
From CO2 to Value-Added Products: A Review about Carbon-Based Materials for Electro-Chemical CO2 Conversion

 ,
, 
 and
and 
Abstract
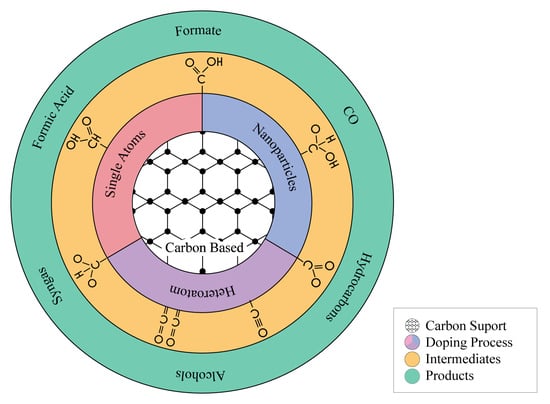
:
1. Introduction
2. Metal-Free Carbon Materials as Catalyst
2.1. N-Doped Carbon-Based Materials
2.1.1. N-Doping Methodology and N-Doped Catalyst Active Sites
2.1.2. CO as Main Product
2.1.3. Syngas Production
2.1.4. Formate and Formic Acid as Main Products
2.1.5. Multi-Carbon Products
2.2. Others-Doped Carbon-Based Materials
2.3. Dual-Doped Carbon-Based Materials
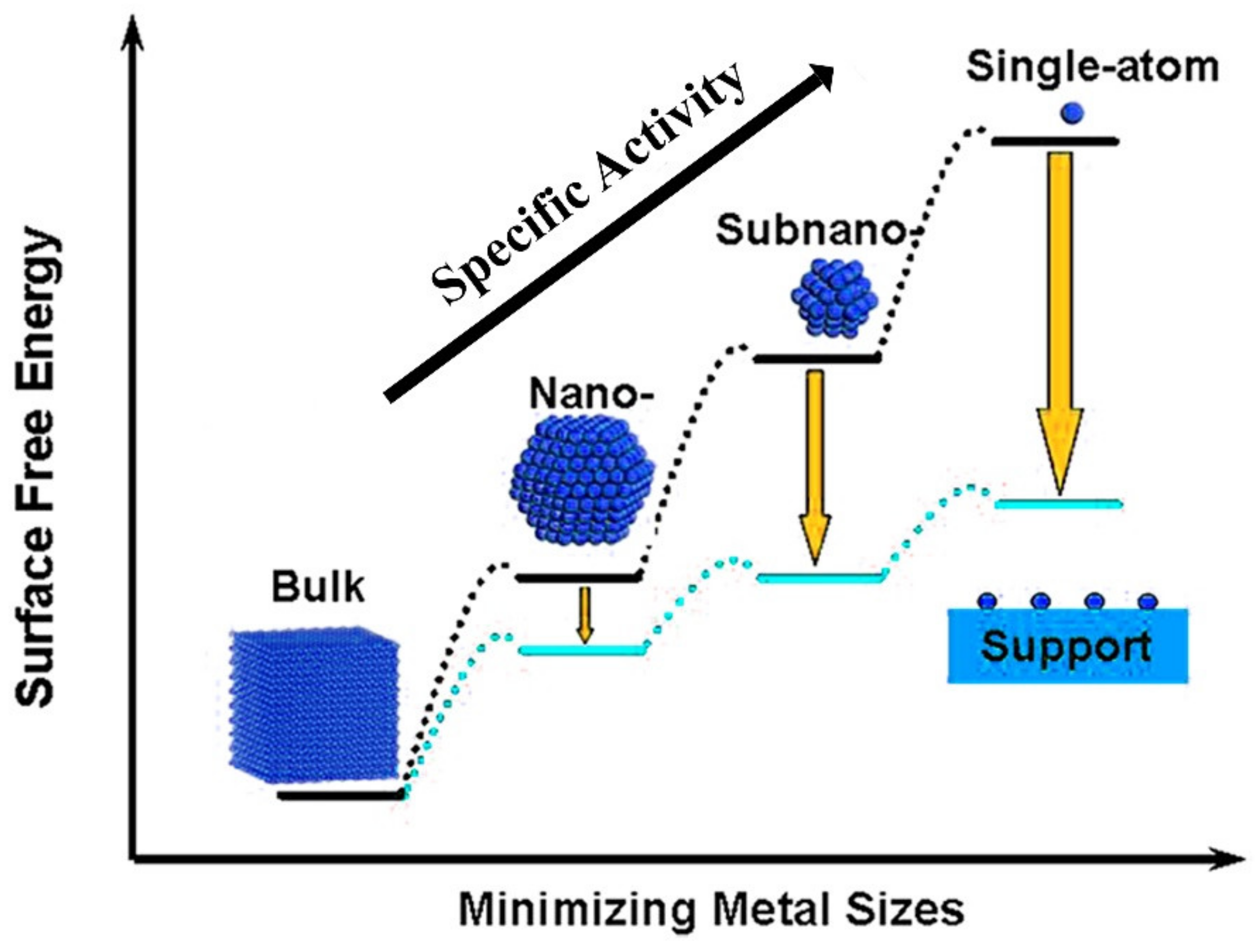
3. Single-Atom Metal-Doped Carbon Catalysts
3.1. Single-Atoms Supported on N-Doped Carbon-Based Materials (SMAs-N-C)
3.1.1. SMAs-N-Graphene
3.1.2. SMAs-N-Other Carbon Materials
4. Metal Nanoparticles Supported on Carbon-Based Materials (M-NPs-C)
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaneco, S.; Kurimoto, H.; Shimizu, Y.; Ohta, K.; Mizuno, T. Photocatalytic reduction of CO2 using TiO2 powders in supercritical fluid CO2. Energy 1999, 24, 21–30. [Google Scholar] [CrossRef]
- Mori, K.; Yamashita, H.; Anpo, M. Photocatalytic reduction of CO2 with H2O on various titanium oxide photocatalysts. RSC Adv. 2012, 2, 3165–3172. [Google Scholar] [CrossRef]
- Xie, S.; Zhang, Q.; Liu, G.; Wang, Y. Photocatalytic and photoelectrocatalytic reduction of CO2 using heterogeneous catalysts with controlled nanostructures. Chem. Commun. 2016, 52, 35–59. [Google Scholar] [CrossRef]
- Wang, C.; Sun, Z.; Zheng, Y.; Hu, Y.H. Recent progress in visible light photocatalytic conversion of carbon dioxide. J. Mater. Chem. A 2019, 7, 865–887. [Google Scholar] [CrossRef]
- Chueh, W.C.; Haile, S.M. Ceria as a Thermochemical Reaction Medium for Selectively Generating Syngas or Methane from H2O and CO2. ChemSusChem 2009, 2, 735–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, M.; Édouard, D.; Roger, A.-C. Optimization of structured cellular foam-based catalysts for low-temperature carbon dioxide methanation in a platelet milli-reactor. C. R. Chim. 2015, 18, 283–292. [Google Scholar] [CrossRef]
- Roy, S.; Cherevotan, A.; Peter, S.C. Thermochemical CO2 Hydrogenation to Single Carbon Products: Scientific and Technological Challenges. ACS Energy Lett. 2018, 3, 1938–1966. [Google Scholar] [CrossRef] [Green Version]
- Ali, N.; Bilal, M.; Nazir, M.S.; Khan, A.; Ali, F.; Iqbal, H.M.N. Thermochemical and electrochemical aspects of carbon dioxide methanation: A sustainable approach to generate fuel via waste to energy theme. Sci. Total Environ. 2020, 712, 136482. [Google Scholar] [CrossRef]
- Grodkowski, J.; Neta, P. Copper-Catalyzed Radiolytic Reduction of CO2 to CO in Aqueous Solutions. J. Phys. Chem. B 2001, 105, 4967–4972. [Google Scholar] [CrossRef]
- Ramirez-Corredores, M.M.; Gadikota, G.; Huang, E.E.; Gaffney, A.M. Radiation-Induced Chemistry of Carbon Dioxide: A Pathway to Close the Carbon Loop for a Circular Economy. Front. Energy Res. 2020, 8, 108. [Google Scholar] [CrossRef]
- Machado, I.M.P.; Atsumi, S. Cyanobacterial biofuel production. J. Biotechnol. 2012, 162, 50–56. [Google Scholar] [CrossRef]
- Rabinovitch-Deere, C.A.; Oliver, J.W.K.; Rodriguez, G.M.; Atsumi, S. Synthetic Biology and Metabolic Engineering Approaches To Produce Biofuels. Chem. Rev. 2013, 113, 4611–4632. [Google Scholar] [CrossRef] [PubMed]
- Rittmann, S.; Seifert, A.; Herwig, C. Essential prerequisites for successful bioprocess development of biological CH4 production from CO2 and H2. Crit. Rev. Biotechnol. 2015, 35, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Scibioh, M.A.; Viswanathan, B. Chapter 6—Biochemical Reduction of CO2. In Carbon Dioxide to Chemicals and Fuels; Scibioh, M.A., Viswanathan, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 255–306. ISBN 978-0-444-63996-7. [Google Scholar]
- Sun, D.; Chen, Y. Electrochemical Reduction of Carbon Dioxide Fundamentals and Technologies; CRC Press: Boca Raton, FL, USA, 2016; ISBN 9781315099316. [Google Scholar]
- Tuci, G.; Filippi, J.; Rossin, A.; Luconi, L.; Pham-Huu, C.; Yakhvarov, D.; Vizza, F.; Giambastiani, G. CO2 electrochemical reduction by exohedral N-pyridine decorated metal-free carbon nanotubes. Energies 2020, 13, 2703. [Google Scholar] [CrossRef]
- Jia, C.; Dastafkan, K.; Ren, W.; Yang, W.; Zhao, C. Carbon-based catalysts for electrochemical CO2 reduction. Sustain. Energy Fuels 2019, 3, 2890–2906. [Google Scholar] [CrossRef]
- Irabien, A.; Alvarez-Guerra, M.; Albo, J.; Dominguez-Ramos, A. Electrochemical Conversion of CO2 to Value-Added Products; Elsevier Inc.: Amsterdam, The Netherlands, 2018; ISBN 9780128131602. [Google Scholar]
- Mondal, B.; Song, J.; Neese, F.; Ye, S. Bio-inspired mechanistic insights into CO2 reduction. Curr. Opin. Chem. Biol. 2015, 25, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, M.; Jørgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Sharma, P.P.; Wu, J.; Yadav, R.M.; Liu, M.; Wright, C.J.; Tiwary, C.S.; Yakobson, B.I.; Lou, J.; Ajayan, P.M.; Zhou, X.-D. Nitrogen-Doped Carbon Nanotube Arrays for High-Efficiency Electrochemical Reduction of CO2: On the Understanding of Defects, Defect Density, and Selectivity. Angew. Chem. 2015, 127, 13905–13909. [Google Scholar] [CrossRef]
- Fernandes, D.M.; Peixoto, A.F.; Freire, C. Nitrogen-doped metal-free carbon catalysts for (electro)chemical CO2 conversion and valorisation. Dalt. Trans. 2019, 48, 13508–13528. [Google Scholar] [CrossRef]
- Navalon, S.; Dhakshinamoorthy, A.; Alvaro, M.; Garcia, H. Carbocatalysis by Graphene-Based Materials. Chem. Rev. 2014, 114, 6179–6212. [Google Scholar] [CrossRef]
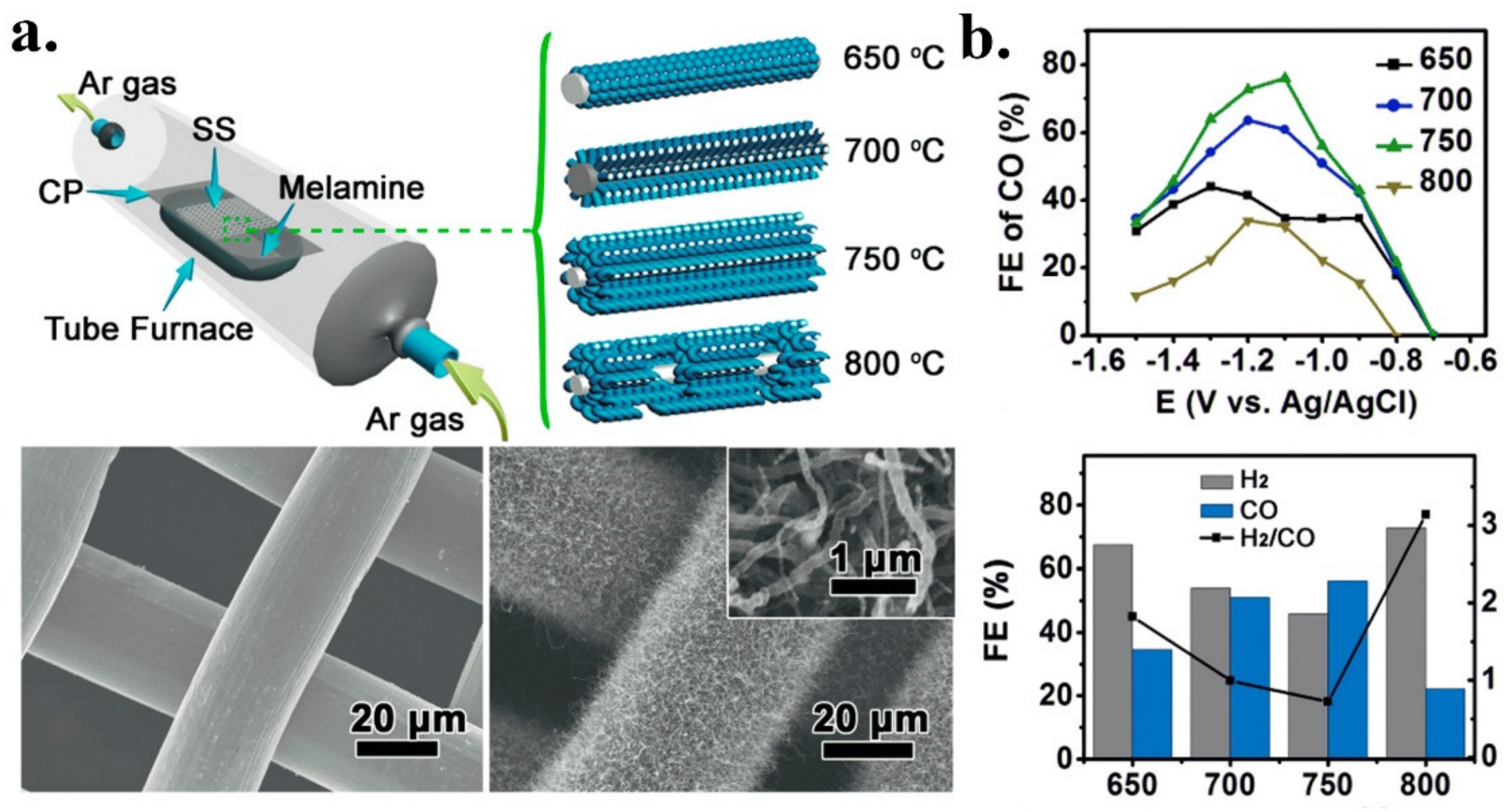
- Ji, Y.; Shi, Y.; Liu, C.; Zhang, B. Plasma-regulated N-doped carbon nanotube arrays for efficient electrosynthesis of syngas with a wide CO/H2 ratio. Sci. China Mater. 2020, 63, 1–7. [Google Scholar] [CrossRef]
- Hu, C.; Dai, L. Doping of Carbon Materials for Metal-Free Electrocatalysis. Adv. Mater. 2019, 31, 1804672. [Google Scholar] [CrossRef]
- Duan, X.; Xu, J.; Wei, Z.; Ma, J.; Guo, S.; Wang, S.; Liu, H.; Dou, S. Metal-Free Carbon Materials for CO2 Electrochemical Reduction. Adv. Mater. 2017, 29, 1701784. [Google Scholar] [CrossRef]
- Liu, W.; Qi, J.; Bai, P.; Zhang, W.; Xu, L. Utilizing spatial confinement effect of N atoms in micropores of coal-based metal-free material for efficiently electrochemical reduction of carbon dioxide. Appl. Catal. B Environ. 2020, 272, 118974. [Google Scholar] [CrossRef]
- Gao, K.; Wang, B.; Tao, L.; Cunning, B.V.; Zhang, Z.; Wang, S.; Ruoff, R.S.; Qu, L. Efficient Metal-Free Electrocatalysts from N-Doped Carbon Nanomaterials: Mono-Doping and Co-Doping. Adv. Mater. 2019, 31, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Tao, L.; Yan, D.; Zou, Y.; Wang, S. Recent Advances on Non-precious Metal Porous Carbon-based Electrocatalysts for Oxygen Reduction Reaction. ChemElectroChem 2018, 5, 1775–1785. [Google Scholar] [CrossRef]
- Inagaki, M.; Toyoda, M.; Soneda, Y.; Morishita, T. Nitrogen-doped carbon materials. Carbon 2018, 132, 104–140. [Google Scholar] [CrossRef]
- Zhu, Y.; Lv, K.; Wang, X.; Yang, H.; Xiao, G.; Zhu, Y. 1D/2D nitrogen-doped carbon nanorod arrays/ultrathin carbon nanosheets: Outstanding catalysts for the highly efficient electroreduction of CO2 to CO. J. Mater. Chem. A 2019, 7, 14895–14903. [Google Scholar] [CrossRef]
- Wu, J.; Yadav, R.M.; Liu, M.; Sharma, P.P.; Tiwary, C.S.; Ma, L.; Zou, X.; Zhou, X.; Yakobson, B.I.; Lou, J.; et al. Achieving Highly Efficient, Selective, and Stable CO2 Reduction on Nitrogen-Doped Carbon Nanotubes. ACS Nano 2015, 9, 5364–5371. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xiao, N.; Hao, M.; Song, X.; Wang, Y.; Ji, Y.; Liu, C.; Li, C.; Guo, Z.; Zhang, F.; et al. Efficient CO2 electroreduction over pyridinic-N active sites highly exposed on wrinkled porous carbon nanosheets. Chem. Eng. J. 2018, 351, 613–621. [Google Scholar] [CrossRef]
- Jhong, H.R.M.; Tornow, C.E.; Smid, B.; Gewirth, A.A.; Lyth, S.M.; Kenis, P.J.A. A Nitrogen-Doped Carbon Catalyst for Electrochemical CO2 Conversion to CO with High Selectivity and Current Density. ChemSusChem 2017, 10, 1094–1099. [Google Scholar] [CrossRef]
- Xu, J.; Kan, Y.; Huang, R.; Zhang, B.; Wang, B.; Wu, K.H.; Lin, Y.; Sun, X.; Li, Q.; Centi, G.; et al. Revealing the Origin of Activity in Nitrogen-Doped Nanocarbons towards Electrocatalytic Reduction of Carbon Dioxide. ChemSusChem 2016, 9, 1085–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wang, Y.; Xiao, N.; Li, H.; Ji, Y.; Guo, Z.; Liu, C.; Qiu, J. Nitrogen-doped porous carbon from coal for high efficiency CO2 electrocatalytic reduction. Carbon 2019, 151, 46–52. [Google Scholar] [CrossRef]
- Wu, J.; Liu, M.; Sharma, P.P.; Yadav, R.M.; Ma, L.; Yang, Y.; Zou, X.; Zhou, X.D.; Vajtai, R.; Yakobson, B.I.; et al. Incorporation of Nitrogen Defects for Efficient Reduction of CO2 via Two-Electron Pathway on Three-Dimensional Graphene Foam. Nano Lett. 2016, 16, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Kuang, M.; Guan, A.; Gu, Z.; Han, P.; Qian, L.; Zheng, G. Enhanced N-doping in mesoporous carbon for efficient electrocatalytic CO2 conversion. Nano Res. 2019, 12, 2324–2329. [Google Scholar] [CrossRef]
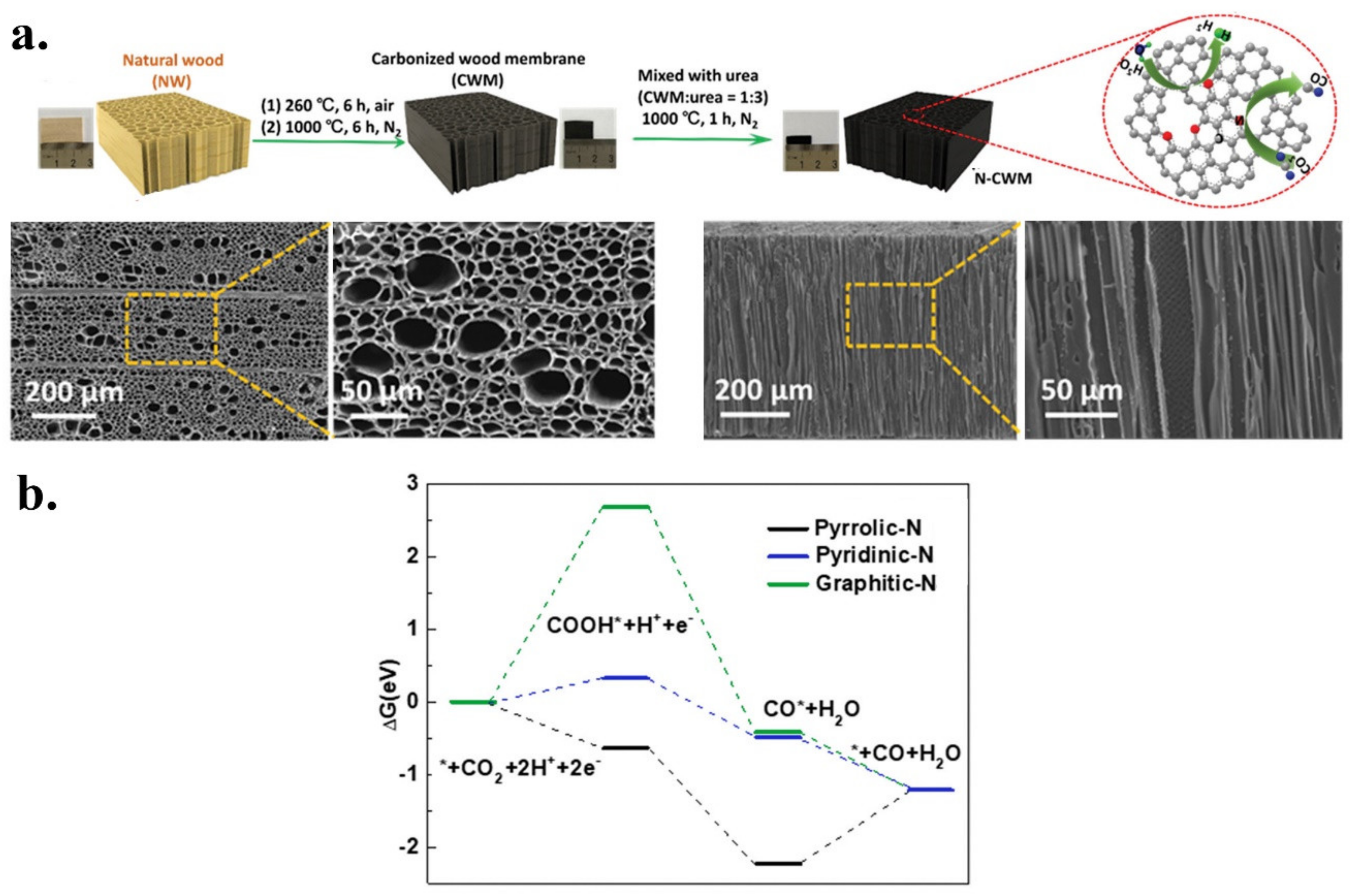
- Zhang, H.; Min, S.; Wang, F.; Zhang, Z.; Kong, C. Efficient electrocatalytic CO2 reduction to CO with high selectivity using a N-doped carbonized wood membrane. New J. Chem. 2020, 44, 6125–6129. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Y.; Hou, X.; Ma, C.; Tan, T. Nitrogen-doped graphenes as efficient electrocatalysts for the selective reduction of carbon dioxide to formate in aqueous solution. Green Chem. 2016, 18, 3250–3256. [Google Scholar] [CrossRef]
- Zhang, S.; Kang, P.; Ubnoske, S.; Brennaman, M.K.; Song, N.; House, R.L.; Glass, J.T.; Meyer, T.J. Polyethylenimine-enhanced electrocatalytic reduction of CO2 to formate at nitrogen-doped carbon nanomaterials. J. Am. Chem. Soc. 2014, 136, 7845–7848. [Google Scholar] [CrossRef]
- Wang, H.; Jia, J.; Song, P.; Wang, Q.; Li, D.; Min, S.; Qian, C.; Wang, L.; Li, Y.F.; Ma, C.; et al. Efficient Electrocatalytic Reduction of CO2 by Nitrogen-Doped Nanoporous Carbon/Carbon Nanotube Membranes: A Step Towards the Electrochemical CO2 Refinery. Angew. Chem. Int. Ed. 2017, 56, 7847–7852. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Mou, K.; Yao, S.; Liu, L. Highly selective electrochemical reduction of CO2 to formate on metal-free nitrogen-doped PC61BM. J. Mater. Chem. A 2018, 6, 11236–11243. [Google Scholar] [CrossRef]
- Li, H.; Xiao, N.; Wang, Y.; Li, C.; Ye, X.; Guo, Z.; Pan, X.; Liu, C.; Bai, J.; Xiao, J.; et al. Nitrogen-doped tubular carbon foam electrodes for efficient electroreduction of CO2 to syngas with potential-independent CO/H2 ratios. J. Mater. Chem. A 2019, 7, 18852–18860. [Google Scholar] [CrossRef]
