
Mayenite-Based Electride C12A7e−: A Reactivity and Stability Study

 ,
,  , , , ,
, , , ,  ,
,  and
and
Abstract
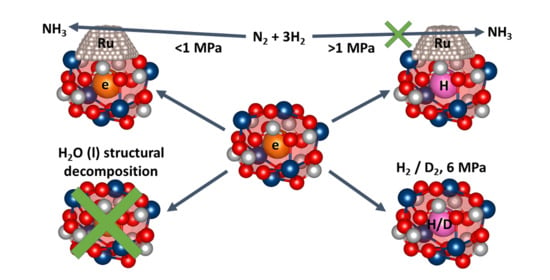
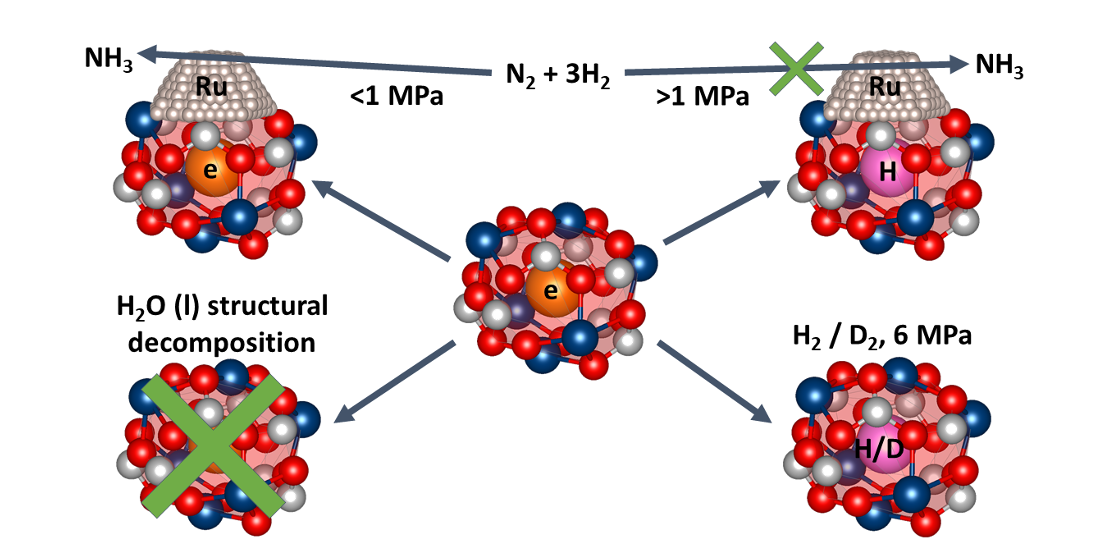
:
1. Introduction
2. Results and Discussion
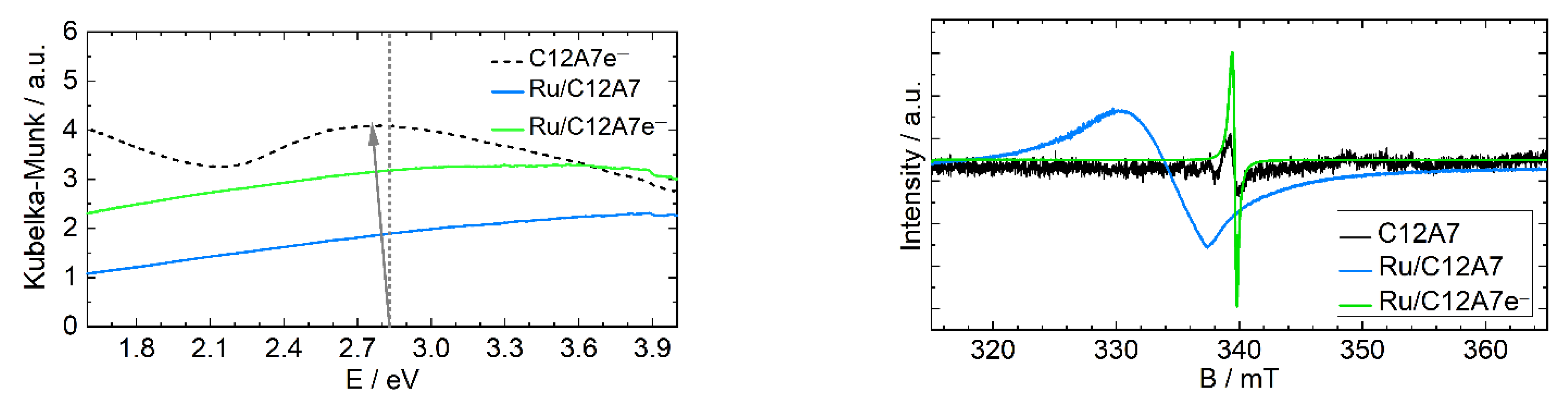
2.1. Ruthenium Deposition
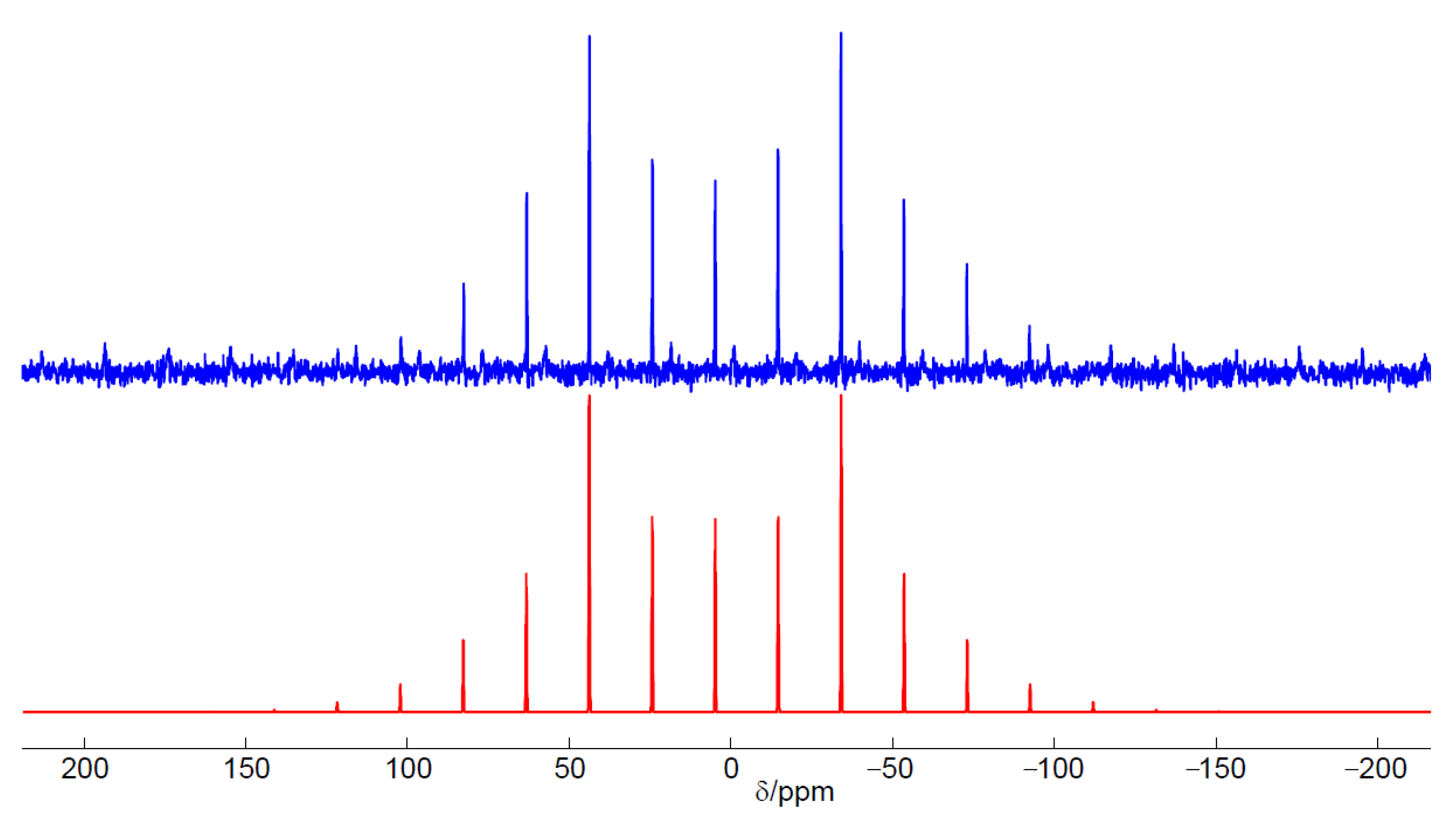
2.2. Reactivity Studies of C12A7 and C12A7e−
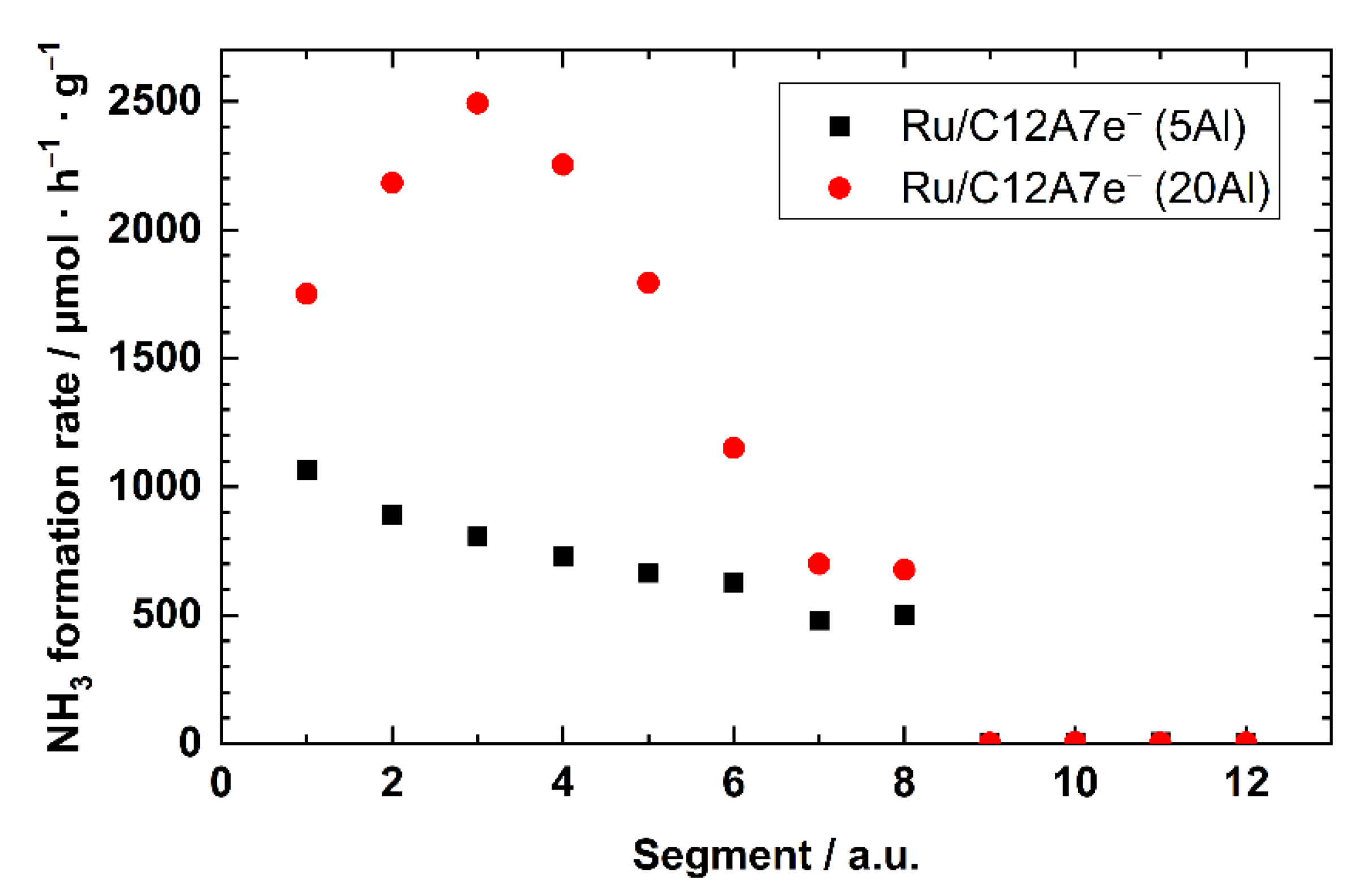
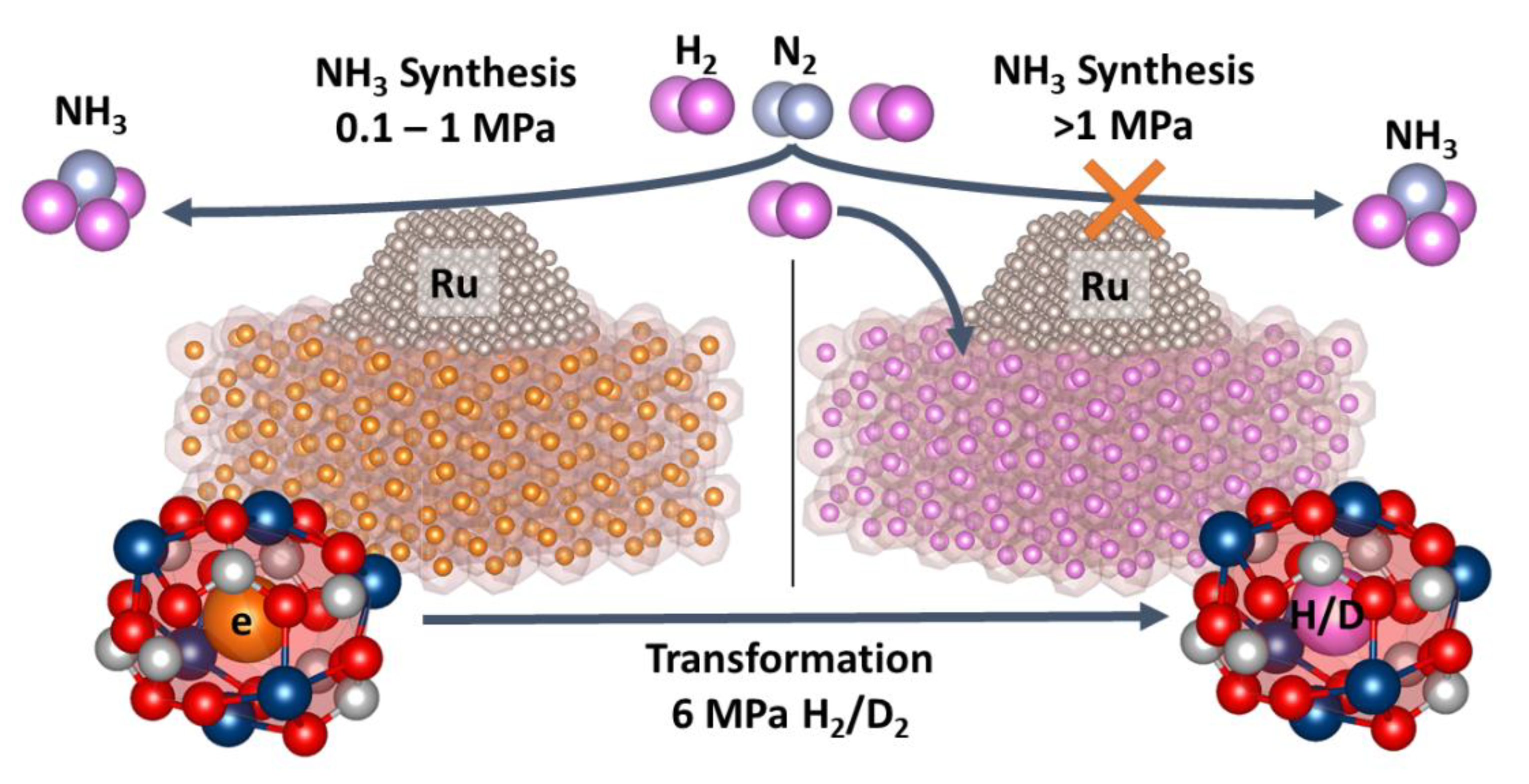
2.2.1. Catalytic Studies of Ru/C12A7e−
2.2.2. Reactivity towards Forming Gas and Hydrogen
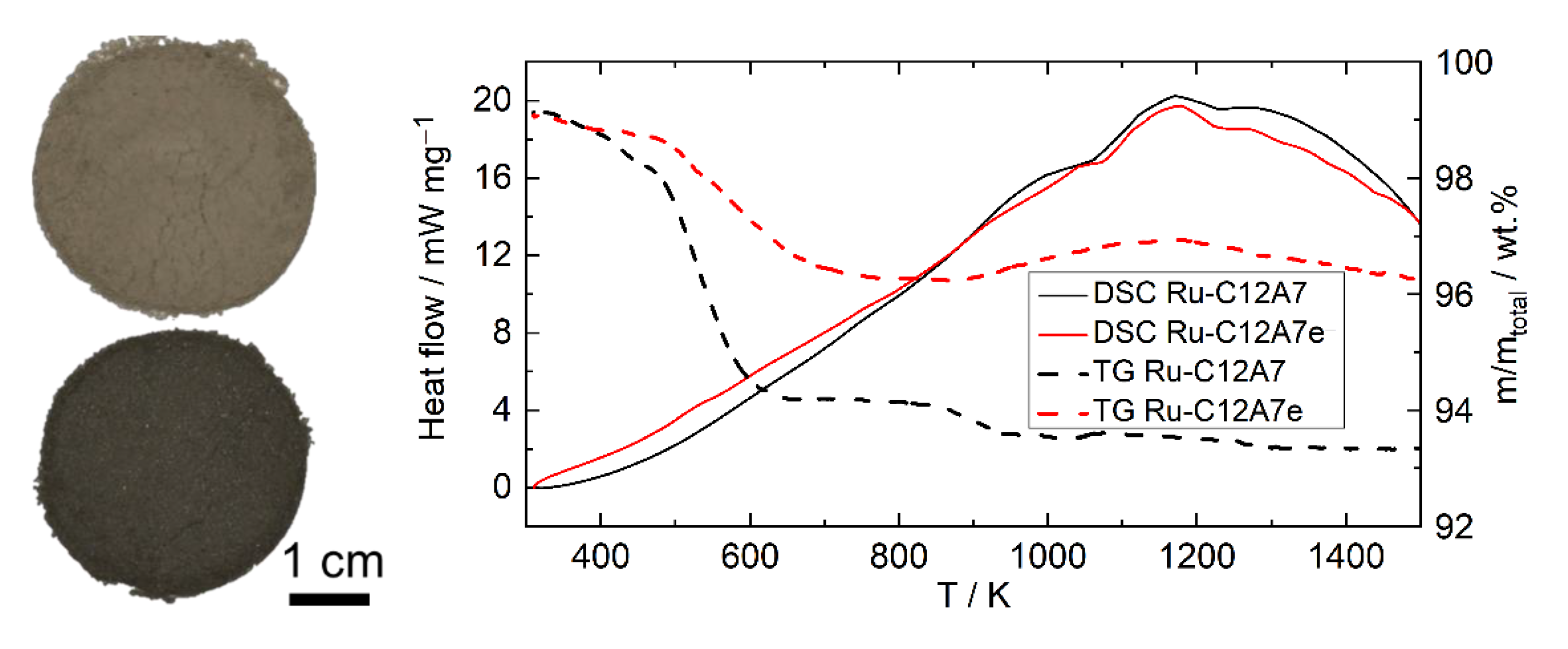
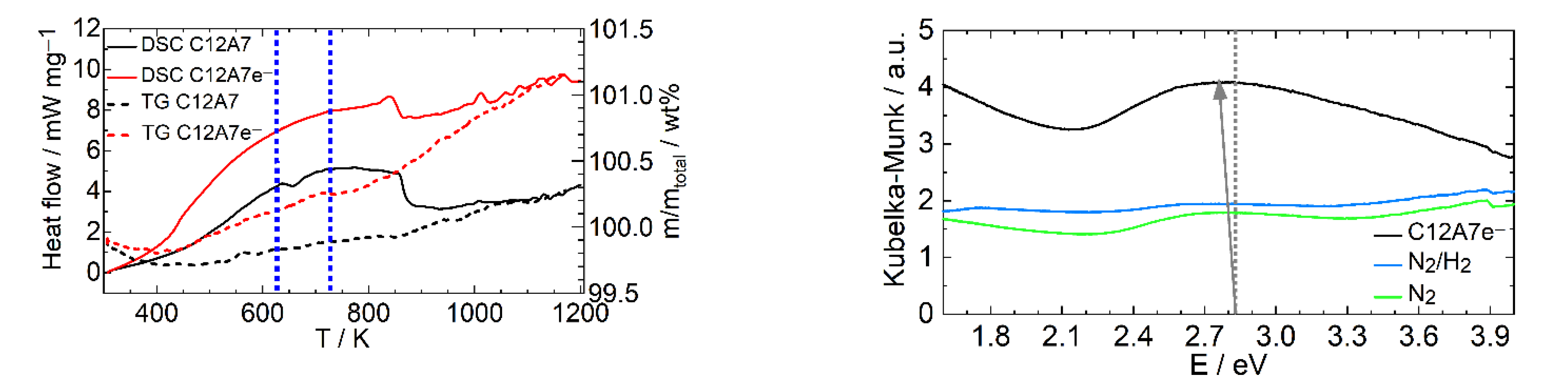
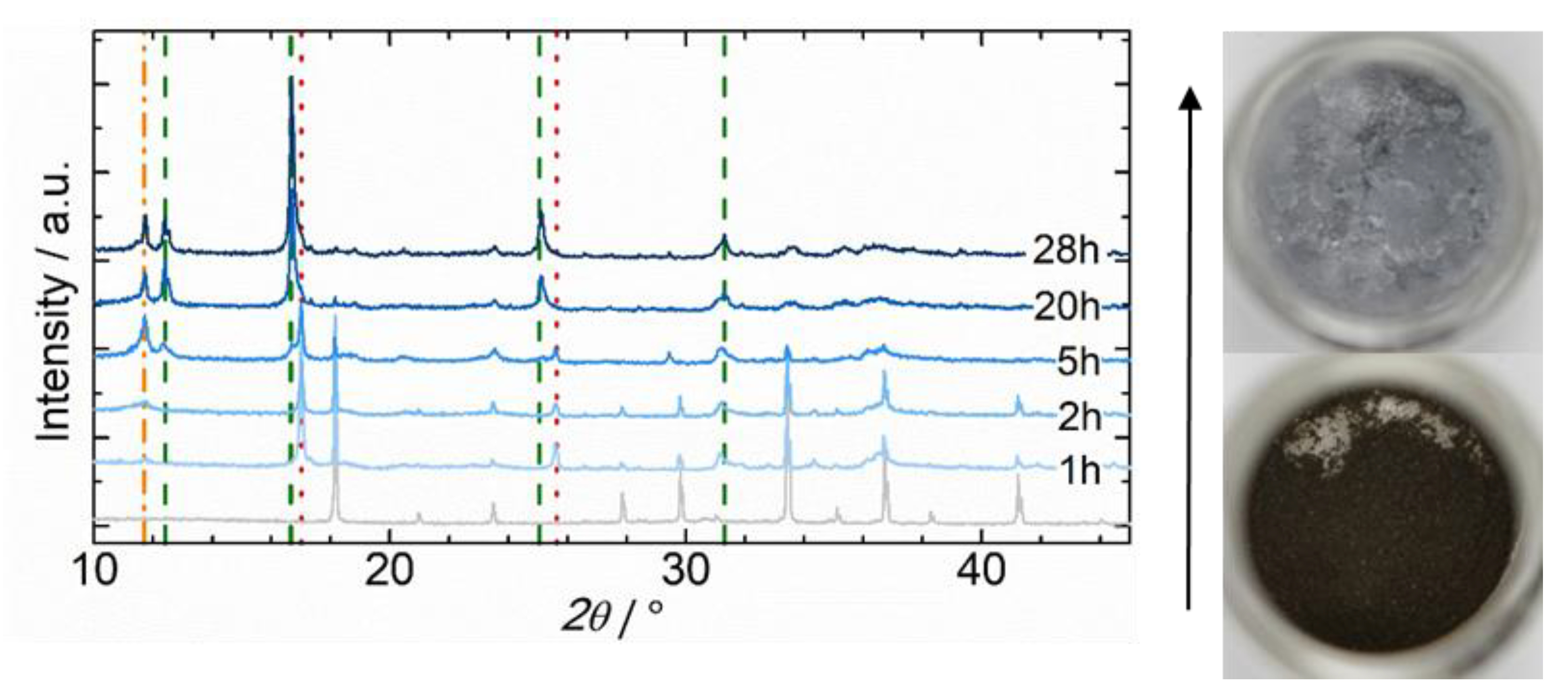
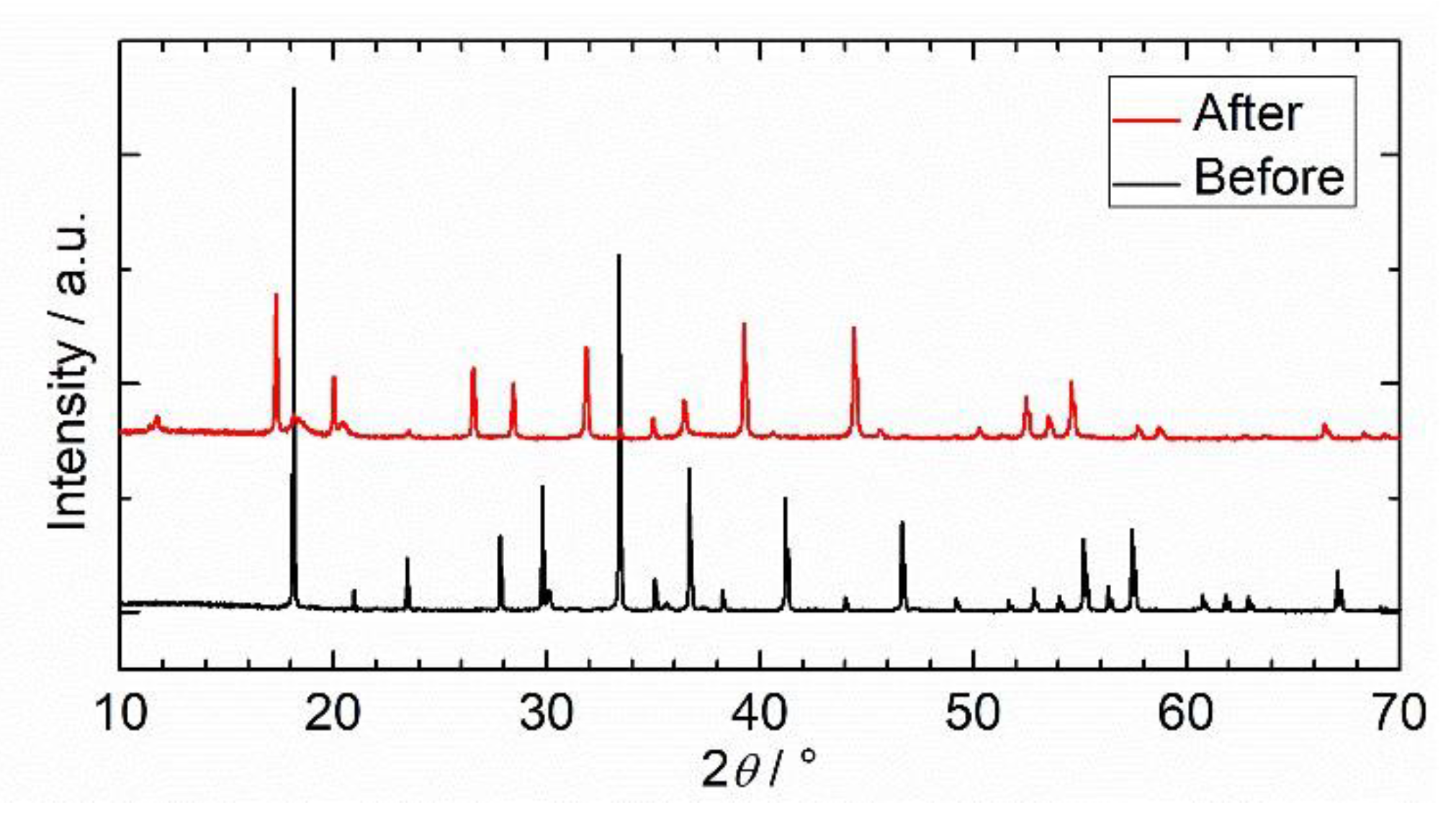
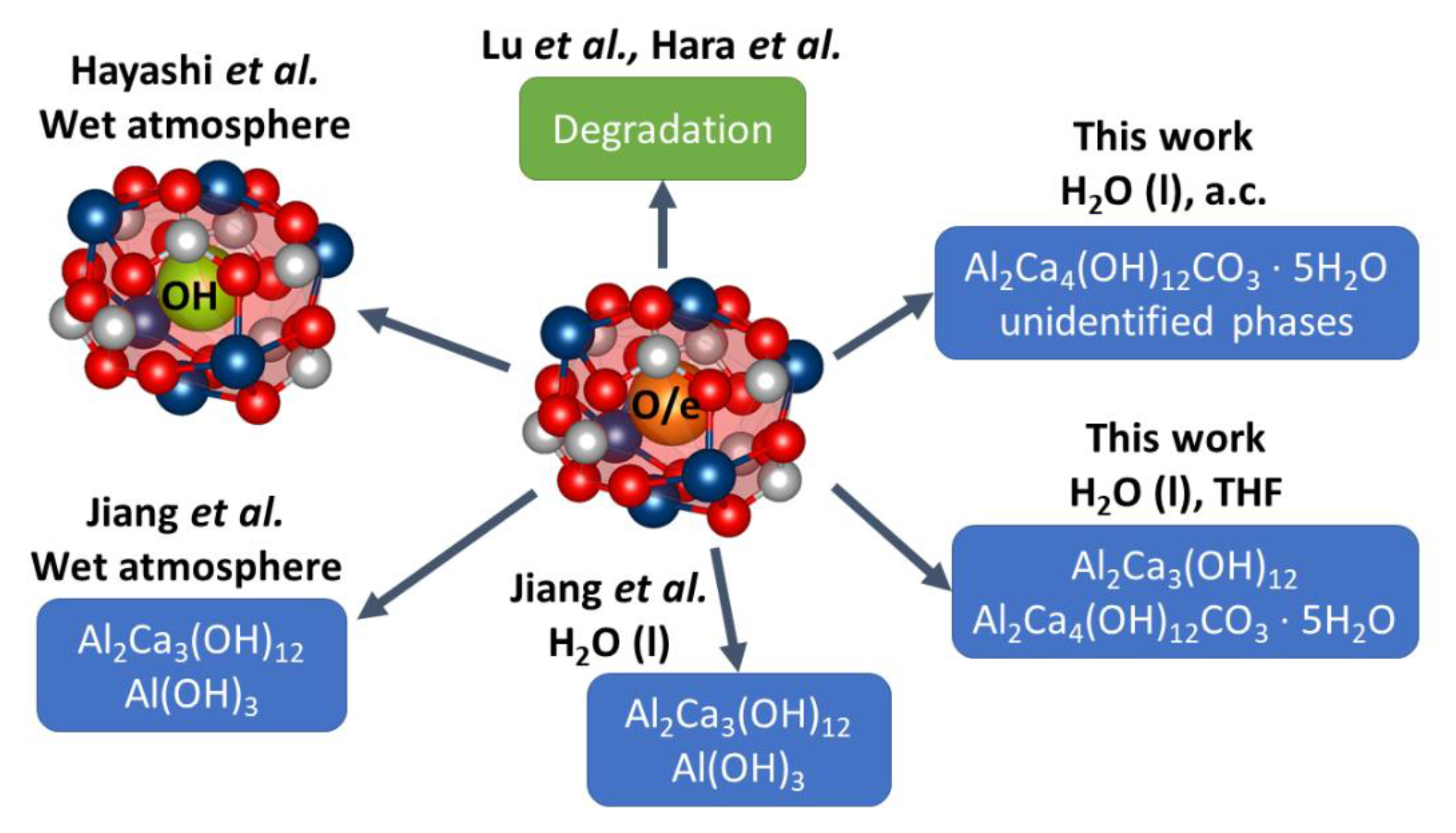
2.2.3. Hydrothermal Reactivity Tests
3. Materials and Methods
3.1. Synthesis
3.1.1. Synthesis of Electrides and Mayenite Hydrides
3.1.2. Ruthenium Deposition
3.2. Characterization
3.3. Reactivity Studies
3.3.1. Catalytic Experiments
3.3.2. Reactivity of Electrides towards Hydrogen
3.3.3. Hydrothermal Reactivity of C12A7 and C12A7e−
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haber, F.; Van Oordt, G. Über die Bildung von Ammoniak den Elementen. Zeitschrift Für Anorganische Und Allgemeine Chemie 1905, 44, 341–378. [Google Scholar] [CrossRef] [Green Version]
- C. Bosch (Inv.). Process of Producing Ammonia. U.S.990,191A, 2 March 1908. [Google Scholar]
- Badische Anilin- & Soda-Fabrik (Inv.). Verfahren zur Synthetischen Darstellung von Ammoniak aus den Elementen. DE235,421, 13 October 1908. [Google Scholar]
- Mittasch, A. Bemerkungen zur Katalyse. Berichte Der Dtsch. Chem. Ges. 1926, 59, 13–36. [Google Scholar] [CrossRef]
