.R.; Hager, L.P.; Sligar, S.G.; Hui Bon Hoa, G.; Rux, J.J.; Dawson, J.H. Probing the Heme Iron Coordination Structure of Pressure-Induced Cytochrome P420cam. Biochemistry 1996, 35, 14530–14536. [Google Scholar] [CrossRef]
- Das, A.; Grinkova, Y.V.; Sligar, S.G. Redox Potential Control by Drug Binding to Cytochrome P450 3A4. J Am. Chem. Soc. 2007, 129, 13778–13779. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, S.; Rusling, J.F. Electrochemically Activated Catalytic Pathways of Human Metabolic Cytochrome P450s in Ultrathin Films. In Electrochemistry of N4 Macrocyclic Metal Complexes; Zagal, J.H., Bedioui, F., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 83–105. ISBN 978-3-319-31330-6. [Google Scholar]
- Munro, A.W.; Girvan, H.M.; Mason, A.E.; Dunford, A.J.; McLean, K.J. What Makes a P450 Tick? Trends Biochem. Sci. 2013, 38, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Munro, A.W.; McLean, K.J.; Grant, J.L.; Makris, T.M. Structure and Function of the Cytochrome P450 Peroxygenase Enzymes. Biochem. Soc. Trans. 2018, 46, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Gilardi, G. Cytochromes P450 Redox Activity. In Encyclopedia of Interfacial Chemistry; Elsevier: Amsterdam, The Netherlands, 2018; pp. 90–109. ISBN 978-0-12-809894-3. [Google Scholar]
- Krishnan, S.; Schenkman, J.B.; Rusling, J.F. Bioelectronic Delivery of Electrons to Cytochrome P450 Enzymes. J. Phys. Chem. B 2011, 115, 8371–8380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheller, F.W.; Yarmana, A.; Wollenberger, U. Cytochrome P450: Electron Transfer and Sensors. In Cytochrome P450 Enzymes: Biochemistry Pharmacology and Health Implications; Jian Wu, Ed.; Nova Science Publishers: New York, NY, USA, 2014; pp. 1–12. ISBN 978-1-61942-209-4. [Google Scholar]
- Hagen, K.D.; Gillan, J.M.; Im, S.-C.; Landefeld, S.; Mead, G.; Hiley, M.; Waskell, L.A.; Hill, M.G.; Udit, A.K. Electrochemistry of Mammalian Cytochrome P450 2B4 Indicates Tunable Thermodynamic Parameters in Surfactant Films. J. Inorg. Biochem. 2013, 129, 30–34. [Google Scholar] [CrossRef] [Green Version]
- Sadeghi, S.J.; Fantuzzi, A.; Gilardi, G. Breakthrough in P450 Bioelectrochemistry and Future Perspectives. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 237–248. [Google Scholar] [CrossRef]
- Udit, A.K.; Hagen, K.D.; Goldman, P.J.; Star, A.; Gillan, J.M.; Gray, H.B.; Hill, M.G. Spectroscopy and Electrochemistry of Cytochrome P450 BM3-Surfactant Film Assemblies. J. Am. Chem. Soc. 2006, 128, 10320–10325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarman, A.; Wollenberger, U.; Scheller, F.W. Sensors Based on Cytochrome P450 and CYP Mimicking Systems. Electrochim. Acta 2013, 110, 63–72. [Google Scholar] [CrossRef]
- Çekiç, S.Z.; Holtmann, D.; Güven, G.; Mangold, K.-M.; Schwaneberg, U.; Schrader, J. Mediated Electron Transfer with P450cin. Electrochem. Commun. 2010, 12, 1547–1550. [Google Scholar] [CrossRef]
- Dodhia, V.R.; Sassone, C.; Fantuzzi, A.; Nardo, G.D.; Sadeghi, S.J.; Gilardi, G. Modulating the Coupling Efficiency of Human Cytochrome P450 CYP3A4 at Electrode Surfaces through Protein Engineering. Electrochem. Commun. 2008, 10, 1744–1747. [Google Scholar] [CrossRef]
- Shumyantseva, V.V.; Kuzikov, A.V.; Masamrekh, R.A.; Bulko, T.V.; Archakov, A.I. From Electrochemistry to Enzyme Kinetics of Cytochrome P450. Biosens. Bioelectron. 2018, 121, 192–204. [Google Scholar] [CrossRef]
- Ducharme, J.; Auclair, K. Use of Bioconjugation with Cytochrome P450 Enzymes. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 32–51. [Google Scholar] [CrossRef] [Green Version]
- Baj-Rossi, C.; Rezzonico Jost, T.; Cavallini, A.; Grassi, F.; De Micheli, G.; Carrara, S. Continuous Monitoring of Naproxen by a Cytochrome P450-Based Electrochemical Sensor. Biosens. Bioelectron. 2014, 53, 283–287. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zheng, Q.; Bai, G.; Dai, Q.; Cao, X.; Yao, Y.; Liu, S.; Yao, C. Polydopamine Functionalized Nanoporous Graphene Foam as Nanoreactor for Efficient Electrode-Driven Metabolism of Steroid Hormones. Biosens. Bioelectron. 2018, 119, 182–190. [Google Scholar] [CrossRef]