- Liu, K.H.; Zhong, H.X.; Yang, X.Y.; Bao, D.; Meng, F.L.; Yan, J.M.; Zhang, X.B. Composition-tunable synthesis of “clean” syngas: Via a one-step synthesis of metal-free pyridinic-N-enriched self-supported CNTs: The synergy of electrocatalyst pyrolysis temperature and potential. Green Chem. 2017, 19, 4284–4288. [Google Scholar] [CrossRef]
- Song, Y.; Chen, W.; Zhao, C.; Li, S.; Wei, W.; Sun, Y. Metal-Free Nitrogen-Doped Mesoporous Carbon for Electroreduction of CO2 to Ethanol. Angew. Chem. Int. Ed. 2017, 56, 10840–10844. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, S.; Chen, W.; Li, S.; Feng, G.; Wei, W.; Sun, Y. Enhanced Ethanol Production from CO2 Electroreduction at Micropores in Nitrogen-Doped Mesoporous Carbon. ChemSusChem 2020, 13, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Zhi, W.Y.; Liu, L.; Yang, M.P.; Wang, H.; Lu, J.X. Electrochemical reduction of CO2 at metal-free N-functionalized graphene oxide electrodes. Electrochim. Acta 2018, 282, 694–701. [Google Scholar] [CrossRef]
- Sun, X.; Kang, X.; Zhu, Q.; Ma, J.; Yang, G.; Liu, Z.; Han, B. Very highly efficient reduction of CO2 to CH4 using metal-free N-doped carbon electrodes. Chem. Sci. 2016, 7, 2883–2887. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Zhou, D.; Sun, Z.; Deng, Y.; Xia, Y.; Zhao, D. A Controllable Synthesis of Rich Nitrogen-Doped Ordered Mesoporous Carbon for CO2 Capture and Supercapacitors. Adv. Funct. Mater. 2013, 23, 2322–2328. [Google Scholar] [CrossRef]
- Cui, X.; Pan, Z.; Zhang, L.; Peng, H.; Zheng, G. Selective Etching of Nitrogen-Doped Carbon by Steam for Enhanced Electrochemical CO2 Reduction. Adv. Energy Mater. 2017, 7, 1701456. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, J.; Cai, Q. Pyrrolic-nitrogen doped graphene: A metal-free electrocatalyst with high efficiency and selectivity for the reduction of carbon dioxide to formic acid: A computational study. Phys. Chem. Chem. Phys. 2016, 18, 5491–5498. [Google Scholar] [CrossRef]
- Daiyan, R.; Saputera, W.H.; Masood, H.; Leverett, J.; Lu, X.; Amal, R. A Disquisition on the Active Sites of Heterogeneous Catalysts for Electrochemical Reduction of CO2 to Value-Added Chemicals and Fuel. Adv. Energy Mater. 2020, 10, 1902106. [Google Scholar] [CrossRef]
- Wu, J.; Ma, S.; Sun, J.; Gold, J.I.; Tiwary, C.; Kim, B.; Zhu, L.; Chopra, N.; Odeh, I.N.; Vajtai, R.; et al. A metal-free electrocatalyst for carbon dioxide reduction to multi-carbon hydrocarbons and oxygenates. Nat. Commun. 2016, 7, 13869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, G.-L.; Guo, Z.-X. Highly effective sites and selectivity of nitrogen-doped graphene/CNT catalysts for CO2 electrochemical reduction. Chem. Sci. 2016, 7, 1268–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, C.; Wang, X.; Chen, W.; Feng, S.; Wen, J.; Wu, Y.A. CO2 transformation to multicarbon products by photocatalysis and electrocatalysis. Mater. Today Adv. 2020, 6, 100071. [Google Scholar] [CrossRef]
- Mohamed, A.G.A.; Huang, Y.; Xie, J.; Borse, R.A.; Parameswaram, G.; Wang, Y. Metal-free sites with multidimensional structure modifications for selective electrochemical CO2 reduction. Nano Today 2020, 33, 100891. [Google Scholar] [CrossRef]
- Hernández, S.; Farkhondehfal, M.A.; Sastre, F.; Makkee, M.; Saracco, G.; Russo, N. Syngas production from electrochemical reduction of CO2: Current status and prospective implementation. Green Chem. 2017, 19, 2326–2346. [Google Scholar] [CrossRef] [Green Version]
- Hiragond, C.B.; Kim, H.; Lee, J.; Sorcar, S.; Erkey, C.; In, S. Il Electrochemical CO2 reduction to CO catalyzed by 2D nanostructures. Catalysts 2020, 10, 98. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Yang, H.; Huang, X.; Liu, L.; Cai, W.; Gao, J.; Li, X.; Zhang, T.; Huang, Y.; Liu, B. Identifying Active Sites of Nitrogen-Doped Carbon Materials for the CO2 Reduction Reaction. Adv. Funct. Mater. 2018, 28, 1800499. [Google Scholar] [CrossRef]
- Siahrostami, S.; Jiang, K.; Karamad, M.; Chan, K.; Wang, H.; Nørskov, J. Theoretical Investigations into Defected Graphene for Electrochemical Reduction of CO2. ACS Sustain. Chem. Eng. 2017, 5, 11080–11085. [Google Scholar] [CrossRef]
- Seredych, M.; Hulicova-Jurcakova, D.; Lu, G.Q.; Bandosz, T.J. Surface functional groups of carbons and the effects of their chemical character, density and accessibility to ions on electrochemical performance. Carbon 2008, 46, 1475–1488. [Google Scholar] [CrossRef]
- Yasuda, S.; Yu, L.; Kim, J.; Murakoshi, K. Selective nitrogen doping in graphene for oxygen reduction reactions. Chem. Commun. 2013, 49, 9627–9629. [Google Scholar] [CrossRef]
- Jia, Y.; Zhang, L.; Du, A.; Gao, G.; Chen, J.; Yan, X.; Brown, C.L.; Yao, X. Defect Graphene as a Trifunctional Catalyst for Electrochemical Reactions. Adv. Mater. 2016, 28, 9532–9538. [Google Scholar] [CrossRef] [PubMed]
- Daiyan, R.; Tan, X.; Chen, R.; Saputera, W.H.; Tahini, H.A.; Lovell, E.; Ng, Y.H.; Smith, S.C.; Dai, L.; Lu, X.; et al. Electroreduction of CO2 to CO on a Mesoporous Carbon Catalyst with Progressively Removed Nitrogen Moieties. ACS Energy Lett. 2018, 3, 2292–2298. [Google Scholar] [CrossRef]
- Hursán, D.; Samu, A.A.; Janovák, L.; Artyushkova, K.; Asset, T.; Atanassov, P.; Janáky, C. Morphological Attributes Govern Carbon Dioxide Reduction on N-Doped Carbon Electrodes. Joule 2019, 3, 1719–1733. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kim, J.; Hyeon, T. Recent Progress in the Synthesis of Porous Carbon Materials. Adv. Mater. 2006, 18, 2073–2094. [Google Scholar] [CrossRef]
- Lee, J.H.; Kattel, S.; Jiang, Z.; Xie, Z.; Yao, S.; Tackett, B.M.; Xu, W.; Marinkovic, N.S.; Chen, J.G. Tuning the activity and selectivity of electroreduction of CO2 to synthesis gas using bimetallic catalysts. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef]
- Sanz, R.; Calleja, G.; Arencibia, A.; Sanz-Pérez, E.S. Development of high efficiency adsorbents for CO2 capture based on a double-functionalization method of grafting and impregnation. J. Mater. Chem. A 2013, 1, 1956–1962. [Google Scholar] [CrossRef]
- Gong, K.; Du, F.; Xia, Z.; Durstock, M.; Dai, L. Nitrogen-Doped Carbon Nanotube Arrays with High Electrocatalytic Activity for Oxygen Reduction. Science 2009, 323, 760–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hu, Y.; Ma, L.; Zhu, G.; Wang, Y.; Xue, X.; Chen, R.; Yang, S.; Jin, Z. Progress and Perspective of Electrocatalytic CO2 Reduction for Renewable Carbonaceous Fuels and Chemicals. Adv. Sci. 2018, 5, 1700275. [Google Scholar] [CrossRef]
- Dutta, S.; Bhaumik, A.; Wu, K.C.W. Hierarchically porous carbon derived from polymers and biomass: Effect of interconnected pores on energy applications. Energy Environ. Sci. 2014, 7, 3574–3592. [Google Scholar] [CrossRef]
- Zou, X.; Liu, M.; Wu, J.; Ajayan, P.M.; Li, J.; Liu, B.; Yakobson, B.I. How Nitrogen-Doped Graphene Quantum Dots Catalyze Electroreduction of CO2 to Hydrocarbons and Oxygenates. ACS Catal. 2017, 7, 6245–6250. [Google Scholar] [CrossRef]
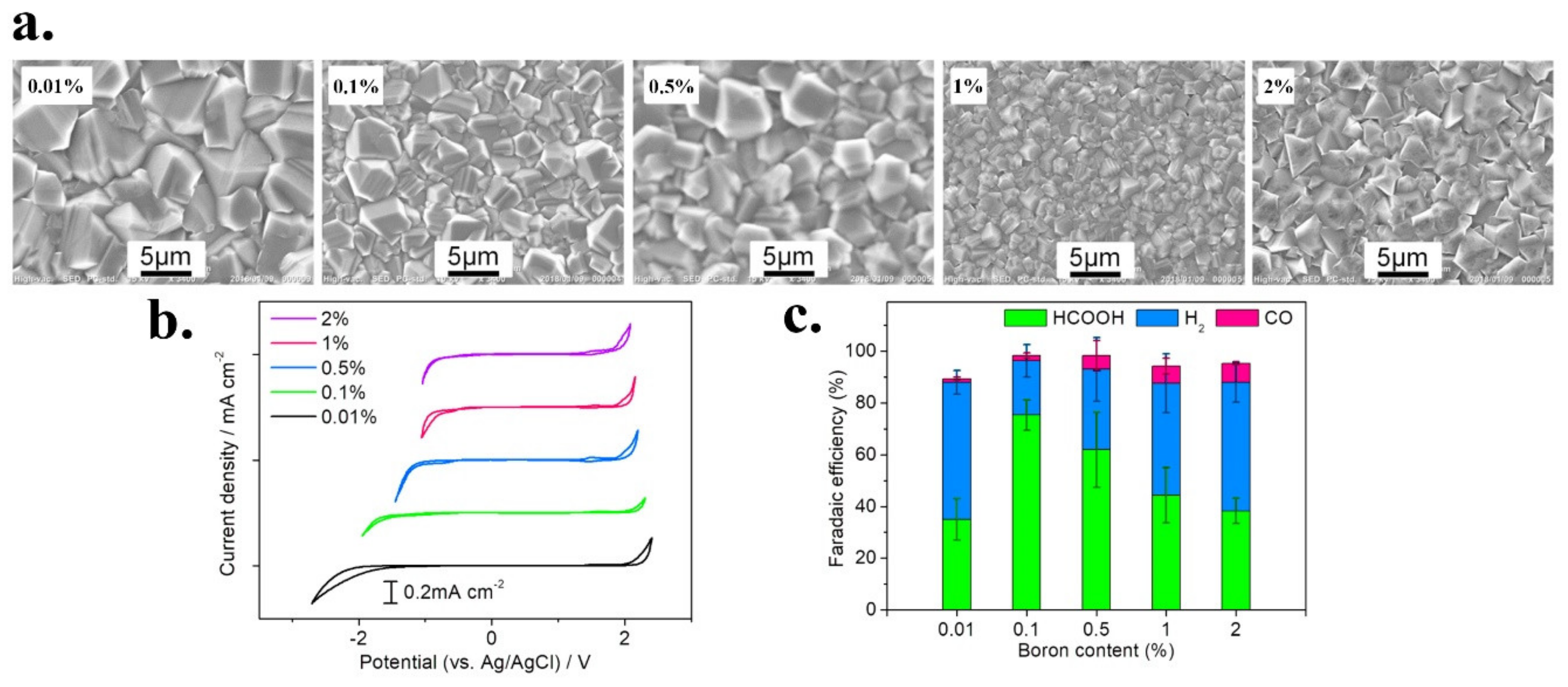
- Sreekanth, N.; Nazrulla, M.A.; Vineesh, T.V.; Sailaja, K.; Phani, K.L. Metal-free boron-doped graphene for selective electroreduction of carbon dioxide to formic acid/formate. Chem. Commun. 2015, 51, 16061–16064. [Google Scholar] [CrossRef]
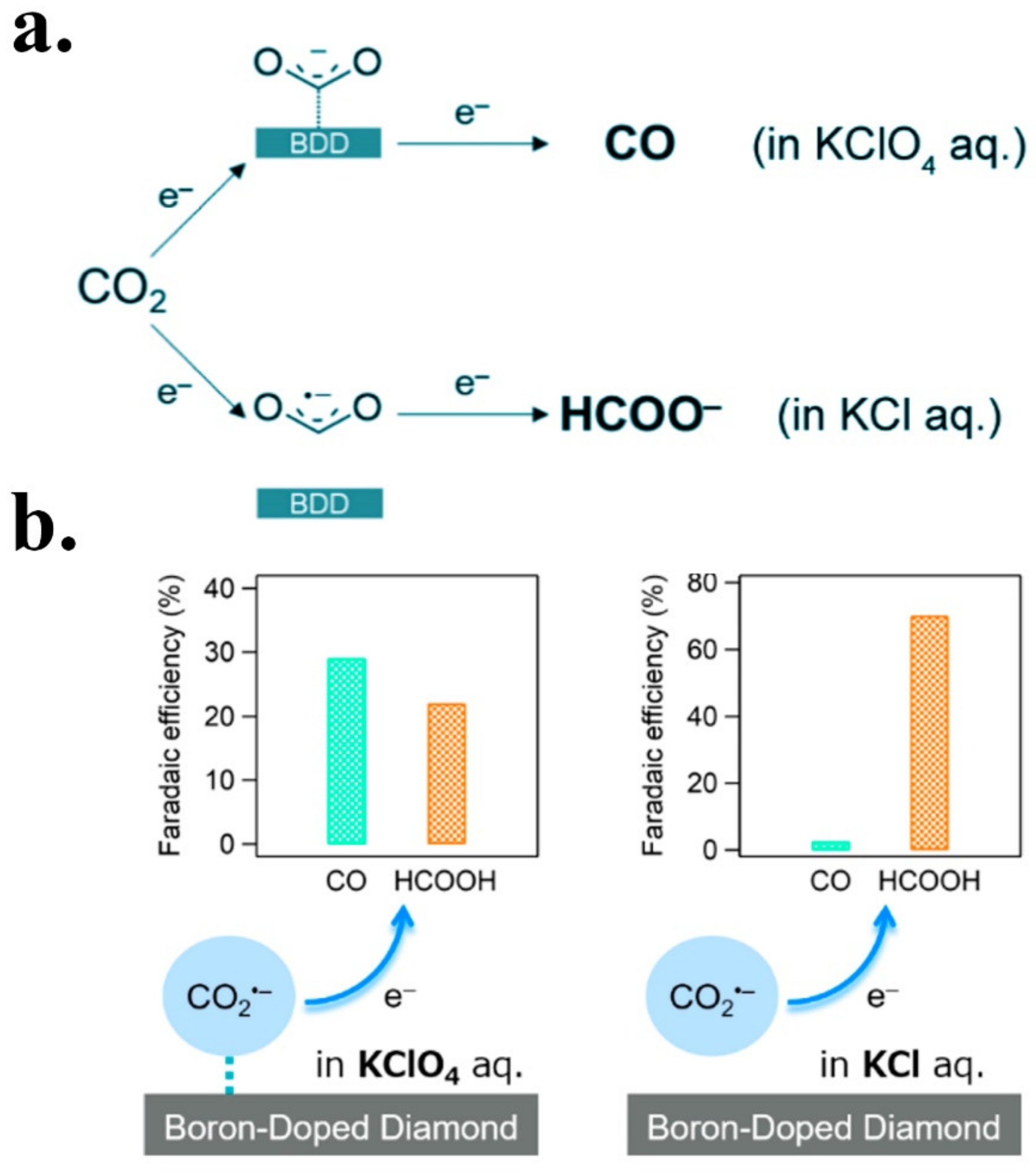
- Einaga, Y. Diamond electrodes for electrochemical analysis. J. Appl. Electrochem. 2010, 40, 1807–1816. [Google Scholar] [CrossRef]
- Ivandini, T.A.; Einaga, Y. Polycrystalline boron-doped diamond electrodes for electrocatalytic and electrosynthetic applications. Chem. Commun. 2017, 53, 1338–1347. [Google Scholar] [CrossRef]
- Nakata, K.; Ozaki, T.; Terashima, C.; Fujishima, A.; Einaga, Y. High-yield electrochemical production of formaldehyde from CO2 and seawater. Angew. Chem. Int. Ed. 2014, 53, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Jiwanti, P.K.; Natsui, K.; Nakata, K.; Einaga, Y. Selective production of methanol by the electrochemical reduction of CO2 on boron-doped diamond electrodes in aqueous ammonia solution. RSC Adv. 2016, 6, 102214–102217. [Google Scholar] [CrossRef]
- Natsui, K.; Iwakawa, H.; Ikemiya, N.; Nakata, K.; Einaga, Y. Stable and Highly Efficient Electrochemical Production of Formic Acid from Carbon Dioxide Using Diamond Electrodes. Angew. Chem. Int. Ed. 2018, 57, 2639–2643. [Google Scholar] [CrossRef]
- Ikemiya, N.; Natsui, K.; Nakata, K.; Einaga, Y. Effect of alkali-metal cations on the electrochemical reduction of carbon dioxide to formic acid using boron-doped diamond electrodes. RSC Adv. 2017, 7, 22510–22514. [Google Scholar] [CrossRef] [Green Version]
- Tomisaki, M.; Natsui, K.; Ikemiya, N.; Nakata, K.; Einaga, Y. Influence of Electrolyte on the Electrochemical Reduction of Carbon Dioxide Using Boron-Doped Diamond Electrodes. ChemistrySelect 2018, 3, 10209–10213. [Google Scholar] [CrossRef]
- Xu, J.; Natsui, K.; Naoi, S.; Nakata, K.; Einaga, Y. Effect of doping level on the electrochemical reduction of CO2 on boron-doped diamond electrodes. Diam. Relat. Mater. 2018, 86, 167–172. [Google Scholar] [CrossRef]
- Tomisaki, M.; Kasahara, S.; Natsui, K.; Ikemiya, N.; Einaga, Y. Switchable Product Selectivity in the Electrochemical Reduction of Carbon Dioxide Using Boron-Doped Diamond Electrodes. J. Am. Chem. Soc. 2019, 141, 7414–7420. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Einaga, Y. Effect of sp2 species in a boron-doped diamond electrode on the electrochemical reduction of CO2. Electrochem. Commun. 2020, 115, 106731. [Google Scholar] [CrossRef]
- Xie, J.; Zhao, X.; Wu, M.; Li, Q.; Wang, Y.; Yao, J. Metal-Free Fluorine-Doped Carbon Electrocatalyst for CO2 Reduction Outcompeting Hydrogen Evolution. Angew. Chem. Int. Ed. 2018, 57, 9640–9644. [Google Scholar] [CrossRef]
- Ni, W.; Xue, Y.; Zang, X.; Li, C.; Wang, H.; Yang, Z.; Yan, Y.-M. Fluorine Doped Cagelike Carbon Electrocatalyst: An Insight into the Structure-Enhanced CO Selectivity for CO2 Reduction at High Overpotential. ACS Nano 2020, 14, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ali, S.; Lian, Z.; Si, C.; Su, D.S.; Li, B. Phosphorus-doped onion-like carbon for CO2 electrochemical reduction: The decisive role of the bonding configuration of phosphorus. J. Mater. Chem. A 2018, 6, 19998–20004. [Google Scholar] [CrossRef]
- Zhao, T.; Tian, Y.; Yan, L.; Su, Z. Heteroatom-doped C3N as a promising metal-free catalyst for a high-efficiency carbon dioxide reduction reaction. New J. Chem. 2020, 44, 11824–11828. [Google Scholar] [CrossRef]
- Mao, X.; Kour, G.; Zhang, L.; He, T.; Wang, S.; Yan, C.; Zhu, Z.; Du, A. Silicon-doped graphene edges: An efficient metal-free catalyst for the reduction of CO2 into methanol and ethanol. Catal. Sci. Technol. 2019, 9, 6800–6807. [Google Scholar] [CrossRef]