- Appl, M. Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2000. [Google Scholar]
- Saadatjou, N.; Jafari, A.; Sahebdelfar, S. Ruthenium Nanocatalysts for Ammonia Synthesis: A Review. Chem. Eng. Commun. 2014, 202, 420–448. [Google Scholar] [CrossRef]
- Boudart, M. Kinetics and Mechanism of Ammonia Synthesis. Catal. Rev. 1981, 23, 1–15. [Google Scholar] [CrossRef]
- Ertl, G. Primary steps in catalytic synthesis of ammonia. J. Vac. Sci. Technol. A 1983, 1, 1247–1253. [Google Scholar] [CrossRef]
- Strongin, D.R.; Carrazza, J.; Bare, S.R.; Somorjai, G.A. The importance of C7 sites and surface roughness in the ammonia synthesis reaction over iron. J. Catal. 1987, 103, 213–215. [Google Scholar] [CrossRef] [Green Version]
- Hinrichsen, K.-O.; Rosowski, F.; Muhler, M.; Ertl, G. The microkinetics of ammonia synthesis catalyzed by caesium-promoted supported ruthenium. Chem. Eng. Sci. 1996, 51, 1683–1690. [Google Scholar] [CrossRef]
- Jacobsen, C.J.; Dahl, S.; Hansen, P.L.; Törnqvist, E.; Jensen, L.; Topsøe, H.; Prip, D.V.; Møenshaug, P.B.; Chorkendorff, I. Structure sensitivity of supported ruthenium catalysts for ammonia synthesis. J. Mol. Catal. A Chem. 2000, 163, 19–26. [Google Scholar] [CrossRef]
- Dahl, S.; Sehested, J.; Jacobsen, C.; Tornqvist, E.; Chorkendorff, I. Surface science based microkinetic analysis of ammonia synthesis over ruthenium catalysts. J. Catal. 2000, 192, 391–399. [Google Scholar] [CrossRef]
- Dahl, S.; Logadottir, A.; Egeberg, R.; Larsen, J.H.; Chorkendorff, I.; Törnqvist, E.; Nørskov, J.K. Role of Steps in N2 Activation on Ru(0001). Phys. Rev. Lett. 1999, 83, 1814–1817. [Google Scholar] [CrossRef] [Green Version]