- Peng, L.; Wollenberger, U.; Hofrichter, M.; Ullrich, R.; Scheibner, K.; Scheller, F.W. Bioelectrocatalytic Properties of Agrocybe Aegerita Peroxygenase. Electrochim. Acta 2010, 55, 7809–7813. [Google Scholar] [CrossRef]
- Peng, L.; Wollenberger, U.; Kinne, M.; Hofrichter, M.; Ullrich, R.; Scheibner, K.; Fischer, A.; Scheller, F.W. Peroxygenase Based Sensor for Aromatic Compounds. Biosens. Bioelectron. 2010, 26, 1432–1436. [Google Scholar] [CrossRef]
- Yarman, A.; Gröbe, G.; Neumann, B.; Kinne, M.; Gajovic-Eichelmann, N.; Wollenberger, U.; Hofrichter, M.; Ullrich, R.; Scheibner, K.; Scheller, F.W. The Aromatic Peroxygenase from Marasmius Rutola—a New Enzyme for Biosensor Applications. Anal. Bioanal. Chem. 2012, 402, 405–412. [Google Scholar] [CrossRef]
- Díaz, A.; Loewen, P.C.; Fita, I.; Carpena, X. Thirty Years of Heme Catalases Structural Biology. Arch. Biochem. Biophys. 2012, 525, 102–110. [Google Scholar] [CrossRef]
- Karakus, Y.Y. Typical Catalases: Function and Structure. In Glutathione System and Oxidative Stress in Health and Disease; Bagatini, M.D., Ed.; IntechOpen: London, UK, 2020. [Google Scholar]
- Lončar, N.; Fraaije, M.W. Catalases as Biocatalysts in Technical Applications: Current State and Perspectives. Appl. Microbiol. Biotechnol. 2015, 99, 3351–3357. [Google Scholar] [CrossRef]
- Prakash, P.A.; Yogeswaran, U.; Chen, S.-M. A Review on Direct Electrochemistry of Catalase for Electrochemical Sensors. Sensors 2009, 9, 1821–1844. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, H.S.; Moosavi-Movahedi, A.A.; Ghourchian, H. Correlation between Biological Activity and Electron Transferring of Bovine Liver Catalase: Osmolytes Effects. Electrochim. Acta 2013, 113, 591–602. [Google Scholar] [CrossRef]
- Akyilmaz, E.; Kozgus, O. Determination of Calcium in Milk and Water Samples by Using Catalase Enzyme Electrode. Food Chem. 2009, 115, 347–351. [Google Scholar] [CrossRef]
- O’Brien, K.B.; Killoran, S.J.; O’Neill, R.D.; Lowry, J.P. Development and Characterization in Vitro of a Catalase-Based Biosensor for Hydrogen Peroxide Monitoring. Biosens. Bioelectron. 2007, 22, 2994–3000. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.-J.; Niu, D.-J.; Liu, X.; Wu, Z.-W.; Fan, Y.; Chang, Y.-F.; Wu, Y.-Y. Direct Electrochemistry of Catalase at Amine-Functionalized Graphene/Gold Nanoparticles Composite Film for Hydrogen Peroxide Sensor. Electrochim. Acta 2011, 56, 2947–2953. [Google Scholar] [CrossRef]
- Alim, S.; Vejayan, J.; Kafi, A.K.M. Direct Electrochemistry of Catalase Immobilized at Polymerized-SnO2 Multiporous Modified Electrode for an Amperometric H2O2 Biosensor. Biomed. J. Sci. Tech. Res. 2018, 3, 3488–3493. [Google Scholar] [CrossRef]
- Fusco, G.; Bollella, P.; Mazzei, F.; Favero, G.; Antiochia, R.; Tortolini, C. Catalase-Based Modified Graphite Electrode for Hydrogen Peroxide Detection in Different Beverages. J. Anal. Methods Chem. 2016, 2016, 8174913. [Google Scholar] [CrossRef] [PubMed]
- Hashemnia, S.; Khayatzadeh, S.; Moosavi-Movahedi, A.A.; Ghourchian, H. Direct Electrochemistry of Catalase in Multiwall Carbon Nanotube/Dodecyl Trimethylammonium Bromide Film Covered With a Layer of Nafion on a Glassy Carbon Electrode. Int. J. Electrochem. Sci. 2011, 6, 581–595. [Google Scholar]
- Castiglione, N.; Rinaldo, S.; Giardina, G.; Stelitano, V.; Cutruzzola, F. Nitrite and Nitrite Reductases: From Molecular Mechanisms to Significance in Human Health and Disease. Antioxid. Redox Signal. 2012, 17, 684–716. [Google Scholar] [CrossRef] [Green Version]
- Almeida, M.G.; Serra, A.; Silveira, C.M.; Moura, J.J. Nitrite Biosensing via Selective Enzymes—A Long but Promising Route. Sensors 2010, 10, 11530–11555. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.; Roig-Zamboni, V.; Cutruzzola’, F.; Arese, M.; Sun, W.; Brunori, M.; Cambillau, C.; Tegoni, M. Domain Swing upon His to Ala Mutation in Nitrite Reductase of Pseudomonas Aeruginosa. J. Mol. Biol. 2001, 312, 541–554. [Google Scholar] [CrossRef]