- Wang, G.; Liu, M.; Jia, J.; Xu, H.; Zhao, B.; Lai, K.; Tu, C.; Wen, Z. Nitrogen and Sulfur Co-doped Carbon Nanosheets for Electrochemical Reduction of CO2. ChemCatChem 2020, 12, 2203–2208. [Google Scholar] [CrossRef]
- Pan, F.; Li, B.; Deng, W.; Du, Z.; Gang, Y.; Wang, G.; Li, Y. Promoting electrocatalytic CO2 reduction on nitrogen-doped carbon with sulfur addition. Appl. Catal. B Environ. 2019, 252, 240–249. [Google Scholar] [CrossRef]
- Xie, J.; Ghausi, M.A.; Wang, J.; Wang, X.; Wang, W.; Yang, R.; Wu, M.; Zhang, Q.; Wang, Y. Low-Energy CO2 Reduction on a Metal-Free Carbon Material. ChemElectroChem 2020, 7, 2145–2150. [Google Scholar] [CrossRef]
- Chen, S.; Liu, T.; Olanrele, S.O.; Lian, Z.; Si, C.; Chen, Z.; Li, B. Boosting electrocatalytic activity for CO2 reduction on nitrogen-doped carbon catalysts by co-doping with phosphorus. J. Energy Chem. 2021, 54, 143–150. [Google Scholar] [CrossRef]
- Jia, C.; Ren, W.; Chen, X.; Yang, W.; Zhao, C. (N, B) Dual Heteroatom-Doped Hierarchical Porous Carbon Framework for Efficient Electroreduction of Carbon Dioxide. ACS Sustain. Chem. Eng. 2020, 8, 6003–6010. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Cheng, K.; Quan, X.; Fan, X.; Su, Y.; Chen, S.; Zhao, H.; Zhang, Y.; Yu, H.; et al. Selective Electrochemical Reduction of Carbon Dioxide to Ethanol on a Boron- and Nitrogen-Co-doped Nanodiamond. Angew. Chem. Int. Ed. 2017, 56, 15607–15611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, G.; Shi, L.; Ye, J. Single-Atom Catalysts: Emerging Multifunctional Materials in Heterogeneous Catalysis. Adv. Energy Mater. 2018, 8, 1701343. [Google Scholar] [CrossRef]
- Yang, X.-F.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef]
- Li, M.; Wang, H.; Luo, W.; Sherrell, P.C.; Chen, J.; Yang, J. Heterogeneous Single-Atom Catalysts for Electrochemical CO2 Reduction Reaction. Adv. Mater. 2020, 32, 1–24. [Google Scholar] [CrossRef]
- Hou, C.-C.; Wang, H.-F.; Li, C.; Xu, Q. From metal–organic frameworks to single/dual-atom and cluster metal catalysts for energy applications. Energy Environ. Sci. 2020, 13, 1658–1693. [Google Scholar] [CrossRef]
- Jeong, H.Y.; Balamurugan, M.; Choutipalli, V.S.K.; Jo, J.; Baik, H.; Subramanian, V.; Kim, M.; Sim, U.; Nam, K.T. Tris(2-benzimidazolylmethyl)amine-Directed Synthesis of Single-Atom Nickel Catalysts for Electrochemical CO Production from CO2. Chem. A Eur. J. 2018, 24, 18444–18454. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Rong, H.; Zhang, J.; Wang, D.; Li, Y. Modulating the local coordination environment of single-atom catalysts for enhanced catalytic performance. Nano Res. 2020, 13, 1842–1855. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Salehi, M.; Van Le, Q.; Seifitokaldani, A.; Dinh, C.T. Fundamentals of Electrochemical CO2 Reduction on Single-Metal-Atom Catalysts. ACS Catal. 2020, 10, 10068–10095. [Google Scholar] [CrossRef]
- Peng, Y.; Lu, B.; Chen, S. Carbon-Supported Single Atom Catalysts for Electrochemical Energy Conversion and Storage. Adv. Mater. 2018, 30, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Romanchuk, A.Y.; Slesarev, A.S.; Kalmykov, S.N.; Kosynkin, D.V.; Tour, J.M. Graphene oxide for effective radionuclide removal. Phys. Chem. Chem. Phys. 2013, 15, 2321–2327. [Google Scholar] [CrossRef]
- Jiang, K.; Siahrostami, S.; Zheng, T.; Hu, Y.; Hwang, S.; Stavitski, E.; Peng, Y.; Dynes, J.; Gangisetty, M.; Su, D.; et al. Isolated Ni single atoms in graphene nanosheets for high-performance CO2 reduction. Energy Environ. Sci. 2018, 11, 893–903. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, S.; Wu, J.; Liu, M.; Yazdi, S.; Ren, M.; Sha, J.; Zhong, J.; Nie, K.; Jalilov, A.S.; et al. Electrochemical CO2 Reduction with Atomic Iron-Dispersed on Nitrogen-Doped Graphene. Adv. Energy Mater. 2018, 1703487, 1–9. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhao, S.; Li, H.; He, S.; Veder, J.P.; Johannessen, B.; Xiao, J.; Lu, S.; Pan, J.; Chisholm, M.F.; et al. Unsaturated edge-anchored Ni single atoms on porous microwave exfoliated graphene oxide for electrochemical CO2. Appl. Catal. B Environ. 2019, 243, 294–303. [Google Scholar] [CrossRef]
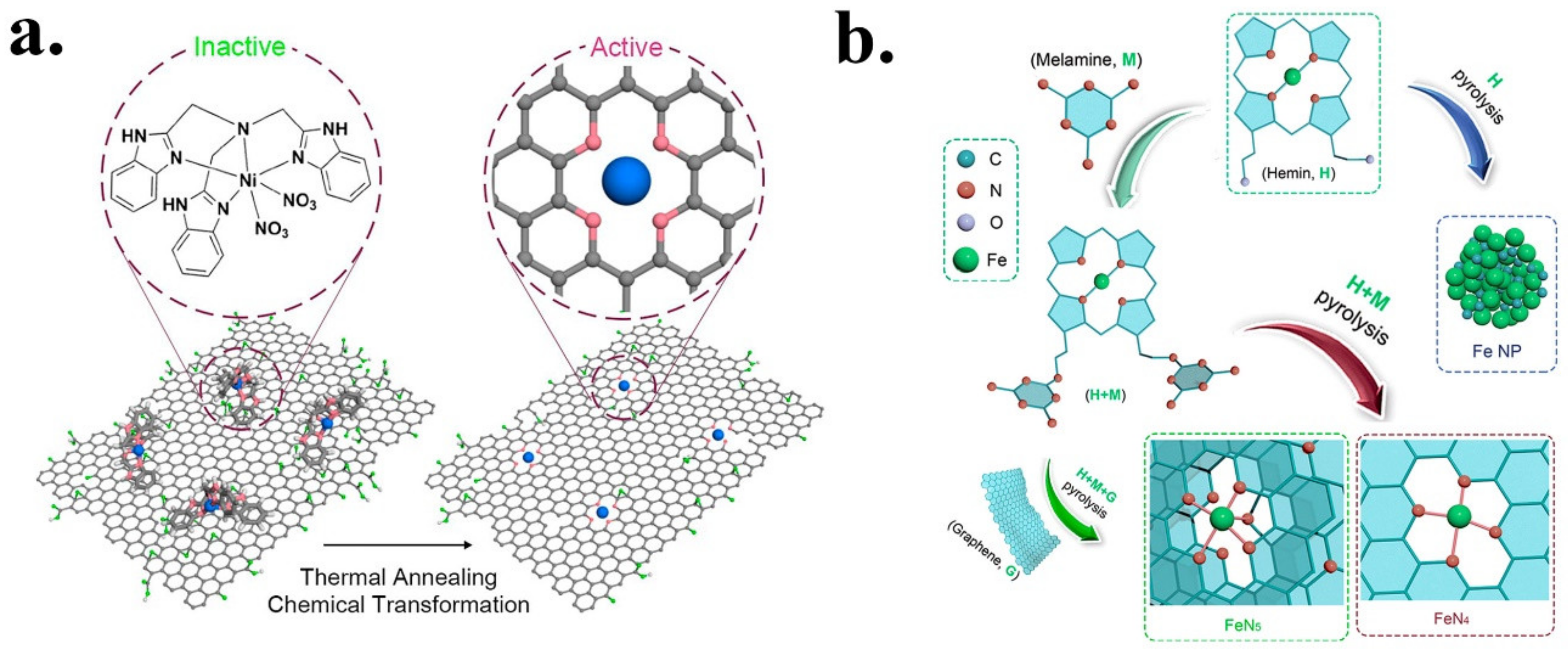
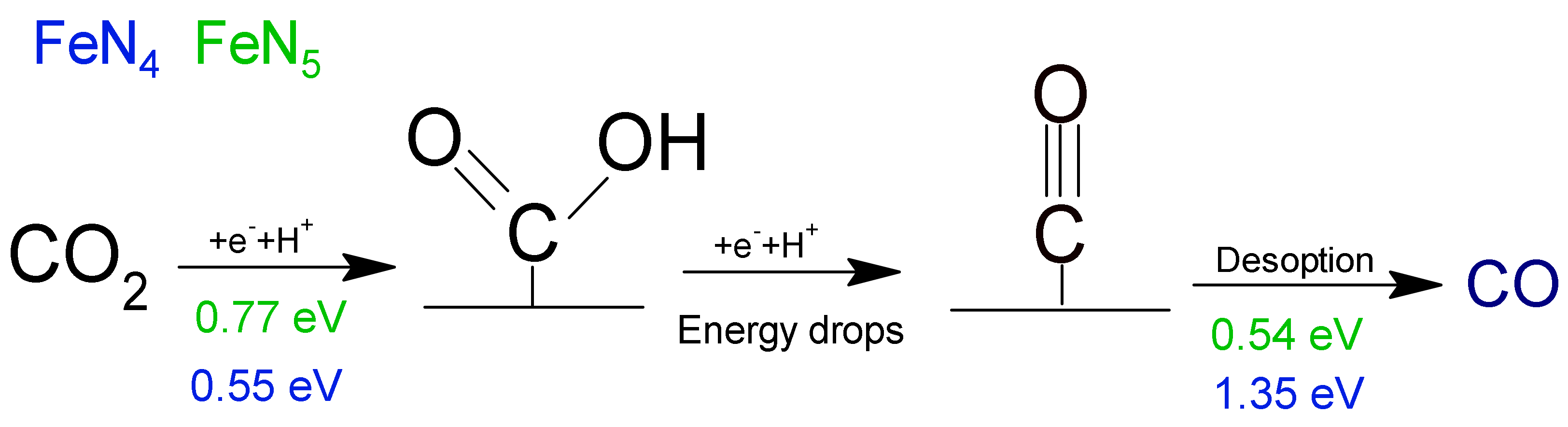
- Zhang, H.; Li, J.; Xi, S.; Du, Y.; Hai, X.; Wang, J.; Xu, H.; Wu, G.; Zhang, J.; Lu, J.; et al. A Graphene-Supported Single-Atom FeN 5 Catalytic Site for Efficient Electrochemical CO2 Reduction. Angew. Chem. Int. Ed. 2019, 58, 14871–14876. [Google Scholar] [CrossRef]
- Ren, X.; Liu, S.; Li, H.; Ding, J.; Liu, L.; Kuang, Z.; Li, L.; Yang, H.; Bai, F.; Huang, Y.; et al. Electron-withdrawing functional ligand promotes CO2 reduction catalysis in single atom catalyst. Sci. China Ser. B Chem. 2020, 63, 1727–1733. [Google Scholar] [CrossRef]
- Li, X.; Chai, G.; Xu, X.; Liu, J.; Zhong, Z.; Cao, A.; Tao, Z.; You, W.; Kang, L. Electrocatalytic reduction of CO2 to CO over iron phthalocyanine-modified graphene nanocomposites. Carbon 2020, 167, 658–667. [Google Scholar] [CrossRef]
- Yang, H.; Hung, S.-F.; Liu, S.; Yuan, K.; Miao, S.; Zhang, L.; Huang, X.; Wang, H.-Y.; Cai, W.; Chen, R.; et al. Atomically dispersed Ni(i) as the active site for electrochemical CO2 reduction. Nat. Energy 2018, 3, 140–147. [Google Scholar] [CrossRef]
- Tuo, J.; Zhu, Y.; Jiang, H.; Shen, J.; Li, C. The Effect of Coordination Environment of Atomically Dispersed Fe and N Co-doped Carbon Nanosheets on CO2 Electroreduction. ChemElectroChem 2020, 7, 4767–4772. [Google Scholar] [CrossRef]
- Back, S.; Lim, J.; Kim, N.Y.; Kim, Y.H.; Jung, Y. Single-atom catalysts for CO2 electroreduction with significant activity and selectivity improvements. Chem. Sci. 2017, 8, 1090–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Jagvaral, Y. Electrochemical reduction of CO2 on graphene supported transition metals-towards single atom catalysts. Phys. Chem. Chem. Phys. 2017, 19, 11436–11446. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, X.; Wang, X.; Lv, K.; Xiao, G.; Feng, J.; Jiang, X.; Fang, M.; Zhu, Y. Single-Atom Iron-Nitrogen Catalytic Site with Graphitic Nitrogen for Efficient Electroreduction of CO2. ChemistrySelect 2020, 5, 1282–1287. [Google Scholar] [CrossRef]
- Yan, C.; Ye, Y.; Lin, L.; Wu, H.; Jiang, Q.; Wang, G.; Bao, X. Improving CO2 electroreduction over ZIF-derived carbon doped with Fe-N sites by an additional ammonia treatment. Catal. Today 2019, 330, 252–258. [Google Scholar] [CrossRef]
- Pan, F.; Zhang, H.; Liu, K.; Cullen, D.; More, K.; Wang, M.; Feng, Z.; Wang, G.; Wu, G.; Li, Y. Unveiling Active Sites of CO2 Reduction on Nitrogen-Coordinated and Atomically Dispersed Iron and Cobalt Catalysts. ACS Catal. 2018, 8, 3116–3122. [Google Scholar] [CrossRef]
- Lu, P.; Yang, Y.; Yao, J.; Wang, M.; Dipazir, S.; Yuan, M.; Zhang, J.; Wang, X.; Xie, Z.; Zhang, G. Facile synthesis of single-nickel-atomic dispersed N-doped carbon framework for efficient electrochemical CO2 reduction. Appl. Catal. B Environ. 2019, 241, 113–119. [Google Scholar] [CrossRef]
- Zhao, C.; Dai, X.; Yao, T.; Chen, W.; Wang, X.; Wang, J.; Yang, J.; Wei, S.; Wu, Y.; Li, Y. Ionic Exchange of Metal-Organic Frameworks to Access Single Nickel Sites for Efficient Electroreduction of CO2. J. Am. Chem. Soc. 2017, 139, 8078–8081. [Google Scholar] [CrossRef]
- Pan, F.; Zhang, H.; Liu, Z.; Cullen, D.; Liu, K.; More, K.; Wu, G.; Wang, G.; Li, Y. Atomic-level active sites of efficient imidazolate framework-derived nickel catalysts for CO2 reduction. J. Mater. Chem. A 2019, 7, 26231–26237. [Google Scholar] [CrossRef]
- Geng, Z.; Cao, Y.; Chen, W.; Kong, X.; Liu, Y.; Yao, T.; Lin, Y. Regulating the coordination environment of Co single atoms for achieving efficient electrocatalytic activity in CO2 reduction. Appl. Catal. B Environ. 2019, 240, 234–240. [Google Scholar] [CrossRef]
- Gu, J.; Hsu, C.S.; Bai, L.; Chen, H.M.; Hu, X. Atomically dispersed Fe3+ sites catalyze efficient CO2 electroreduction to CO. Science 2019, 364, 1091–1094. [Google Scholar] [CrossRef]
- Chen, X.; Ma, D.D.; Chen, B.; Zhang, K.; Zou, R.; Wu, X.T.; Zhu, Q.L. Metal–organic framework-derived mesoporous carbon nanoframes embedded with atomically dispersed Fe–Nx active sites for efficient bifunctional oxygen and carbon dioxide electroreduction. Appl. Catal. B Environ. 2020, 267, 118720. [Google Scholar] [CrossRef]
- Zhang, C.; Fu, Z.; Zhao, Q.; Du, Z.; Zhang, R.; Li, S. Single-atom-Ni-decorated, nitrogen-doped carbon layers for efficient electrocatalytic CO2 reduction reaction. Electrochem. Commun. 2020, 116, 106758. [Google Scholar] [CrossRef]
- Wang, T.; Sang, X.; Zheng, W.; Yang, B.; Yao, S.; Lei, C.; Li, Z.; He, Q.; Lu, J.; Lei, L.; et al. Gas Diffusion Strategy for Inserting Atomic Iron Sites into Graphitized Carbon Supports for Unusually High-Efficient CO2 Electroreduction and High-Performance Zn–CO2 Batteries. Adv. Mater. 2020, 32, e2002430. [Google Scholar] [CrossRef]
- Hou, Y.; Liang, Y.L.; Shi, P.C.; Huang, Y.B.; Cao, R. Atomically dispersed Ni species on N-doped carbon nanotubes for electroreduction of CO2 with nearly 100% CO selectivity. Appl. Catal. B Environ. 2020, 271, 118929. [Google Scholar] [CrossRef]
- Pan, F.; Li, B.; Sarnello, E.; Fei, Y.; Gang, Y.; Xiang, X.; Du, Z.; Zhang, P.; Wang, G.; Nguyen, H.T.; et al. Atomically Dispersed Iron-Nitrogen Sites on Hierarchically Mesoporous Carbon Nanotube and Graphene Nanoribbon Networks for CO2 Reduction. ACS Nano 2020, 14, 5506–5516. [Google Scholar] [CrossRef]
- Li, X.; Bi, W.; Chen, M.; Sun, Y.; Ju, H.; Yan, W.; Zhu, J.; Wu, X.; Chu, W.; Wu, C.; et al. Exclusive Ni-N4 Sites Realize Near-Unity CO Selectivity for Electrochemical CO2 Reduction. J. Am. Chem. Soc. 2017, 139, 14889–14892. [Google Scholar] [CrossRef]
- Zhao, S.; Cheng, Y.; Veder, J.; Johannessen, B.; Saunders, M.; Zhang, L.; Liu, C.; Chisholm, M.F.; De Marco, R.; Liu, J.; et al. One-Pot Pyrolysis Method to Fabricate Carbon Nanotube Supported Ni Single-Atom Catalysts with Ultrahigh Loading. ACS Appl. Energy Mater. 2018, 1, 5286–5297. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhao, S.; Johannessen, B.; Veder, J.; Saunders, M.; Rowles, M.R.; Cheng, M.; Liu, C.; Chisholm, M.F.; De Marco, R.; et al. Atomically Dispersed Transition Metals on Carbon Nanotubes with Ultrahigh Loading for Selective Electrochemical Carbon Dioxide Reduction. Adv. Mater. 2018, 30, e1706287. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, H.; Yu, P.; Yuan, Y.; Shahbazian-Yassar, R.; Sheng, Y.; Wu, S.; Tu, W.; Liu, G.; Kraft, M.; et al. Isolated Ni single atoms in nitrogen doped ultrathin porous carbon templated from porous g-C3N4 for high-performance CO2 reduction. Nano Energy 2020, 77, 105158. [Google Scholar] [CrossRef]