- Dahl, S.; Törnqvist, E.; Chorkendorff, I. Dissociative adsorption of N on Ru(0001): A surface reaction totally dominated by steps. J. Catal. 2000, 192, 381–390. [Google Scholar] [CrossRef]
- Rosowski, F.; Hornung, A.; Hinrichsen, O.; Herein, D.; Muhler, M.; Ertl, G. Ruthenium catalysts for ammonia synthesis at high pressures: Preparation, characterization, and power-law kinetics. Appl. Catal. A Gen. 1997, 151, 443–460. [Google Scholar] [CrossRef]
- Aika, K.-I. Role of alkali promoter in ammonia synthesis over ruthenium catalysts—Effect on reaction mechanism. Catal. Today 2017, 286, 14–20. [Google Scholar] [CrossRef]
- Hara, M.; Kitano, M.; Hosono, H. Ru-Loaded C12A7:e– Electride as a Catalyst for Ammonia Synthesis. ACS Catal. 2017, 7, 2313–2324. [Google Scholar] [CrossRef]
- Kitano, M.; Inoue, Y.; Yamazaki, Y.; Hayashi, F.; Kanbara, S.; Matsuishi, S.; Yokoyama, T.; Kim, S.-W.; Hara, M.; Hosono, H. Ammonia synthesis using a stable electride as an electron donor and reversible hydrogen store. Nat. Chem. 2012, 4, 934–940. [Google Scholar] [CrossRef]
- Hayashi, K.; Matsuishi, S.; Kamiya, T.; Hirano, M.; Hosono, H. Light-induced conversion of an insulating refractory oxide into a persistent electronic conductor. Nat. Cell Biol. 2002, 419, 462–465. [Google Scholar] [CrossRef]
- Kim, S.W.; Hosono, H. Synthesis and properties of 12CaO · 7Al2O3 electride: Review of single crystal and thin film growth. Philos. Mag. 2012, 92, 2596–2628. [Google Scholar] [CrossRef]
- Matsuishi, S.; Toda, Y.; Miyakawa, M.; Hayashi, K.; Kamiya, T.; Hirano, M.; Tanaka, I.; Hosono, H. High-Density Electron Anions in a Nanoporous Single Crystal: [Ca24Al28O64]4+(4e–). Science 2003, 301, 626–629. [Google Scholar] [CrossRef]