- Maia, L.B.; Moura, J.J.G. How Biology Handles Nitrite. Chem. Rev. 2014, 114, 5273–5357. [Google Scholar] [CrossRef] [PubMed]
- Silveira, C.M.; Quintas, P.O.; Moura, I.; Moura, J.J.G.; Hildebrandt, P.; Almeida, M.G.; Todorovic, S. SERR Spectroelectrochemical Study of Cytochrome Cd1 Nitrite Reductase Co-Immobilized with Physiological Redox Partner Cytochrome C552 on Biocompatible Metal Electrodes. PLoS ONE 2015, 10, e0129940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besson, S.; Carneiro, C.; Moura, J.J.G.; Moura, I.; Fauque, G. A Cytochrome Cd1-Type Nitrite Reductase Isolated from the Marine Denitrifier Pseudomonas Nautica 617: Purification and Characterization. Anaerobe 1995, 1, 219–226. [Google Scholar] [CrossRef]
- Koppenhöfer, A.; Turner, K.L.; Allen, J.W.A.; Chapman, S.K.; Ferguson, S.J. Cytochrome Cd1 from Paracoccus Pantotrophus Exhibits Kinetically Gated, Conformationally Dependent, Highly Cooperative Two-Electron Redox Behavior. Biochemistry 2000, 39, 4243–4249. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Fülöp, V.; Garman, E.F.; Saunders, N.F.W.; Ferguson, S.J.; Hajdu, J. Haem-Ligand Switching during Catalysis in Crystals of a Nitrogen-Cycle Enzyme. Nature 1997, 389, 406–412. [Google Scholar] [CrossRef]
- Moura, I.; Pauleta, S.R.; Moura, J.J.G. Enzymatic Activity Mastered by Altering Metal Coordination Spheres. JBIC J. Biol. Inorg. Chem. 2008, 13, 1185–1195. [Google Scholar] [CrossRef] [Green Version]
- Cunha, C.A.; Macieira, S.; Dias, J.M.; Almeida, G.; Gonçalves, L.L.; Costa, C.; Lampreia, J.; Huber, R.; Moura, J.J.G.; Moura, I.; et al. Cytochrome c Nitrite Reductase from Desulfovibrio Desulfuricans ATCC 27774: The Relevance of The Two Calcium Sites In The Structure Of The Catalytic Subunit (NrfA). J. Biol. Chem. 2003, 278, 17455–17465. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.L.; Oliveira, T.F.; Pereira, I.A.C.; Archer, M. X-Ray Structure of the Membrane-Bound Cytochrome c Quinol Dehydrogenase NrfH Reveals Novel Haem Coordination. EMBO J. 2006, 25, 5951–5960. [Google Scholar] [CrossRef] [Green Version]
- Costa, C.; Moura, J.J.G.; Moura, I.; Wang, Y.; Huynh, B.H. Redox Properties of Cytochrome c Nitrite Reductase from Desulfovibrio Desulfuricans ATCC 27774. J. Biol. Chem. 1996, 271, 23191–23196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, M.G.; Silveira, C.M.; Guigliarelli, B.; Bertrand, P.; Moura, J.J.G.; Moura, I.; Léger, C. A Needle in a Haystack: The Active Site of the Membrane-Bound Complex Cytochrome c Nitrite Reductase. FEBS Lett. 2007, 581, 284–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todorovic, S.; Rodrigues, M.L.; Matos, D.; Pereira, I.A.C. Redox Properties of Lysine- and Methionine-Coordinated Hemes Ensure Downhill Electron Transfer in NrfH2A4 Nitrite Reductase. J. Phys. Chem. B 2012, 116, 5637–5643. [Google Scholar] [CrossRef]
- Dutt, J.; Davis, J. Current Strategies in Nitrite Detection and Their Application to Field Analysis. J. Environ. Monit. 2002, 4, 465–471. [Google Scholar] [CrossRef]
- Delgado, J.A.; Follett, R.F. Advances in Nitrogen Management for Water Quality. J. Soil Water Conserv. 2011, 66, 25A. [Google Scholar] [CrossRef]
- Lundberg, J.O.; Gladwin, M.T.; Ahluwalia, A.; Benjamin, N.; Bryan, N.S.; Butler, A.; Cabrales, P.; Fago, A.; Feelisch, M.; Ford, P.C.; et al. Nitrate and Nitrite in Biology, Nutrition and Therapeutics. Nat. Chem. Biol. 2009, 5, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.G.; Silveira, C.M.; Moura, J.J.G. Biosensing Nitrite Using the System Nitrite Redutase/Nafion/Methyl Viologen—A Voltammetric Study. Biosens. Bioelectron. 2007, 22, 2485–2492. [Google Scholar] [CrossRef] [PubMed]
- Silveira, C.M.; Baur, J.; Holzinger, M.; Moura, J.J.G.; Cosnier, S.; Almeida, M.G. Enhanced Direct Electron Transfer of a Multihemic Nitrite Reductase on Single-Walled Carbon Nanotube Modified Electrodes. Electroanalysis 2010, 22, 2973–2978. [Google Scholar] [CrossRef]
- Gwyer, J.D.; Richardson, D.J.; Butt, J.N. Diode or Tunnel-Diode Characteristics? Resolving the Catalytic Consequences of Proton Coupled Electron Transfer in a Multi-Centered Oxidoreductase. J. Am. Chem. Soc. 2005, 127, 14964–14965. [Google Scholar] [CrossRef] [PubMed]