- Ni, W.; Liu, Z.; Zhang, Y.; Ma, C.; Deng, H.; Zhang, S.; Wang, S. Electroreduction of Carbon Dioxide Driven by the Intrinsic Defects in the Carbon Plane of a Single Fe–N4 Site. Adv. Mater. 2020, 33, e2003238. [Google Scholar] [CrossRef]
- Tuo, J.; Zhu, Y.; Cheng, L.; Li, Y.; Yang, X.; Shen, J.; Li, C. Layered Confinement Reaction: Atomic-level Dispersed Iron–Nitrogen Co-Doped Ultrathin Carbon Nanosheets for CO2 Electroreduction. ChemSusChem 2019, 12, 2644–2650. [Google Scholar] [CrossRef]
- Wu, S.; Lv, X.; Ping, D.; Zhang, G.; Wang, S.; Wang, H.; Yang, X.; Guo, D.; Fang, S. Highly exposed atomic Fe–N active sites within carbon nanorods towards electrocatalytic reduction of CO2 to CO. Electrochim. Acta 2020, 340, 135930. [Google Scholar] [CrossRef]
- He, Q.; Lee, J.H.; Liu, D.; Liu, Y.; Lin, Z.; Xie, Z.; Hwang, S.; Kattel, S.; Song, L.; Chen, J.G. Accelerating CO2 Electroreduction to CO Over Pd Single-Atom Catalyst. Adv. Funct. Mater. 2020, 30, 1–8. [Google Scholar] [CrossRef]
- Han, S.G.; Ma, D.D.; Zhou, S.H.; Zhang, K.; Wei, W.B.; Du, Y.; Wu, X.T.; Xu, Q.; Zou, R.; Zhu, Q.L. Fluorine-tuned single-atom catalysts with dense surface Ni-N4 sites on ultrathin carbon nanosheets for efficient CO2 electroreduction. Appl. Catal. B Environ. 2021, 283, 119591. [Google Scholar] [CrossRef]
- Chen, Y.; Ma, L.; Chen, C.; Hu, W.; Zou, L.; Zou, Z.; Yang, H. Fe and N co-doped carbon with High doping content of sulfur and nitrogen for efficient CO2 electro-reduction. J. CO2 Util. 2020, 42, 101316. [Google Scholar] [CrossRef]
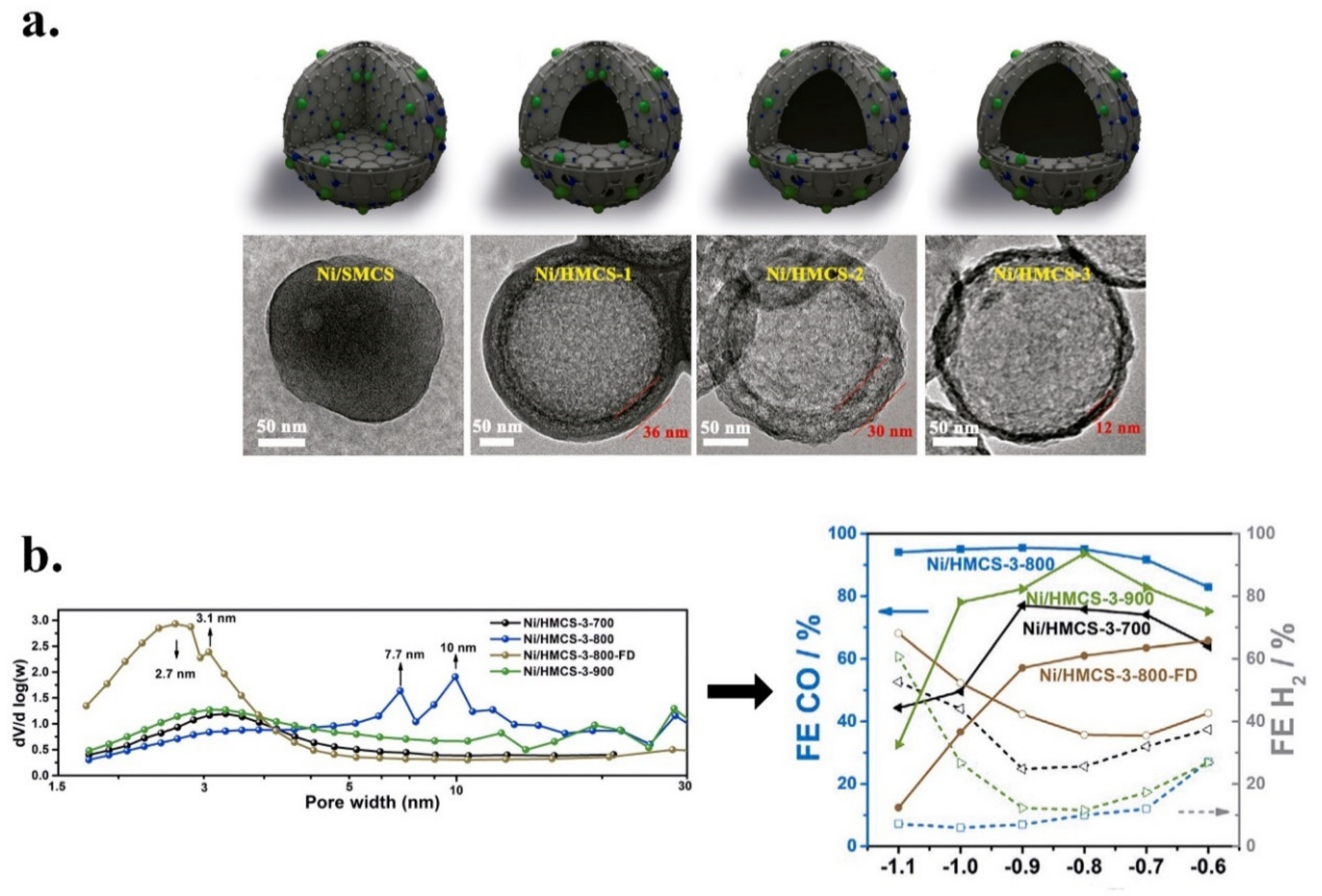
- Xiong, W.; Li, H.; Wang, H.; Yi, J.; You, H.; Zhang, S.; Hou, Y.; Cao, M.; Zhang, T.; Cao, R. Hollow Mesoporous Carbon Sphere Loaded Ni–N4 Single-Atom: Support Structure Study for CO2 Electrocatalytic Reduction Catalyst. Small 2020, 16, 2003943. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liu, Z.; Ma, J.; Liu, J.; Zhou, P.; Chao, Y.; Ma, C.; Bo, X.; Liu, J.; Hei, Y.; et al. Sustainability Perspective-Oriented Synthetic Strategy for Zinc Single-Atom Catalysts Boosting Electrocatalytic Reduction of Carbon Dioxide and Oxygen. ACS Sustain. Chem. Eng. 2020, 8, 13813–13822. [Google Scholar] [CrossRef]
- Abbas, S.A.; Song, J.T.; Tan, Y.C.; Nam, K.M.; Oh, J.; Jung, K. Synthesis of a Nickel Single-Atom Catalyst Based on Ni−N4−xCx Active Sites for Highly Efficient CO2 Reduction Utilizing a Gas Diffusion Electrode. ACS Appl. Energy Mater. 2020, 3, 8739–8745. [Google Scholar] [CrossRef]
- Ni, Y.; Miao, L.; Wang, J.; Liu, J.; Yuan, M.; Chen, J. Pore size effect of graphyne supports on CO2 electrocatalytic activity of Cu single atoms. Phys. Chem. Chem. Phys. 2020, 22, 1181–1186. [Google Scholar] [CrossRef]
- Xu, H.; Cheng, D.; Cao, D.; Zeng, X.C. Publisher Correction: A universal principle for a rational design of single-atom electrocatalysts. Nat. Catal. 2018, 1, 632. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Qiao, S.Z.; Liu, H.; Chen, J.; Orpe, A.; Zhao, D.; Lu, G.Q. (Max) Extension of The Stöber Method to the Preparation of Monodisperse Resorcinol–Formaldehyde Resin Polymer and Carbon Spheres. Angew. Chemie Int. Ed. 2011, 50, 5947–5951. [Google Scholar] [CrossRef]
- Pan, Y.; Lin, R.; Chen, Y.; Liu, S.; Zhu, W.; Cao, X.; Chen, W.; Wu, K.; Cheong, W.C.; Wang, Y.; et al. Design of Single-Atom Co-N5 Catalytic Site: A Robust Electrocatalyst for CO2 Reduction with Nearly 100% CO Selectivity and Remarkable Stability. J. Am. Chem. Soc. 2018, 140, 4218–4221. [Google Scholar] [CrossRef]
- Ma, S.; Su, P.; Huang, W.; Jiang, S.P.; Bai, S.; Liu, J. Atomic Ni Species Anchored N-Doped Carbon Hollow Spheres as Nanoreactors for Efficient Electrochemical CO2 Reduction. ChemCatChem 2019, 11, 6092–6098. [Google Scholar] [CrossRef]
- Jeong, H.Y.; Balamurugan, M.; Choutipalli, V.S.K.; Jeong, E.S.; Subramanian, V.; Sim, U.; Nam, K.T. Achieving highly efficient CO2 to CO electroreduction exceeding 300 mA cm-2 with single-atom nickel electrocatalysts. J. Mater. Chem. A 2019, 7, 10651–10661. [Google Scholar] [CrossRef]
- Chen, Y.; Zou, L.; Liu, H.; Chen, C.; Wang, Q.; Gu, M.; Yang, B.; Zou, Z.; Fang, J.; Yang, H. Fe and N Co-Doped Porous Carbon Nanospheres with High Density of Active Sites for Efficient CO2 Electroreduction. J. Phys. Chem. C 2019, 123, 16651–16659. [Google Scholar] [CrossRef]
- Pan, F.; Li, B.; Sarnello, E.; Fei, Y.; Feng, X.; Gang, Y.; Xiang, X.; Fang, L.; Li, T.; Hu, Y.H.; et al. Pore-Edge Tailoring of Single-Atom Iron−Nitrogen Sites on Graphene for Enhanced CO2 Reduction. ACS Catal. 2020, 10, 10803–10811. [Google Scholar] [CrossRef]
- Fang, M.; Wang, X.; Li, X.; Zhu, Y.; Xiao, G.; Feng, J.; Jiang, X.; Lv, K.; Zhu, Y.; Lin, W.F. Curvature-induced Zn3d Electron Return on Zn−N4 Single-atom Carbon Nanofibers for Boosting Electroreduction of CO2. ChemCatChem 2020, 13, 603–609. [Google Scholar] [CrossRef]
- Mou, K.; Chen, Z.; Zhang, X.; Jiao, M.; Zhang, X.; Ge, X.; Zhang, W.; Liu, L. Highly Efficient Electroreduction of CO2 on Nickel Single-Atom Catalysts: Atom Trapping and Nitrogen Anchoring. Small 2019, 15, e1903668. [Google Scholar] [CrossRef]
- Ma, Z.; Zhang, X.; Wu, D.; Han, X.; Zhang, L.; Wang, H.; Xu, F.; Gao, Z.; Jiang, K. Ni and nitrogen-codoped ultrathin carbon nanosheets with strong bonding sites for efficient CO2 electrochemical reduction. J. Colloid Interface Sci. 2020, 570, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Mao, X.; Ma, M.; Jiang, C.; Zhang, P.; Wang, J.; Deng, Q.; Zeng, Z.; Deng, S. Scalable strategy to fabricate single Cu atoms coordinated carbons for efficient electroreduction of CO2 to CO. Carbon 2020, 168, 528–535. [Google Scholar] [CrossRef]
- Takele Menisa, L.; Cheng, P.; Long, C.; Qiu, X.; Zheng, Y.; Han, J.; Zhang, Y.; Gao, Y.; Tang, Z. Insight into atomically dispersed porous M-N-C single-site catalysts for electrochemical CO2 reduction. Nanoscale 2020, 12, 16617–16626. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Song, P.; Liu, X.; Mei, B.; Xing, W.; Jiang, Z.; Gu, L.; Xu, W. Highly Efficient CO2 Electroreduction on ZnN4-based Single-Atom Catalyst. Angew. Chem. Int. Ed. 2018, 57, 12303–12307. [Google Scholar] [CrossRef]
- Li, F.; Hong, S.; Wu, T.S.; Li, X.; Masa, J.; Soo, Y.L.; Sun, Z. Atomically Dispersed Nickel Sites for Selective Electroreduction of CO2. ACS Appl. Energy Mater. 2019, 2, 8836–8842. [Google Scholar] [CrossRef]
- He, Y.; Li, Y.; Zhang, J.; Wang, S.; Huang, D.; Yang, G.; Yi, X.; Lin, H.; Han, X.; Hu, W.; et al. Low-temperature strategy toward Ni-NC@Ni core-shell nanostructure with Single-Ni sites for efficient CO2 electroreduction. Nano Energy 2020, 77, 105010. [Google Scholar] [CrossRef]
- Song, X.; Zhang, H.; Yang, Y.; Zhang, B.; Zuo, M.; Cao, X.; Sun, J.; Lin, C.; Li, X.; Jiang, Z. Bifunctional Nitrogen and Cobalt Codoped Hollow Carbon for Electrochemical Syngas Production. Adv. Sci. 2018, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Liu, D.; Lee, J.H.; Liu, Y.; Xie, Z.; Hwang, S.; Kattel, S.; Song, L.; Chen, J.G. Electrochemical Conversion of CO2 to Syngas with Controllable CO/H2 Ratios over Co and Ni Single-Atom Catalysts. Angew. Chem. Int. Ed. 2020, 59, 3033–3037. [Google Scholar] [CrossRef]
- Zhu, W.; Fu, J.; Liu, J.; Chen, Y.; Li, X.; Huang, K.; Cai, Y.; He, Y.; Zhou, Y.; Su, D.; et al. Tuning single atom-nanoparticle ratios of Ni-based catalysts for synthesis gas production from CO2. Appl. Catal. B Environ. 2020, 264, 118502. [Google Scholar] [CrossRef]
- Daiyan, R.; Chen, R.; Kumar, P.; Bedford, N.M.; Qu, J.; Cairney, J.M.; Lu, X.; Amal, R. Tunable Syngas Production through CO2 Electroreduction on Cobalt-Carbon Composite Electrocatalyst. ACS Appl. Mater. Interfaces 2020, 12, 9307–9315. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, Z.; Gu, L.; Zhang, Q.; Zhang, L.; Song, Q.; Zhou, W.; Hu, S. Electrochemical conversion of CO2 to syngas with a wide range of CO/H2 ratio over Ni/Fe binary single-atom catalysts. Nano Res. 2020, 13, 3206–3211. [Google Scholar] [CrossRef]
- Yang, H.; Wu, Y.; Li, G.; Lin, Q.; Hu, Q.; Zhang, Q.; Liu, J.; He, C. Scalable Production of Efficient Single-Atom Copper Decorated Carbon Membranes for CO2 Electroreduction to Methanol. J. Am. Chem. Soc. 2019, 141, 12717–12723. [Google Scholar] [CrossRef]
- Karapinar, D.; Huan, N.T.; Ranjbar Sahraie, N.; Li, J.; Wakerley, D.; Touati, N.; Zanna, S.; Taverna, D.; Galvão Tizei, L.H.; Zitolo, A.; et al. Electroreduction of CO2 on Single-Site Copper-Nitrogen-Doped Carbon Material: Selective Formation of Ethanol and Reversible Restructuration of the Metal Sites. Angew. Chem. Int. Ed. 2019, 58, 15098–15103. [Google Scholar] [CrossRef]
- Zhao, K.; Nie, X.; Wang, H.; Chen, S.; Quan, X.; Yu, H.; Choi, W.; Zhang, G.; Kim, B.; Chen, J.G. Selective electroreduction of CO2 to acetone by single copper atoms anchored on N-doped porous carbon. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Shang, H.; Wang, T.; Pei, J.; Jiang, Z.; Zhou, D.; Wang, Y.; Li, H.; Dong, J.; Zhuang, Z.; Chen, W.; et al. Design of a Single-Atom Indiumδ+–N4 Interface for Efficient Electroreduction of CO2 to Formate. Angew. Chem. 2020, 132, 22651–22655. [Google Scholar] [CrossRef]
- Jiang, Z.; Wang, T.; Pei, J.; Shang, H.; Zhou, D.; Li, H.; Dong, J.; Wang, Y.; Cao, R.; Zhuang, Z.; et al. Discovery of main group single Sb-N4 active sites for CO2 electroreduction to formate with high efficiency. Energy Environ. Sci. 2020, 13, 2856–2863. [Google Scholar] [CrossRef]
- Yang, X.; Chen, Y.; Qin, L.; Wu, X.; Wu, Y.; Yan, T.; Geng, Z.; Zeng, J. Boost Selectivity of HCOO− Using Anchored Bi Single Atoms towards CO2 Reduction. ChemSusChem 2020, 13, 6307–6311. [Google Scholar] [CrossRef]
- Han, L.; Song, S.; Liu, M.; Yao, S.; Liang, Z.; Cheng, H.; Ren, Z.; Liu, W.; Lin, R.; Qi, G.; et al. Stable and Efficient Single-Atom Zn Catalyst for CO2 Reduction to CH4. J. Am. Chem. Soc. 2020, 142, 12563–12567. [Google Scholar] [CrossRef]
- Lu, X.L.; Rong, X.; Zhang, C.; Lu, T.B. Carbon-based single-atom catalysts for CO2 electroreduction: Progress and optimization strategies. J. Mater. Chem. A 2020, 8, 10695–10708. [Google Scholar] [CrossRef]
- Beniya, A.; Higashi, S. Towards dense single-atom catalysts for future automotive applications. Nat. Catal. 2019, 2, 590–602. [Google Scholar] [CrossRef]
- Soloducho, J.; Cabaj, J.; Zajac, D. Advances in Electrode Materials; Scrivener Publishing LLC: Beverly, MA, USA, 2016; pp. 1–26. [Google Scholar] [CrossRef]
- Nursanto, E.B.; Jeon, H.S.; Kim, C.; Jee, M.S.; Koh, J.H.; Hwang, Y.J.; Min, B.K. Gold catalyst reactivity for CO2 electro-reduction: From nano particle to layer. Catal. Today 2016, 260, 107–111. [Google Scholar] [CrossRef]
- Kemp, R.; Dickie, D.; Donovan, E.S.; Barry, B. Carbon dioxde transformation facilitated by earth abundant metals. U.S. Patent 10,131,996, 20 November 2018. Available online: https://patents.google.com/patent/US20170016126A1/en (accessed on 19 October 2020).