- Salasin, J.R.; Rawn, C. Structure Property Relationships and Cationic Doping in [Ca24Al28O64]4+ Framework: A Review. Cryst. 2017, 7, 143. [Google Scholar] [CrossRef] [Green Version]
- Boysen, H.; Lerch, M.; Stys, A.; Senyshyn, A. Structure and oxygen mobility in mayenite (Ca12Al14O33): A high-temperature neutron powder diffraction study. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2007, 63, 675–682. [Google Scholar] [CrossRef]
- Palacios, L.; De La Torre, Á.G.; Bruque, S.; García-Muñoz, J.L.; García-Granda, S.; Sheptyakov, A.D.; Aranda, M.A.G. Crystal Structures and in-Situ Formation Study of Mayenite Electrides. Inorg. Chem. 2007, 46, 4167–4176. [Google Scholar] [CrossRef]
- Jeevaratnam, J.; Glasser, L.S.D.; Glasser, F.P. Structure of Calcium Aluminate, 12CaO·7Al2O3. Nat. Cell Biol. 1962, 194, 764–765. [Google Scholar] [CrossRef]
- Hosono, H.; Abe, Y. Occurrence of superoxide radical ion in crystalline calcium aluminate 12CaO·7Al2O3 prepared via solid-state reactions. Inorg. Chem. 1987, 26, 1192–1195. [Google Scholar] [CrossRef]
- Bartl, H.B.; Scheller, T. On the Structure of 12CaO⋅7Al2O3, Neues Jahrb. Miner. Monatsh. 1970, 35, 547–552. [Google Scholar]
- Hayashi, K.; Hirano, A.M.; Hosono†, H. Thermodynamics and Kinetics of Hydroxide Ion Formation in 12CaO·7Al2O3. J. Phys. Chem. B 2005, 109, 11900–11906. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Sushko, P.V.; Hashimoto, Y.; Shluger, A.L.; Hosono, H. Hydride ions in oxide hosts hidden by hydroxide ions. Nat. Commun. 2014, 5, 3515. [Google Scholar] [CrossRef] [PubMed]
- Imlach, J.; Glasser, L.D.; Glasser, F. Excess oxygen and the stability of “12CaO·7A12O3”. Cem. Concr. Res. 1971, 1, 57–61. [Google Scholar] [CrossRef]