- Angove, H.C.; Cole, J.A.; Richardson, D.J.; Butt, J.N. Protein Film Voltammetry Reveals Distinctive Fingerprints of Nitrite and Hydroxylamine Reduction by a Cytochrome c Nitrite Reductase. J. Biol. Chem. 2002, 277, 23374–23381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, T.; Rodrigues, P.R.; Goncalves, A.L.; Moura, J.J.; Jubete, E.; Anorga, L.; Piknova, B.; Schechter, A.N.; Silveira, C.M.; Almeida, M.G. Construction of Effective Disposable Biosensors for Point of Care Testing of Nitrite. Talanta 2015, 142, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Silveira, C.M.; Pimpão, M.; Pedroso, H.A.; Rodrigues, P.R.S.; Moura, J.J.G.; Pereira, M.F.R.; Almeida, M.G. Probing the Surface Chemistry of Different Oxidized MWCNT for the Improved Electrical Wiring of Cytochrome c Nitrite Reductase. Electrochem. Commun. 2013, 35, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Lojou, E.; Cutruzzolà, F.; Tegoni, M.; Bianco, P. Electrochemical Study of the Intermolecular Electron Transfer to Pseudomonas Aeruginosa Cytochrome Cd1 Nitrite Reductase. Electrochim. Acta 2003, 48, 1055–1064. [Google Scholar] [CrossRef]
- Pedroso, H.A.; Silveira, C.M.; Almeida, R.M.; Almeida, A.; Besson, S.; Moura, I.; Moura, J.J.G.; Almeida, M.G. Electron Transfer and Docking between Cytochrome Cd1 Nitrite Reductase and Different Redox Partners—A Comparative Study. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1412–1421. [Google Scholar] [CrossRef]
- Strehlitz, B.; Gründig, B.; Schumacher, W.; Kroneck, P.M.H.; Vorlop, K.-D.; Kotte, H. A Nitrite Sensor Based on a Highly Sensitive Nitrite Reductase Mediator-Coupled Amperometric Detection. Anal. Chem. 1996, 68, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Strehlitz, B.; Gründig, B.; Vorlop, K.D.; Bartholmes, P.; Kotte, H.; Stottmeister, U. Artificial Electron Donors for Nitrate and Nitrite Reductases Usable as Mediators in Amperometric Biosensors. Fresenius J. Anal. Chem. 1994, 349, 676–678. [Google Scholar] [CrossRef]
- Karunakaran, C.; Pandiaraj, M.; Santharaman, P. Immunosensors. In Biosensors and Bioelectronics; Karunakaran, C., Bhargava, K., Benjamin, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Chapter 4; pp. 205–245. ISBN 978-0-12-803100-1. [Google Scholar]
- Batra, B.; Lata, S.; Rani, S.; Pundir, C.S. Fabrication of a Cytochrome c Biosensor Based on Cytochrome Oxidase/NiO-NPs/CMWCNT/PANI Modified Au Electrode. J. Biomed. Nanotechnol. 2013, 9, 409–416. [Google Scholar] [CrossRef]
- Katz, E.; Lioubashevski, O.; Willner, I. Magnetic Field Effects on Bioelectrocatalytic Reactions of Surface-Confined Enzyme Systems: Enhanced Performance of Biofuel Cells. J. Am. Chem. Soc. 2005, 127, 3979–3988. [Google Scholar] [CrossRef]
- Santucci, R.; Sinibaldi, F.; Cozza, P.; Polticelli, F.; Fiorucci, L. Cytochrome c: An Extreme Multifunctional Protein with a Key Role in Cell Fate. Int. J. Biol. Macromol. 2019, 136, 1237–1246. [Google Scholar] [CrossRef]
- Hannibal, L.; Tomasina, F.; Capdevila, D.A.; Demicheli, V.; Tórtora, V.; Alvarez-Paggi, D.; Jemmerson, R.; Murgida, D.H.; Radi, R. Alternative Conformations of Cytochrome c: Structure, Function, and Detection. Biochemistry 2016, 55, 407–428. [Google Scholar] [CrossRef]
- Silveira, C.M.; Castro, M.A.; Dantas, J.M.; Salgueiro, C.; Murgida, D.H.; Todorovic, S. Structure, Electrocatalysis and Dynamics of Immobilized Cytochrome PccH and Its Microperoxidase. Phys. Chem. Chem. Phys. 2017, 19, 8908–8918. [Google Scholar] [CrossRef] [PubMed]
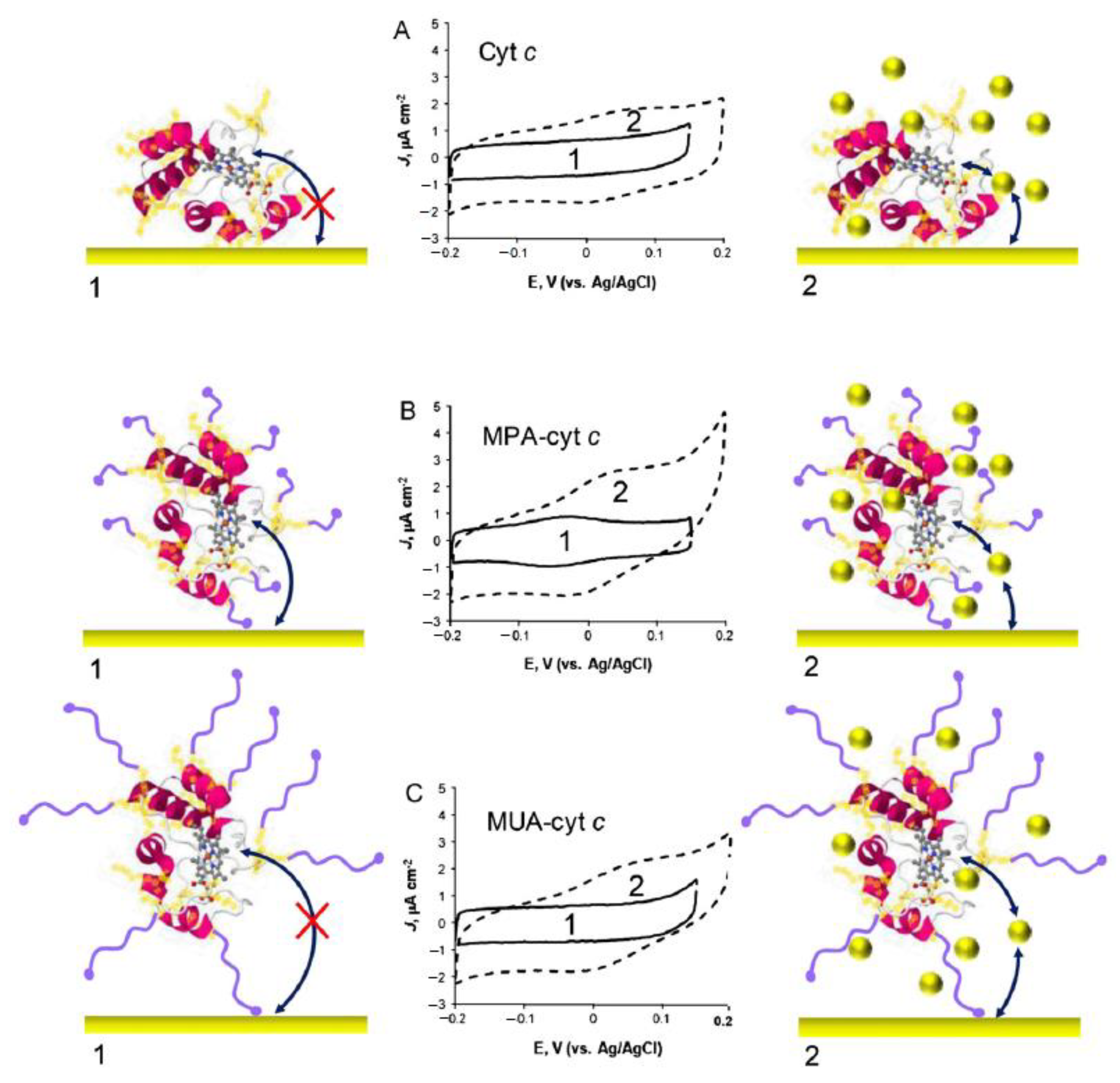
- Eddowes, M.J.; Hill, H.A.O. Electrochemistry of horse heart cytochrome c. J. Am. Chem. Soc. 1979, 101, 4461–4464. [Google Scholar] [CrossRef]
- Murgida, D.H.; Hildebrandt, P. Disentangling Interfacial Redox Processes of Proteins by SERR Spectroscopy. Curr. Soc. Rev. 2008, 37, 937–945. [Google Scholar] [CrossRef]