- Yang, Z.; Oropeza, F.E.; Zhang, K.H.L. P-block metal-based (Sn, In, Bi, Pb) electrocatalysts for selective reduction of CO2 to formate. APL Mater. 2020, 8, 60901. [Google Scholar] [CrossRef]
- Zhang, D.; Tao, Z.; Feng, F.; He, B.; Zhou, W.; Sun, J.; Xu, J.; Wang, Q.; Zhao, L. High efficiency and selectivity from synergy: Bi nanoparticles embedded in nitrogen doped porous carbon for electrochemical reduction of CO2 to formate. Electrochim. Acta 2020, 334. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S.; Winè, G.; Gangeri, M. Electrocatalytic conversion of CO2 to long carbon-chain hydrocarbons. Green Chem. 2007, 9, 671–678. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, S.; Pastor, E.; Lázaro, M.J. Noble metal-free catalysts supported on carbon for CO2 electrochemical reduction. J. CO2 Util. 2017, 18, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; Kattel, S.; Yao, S.; Yan, B.; Liang, Z.; Hawxhurst, C.J.; Wu, Q.; Chen, J.G. Electrochemical reduction of CO2 to synthesis gas with controlled CO/H2 ratios. Energy Environ. Sci. 2017, 10, 1180–1185. [Google Scholar] [CrossRef]
- Xie, H.; Chen, S.; Ma, F.; Liang, J.; Miao, Z.; Wang, T.; Wang, H.L.; Huang, Y.; Li, Q. Boosting Tunable Syngas Formation via Electrochemical CO2 Reduction on Cu/In2O3 Core/Shell Nanoparticles. ACS Appl. Mater. Interfaces 2018, 10, 36996–37004. [Google Scholar] [CrossRef]
- Silva, W.O.; Silva, G.C.; Webster, R.F.; Benedetti, T.M.; Tilley, R.D.; Ticianelli, E.A. Electrochemical Reduction of CO2 on Nitrogen-Doped Carbon Catalysts With and Without Iron. ChemElectroChem 2019, 6, 4626–4636. [Google Scholar] [CrossRef]
- Franco, F.; Rettenmaier, C.; Jeon, H.S.; Roldan Cuenya, B. Transition metal-based catalysts for the electrochemical CO2 reduction: From atoms and molecules to nanostructured materials. Chem. Soc. Rev. 2020, 49, 6884–6946. [Google Scholar] [CrossRef]
- Kim, C.; Jeon, H.S.; Eom, T.; Jee, M.S.; Kim, H.; Friend, C.M.; Min, B.K.; Hwang, Y.J. Achieving Selective and Efficient Electrocatalytic Activity for CO2 Reduction Using Immobilized Silver Nanoparticles. J. Am. Chem. Soc. 2015, 137, 13844–13850. [Google Scholar] [CrossRef]
- Jia, M.; Choi, C.; Wu, T.S.; Ma, C.; Kang, P.; Tao, H.; Fan, Q.; Hong, S.; Liu, S.; Soo, Y.L.; et al. Carbon-supported Ni nanoparticles for efficient CO2 electroreduction. Chem. Sci. 2018, 9, 8775–8780. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Mao, F.; Zheng, L.R.; Wang, H.F.; Yang, X.H.; Yang, H.G. Tuning Metal Catalyst with Metal-C3N4 Interaction for Efficient CO2 Electroreduction. ACS Catal. 2018, 8, 11035–11041. [Google Scholar] [CrossRef]
- Zhao, S.; Tang, Z.; Guo, S.; Han, M.; Zhu, C.; Zhou, Y.; Bai, L.; Gao, J.; Huang, H.; Li, Y.; et al. Enhanced Activity for CO2 Electroreduction on a Highly Active and Stable Ternary Au-CDots-C3N4 Electrocatalyst. ACS Catal. 2018, 8, 188–197. [Google Scholar] [CrossRef]
- Zhang, S.; Kang, P.; Meyer, T.J. Nanostructured Tin Catalysts for Selective Electrochemical Reduction of Carbon Dioxide to Formate. J. Am. Chem. Soc. 2014, 136, 1734–1737. [Google Scholar] [CrossRef]
- Song, Y.; Peng, R.; Hensley, D.K.; Bonnesen, P.V.; Liang, L.; Wu, Z.; Meyer, H.M.; Chi, M.; Ma, C.; Sumpter, B.G.; et al. High-Selectivity Electrochemical Conversion of CO2 to Ethanol using a Copper Nanoparticle/N-Doped Graphene Electrode. ChemistrySelect 2016, 1, 6055–6061. [Google Scholar] [CrossRef] [Green Version]
- Legrand, U.; Boudreault, R.; Meunier, J.L. Decoration of N-functionalized graphene nanoflakes with copper-based nanoparticles for high selectivity CO2 electroreduction towards formate. Electrochim. Acta 2019, 318, 142–150. [Google Scholar] [CrossRef]
- Rogers, C.; Perkins, W.S.; Veber, G.; Williams, T.E.; Cloke, R.R.; Fischer, F.R. Synergistic Enhancement of Electrocatalytic CO2 Reduction with Gold Nanoparticles Embedded in Functional Graphene Nanoribbon Composite Electrodes. J. Am. Chem. Soc. 2017, 139, 4052–4061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Yang, M.P.; Zhi, W.Y.; Wang, H.; Wang, H.; Lu, J.X. Efficient electrochemical reduction of CO2 to ethanol on Cu nanoparticles decorated on N-doped graphene oxide catalysts. J. CO2 Util. 2019, 33, 452–460. [Google Scholar] [CrossRef]
- Ning, H.; Mao, Q.; Wang, W.; Yang, Z.; Wang, X.; Zhao, Q.; Song, Y.; Wu, M. N-doped reduced on graphene oxide supported Cu2O nanocubes as high active catalyst for CO2 electroreduction to C2H4. J. Alloys Compd. 2019, 785, 7–12. [Google Scholar] [CrossRef]
- Zeng, L.; Shi, J.; Luo, J.; Chen, H. Silver sulfide anchored on reduced graphene oxide as a high -performance catalyst for CO2 electroreduction. J. Power Sources 2018, 398, 83–90. [Google Scholar] [CrossRef]
- Sun, J.; Zheng, W.; Lyu, S.; He, F.; Yang, B.; Li, Z.; Lei, L.; Hou, Y. Bi/Bi2O3 nanoparticles supported on N-doped reduced graphene oxide for highly efficient CO2 electroreduction to formate. Chin. Chem. Lett. 2020, 31, 1415–1421. [Google Scholar] [CrossRef]
- Hossain, M.N.; Wen, J.; Chen, A. Unique copper and reduced graphene oxide nanocomposite toward the efficient electrochemical reduction of carbon dioxide. Sci. Rep. 2017, 7, 3184. [Google Scholar] [CrossRef] [Green Version]
- Genovese, C.; Ampelli, C.; Perathoner, S.; Centi, G. Electrocatalytic conversion of CO2 on carbon nanotube-based electrodes for producing solar fuels. J. Catal. 2013, 308, 237–249. [Google Scholar] [CrossRef]
- Xu, C.; Vasileff, A.; Jin, B.; Wang, D.; Xu, H.; Zheng, Y.; Qiao, S.Z. Graphene-encapsulated nickel-copper bimetallic nanoparticle catalysts for electrochemical reduction of CO2 to CO. Chem. Commun. 2020, 56, 11275–11278. [Google Scholar] [CrossRef]
- Wei, B.; Xiong, Y.; Zhang, Z.; Hao, J.; Li, L.; Shi, W. Efficient electrocatalytic reduction of CO2 to HCOOH by bimetallic In-Cu nanoparticles with controlled growth facet. Appl. Catal. B Environ. 2021, 283, 119646. [Google Scholar] [CrossRef]
- Safdar Hossain, S.; Rahman, S.U.; Ahmed, S. Electrochemical reduction of carbon dioxide over CNT-supported nanoscale copper electrocatalysts. J. Nanomater. 2014, 2014, 374318. [Google Scholar] [CrossRef]
- Zhang, R.; Lv, W.; Li, G.; Lei, L. Electrochemical reduction of CO2 on SnO2/nitrogen-doped multiwalled carbon nanotubes composites in KHCO3 aqueous solution. Mater. Lett. 2015, 141, 63–66. [Google Scholar] [CrossRef]
- Zheng, W.; Guo, C.; Yang, J.; He, F.; Yang, B.; Li, Z.; Lei, L.; Xiao, J.; Wu, G.; Hou, Y. Highly active metallic nickel sites confined in N-doped carbon nanotubes toward significantly enhanced activity of CO2 electroreduction. Carbon 2019, 150, 52–59. [Google Scholar] [CrossRef]
- Wang, T.; Yang, J.; Chen, J.; He, Q.; Li, Z.; Lei, L.; Lu, J.; Leung, M.K.H.; Yang, B.; Hou, Y. Nitrogen-doped carbon nanotube-encapsulated nickel nanoparticles assembled on graphene for efficient CO2 electroreduction. Chin. Chem. Lett. 2020, 31, 1438–1442. [Google Scholar] [CrossRef]
- Lee, M.Y.; Ringe, S.; Kim, H.; Kang, S.; Kwon, Y. Electric field mediated selectivity switching of electrochemical CO2 reduction from formate to CO on carbon supported Sn. ACS Energy Lett. 2020, 5, 2987–2994. [Google Scholar] [CrossRef]
- Maldonado-Hódar, F.J.; Moreno-Castilla, C.; Pérez-Cadenas, A.F. Surface morphology, metal dispersion, and pore texture of transition metal-doped monolithic carbon aerogels and steam-activated derivatives. Microporous Mesoporous Mater. 2004, 69, 119–125. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; Maldonado-Hódar, F.J.; Pérez-Cadenas, A.F. Physicochemical Surface Properties of Fe, Co, Ni, and Cu-Doped Monolithic Organic Aerogels. Langmuir 2003, 19, 5650–5655. [Google Scholar] [CrossRef]
- Maldonado-Hódar, F.J.; Moreno-Castilla, C.; Rivera-Utrilla, J.; Hanzawa, Y.; Yamada, Y. Catalytic Graphitization of Carbon Aerogels by Transition Metals. Langmuir 2000, 16, 4367–4373. [Google Scholar] [CrossRef]
- Gallegos-Suárez, E.; Pérez-Cadenas, A.F.; Maldonado-Hódar, F.J.; Carrasco-Marín, F. On the micro- and mesoporosity of carbon aerogels and xerogels. The role of the drying conditions during the synthesis processes. Chem. Eng. J. 2012, 181–182, 851–855. [Google Scholar] [CrossRef]
- Pérez-Cadenas, A.F.; Moreno-Castilla, C.; Carrasco-Marin, F.; Maldonado-Hódar, F.; Morales-Torres, S.; Kapteijn, F. Dodep Carbon Material for the Electrocatalytic Conversion of CO2 into Hydrocarbons, Uses pf the Material and Conversion Method Using Said. Material. Patent No. WO2013004882, 10 January 2013. [Google Scholar]
- Pérez-Cadenas, A.F.; Ros, C.H.; Morales-Torres, S.; Pérez-Cadenas, M.; Kooyman, P.J.; Moreno-Castilla, C.; Kapteijn, F. Metal-doped carbon xerogels for the electro-catalytic conversion of CO2 to hydrocarbons. Carbon 2013, 56, 324–331. [Google Scholar] [CrossRef]
- Abdelwahab, A.; Castelo-Quibén, J.; Pérez-Cadenas, M.; Elmouwahidi, A.; Maldonado-Hódar, F.J.; Carrasco-Marín, F.; Pérez-Cadenas, A.F. Cobalt-doped carbon gels as electro-catalysts for the reduction of CO2 to hydrocarbons. Catalysts 2017, 7, 25. [Google Scholar] [CrossRef] [Green Version]
- Castelo-Quibén, J.; Abdelwahab, A.; Pérez-Cadenas, M.; Morales-Torres, S.; Maldonado-Hódar, F.J.; Carrasco-Marín, F.; Pérez-Cadenas, A.F. Carbon-iron electro-catalysts for CO2 reduction. The role of the iron particle size. J. CO2 Util. 2018, 24, 240–249. [Google Scholar] [CrossRef]
- Castelo-Quibén, J.; Elmouwahidi, A.; Maldonado-Hódar, F.J.; Carrasco-Marín, F.; Pérez-Cadenas, A.F. Metal-carbon-CNF composites obtained by catalytic pyrolysis of urban plastic residues as electro-catalysts for the reduction of CO2. Catalysts 2018, 8, 198. [Google Scholar] [CrossRef] [Green Version]