- Kitano, M.; Kanbara, S.; Inoue, Y.F.; Kuganathan, N.; Sushko, P.V.; Yokoyama, T.; Hara, M.; Hosono, H. Electride support boosts nitrogen dissociation over ruthenium catalyst and shifts the bottleneck in ammonia synthesis. Nat. Commun. 2015, 6, 6731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kammert, J.; Moon, J.; Cheng, Y.; Daemen, L.L.; Irle, S.; Fung, V.; Liu, J.; Page, K.; Ma, X.; Phaneuf, V.; et al. Nature of Reactive Hydrogen for Ammonia Synthesis over a Ru/C12A7 Electride Catalyst. J. Am. Chem. Soc. 2020, 142, 7655–7667. [Google Scholar] [CrossRef]
- Jiang, D.; Zhao, Z.; Mu, S.; Phaneuf, V.; Tong, J. Insights into the dynamic hydrogenation of mayenite [Ca24Al28O64]4+(O2−)2: Mixed ionic and electronic conduction within the sub-nanometer cages. Int. J. Hydrog. Energy 2019, 44, 18360–18371. [Google Scholar] [CrossRef]
- Hayashi, K. Heavy doping of H− ion in 12CaO·7Al2O3. J. Solid State Chem. 2011, 184, 1428–1432. [Google Scholar] [CrossRef]
- Polfus, J.M.; Toyoura, K.; Hervoches, C.H.; Sunding, M.F.; Tanaka, I.; Haugsrud, R. Nitrogen and hydrogen defect equilibria in Ca12Al14O33: A combined experimental and computational study. J. Mater. Chem. 2012, 22, 15828. [Google Scholar] [CrossRef]
- Boysen, H.; Kaiser-Bischhoff, I.; Lerch, M. Anion Diffusion Processes in O- and N-Mayenite Investigated by Neutron Powder Dif-fraction. Diffus. Fundam. 2008, 8, 2.1–2.8. [Google Scholar]
- Hayashi, F.; Tomota, Y.; Kitano, M.; Toda, Y.; Yokoyama, T.; Hosono, H. NH2– Dianion Entrapped in a Nanoporous 12CaO·7Al2O3 Crystal by Ammonothermal Treatment: Reaction Pathways, Dynamics, and Chemical Stability. J. Am. Chem. Soc. 2014, 136, 11698–11706. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.; Tareen, A.K.; Aslam, M.; Thebo, K.H.; Khan, U.; Wang, R.; Shams, S.S.; Han, Z.; Ouyang, Z. A comprehensive review on synthesis of pristine and doped inorganic room temperature stable mayenite electride, [Ca24Al28O64]4+(e−)4 and its applications as a catalyst. Prog. Solid State Chem. 2019, 54, 1–19. [Google Scholar] [CrossRef]
- Toda, Y.; Yanagi, H.; Ikenaga, E.; Kim, J.J.; Kobata, M.; Ueda, S.; Kamiya, T.; Hirano, M.; Kobayashi, K.; Hosono, H. Work Function of a Room-Temperature, Stable Electride [Ca24Al28O64]4+(e–)4. Adv. Mater. 2007, 19, 3564–3569. [Google Scholar] [CrossRef]
- Kanbara, S.; Kitano, M.; Inoue, Y.; Yokoyama, T.; Hara, M.; Hosono, H. Mechanism Switching of Ammonia Synthesis over Ru-Loaded Electride Catalyst at Metal–Insulator Transition. J. Am. Chem. Soc. 2015, 137, 14517–14524. [Google Scholar] [CrossRef]
- Jiang, D.; Zhao, Z.; Mu, S.; Qian, H.; Tong, J. Facile and Massive Aluminothermic Synthesis of Mayenite Electrides from Cost-Effective Oxide and Metal Precursors. Inorg. Chem. 2018, 58, 960–967. [Google Scholar] [CrossRef]
- Weber, S.; Schäfer, S.; Saccoccio, M.; Seidel, K.; Kohlmann, H.; Gläser, R.; Schunk, S.A. Mayenite-based electride C12A7e−: An innovative synthetic method via plasma arc melting. Mater. Chem. Front. 2021, 5, 1301–1314. [Google Scholar] [CrossRef]
- Jiang, D.; Zhao, Z.; Mu, S.; Phaneuf, V.; Tong, J. Simple and Efficient Fabrication of Mayenite Electrides from a Solution-Derived Precursor. Inorg. Chem. 2017, 56, 11702–11709. [Google Scholar] [CrossRef]
- Lu, Y.; Li, J.; Tada, T.; Toda, Y.; Ueda, S.; Yokoyama, T.; Kitano, M.; Hosono, H. Water Durable Electride Y5Si3: Electronic Structure and Catalytic Activity for Ammonia Synthesis. J. Am. Chem. Soc. 2016, 138, 3970–3973. [Google Scholar] [CrossRef] [PubMed]