- Tavagnacco, C.; Monari, S.; Ranieri, A.; Bortolotti, C.A.; Peressini, S.; Borsari, M. Immobilized Unfolded Cytochrome c Acts as a Catalyst for Dioxygen Reduction. Chem. Commun. 2011, 47, 11122. [Google Scholar] [CrossRef]
- Wisitruangsakul, N.; Zebger, I.; Ly, K.H.; Murgida, D.H.; Ekgasit, S.; Hildebrandt, P. Redox-Linked Protein Dynamics of Cytochrome c Probed by Time-Resolved Surface Enhanced Infrared Absorption Spectroscopy. Phys. Chem. Chem. Phys. 2008, 10, 5276–5286. [Google Scholar] [CrossRef]
- Ly, H.K.; Sezer, M.; Wisitruangsakul, N.; Feng, J.-J.; Kranich, A.; Millo, D.; Weidinger, I.M.; Zebger, I.; Murgida, D.H.; Hildebrandt, P. Surface-Enhanced Vibrational Spectroscopy for Probing Transient Interactions of Proteins with Biomimetic Interfaces: Electric Field Effects on Structure, Dynamics and Function of Cytochrome c. FEBS J. 2011, 1382–1390. [Google Scholar] [CrossRef]
- Ranieri, A.; Bernini, F.; Bortolotti, C.A.; Castellini, E. The Met80Ala Point Mutation Enhances the Peroxidase Activity of Immobilized Cytochrome c. Catal. Sci. Technol. 2012, 2, 2206. [Google Scholar] [CrossRef]
- Casalini, S.; Battistuzzi, G.; Borsari, M.; Bortolotti, C.A.; Ranieri, A.; Sola, M. Electron Transfer and Electrocatalytic Properties of the Immobilized Methionine80Alanine Cytochrome c Variant. J. Phys. Chem. B 2008, 112, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-H.; Lin, Y.-W.; Rosell, F.I.; Ni, F.-Y.; Lu, H.-J.; Yang, P.-Y.; Tan, X.-S.; Li, X.-Y.; Huang, Z.-X.; Mauk, A.G. Converting Cytochrome c into a Peroxidase-Like Metalloenzyme by Molecular Design. ChemBioChem 2007, 8, 607–609. [Google Scholar] [CrossRef]
- Casalini, S.; Battistuzzi, G.; Borsari, M.; Bortolotti, C.A.; Di Rocco, G.; Ranieri, A.; Sola, M. Electron Transfer Properties and Hydrogen Peroxide Electrocatalysis of Cytochrome c Variants at Positions 67 and 80. J. Phys. Chem. B 2010, 114, 1698–1706. [Google Scholar] [CrossRef]
- Aghamiri, Z.S.; Mohsennia, M.; Rafiee-Pour, H.-A. Immobilization of Cytochrome c and Its Application as Electrochemical Biosensors. Talanta 2018, 176, 195–207. [Google Scholar] [CrossRef]
- Bathinapatla, A.; Kanchi, S.; Singh, P.; Sabela, M.I.; Bisetty, K. An Ultrasensitive Performance Enhanced Novel Cytochrome c Biosensor for the Detection of Rebaudioside A. Biosens. Bioelectron. 2016, 77, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zheng, J.; Wang, J.; Xu, J.; Hayat, T.; Alharbi, N.S. Direct Electrochemistry of Cytochrome c Immobilized on One Dimensional Au Nanoparticles Functionalized Magnetic N-Doped Carbon Nanotubes and Its Application for the Detection of H2O2. Sens. Actuators B 2019, 282, 85–95. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, Y.; Hou, J.; Jia, Z.; Zhong, D.; Zhou, S.; Huo, D.; Yang, M.; Hou, C. Electrochemical Biointerface Based on Electrodeposition AuNPs on 3D Graphene Aerogel: Direct Electron Transfer of Cytochrome c and Hydrogen Peroxide Sensing. J. Electroanal. Chem. 2019, 842, 16–23. [Google Scholar] [CrossRef]
- Aghamiri, Z.S.; Mohsennia, M.; Rafiee-Pour, H.-A. Immobilization of Cytochrome c on Polyaniline/Polypyrrole/Carboxylated Multi-Walled Carbon Nanotube/Glassy Carbon Electrode: Biosensor Fabrication. J. Solid State Electrochem. 2019, 23, 2233–2242. [Google Scholar] [CrossRef]
- Guo, C.; Wang, J.; Chen, X.; Li, Y.; Wu, L.; Zhang, J.; Tao, C. Construction of a Biosensor Based on a Combination of Cytochrome c, Graphene, and Gold Nanoparticles. Sensors 2019, 19, 40. [Google Scholar] [CrossRef] [Green Version]
- Eguílaz, M.; Gutiérrez, A.; Rivas, G. Non-Covalent Functionalization of Multi-Walled Carbon Nanotubes with Cytochrome c: Enhanced Direct Electron Transfer and Analytical Applications. Sens. Actuators B 2016, 225, 74–80. [Google Scholar] [CrossRef]
- Suárez, G.; Santschi, C.; Martin, O.J.F.; Slaveykova, V.I. Biosensor Based on Chemically-Designed Anchorable Cytochrome c for the Detection of H2O2 Released by Aquaticcells. Biosens. Bioelectron. 2013, 42, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Molinas, M.F.; Benavides, L.; Castro, M.A.; Murgida, D.H. Stability, Redox Parameters and Electrocatalytic Activity of a Cytochrome Domain from a New Subfamily. Bioelectrochemistry 2015, 105, 25–33. [Google Scholar] [CrossRef]