- Castelo-Quibén, J.; Bailón-García, E.; Pérez-Fernández, F.J.; Carrasco-Marín, F.; Pérez-Cadenas, A.F. Mesoporous carbon nanospheres with improved conductivity for electro-catalytic reduction of O2 and CO2. Carbon 2019, 155, 88–99. [Google Scholar] [CrossRef]
- Ripatti, D.S.; Veltman, T.R.; Kanan, M.W. Carbon Monoxide Gas Diffusion Electrolysis that Produces Concentrated C2 Products with High Single-Pass Conversion. Joule 2019, 3, 240–256. [Google Scholar] [CrossRef] [Green Version]
- Messias, S.; da Ponte, M.; Reis-Machado, S.A. Carbon Materials as Cathode Constituents for Electrochemical CO2 Reduction—A Review. J. Carbon Res. 2019, 5, 83. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Luo, R.; Gold, J.I.; Yu, A.Z.; Kim, B.; Kenis, P.J.A. Carbon nanotube containing Ag catalyst layers for efficient and selective reduction of carbon dioxide. J. Mater. Chem. A 2016, 4, 8573–8578. [Google Scholar] [CrossRef]
- Daiyan, R.; Lu, X.; Tan, X.; Zhu, X.; Chen, R.; Smith, S.C.; Amal, R. Antipoisoning nickel-carbon electrocatalyst for practical electrochemical CO2 reduction to CO. ACS Appl. Energy Mater. 2019, 2, 8002–8009. [Google Scholar] [CrossRef]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reduction Reactions | ||
|---|---|---|
| 2CO2(g) + 2H+ + 2e−H2C2O4 | +91.8 | −0.475 |
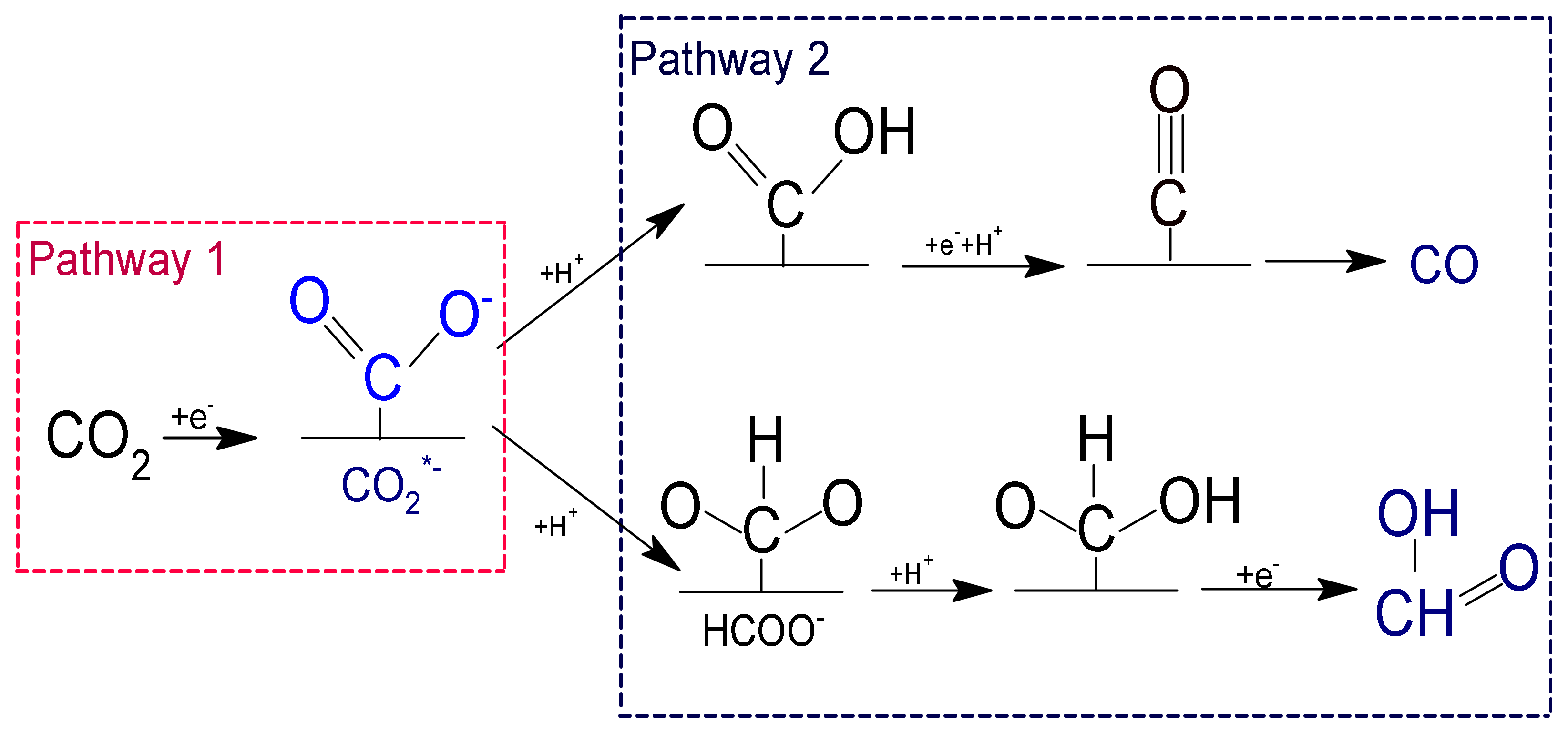
| CO2(g) + 2H+ + 2e−HCOOH(aq) | +38.4 | −0.199 |
| CO2(g) + 2H+ + 2e−CO(g) + H2O | +19.9 | −0.103 |
| CO2(g) + 4H+ + 4e−HCHO(aq) + H2O | +27.5 | −0.071 |
| CO2(g) + 6H+ + 6e−CH3OH(aq) + H2O | −17.3 | +0.030 |
| CO2(g) + 8H+ + 8e−CH4(g) + 2H2O | −130.8 | +0.169 |
| Sample | Main Product | Carbon Precursor | N Precursor | Type a | Synthesis Method | Nitrogen Species (% atomic) XPS | Active site | Ref | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N b | Pyri c | Pyrr d | G e | O f | ||||||||
| NR/CS-900 | CO | CS (porous carbon nanosheets) | Polymerized Aniline with Ammonium Persistence to Polyaniline (Solid) | Post | Activation (ZnCl2) and pyrolysis | 5.30 | 1.45 | 1.05 | 2 | 0.8 | Pyridinic | [31] |
| NCNTs | CO | CNTs | Acetonitrile- dicyandiamide | In situ | Liquid vapor deposition (CVD) | 5.0 | 1.5 | 1.1 | 2.4 | n.d. | Pyridinic | [32] |
| WNCNs-1000 | CO | Coal, NaCl template method C-700 | NH3 | Post | Pyrolysis | 4.3 | 2.61 | 1.45 | 0.16 | 0.08 | Pyridinic | [33] |
| NCNTs-ACN-850 | CO | CNTs (Acetonitrile) | Dicyandiamide | In situ | Liquid chemical vapor deposition | 4.9 | 1 | 0.5 | 3.4 | n.d. | Pyridinic | [21] |
| CN/MWCNT | CO | MWCNT | NaN3 reacts with C3N2Cl3 to form g-C3N4 | Post | Pyrolysis | 0.12 | n.d. | n.d. | n.d. | n.d. | n.d. | [34] |
| NCNT-3-700 | CO | CNTs | Poly(diallyldimethylammonium chloride) (PDDA) | Post | Pyrolysis | 1.75 | 0.5 | 0.625 | 0.5 | 0.125 | Graphitic | [35] |
| CNPC-1100 | CO | Coal | NH3 | Post | Ammonia etching/pyrolysis | 4 | 1 | 1 | 1.5 | 0.5 | n.d. | [36] |
| NG-800 | CO | GF (Graphene foam) Ni-foam vapor deposition | gC3N4 (Solid) | Post | Pyrolysis | 6.6 | 4.5 | 1.5 | 0.6 | n.d. | Pyridinic | [37] |
| MNC-D | CO | ZIF-8 | Dimethyformamide DMF | Post | Heat-treated | 16.97 | 8 | 5 | 4 | n.d. | Pyridinic | [38] |
| NPC-900 | CO | Coal arantracite | Dicyandiamide (DICY) | In situ | Activation (KOH)/Pyrolysis | 1.92 | 0.60 | 0.50 | 0.83 | n.d. | n.d. | [27] |
| N-CWM | CO | Natural wood | Urea | Post | Pyrolysis | 4.07 | 0.54 | 2.90 | 0.55 | 0.09 | Pyridinic | [39] |
| N-graphene | Formate | Graphene oxide | Melamine (Solid) | In situ | Pyrolysis | 5.5 | 3 | 0.9 | 1.6 | n.d. | Pyridinic | [40] |
| PEI-NCNT/GC | Formate | Multiwalled CNT | Ammonia plasma/co-catalyst (PEI) | Post | Ammonia plasma enhanced chemical vapor deposition (PECVD) | 11.3 | n.d. | n.d. | n.d. | n.d. | n.d. | [41] |
| HNCM/CNT | Formate | CNT | Dimethyformamide DMF | In situ | Pyrolysis | 8.26 | 3.5 | n.d. | 4.8 | n.d. | Pyridinic | [42] |
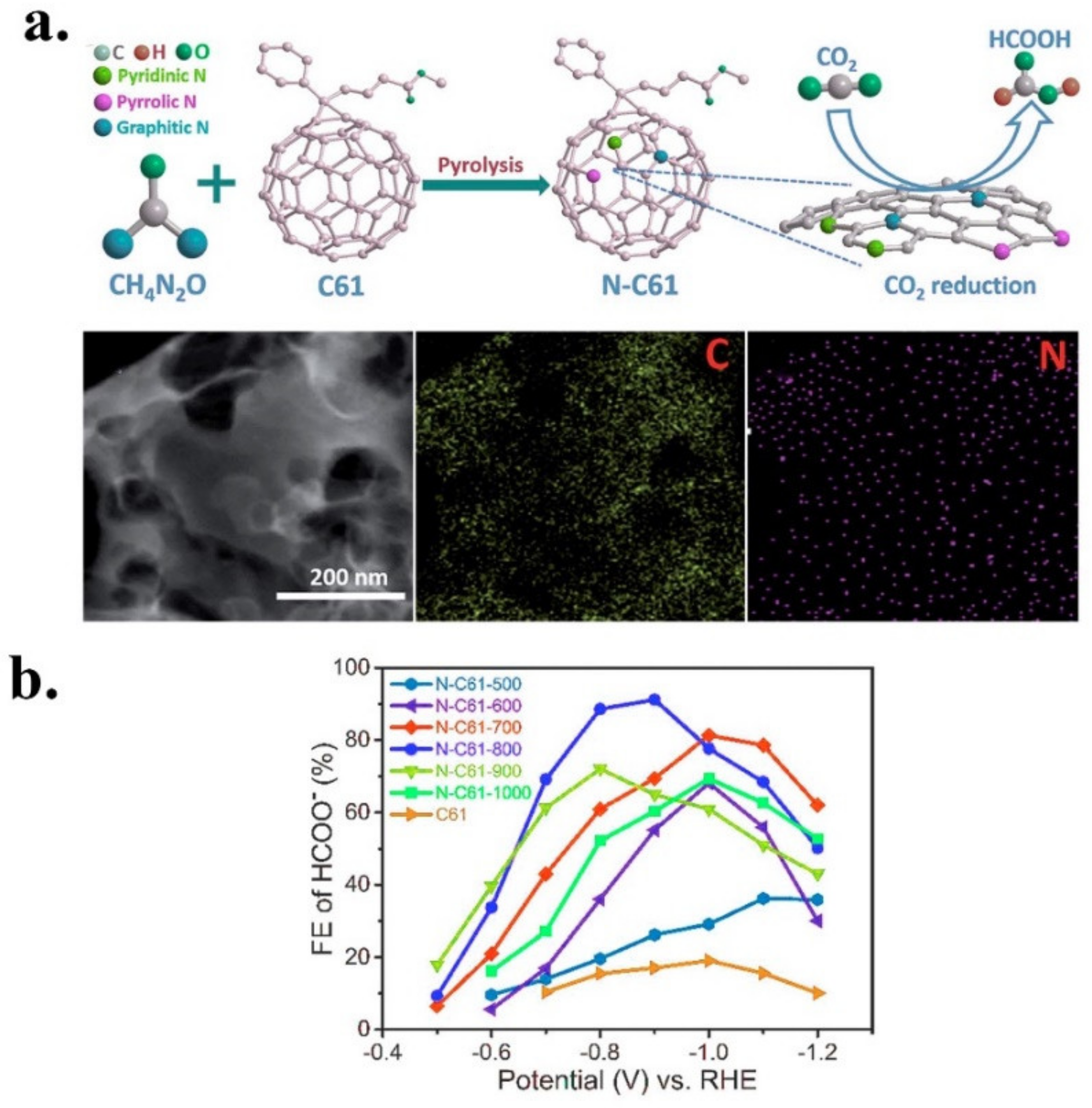
| N-C61-800 | Formate | Fullerene PC61BM | Urea | In situ | Pyrolysis | 2.57 | 0.39 | 0.54 | 1.64 | n.d | Graphitic | [43] |
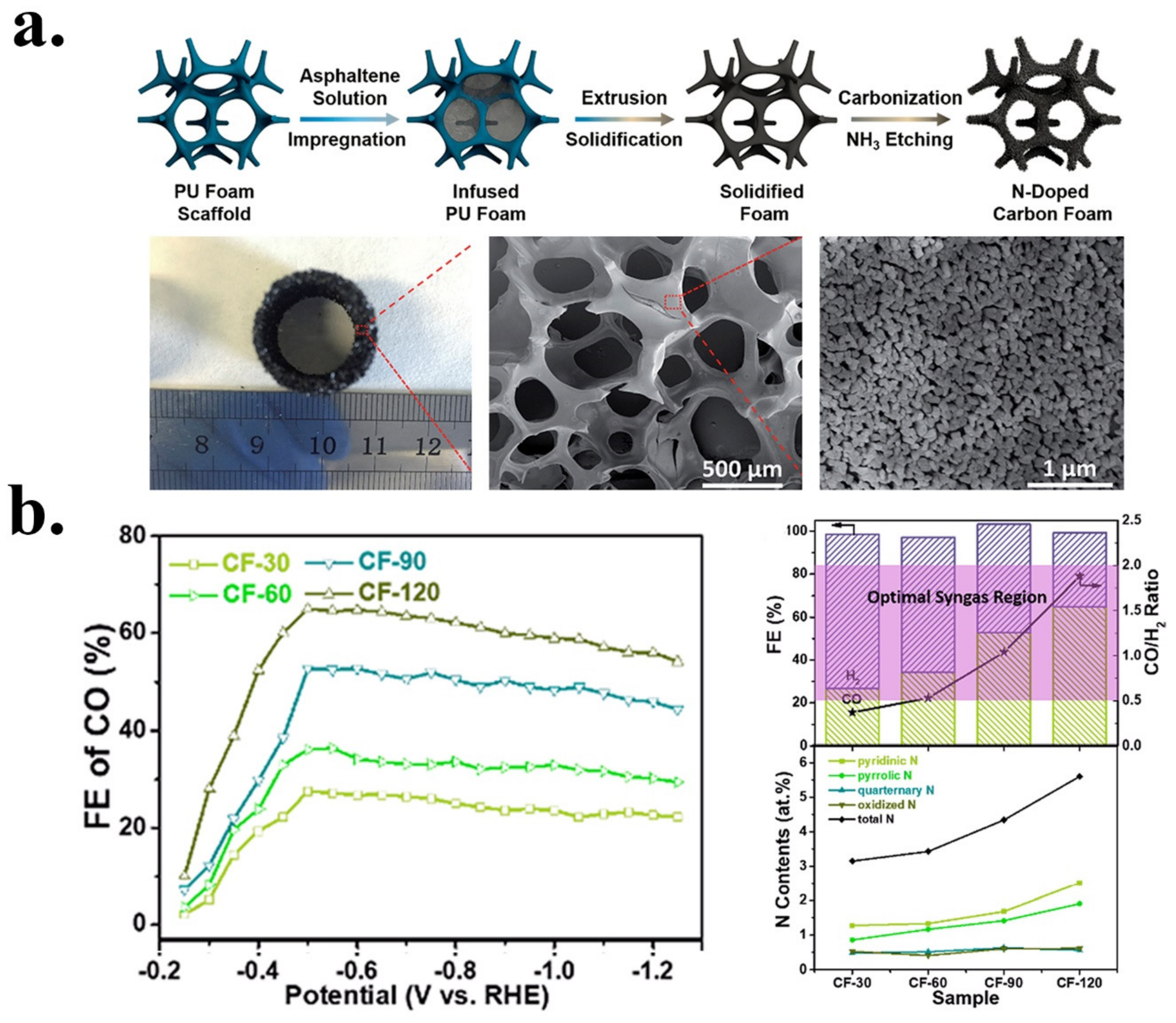
| CF-120 | Syngas | CF (carbon foam)PU | NH3 | Post | Ammonia etching/pyrolysis | 5.5 | 2.5 | 1.8 | 0.6 | 0.6 | Pyridinic/Pyrrolic | [44] |
| 3D N-CNTs/SS-750 | Syngas | Melamine | Melamine | In situ | Pyrolysis | 6.8 | 3.90 | 0.50 | 2.50 | n.d. | n.d. | [45] |
| c-NC | Ethanol | Resol | Dicyandiamide | In situ | Pyrolysis/Template method-mesoporous materials | 7 | 2.6 | 2.8 | 1.6 | n.d. | Pyridinic/Pyrrolic | [46] |
| MNCs-5 | Ethanol | Resol | Dicyandiamide | In situ | Pyrolysis/triblock-copolymer-templating method. | ~6.2 | 2.1 | 2.8 | 1.3 | n.d. | Pyridinic/Pyrrolic | [47] |
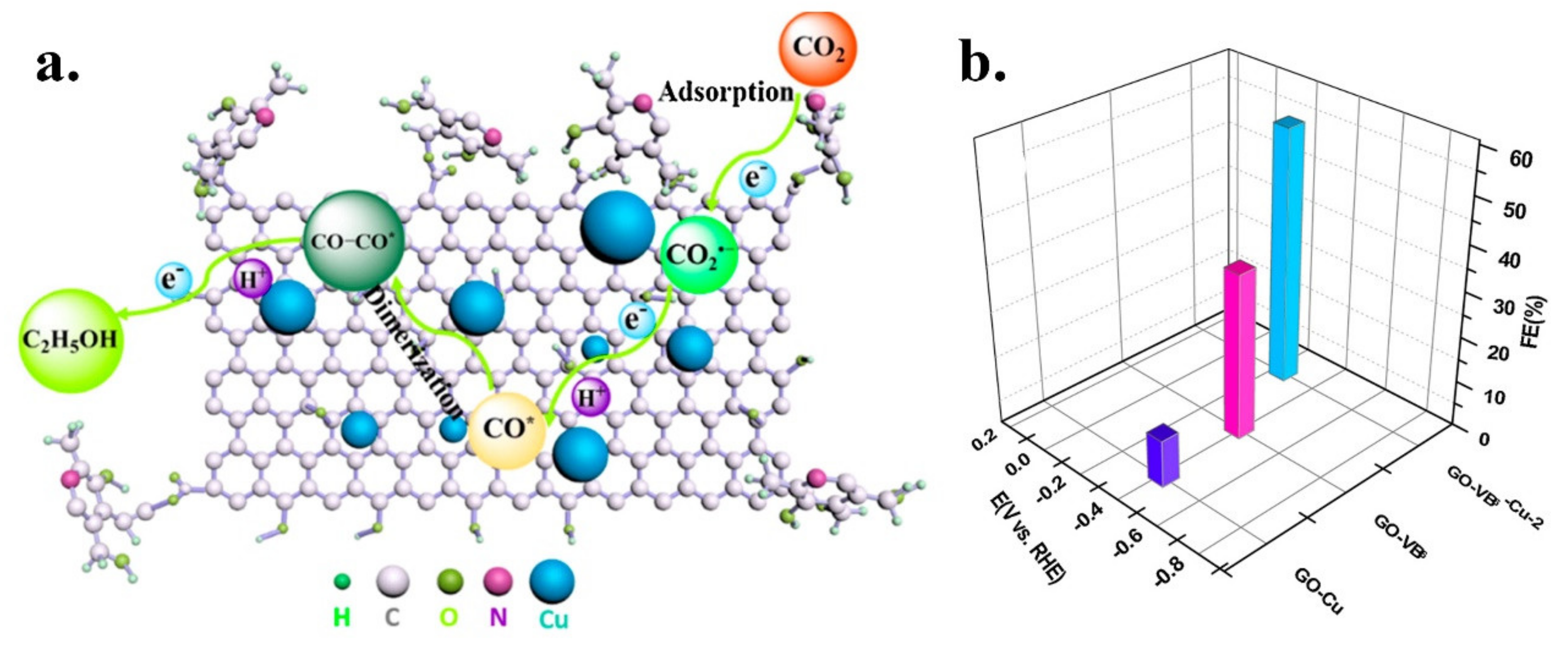
| GO-VB6-4 | Ethanol-Acetone | Graphene oxide | B6 Vitamin | Post | heat treatment | 2.3 | 2.3 | n.d. | n.d. | n.d. | Pyridinic | [48] |
| NGM-1/CP | CH4 | Graphene | 3-Pyridinecarbonitrile | In situ | Pyrolysis | 6.52 | 1.45 | 3.35 | 1.72 | n.d. | Pyridinic/Pyrrolic | [49] |
| Sample | Catalysts Material | Pore Structure | FE(co) % | Jco [mA/cm2] | Nitrogen Species (% atomic) XPS | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| N Total | Pyridinic | Pyrrolic | Graphitic | Oxide | ||||||
| CNPC-1100 | Carbon Coal | Microporous-Mesoporous-Macro porous | 92 at −0.6 V vs. RHE | 0.3 at −0.6 V vs. RHE | 3.90 | 0.95 | 1.13 | 1.27 | 0.55 | [36] |
| MNC-D | ZIF-8 | Mesoporous | 92 at −0.58 V vs. RHE | −6.8 at −0.58 V vs. RHE | 16.97 | 8.00 | 5.00 | 4.00 | n.d. | [38] |
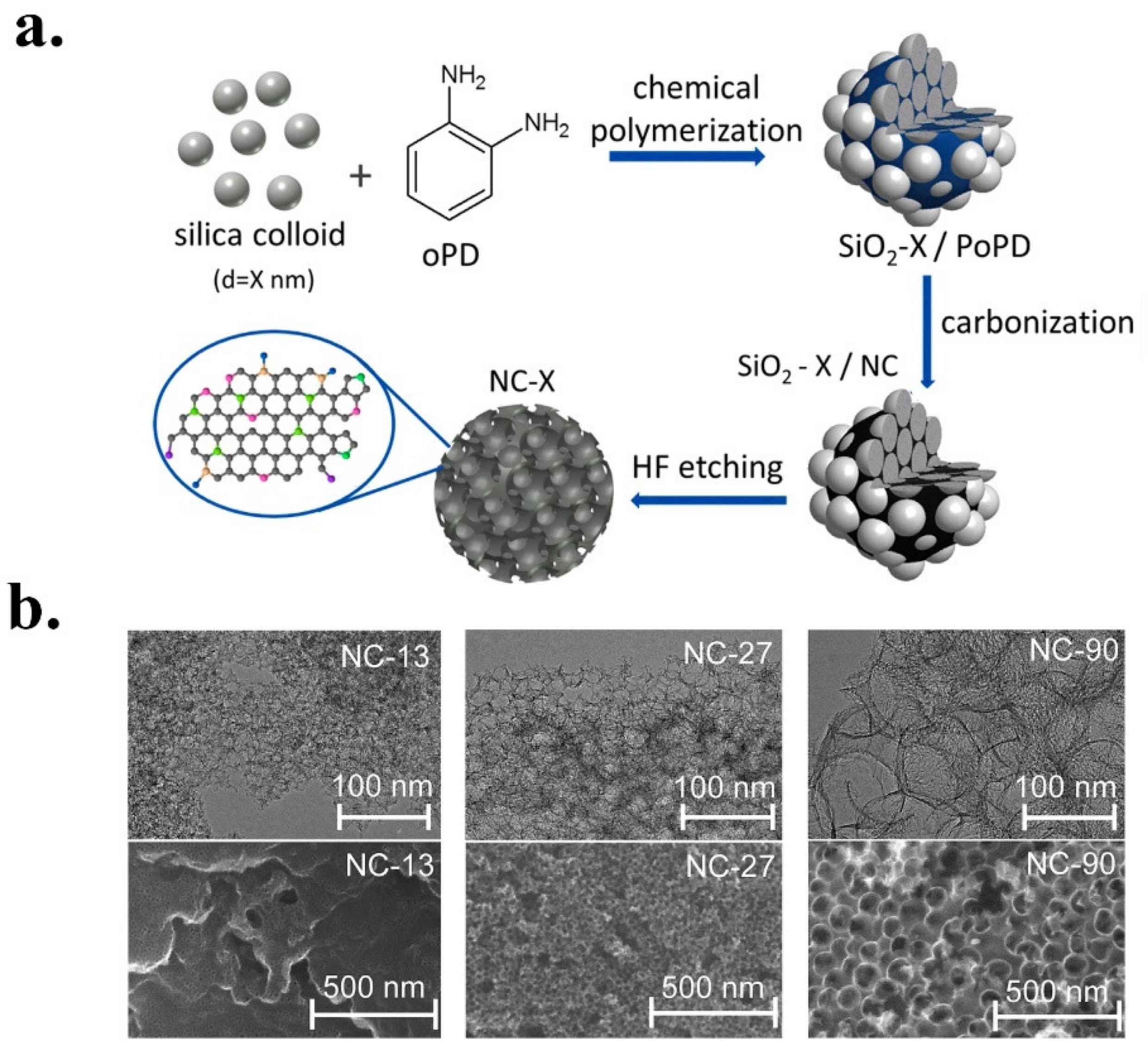
| NC-27 | oPD (o-phenylenediamine) | Mesoporous- Macro pores | 75 at −0.71 V vs. RHE | −1.53 at 0.71 V vs. RHE | 8.43 | 2.25 | n.d. | n.d. | n.d. | [66] |
| NPC-900 | Carbon Coal | Microporous | 95 at −0.67 V vs. RHE | −2.3 at −0.67 V vs. RHE | 1.92 | 0.60 | 0.50 | 0.83 | n.d. | [27] |
| Sample | Main Product | DopingAtom | Carbon Precursor | Heteroatom Precursor | Doping Type | Synthesis Method | Active Site/Heteroatom Content | Ref |
|---|---|---|---|---|---|---|---|---|
| BG | Formate | B | graphene oxide | Boric acid | In situ | Direct solid state/thermal annealing | B y C atoms/4.1 at % (XPS) | [75] |
| BDD | Methanol | B | Acetone | Trimethosyborane B(OCH3)3 | In situ | Plasma assisted chemical vapor deposition | Bicarbonate ions/B/C ratio 1% | [79] |
| BDD | Formaldehyde | B | Acetone | B(OCH3) | In situ | Microwave plasma assisted chemical vapor deposition (MPVCD) | Sp3- bonded carbon/B/C atomic ratio 1.0 w/w | [78] |
| BDD | HCOOH | B | Acetone | Trimethosyborane B(OCH3)3 | In situ | Microwave plasma assisted chemical vapor deposition (MPVCD) | n.d. | [80] |
| BDD | HCOOH | B | Acetone | Trimethosyborane B(OCH3)3 | In situ | Microwave plasma assisted chemical vapor deposition (MPVCD) | B/C atomic ratio 1 w/w | [81] |
| BDD | HCOOH | B | CH4 | Trimethylboron | In situ | Microwave plasma assisted chemical vapor deposition (MPVCD) | n.d. | [82] |
| BDD-0.01% | HCOOH | B | CH4 | Trimethosyborane B(OCH3)3 | In situ | Microwave plasma assisted chemical vapor deposition (MPVCD) | B atoms/B/C ratio 0.01% | [83] |
| BDD | HCOOH | B | CH4 | Trimethylboron | In situ | Microwave plasma assisted chemical vapor deposition (MPVCD) | B atoms/B/C ratio 1% and 0.1% | [84] |
| BDD | HCOOH | B | CH4 | Trimethosyborane B(OCH3)3 | In situ | Microwave plasma assisted chemical vapor deposition (MPVCD) | B/C ratio 0.07% | [85] |
| FC | CO | F | BP 2000 (commercial carbon nanomaterial) | Polytetrafluoroethylene (PTFE) | In situ | Pyrolysis | C atoms close to CF2 bonds/0.32% Atomic | [86] |
| F-CPC | CO | F | Resorcinol and formaldehyde (Aldol reaction) | Polytetrafluoroethylene (PTFE) | In situ | Template method (SiO2) | Semi-ionic C-F bond/0.65% atomic | [87] |
| P-OLC-IP | CO | P | Onion-like carbon (nanodiamond power) | Diammonium hydrogen Phosphate (NH4)2HPO4 | Post-doping | Impregnation | P-O/2.8% atomic | [88] |
| P-OLC-CVD | CO | P | Onion-like carbon (nanodiamond power) | Triphenylphosphine (TPP) | Post-doping | Chemical vapor deposition CVD | P-C/3.8% atomic | [88] |
| Sample | Main Product | Heteroatoms | Carbon Precursor | Heteroatom Precursor | Synthesis Method | Active Site | Ref |
|---|---|---|---|---|---|---|---|
| NS-CNSs | CO | N, S | Iron oleate | N (urea), S (Na2SO4) | Pyrolysis | N-pyridinic | [91] |
| NS-C | CO | N, S | Citric acid | N, S (Thiourea) | Pyrolysis | N-pyridinic | [92] |
| NPC | CO | N, P | Glucose anhydrous | N(urea), P(phytic acid) | Pyrolysis | P-N-C- | [93] |
| NPCM | CO | N, P | Aniline monomer | N (Aniline monomer), P (Phytic acid) | Pyrolysis | N-pyridinic N-Pyrrolic | [94] |
| NBPC | CO | N, B | Glucose | N (ammonia chloride NH4Cl), B (boric acid H3BO3) | nitrogen-assisted freeze/Pyrolysis | N-pyridinic | [95] |
| BND | Ethanol | B, N | CH4 | B2H6/N2 | Chemical vapor deposition | n.d. | [96] |
| Electrode | Hetero-Atom | Sample | Electrolyte | Product | FE % | J [mA/cm2] | Potential Cathode | Onset Potential/Overpotential | Stability vs. (RHE) | Tafel Slope [mV/dec] | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1D/2D N-doped carbon nanorod arrays/ultrathin carbon nanosheets | N | NR/CS-900 | 0.5 M KHCO3 | CO | 94.2 | 3.78 | −0.45 V vs. RHE | −0.207 V vs. RHE/97 mV | 30 h at −0.45 V | 65 | [31] |
| Metal-free N-doped graphene | N | N-graphene | 0.5 M KHCO3 | Formate | 73 | 7.50 | −0.84 V vs. RHE | −0.3 V vs. RHE/0.78 V | 12 h at −0.84 V | 135 | [40] |
| N-doped carbon nanotubes | N | PEI-NCNT | 0.1 M KHCO3 | Formate | 85 | 7.20 | −1.85 V vs. SCE | −1.2 V vs. SCE | 24 h at −1.8 V | 134 | [41] |
| N-doped carbon nanotubes | N | NCNTs | 0.1 M KHCO3 | CO | 80 | ~1.0 | −0.78 V vs. SHE | −0.70 V vs. SHE/0.18V | 10 h at −0.8 V | 203 | [32] |
| 2D N-doped porous carbon nanosheets (coal tar pitch) | N | WNCNs-1000 | 0.1 M KHCO3 | CO | 84 | 1.15 | −0.6 V vs. RHE | −0.3 V vs. RHE/0.19 V | 8 h at −0.6 V | 132 | [33] |