- Trofymluk, O.; Toda, Y.; Hosono, H.; Navrotsky, A. Energetics of Formation and Oxidation of Microporous Calcium Aluminates: A New Class of Electrides and Ionic Conductors. Chem. Mater. 2005, 17, 5574–5579. [Google Scholar] [CrossRef]
- Yoshizumi, T.; Matsuishi, S.; Kim, S.-W.; Hosono, H.; Hayashi, K. Iodometric Determination of Electrons Incorporated into Cages in 12CaO·7Al2O3 Crystals. J. Phys. Chem. C 2010, 114, 15354–15357. [Google Scholar] [CrossRef]
- Matsuishi, S.; Nomura, T.; Hirano, M.; Kodama, K.; Shamoto, S.-I.; Hosono, H. Direct Synthesis of Powdery Inorganic Electride [Ca24Al28O64]4+(e−)4 and Determination of Oxygen Stoichiometry. Chem. Mater. 2009, 21, 2589–2591. [Google Scholar] [CrossRef]
- Gražulis, S.; Chateigner, D.; Downs, R.T.; Yokochi, A.F.T.; Quirós, M.; Lutterotti, L.; Manakova, E.; Butkus, J.; Moeck, P.; Le Bail, A. Crystallography Open Database–an open-access collection of crystal structures. J. Appl. Crystallogr. 2009, 42, 726–729. [Google Scholar] [CrossRef]
- Allen, F.H. The Development, Status and Scientific Impact of Crystallographic Databases. Acta Crystallogr. Sect. A Found. Crystallogr. 1998, 54, 758–771. [Google Scholar] [CrossRef]
- Rodríguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Phys. B Condens. Matter 1993, 192, 55–69. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample 1 | Color of Powder | Solid Reductant |
|---|---|---|
| (a) C12A7 | colorless | - |
| (b) C12A7e− (5Al) | light green | 5 wt.% Al |
| (c) C12A7e− (20Al) | dark green | 20 wt.% Al |
| (d) C12A7e− (3C) | dark green | 3 wt.% graphite |