- Harbury, H.A.; Cronin, J.R.; Fanger, M.W.; Hettinger, T.P.; Murphy, A.J.; Myer, Y.P.; Vinogradov, S.N. Complex Formation between Methionine and a Heme Peptide from Cytochrome c. Proc. Natl. Acad. Sci. USA 1965, 54, 1658–1664. [Google Scholar] [CrossRef] [Green Version]
- Osman, A.M.; Koerts, J.; Boersma, M.G.; Boeren, S.; Veeger, C.; Rietjens, I.M.C.M. Microperoxidase/H2O2-Catalyzed Aromatic Hydroxylation Proceeds by a Cytochrome-P-450-Type Oxygen-Transfer Reaction Mechanism. Eur. J. Biochem. 1996, 240, 232–238. [Google Scholar] [CrossRef]
- Rusvai, E.; Végh, M.; Kramer, M.; Horváth, I. Hydroxylation of Aniline Mediated by Heme-Bound Oxy-Radicals in a Heme Peptide Model System. Biochem. Pharmacol. 1988, 37, 4574–4577. [Google Scholar] [CrossRef]
- Colonna, S.; Gaggereo, N.; Carrea, G.; Pasta, P. The Microperoxidase-11 Catalyzed Oxidation of Sulfides Is Enantioselective. Tetrahedron Lett. 1994, 35, 9103–9104. [Google Scholar] [CrossRef]
- Boersma, M.G.; Primus, J.-L.; Koerts, J.; Veeger, C.; Rietjens, I.M.C.M. Heme-(Hydro)Peroxide Mediated O- and N-Dealkylation. Eur. J. Biochem. 2000, 267, 6673–6678. [Google Scholar] [CrossRef] [PubMed]
- Verbaro, D.; Hagarman, A.; Soffer, J.; Schweitzer-Stenner, R. The PH Dependence of the 695 Nm Charge Transfer Band Reveals the Population of an Intermediate State of the Alkaline Transition of Ferricytochrome c at Low Ion Concentrations. Biochemistry 2009, 48, 2990–2996. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Van Wart, H.E. Resonance Raman Characterization of the Heme c Group in N-Acetyl Microperoxidase-8: A Thermal Intermediate Spin-High Spin State Mixture. J. Phys. Chem. 1989, 93, 7925–7931. [Google Scholar] [CrossRef]
- Zhu, X.; Yuri, I.; Gan, X.; Suzuki, I.; Li, G. Electrochemical Study of the Effect of Nano-Zinc Oxide on Microperoxidase and Its Application to More Sensitive Hydrogen Peroxide Biosensor Preparation. Biosens. Bioelectron. 2007, 22, 1600–1604. [Google Scholar] [CrossRef]
- Astuti, Y.; Topoglidis, E.; Durrant, J.R. Use of Microperoxidase-11 to Functionalize Tin Dioxide Electrodes for the Optical and Electrochemical Sensing of Hydrogen Peroxide. Anal. Chim. Acta 2011, 686, 126–132. [Google Scholar] [CrossRef]
- Ioannidis, L.A.; Nikolaou, P.; Panagiotopoulos, A.; Vassi, A.; Topoglidis, E. Microperoxidase-11 Modified Mesoporous SnO 2 Film Electrodes for the Detection of Antimalarial Drug Artemisinin. Anal. Methods 2019, 11, 3117–3125. [Google Scholar] [CrossRef]
- Graça, J.S.; Oliveira, R.F.; Moraes, M.L.; Ferreira, M. Amperometric Glucose Biosensor Based on Layer-by-Layer Films of Microperoxidase-11 and Liposome-Encapsulated Glucose Oxidase. Bioelectrochemistry 2014, 96, 37–42. [Google Scholar] [CrossRef]
- Kogikoski, S.; Sousa, C.P.; Liberato, M.S.; Andrade-Filho, T.; Prieto, T.; Ferreira, F.F.; Rocha, A.R.; Guha, S.; Alves, W.A. Multifunctional Biosensors Based on Peptide–Polyelectrolyte Conjugates. Phys. Chem. Chem. Phys. 2016, 18, 3223–3233. [Google Scholar] [CrossRef]
- Miyazaki, C.M.; Shimizu, F.M.; Mejía-Salazar, J.R.; Oliveira, O.N.; Ferreira, M. Surface Plasmon Resonance Biosensor for Enzymatic Detection of Small Analytes. Nanotechnology 2017, 28, 145501. [Google Scholar] [CrossRef]
- Nastri, F.; Lista, L.; Ringhieri, P.; Vitale, R.; Faiella, M.; Andreozzi, C.; Travascio, P.; Maglio, O.; Lombardi, A.; Pavone, V. A Heme–Peptide Metalloenzyme Mimetic with Natural Peroxidase-Like Activity. Chem. Eur. J. 2011, 17, 4444–4453. [Google Scholar] [CrossRef]
- Vitale, R.; Lista, L.; Cerrone, C.; Caserta, G.; Chino, M.; Maglio, O.; Nastri, F.; Pavone, V.; Lombardi, A. An Artificial Heme-Enzyme with Enhanced Catalytic Activity: Evolution, Functional Screening and Structural Characterization. Org. Biomol. Chem. 2015, 13, 4859–4868. [Google Scholar] [CrossRef] [PubMed]