| Metal-free N functionalized graphene oxide | N | GO-VB6-4 | 0.1 M KHCO3 | Ethanol/Acetone | 36.4/8.9 | 0.75 | −0.4 V vs. RHE | n.d. | n.d. | n.d. | [48] |
| N-doped carbon nanotubes (NCNTs) | N | NCNTs-ACN-850 | 0.1 M KHCO3 | CO | 80 | ~ 4.2 | −1.05 V | −0.7 V/0.18 V | n.d. | 160 | [21] |
| N-doped multiwalled carbon nanotubes (MWCNTs) | N | CN/MWCNT | 1 M KCl | CO | 98 | 70 | −1.46 V vs. Ag/AgCl | ~−1.3 V vs. Ag/AgCl | n.d. | n.d. | [34] |
| NCNT | N | NCNT-3-700 | 0.5 M KHCO3 | CO | 90 | ~5.0 | −0.90 V vs. RHE | −0.40 V vs. RHE | 60h at 0.90 V | ~144.5 | [35] |
| N-doped carbon | N | CNPC-1100 | 0.1 M KHCO3 | CO | 92 | ~0.3 | −0.6 V vs. RHE | −0.4 V vs. RHE | 8 h at −0.6 V | ~163 | [36] |
| Carbon foam | N | CF -120 | 0.1 M KHCO3 | Syngas | ~60 CO35 H2 | ~10 | −0.6 V vs. RHE | −0.3 V vs. RHE/0.19V | 8 h at −0.6 V | 103 | [44] |
| Graphene foam | N | NG-800 | 0.1 M KHCO3 | CO | ~85 | ~1.8 | −0.58 V vs. RHE | −0.30 V vs. RHE/0.19V | 5 h at −0.58 V | 222 | [37] |
| Mesoporous carbon | N | MNC-D | 0.1 M KHCO3 | CO | 92 | 6.8 | −0.58 V vs. RHE | −0.28 V vs. RHE | 16 h at −0.58 V | 138 | [38] |
| Microporous carbon | N | NPC-900 | 0.5 M KHCO3 | CO | 95 | 2.3 | −0.67 V vs. RHE | −0.27 V vs. RHE | 10 h at −0.67 V | 198 | [27] |
| Hierarchically wood carbon | N | N-CWM | 0.1 M KHCO3 | CO | 78 | ~1.6 | −0.68 V vs. RHE | −0.43 V vs. RHE | 10 h at −0.68 V | 144 | [39] |
| 3D integrated N-CNTs on SS | N | N-CNTs/SS | 0.1 M KHCO3 | Syngas | ~75% CO ~25 H2 | ~1 | −1.1 V vs. RHE | −0.2 V vs. RHE/90mV | 8 h at −1.1 V | 160 | [45] |
| Plasma-treated CNTA (N-Doped Carbon Nanotube Arrays) | N | pCNTA10 | 0.5M NaHCO3 | Syngas | ~75% CO ~25 H2 | n.d. | −0.82 V vs. RHE | −0.4 V vs. RHE/0.29V | 10 h at −0.82 V | n.d. | [24] |
| N-doped carbon membrane/CNT carbon nanotubes | N | HNDCM/CNT | 0.1M KHCO3 | HCOO | 81 | ~11 | −0.8 V vs. RHE | −0.18 V vs. RHE | 36 h | 138 | [42] |
| PC61BM- N-C61 | N | N-C61-800 | 0.5M KHCO3 | HCOO | 91.2 | 11.6 | −0.9 V vs. RHE | −0.42 V vs. RHE | 12 h at −0.9 V vs. RHE | 146 | [43] |
| N-doped Meso-porous material | N | c-CN | 0.1M KHCO3 | Ethanol | 77 | n.d. | −0.56 V vs. RHE | n.d. | 6–24 h at −0.56 V vs. RHE | n.d. | [46] |
| N-doped Micro/Meso-porous material | N | MNCs-5 | 0.1M KHCO3 | Ethanol | 78 | n.d. | −0.56 V vs. RHE | −0.4 V vs. RHE | 24 h at −0.56 V vs. RHE | n.d. | [47] |
| N-doped graphene | N | NGM-1/CP | [Bmim]BF4 | CH4 | 90.1 | 3.26 | −1.4 V vs. SHE | n.d. | n.d. | n.d. | [49] |
| B-doped graphene | B | BG | 0.1M KHCO3 | HCOO | 66 | n.d. | −1.4 V vs. SCE | −1.4 V vs. SCE | n.d. | n.d. | [75] |
| Diamond doped with B | B | BDD | 0.1M NH3 | Methanol | 24.3 | n.d. | −1.3 V vs. Ag/AgCl | n.d. | n.d. | n.d. | [79] |
| Diamond doped with B | B | BDD | 0.1M MeOH | Formaldehyde | 74 | n.d. | −1.7 V vs. Ag/AgCl | n.d. | n.d. | n.d. | [78] |
| Diamond doped with B | B | BDD | 0.5M KCl | HCOOH | 94.7 | 2 | ~−2.3 V vs. Ag/AgCl | n.d. | 24 h | - | [80] |
| Diamond doped with B | B | BDD | 0.075M RbOH | HCOOH | 71% | 2 | ~−2.5 V vs. Ag/AgCl | n.d. | n.d. | n.d. | [81] |
| Diamond doped with B | B | BDD | 0.5M RbBr | HCOOH | 95% | 2 | n.d. | n.d. | n.d. | n.d. | [82] |
| Diamond doped with B | B | BDD-0.01% | 0.5M KCl | HCOOH | 75 | 2 | ~−2.3 V vs. Ag/AgCl | n.d. | n.d. | n.d. | [83] |
| Diamond doped with B | B | BDD | KClO4/KCl | CO/HCOOH | 36/87 | n.d. | −1.8 V vs. Ag/AgCl/−2.5 V vs. Ag/AgCl | n.d. | n.d. | n.d. | [84] |
| Diamond doped with B | B | BDD | 0.5 M KCl | HCOOH | ~80% | 2 | −2.2 V vs. Ag/AgCl | n.d. | n.d. | n.d. | [85] |
| Fluorine-doped Cage-like Carbon | F | F-CPC | n.d. | CO | 83.3 | 37.5 | −1.0 V vs. RHE | n.d. | 12 at −0.9 V vs. RHE | 130 | [87] |
| Fluorine-doped BD-2000 | F | FC | 0.1 M NaClO4 | CO | 89.6 | ~0.224 | −0.62 V vs. RHE | n.d. | n.d. | 81 | [86] |
| P-doped Onion-like carbon | P | P-OLC-IP | 0.5 NaHCO3 | CO | 60 | 0.5 | −1.0 V vs. RHE | −1.0 V vs. RHE | n.d. | n.d. | [88] |
| P-doped Onion-like carbon | P | P-OLC-CVD | 0.5 NaHCO3 | CO | 81 | 4.9 | −0.9 V vs. RHE | −0.65 V vs. RHE | 27 h at −0.9 V | 131 | [88] |
| N, S-doped Nanosheets | N,S | NS-CNSs | 0.5M KHCO3 | CO | 85 | 5.36 | −0.55 V vs. RHE | − | 20 h at −0.55 V | 260 | [91] |
| N, S-doped carbon/carbon paper | N,S | NS-C-900 | 0.1M KHCO3 | CO | 92 | 2.63 | −0.6 V vs. RHE | −0.4 V vs. RHE/290 mV | 21 h at −0.6 V | 94 | [92] |
| N, P-doped carbon/carbon paper | N,P | NPC | 0.1M KHCO3 | CO | 87 | 0.81 | −0.45 V vs. RHE | −0.25 V vs. RHE/140 mV | 20 h at −0.45 V | 72 | [93] |
| N, P-doped carbon/carbon paper | N,P | NPCM | 0.5 M NaHCO3 | CO | 92 | n.d. | −0.55 V vs. RHE | −0.38V vs. RHE/270mV | 24 h at −0.55 V | 122 | [94] |
| N, B-doped 3D hierarchical porous carbon/glassy carbon plate | N,B | NBPC | 0.5 M KHCO3 | CO | 83 | n.d. | −0.4 V vs. RHE | n.d. | 20 h at −0.4 V | n.d. | [95] |
| B,N-doped nanodiamond | B,N | BND3 | 0.1 M NaHCO3 | Ethanol | 93.2 | n.d. | −1.0 V vs. RHE | −0.6 V vs. RHE | 3 h at −1.0 V | n.d. | [96] |
| Electrode | Electrolyte/Main Product | Precursor/Matrix | N Precursor | SMAs | MC a (wt. %) | M-N b | FE % | JCO mA/cm2 | Potential Cathode | Onset Potential/η [mV] | Stability | Experimental | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ni-N4-C/Glassy carbon | 0.5 M KHCO3/CO | g-C3N4/Carbon sheets | n.d. | Ni | n.d. | Ni-N4 | 99.0 | 28.6 | −0.8 V vs. RHE | n.d./~700 | 30 h at −0.81 V | Unvided cell/three-electrode system at an electrochemical station (CHI760E) | [129] |
| Ni SAs/N-C/carbon fiber paper | 0.5 M KHCO3/CO | ZIF-8/Porous carbon | n.d. | Ni | 1.53 | Ni-C/Ni-N3 | 71.9 | 7.37 at −1.0 V | −0.9 V vs. RHE | −0.57 V vs. RHE/890 | 60 h at −1.0 V | Gas-tighttwo-compartment cell, Nafion 115 membrane | [120] |
| Ni- and N-doped reduced graphene oxide (Ni-N-rGO)/Carbon paper | 0.5 M KHCO3/CO | Graphene oxide (GO)-Graphene (Hummer´s method) | Tris (2benzimidazolylmethyl) amine (NTB) | Ni | 0.39 | Ni-N4/Ni-O | 97.0 | ~25 at −0.8 V | −8 V vs. RHE | −0.60 V vs. RHE/490 | n.d. | Two-compartment electrochemical cell (H-type),Nafion 117 membrane | [101] |
| Fe-N-doped graphene/Carbon fiber paper | 0.1 M KHCO3/CO | Graphene oxide (GO)-Graphene (Hummer´s method) | NH3 | Fe | 0.52 | Fe-N4 (Pyridinic N) | ~80.0 | ~1.75 at −0.6V | − 0.6 V vs. RHE | −0.30V vs. RHE/190 | 10 h at −0.60 V | One-compartment | [105] |
| Ni-NG/Glassy carbon | 0.5 M KHCO3 CO | Graphene oxide (GO)-Graphene | NH3 | Ni | 0.44 | Ni-N (Pyridinic N, Pyrrolic) Ni-C | ~95.0 | ~11 at 0.73 V | −0.73 V vs. RHE | −0.31 V vs. RHE/~190 | 20 h at −0.75 V | H-Type glass cell, Nafion 117 membrane, Counter electrode of platinum foil, reference electrode SCE | [106] |
| Ni-NMEGO/Carbon paper | 0.5 M KHCO3/CO | Exfoliated graphene oxide (MEGO) | Urea/NH3 | Ni | ~6.9 | Ni-N edge (Pyridinic Graphitic N) Ni-C | 92.1 | 26.8 at 0.7 V | −0.7 V vs. RHE | −0.29 V vs. RHE/180 | 21 h at −0.55 V | One-compartment, Counter electrode of Ir black carbon on carbon paper | [109] |
| A-Ni-NG/Glassy carbon | 0.5 M KHCO3/CO | Graphene/2D Graphene | Melamine (N)/L-Cysteine(S) | Ni | 4.0 | Ni(I)-N-C | 97 at ~−0.7 | 31.5 | −0.72V vs. RHE | ~−0.35 V vs. RHE/n.d. | n.d. | H-type electrochemical cell, Nafion 117 membrane | [112] |
| A-Ni-NSG/Glassy carbon | 2.5 | Ni(I)-S-C | 97 at ~−0.5V | 36.5 | ~−0.18 V vs. RHE/n.d. | ||||||||
| NiSA-N-CNT/Carbon paper | 0.5 M KHCO3/CO | Dicyandiamide/Bamboo-like CNTs (multiwalled) | Dicyandiamide | Ni | 20.3 | Ni-N4 | 90.3–91.3 | 23.5 at −0.7 V | −0.55 V to −0.7 V vs. RHE | (0.275 V vs. RHE/165 | n.d. | Unvided cell/three-electrode system | [130] |
| Co-HNC/Carbon fiber paper | 0.1 M KHCO3/Syngas | Zeolite imidazolate framework ZIF/3D hollow carbon structure | Zeolite imidazolate framework ZIF | Co | 3.4 | Co-C2N2 | ~100 | n.d. | >−0.7V vs. RHE | n.d. | 24 h at −0.8 V | Gas-tight full electrochemical cell (Typical threeelectrode configuration) | [158] |
| ZnNx/C/Glassy carbon totating disk electrode | 0.5 M KHCO3/CO | Carbon black BP 2000/Micro and mesoporous carbon | Urea | Zn | 0.1 | Zn-N4 | 95 | 4.8 at −0.43 V | −0.43 V vs. RHE | −0.13V vs. RHE/320 | 75 h at −0.43 V | Three-electrode system with a gas-tight twocompartments H-cell. | [155] |
| Co-N5/HNPCSs/carbon paper | 0.2 M NaHCO3/CO | Melamine/Hollow carbon spheres | Melamine | Co | 3.5 | Co-N5 | >90 | ~4.5 at −0.73 V | −0.57 V to −0.88 V vs. RHE | n.d. | 10 h at −0.73 V | H-type cell, Nafion 117 membrane | [145] |
| H-M-G/Carbon paper | 0.1 M KHCO3/CO | Graphene | Melamine | Fe | 1.4 | Fe-N5 (Pyridinic -Pyrrolic N)/Fe-N4 | 97 | ~1.8 at −0.46 V | −0.46 V vs. RHE | −0.26 V vs. RHE/150 | 24 h at −0.46 V | Two-compartment electrochemical cell (H-type),Nafion 117 membrane | [110] |
| Ni-N/C-1/4/Carbon paper | 0.5 M KHCO3/CO | ZIF-8/Carbon nanotubes | ZIF-8 | Ni | 6.6 | Ni-N | ~ 80 | ~6.77 at −0.98 V | −0.78 V vs. RHE | n.d. | 4.5 h at −0.78 V | H shaped electrochemical cellNafion 115 membrane | [146] |
| NiSA-NGA-900/Carbon cloth | 0.5 M KHCO3/CO | Melamine foam/Sponge templated graphene oxide aerogel | Melamine | Ni | ~2.6 | Ni-N | 90.2 | ~8.0 | −0.8 V vs. RHE | −0.4 V vs. RHE/690 | 6 h at −0.8 V | Gas tight H-cell, Nafion membrane N117 | [151] |
| Ni/NC_950/Glassy carbon | 0.1 M KHCO3/CO | Carbon black/Micro-mesoporous carbon | Ni(NH3)6I2 | Ni | n.d. | Ni-N | 90.7 | ~2.5 | −0.8 V vs. RHE | −0.6V vs. RHE/690 | 10 h at −0.8 V | H-type cell, Nafion 117 membrane | [156] |
| Self-supported CuSAs/TCNFs | 0.1 M KHCO3/CH3OH | ZIF-8/Through-hole carbon nanofibers | ZIF-8 | Cu | 1.3 | Cu-N4 | 44.0 | 93.0 | −0.9 V vs. RHE | −0.41 V vs. RHE/n.d. | 50 h at −0.9 V | Two-compartment electrochemical cell, Nafion 117 | [163] |
| Fe-N-G-p/Carbon paper | 0.1 M KHCO3/CO | Graphene oxide(GO)-Graphene (Hummer´s method) | Urea | Fe | 0.36 | Fe+3-N4/Fe-O | 94.0 | 4.5 at −0.58 V | −0.58 V vs. RHE | n.d./470 | 9 h at −0.58 V | Two-compartment electrochemical cell (H-type), Nafion 117 membrane | [149] |