| (e) C12A7e− (SSR) | dark green | - |
| (f) Ru/C12A7 | gray | - |
| (g) Ru/C12A7e− (5Al) | gray | 5 wt.% Al |
| (h) Ru/C12A7e− (20Al) | dark gray | 20 wt.% Al |
| (i) Ru/C12A7e− (3C) | dark gray | 3 wt.% graphite |
| (j) C12A7:H | colorless | - |
| (k) C12A7:D | colorless | - |
| Segment | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T/K | 673 | 653 | 633 | 623 | 613 | 603 | 593 | 633 | 633 | 633 | 633 | 633 |
| p/Mpa | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 1.1 | 3.1 | 6.1 | 7.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weber, S.; Schäfer, S.; Saccoccio, M.; Ortner, N.; Bertmer, M.; Seidel, K.; Berendts, S.; Lerch, M.; Gläser, R.; Kohlmann, H.; et al. Mayenite-Based Electride C12A7e−: A Reactivity and Stability Study. Catalysts 2021, 11, 334. https://doi.org/10.3390/catal11030334
Weber S, Schäfer S, Saccoccio M, Ortner N, Bertmer M, Seidel K, Berendts S, Lerch M, Gläser R, Kohlmann H, et al. Mayenite-Based Electride C12A7e−: A Reactivity and Stability Study. Catalysts. 2021; 11(3):334. https://doi.org/10.3390/catal11030334
Chicago/Turabian StyleWeber, Sebastian, Sebastian Schäfer, Mattia Saccoccio, Nils Ortner, Marko Bertmer, Karsten Seidel, Stefan Berendts, Martin Lerch, Roger Gläser, Holger Kohlmann, and et al. 2021. "Mayenite-Based Electride C12A7e−: A Reactivity and Stability Study" Catalysts 11, no. 3: 334. https://doi.org/10.3390/catal11030334
APA StyleWeber, S., Schäfer, S., Saccoccio, M., Ortner, N., Bertmer, M., Seidel, K., Berendts, S., Lerch, M., Gläser, R., Kohlmann, H., & Schunk, S. A. (2021). Mayenite-Based Electride C12A7e−: A Reactivity and Stability Study. Catalysts, 11(3), 334. https://doi.org/10.3390/catal11030334