- Caserta, G.; Chino, M.; Firpo, V.; Zambrano, G.; Leone, L.; D’Alonzo, D.; Nastri, F.; Maglio, O.; Pavone, V.; Lombardi, A. Enhancement of Peroxidase Activity in Artificial Mimochrome VI Catalysts through Rational Design. ChemBioChem 2018, 19, 1823–1826. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, G.; Nastri, F.; Pavone, V.; Lombardi, A.; Chino, M. Use of an Artificial Miniaturized Enzyme in Hydrogen Peroxide Detection by Chemiluminescence. Sensors 2020, 20, 3793. [Google Scholar] [CrossRef] [PubMed]
- Neumann, B.; Götz, R.; Wrzolek, P.; Scheller, F.W.; Weidinger, I.M.; Schwalbe, M.; Wollenberger, U. Enhancement of the Electrocatalytic Activity of Thienyl-Substituted Iron Porphyrin Electropolymers by a Hangman Effect. ChemCatChem 2018, 10, 4353–4361. [Google Scholar] [CrossRef]
- Li, T.; Hu, P.; Li, J.; Huang, P.; Tong, W.; Gao, C. Enhanced Peroxidase-like Activity of Fe@PCN-224 Nanoparticles and Their Applications for Detection of H2O2and Glucose. Colloids Surf. A 2019, 577, 456–463. [Google Scholar] [CrossRef]
- Palanisamy, S.; Velusamy, V.; Chen, S.-W.; Yang, T.C.K.; Balu, S.; Banks, C.E. Enhanced Reversible Redox Activity of Hemin on Cellulose Microfiber Integrated Reduced Graphene Oxide for H2O2 Biosensor Applications. Carbohydr. Polym. 2019, 204, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Tang, H.; Wu, Y.; Zhang, Y.; Li, Z. Highly Electrocatalytic Biosensor Based on Hemin@AuNPs/Reduced Graphene Oxide/Chitosan Nanohybrids for Non-Enzymatic Ultrasensitive Detection of Hydrogen Peroxide in Living Cells. Biosens. Bioelectron. 2019, 132, 217–223. [Google Scholar] [CrossRef]
- Zhao, P.; Chen, S.; Zhou, J.; Zhang, S.; Huo, D.; Hou, C. A Novel Fe-Hemin-Metal Organic Frameworks Supported on Chitosan-Reduced Graphene Oxide for Real-Time Monitoring of H2O2 Released from Living Cells. Anal. Chim. Acta 2020, 1128, 90–98. [Google Scholar] [CrossRef]
- Ye, J.; Baldwin, R.P. Catalytic Reduction of Myoglobin and Hemoglobin at Chemically Modified Electrodes Containing Methylene Blue. Anal. Chem. 1988, 60, 2263–2268. [Google Scholar] [CrossRef]
- Reeder, B.J. The Redox Activity of Hemoglobins: From Physiologic Functions to Pathologic Mechanisms. Antioxid. Redox Signal. 2010, 13, 1087–1123. [Google Scholar] [CrossRef] [PubMed]
- Zu, X.; Lu, Z.; Zhang, Z.; Schenkman, J.B.; Rusling, J.F. Electroenzyme-Catalyzed Oxidation of Styrene and Cis -β-Methylstyrene Using Thin Films of Cytochrome P450cam and Myoglobin. Langmuir 1999, 15, 7372–7377. [Google Scholar] [CrossRef]
- Keilin, D.; Hartree, E.F. Reaction of Methæmoglobin with Hydrogen Peroxide. Nature 1950, 166, 513–514. [Google Scholar] [CrossRef]
- Onuoha, A.C.; Zu, X.; Rusling, J.F. Electrochemical Generation and Reactions of Ferrylmyoglobins in Water and Microemulsions. J. Am. Chem. Soc. 1997, 119, 3979–3986. [Google Scholar] [CrossRef]
- Wen, Y.; Wen, W.; Zhang, X.; Wang, S. Highly Sensitive Amperometric Biosensor Based on Electrochemically-Reduced Graphene Oxide-Chitosan/Hemoglobin Nanocomposite for Nitromethane Determination. Biosens. Bioelectron. 2016, 79, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, J.; Ni, X.; Cao, Y. A Biosensor Based on Hemoglobin Immobilized with Magnetic Molecularly Imprinted Nanoparticles and Modified on a Magnetic Electrode for Direct Electrochemical Determination of 3-Chloro-1,2-Propandiol. J. Electroanal. Chem. 2019, 834, 233–240. [Google Scholar] [CrossRef]
- Niu, Y.; Xie, H.; Luo, G.; Weng, W.; Ruan, C.; Li, G.; Sun, W. Electrochemical Performance of Myoglobin Based on TiO 2 -Doped Carbon Nanofiber Decorated Electrode and Its Applications in Biosensing. RSC Adv. 2019, 9, 4480–4487. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Weng, W.; Xie, H.; Luo, G.; Li, G.; Sun, W.; Ruan, C.; Wang, X. Myoglobin- and Hydroxyapatite-Doped Carbon Nanofiber-Modified Electrodes for Electrochemistry and Electrocatalysis. ACS Omega 2019, 4, 15653–15659. [Google Scholar] [CrossRef] [Green Version]
- Brissos, V.; Ferreira, M.; Grass, G.; Martins, L.O. Turning a Hyperthermostable Metallo-Oxidase into a Laccase by Directed Evolution. ACS Catal. 2015, 5, 4932–4941. [Google Scholar] [CrossRef]
- Packer, M.S.; Liu, D.R. Methods for the Directed Evolution of Proteins. Nat. Rev. Genet. 2015, 16, 379–394. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biosensor Configuration/Electrode | Target Analyte | Detection Mode (Mediator) | Ew (V) vs. NHE | Sensitivity (A M−1 cm−2) | Linear Range (µM) | Limit of Detection (µM) | Stability (% Initial Activity/Time) | [Ref] |
|---|---|---|---|---|---|---|---|---|
| HRP + PMN/GC | H2O2 | DET | Amp −0.006 | 0.620 | 0.19–1.35 | 0.0475 | 92%/1 month | [115] |
| HRP/PEGDGE/[Os(dmp)PVI]+/2+/G | MET ([Os(dmp)PVI]+/2) | Amp +0.188 | 0.300 | 1–500 | 0.3 | 50%/1 month | [95] | |
| HRP/MWCNT/thionine/AuNP/GC | DET | Amp +0.199 | 6.87 × 10−5 a | 640–7000 | 0.1 | n.d. | [37] | |
| HRP + HA + CdS-IL/CILE | DET | CV n.d. | 0.040 a | 10–170 | 3.30 | 95.6%/2 weeks | [81] | |
| TCA | DET | CV ~−0.060 c | 0.00208 a | 1600–18,000 | 530 | 95.6%/2 weeks | ||
| TOP/PEGDGE/[Os(dmp)PVI]+/2+/G | H2O2 | MET ([Os(dmp)PVI]+/2) | Amp +0.188 | 0.470 | 1–500 | 0.3 | 83%/1 month | [95] |
| DyP/SAM/Ag | H2O2 | DET | Amp +0.100 | 1.31 | 1–200 | 3.60 | 85%/1 month | [67] |
| P450-2C19/CS/CeNP/RGO/GC | Omeprazole | DET | Amp −0.276 | 1.440 a | 2–50 | 0.42 | n.d. | [117] |
| P450-3A4/P450-1A2/GA/AuNP/CS/RGO /GC | Clopidogel | DET | CV ~−0.360 c | 0.730 a | 2–50 | 0.63 | n.d. | [118] |
| Cat + NiO/GC | H2O2 | DET | Amp −0.059 | 0.478 a | 1–10 | 0.60 | 93%/2 weeks | [119] |
| [(Cat + NH2-IL)]7 + TiN NP/GC | DET | Amp −0.150 | 0.380 | 1–2100 | 0.100 | n.d. | [120] | |
| Cat/[BMIM]BF4-IL/NH2-MWCNT/GC | DET | Amp −0.345 | 69.3 a | 0.0086–0.140 | 0.0037 | n.d. | [121] | |
| Cat/PLL/f-MWCNT/GC | DET | Amp −0.250 | 0.392 | 0.001–3.6 | 0.008 | 89%/14 days | [122] | |
| ccNiR/[ZnCr-AQS] LDH/GC | NO2− | MET (ZnCr-AQS) | Amp −0.400 | 1.8 | 0.015–2.35 | 0.004 | 60%/32 days | [123] |
| ccNiR/sol-gel/PG | DET | Amp −0.700 | 0.430 | 0.25–50 | 0.120 | >90%/2 weeks | [124] | |
| ccNiR + WO3NP/ITO | DET | CV −0.600 | 2.143 | 5–50 | 5 | n.d. | [125] | |
| CcO/DDAB/Au | Cyt c | DET | SQW ~+0.550 c | n.d. | 0.2–4 | 0.2 | n.d. | [126] |
| CcO/DOPE-DOPC/Au | DET | Amp +0.485 | n.d. | 0–10 | 0.1 | n.d. | [127] | |
| Cyt c/l-Cys/P3MT/MWCNT/GC | H2O | DET | Amp +0.210 | 0.0178 a | 0.7–400 | 0.23 | n.d. | [128] |
| Cyt c-BDND | DET | Amp +0.149 | 0.0756 | 1–450 | 0.7 | 85%/6 weeks | [129] | |
| Nafion/Cyt c-MPA-AuNP-CS/SAM/Au | NO | DET | Amp −0.500 | 0.443 b | 10–215 | 4.5 | 96.3%/1 month | [130] |
| MP-11/PDADMAC-mpATO/ITO | H2O2 | DET | Amp +0.210 | 0.00387 | 10–750 | n.d. | n.d. | [131] |
| MP-11 @ PCN-333 (Al)/3D-KSC | H2O2 | DET | Amp −0.056 | 0.168 | 0.4–1725 | 0.127 | 89.5%/1 month | [132] |
| MP/Cat/SOD/MWCNT-PTTCA/AuNP/GC | NO | DET | Amp −0.600 | 15.7 a | 1.0–40 | 0.0043 | 97%/1 month | [133] |
| Nafion/Hb/H-TiO2-rGOMS/GC | H2O2 | DET | Amp −0.140 | n.d. | 0.1–145 | 0.01 | 95%/21 days | [134] |
| Nafion/Hb+CS+bBi2S3/GC | DET | Amp −0.203 | 14.1 a | 0.4–4.8 | 0.096 | 95.7%/15 days | [135] | |
| Hb/CNF/GC | ART | DET | CV ~−0.200 c | 4.70 a | 0–200 | n.d. | n.d. | [136] |
| Mb/AuNP-PTy-f-MWCNT/GC | H2O2 | DET | Amp −0.100 | 0.140 | 2–5000 | 0.01 | n.d. | [137] |
| Nafion/Mb/AuNP/Mg-MOF-74/CILE | NO2− | DET | CV −0.335 c | 0.1157 AM−1 | 800–18,000 | 267 | n.d. | [138] |
| TCA | DET | CV −0.098 c | 6.942 × 10−3 AM−1 | 1000–200,000 | 333 | n.d. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuccarello, L.; Barbosa, C.; Todorovic, S.; Silveira, C.M. Electrocatalysis by Heme Enzymes—Applications in Biosensing. Catalysts 2021, 11, 218. https://doi.org/10.3390/catal11020218
Zuccarello L, Barbosa C, Todorovic S, Silveira CM. Electrocatalysis by Heme Enzymes—Applications in Biosensing. Catalysts. 2021; 11(2):218. https://doi.org/10.3390/catal11020218
Chicago/Turabian StyleZuccarello, Lidia, Catarina Barbosa, Smilja Todorovic, and Célia M. Silveira. 2021. "Electrocatalysis by Heme Enzymes—Applications in Biosensing" Catalysts 11, no. 2: 218. https://doi.org/10.3390/catal11020218
APA StyleZuccarello, L., Barbosa, C., Todorovic, S., & Silveira, C. M. (2021). Electrocatalysis by Heme Enzymes—Applications in Biosensing. Catalysts, 11(2), 218. https://doi.org/10.3390/catal11020218