| SA-NI@NC/Glassy carbon | 0.1 M KHCO3/CO | zeolite imidazolate framework ZIF/2D carbon layers | Zeolite imidazolate framework | Ni | ~1.61 | Ni+2-N3 | 86.2 | n.d. | −0.6 V vs. RHE | −0.3 V vs. RHE/n.d. | 1 0 h at −0.6 V | Two-compartment electrochemical cell (H-type), Nafion 115 membrane | [125] |
| Fe1NC/S1-1000/Carbon paper | 0.5 M KHCO3/CO | ZIF-8/Porous Carbon | ZIF-8 | Fe | 0.38 | Fe-N3 (graphitic N) | 96.0 | 6.8 at −0.6 V | −0.5 V vs. RHE | −0.3 V vs. RHE/190 | 45 h at −0.45 V | Two-compartment electrochemical cell (H-type), Nafion 117 membrane, | [126] |
| Fe-SA/NCS-700/Carbon paper | 0.5 M KHCO3/CO | Hemin/Carbon nanosheets | polyaniline | Fe | 0.89 | Fe-N4 (graphitic N) | 87.0 | 6.5 at −0.65 V | −0.47 V vs. RHE | −0.215 V vs. RHE/105 | 10 h at −0.47 V vs. RHE | Two-compartment electrochemical cell (H-type), Nafion 117 membrane | [116] |
| Ni/HMCS-3-800/Glassy carbon disk | 0.5 M KHCO3/CO | Dopamine/Hollow mesoporous carbon spheres (HMCS) | n.d. | Ni | 0.26 | Ni-N4-C (Pyridinic N, graphitic N) | 95 | 10.5 at 1.1 V | −0.7 to −1.1 V vs. RHE | n.d. | 10 h a −0.9 V vs. RHE | Two-compartment electrochemical cell (H-type), Nafion 117 membrane. | [139] |
| SA-Zn-NHPC/Carbon paper | 0.5 M KHCO3/CO | Apples/Hierarchically porouscarbon | Egg whites | Zn | 0.34 | Zn-N4 | 96 | ~ 5 at −0.44 V | −0.44 V vs. RHE | −0.16 V vs. RHE/50 | 20 h at −0.44 V vs. RHE | Two-compartment electrochemical cell (H-type), Nafion 115 membrane. | [140] |
| C800NiA/GDE composed of a packed carbon microporous layer (MPL). | 1.0M KOH/CO | Adenine/Porous Carbon | Adenine | Ni | 2.6 | Ni-N4-C | 99 | 199 at −0.31 V | −0.31 V vs. RHE | n.d./235 | 6 h at 0.31 V vs. RHE | Flow cell, anion exchange membrane | [141] |
| CoNi-NC/Carbon paper | 0.5 M KHCO3/Syngas | Glucose/Carbon | Picyandiamide | Ni, Co | Ratio Co/Ni=1 | Ni-N, Co-N (pyridinic N) | CO:53% CO/H2 ratio: 0.8–1.3 | 51 at −0.9 V | −0.9 V vs. RHE | n.d. | 7 h at 0.9 V vs. RHE | Two-compartment electrochemical cell (H-type), Nafion 117 membrane | [159] |
| Ni SAC/Carbon paper | 0.5 M KHCO3/Syngas | Dimethylglyoxime/mesoporous carbon nanorods | dimethylglyoxime | Ni | n.d. | Ni-N | CO/H2 ratio: 1:9 to 19:1 | 15 at −0.65 V | −0.65 V vs. RHE | n.d. | >8 h at 0.65 V | Two-compartment electrochemical cell (H-type), Nafion 115 membrane. | [160] |
| Cu SAs/NC/Carbon paper | 0.1 M KHCO3/CO | Chitosan/Microporous carbon | Chitosan | Cu | 0.32 | Cu-N5 | 92 | 8.9 | −0.7 V vs. RHE | −0.23 V vs. RHE/n.d. | 30 h at −0.7 V | Gastight two-compartment electrochemical cell, Nafion 212 membrane | [153] |
| NiSA/N-C/Carbon paper | 0.5 M KHCO3/CO | g-C3N4/ultrathin porous carbon nanosheets | Polydopamine(PDA) | Ni | 0.86 | Ni-N4 | 96 | 26.4 | −0.86 V vs. RHE | n.d./750 | 22 h at −0.86 V | H-type electrochemical cell, Nafion 117 membrane | [132] |
| SAs/NC/Carbon paper | 0.5 M KHCO3/Formate | ZIF-8/Porous Carbon | ZIF-8 | In | 1.25 | In-N4 | 96 | 8.87 | −0.65 V vs. RHE | n.d. | 60 h at −0.65 V | Gas-tight standard three-electrode H-type cell | [166] |
| DNG-SAFe/Carbon paper | 0.1M KHCO3/CO | C-g-C3N4/Graphene likeporous carbon | C-g-C3N4 | Fe | 0.74 | Fe-N4 | 90 | 33 | −0.75 V vs. RHE | n.d. | 17 h at −0.75 V | Gas-tight H-cell, Nafion 117 membrane | [133] |
| Sb SA/NC/Glassy carbon | 0.5M KHCO3/Formate | Trimesic acid/ultrathin porous carbon nanosheets | Dicyandimide | Sb | 2.86 | Sb-N4 | 94 | ~20 | −0.8 V vs. RHE | n.d. | 10 h at −0.8 V | Gas-tight H-cell,Nafion 117 membrane | [167] |
| Bi Sas/C/Carbon fiber paper | 0.1M KHCO3/Formate | Carbon black | n.d. | Bi | n.d. | n.d. | 83.6 | 12 | −1.1 V vs. RHE | 10 h at −1.1 V | H-cell system, Nafion 115 membrane | [168] | |
| Ni-NC@Ni/Carbon fiber paper | 0.1M KHCO3/CO | Urea/sheet-like open nanostructure | Urea | Ni | 11.05 | Ni-N | ~87 | 14.8 | −0.78 V vs. RHE | n.d./670 | 150 h at −0.78 V | Gas-tight H-cell, Nafion 117 membrane | [157] |
| SA-Zn/MNC/Carbon fiber cloth | 0.1M KHCO3/CO | Glucose/Microporous N-doped carbon | Hydroxyl ammonium chloride ((NH3OH)Cl) | Zn | 2.66 | Zn-N4 | 85 | −31.8 | −1.8 V vs. SCE | n.d. | 35 h at −1.8 V | Two-compartment batch cell | [169] |
| ZnO@ZIF-NiZn50/Carbon fiber paper | 0.5M KHCO3/CO | ZIF-8/Carbon nanotubes | ZIF-8 | Ni | 0.46 | Ni-N4 | 98 | 34.3 | −1.0 V vs. RHE | −0.32 V vs. RHE/n.d. | 20 h at −0.8 V | H-type cell with two-compartments, anion exchange membrane (Nafion-117) | [127] |
| Fe-N/CNT@GNR-2/Carbon paper | 0.1 M KHCO3/CO | Commercial carbon nanotube/Hierarchical carbon nanotube and graphene nanoribbon networks | Urea | Fe | 0.74 | Fe-N4 | 96 | 22.6 | −0.76 V vs. RHE | n.d./650 | 5 h at −0.76 V | Two-chamber cell separated, nafion 115 membrane | [128] |
| Cu-SA/NPC/Carbon paper | 0.1M KHCO3/CH3COCH3 | ZIF-8/Porous Carbon | ZIF-8 | Cu | 0.59 | Fe-N4 (Pyrrolic) | 36.7 | n.d. | −0.36 V vs. RHE | −0.16 V vs. RHE/250 | n.d. | Airtight double-cell, Nafion 117 membrane | [152] |
| Fe-N-C/Glassy carbon | 0.5M KHCO3/CO | Polyaniline (PANI)/Carbon nanoroads | Urea | Fe | n.d. | Fe-N | 95 | 1.9 | −0.64 V vs. RHE | n.d./530 | n.d. | Gas-tight classical H-cell | [135] |
| Ni-Sas@FNC/Carbon paper | 0.5M KHCO3/CO | Glucose/Carbon nanosheets | Melamine | Ni | 5.92 | Ni-N4 | 95 | 25 | −0.77 V vs. RHE | n.d. | 10 h at −0.77 V | Two-compartment tight electrolytic-cell, Nafion 117 membrane | [137] |
| Electrode | Electrolyte/Main Product | Precursor/Matrix | Nanoparticles | Metal Contents (wt. %) | FE % | JCO [mA/cm2] | Onset Potential | Stability | Experimental | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| Ni-NC-ATPA@C/Glassy carbon | 0.5 M KHCO3/CO | Carbon black Vulcan XC72R | Ni (10–100 nm) | ~3.6 | 93.7 at −0.7 vs. RHE | 22.7 at −1.1V | −0.5 V vs. RHE | 54 h at −0.7 V | Gas-tight three electrode | [184] |
| Au-10/carbon paper | 0.5 M KHCO3/CO, syngas | Carbon paper TGP-H, Toray | Au (0.5–30 nm) | n.d. | 78.08 at −0.59 V vs. RHE | 0.8 at −0.59 V | n.d. | n.d. | Two-compartment electrochemical cell separated by a proton exchange membrane (Nafion®,117) | [173] |
| Fe/NC(NH3)/Carbon cloth | 0.5 M KHCO3/CO, syngas | Carbon black Black Pearls | Fe | 20 | 66 at −0.6 V vs. RHE | n.d. | n.d. | n.d. | Undivided cell, three electrodes | [181] |
| Nano-SnO2/graphene/glassy carbon | 0.1M NaHCO3/Formate | Graphene | Sn (5 nm) | ~30 | 93.6% at −1.8 V vs. SCE | 13.1 at −1.8 V vs. SCE | −0.34 V vs. RHE. | 18 h at −1.8 V | gas-tight two compartment electrochemical cell | [178] |
| Cu/CNS | 0.1 M KHCO3/Ethanol | N-doped Graphene | Cu (39.18 nm) | n.d. | 63% at −1.2 V vs. RHE | 2 at −1.2 V vs. RHE | −0.3 V vs. RHE. | 6 h at −1.2 V | Two-compartment electrochemical cell separated by an anion exchange membrane (Selemion AMV, AGC) | [187] |
| Cu/GNFs/carbon cloth | 0.1 M KHCO3/Formate | Graphene nanoflakes | Cu | 20–50 | 40% at −0.6 V vs. RHE | 15.9 at −0.9 V vs. RHE | −0.40 V vs. RHE | n.d. | Two-compartment electrochemical cell separated by a proton exchange membrane (Nafion®) | [189] |
| GO-VB6-Cu-2/Carbon paper | 0.1 M KHCO3/Ethanol | N-doped GO | Cu (2–12 nm) | 10 | 5.3% at −0.25 V vs. RHE | 4.571 at −0.25 vs. RHE | 0.140 V vs. RHE. | 10 h at −0.25 V | H-type cell separated by a Nafion 117 ion exchange membrane | [191] |
| Cu2O/NRGO/glassy carbon | 0.1 M KHCO3/C2H4 | N-doped rGO | Cu2O (90 nm) | 5.94 | 19.7 at −1.3 V vs. RHE | 10 at −1.3 V vs. RHE | n.d. | 1000 s at −1.4 V | H-type cell with two compartments separated by a proton exchange membrane (Nafion 117, Dupont). | [191] |
| Cu-rGO | 0.1 M NaHCO3/CO,CH4, Liquid products | rGO | Cu (10 nm) | n.d. | 76.6 at −0.4 V vs. RHE | n.d. | n.d. | 14 h at −0.5 V | Gas-tight two-compartment electrochemical cell cationic exchange membrane (CMI-7000S) | [195] |
| Ag2S/N-S-doped rGO/Glassy carbon | 0.1 M KHCO3/CO | N-S doped rGO | Ag2S (50 nm) | ~70 | 87.4 at −0.76 V vs. RHE | ~70 μA cm−2 | −0.34 V vs. RHE | 40 h at −0.76 V | Double-chamber electrochemical cell partitioned by a Nafion 117 membrane | [193] |
| Bi/Bi2O3/NrGO-700 | 0.5 M KHCO3/Formate | N-doped rGO | Bi/Bi2O3 (20 nm) | 17.6 | 85 at −0.9 V vs. RHE | 18 at −0.9 V vs. RHE | −0.5 V vs. RHE | 10 h at −0.9 V | H-type three electrodecell. | [194] |
| NiCu0.25/glassy carbon | 0.1 M KHCO3/CO | Graphene | Cu/Ni (12.5 nm) | ratio Cu/Ni 0.25 | 88.5 at −1.0 V vs. RHE | n.d. | −0.7 V vs. RHE | 3000 s at −1.0 V | Three-electrode H-cell separated by a proton exchange membrane (Nafion 117) | [197] |
| N/NiNPs@CNT/G | 0.5 M KHCO3/CO | N-doped nanotube (CNT) assembled on the surface of graphene | Ni (50–75 nm) | n.d. | 97.7 at −0.7 V vs. RHE | 7.9 at −0.7 V | −0.3 V vs. RHE | 10 h at −0.7V | Three-electrode H-type electrolytic cell with Nafion membrane | [202] |
| 20%-Cu/CNTs/Carbon paper | 0.5 M NaHCO3/Methanol | Nanotubes CNTs | Cu (3–60 nm) | 20% | 38.4 at −1.7 V vs. SCE | 17.5 at −1.5 V | −0.5 V vs. SCE | 6000 s at −1.7 V | H-type cell with anode and cathode compartments separated by proton conducting Nafion 117 membrane. | [199] |
| SnO2/N-MWCNTs/Glassy carbon | 0.1 M KHCO3/Formate | N-doped multiwalled carbon nanotubes (N-MWCNTs) | SnO2 | 42.5% | 46 at −0.9 V vs. Ag/AgCl | ~5 at −1.3 V | n.d. | 10 h at −1.3 V | Undivided three-electrode cell | [200] |
| Ni@NCNTs/Carbon paper | 0.5 M KHCO3/CO | N- doped nanotubes | Ni (50–100 nm) | 26% | 99.1 at −0.8 V vs. RHE | 8.01 at −0.72 V | −0.3 V vs. RHE | 10 h at −0.8 V | H-typethree-electrode cell, separated by a proton membrane (Nafion 117) | [201] |
| Bi@NPC/Glassy carbon | 0.1 M KHCO3/Formate | Metal organic framework (MOF)-derived porous carbon material | Bi (~20 nm) | 22.6% | 92 at −1.5 V SCE | 14.4 at −1.5 V | −1.2 V vs. SCE | 20 h at −1.5 V | Gastight electrolytic Three electrode cell | [176] |
| AgNP/MWCNTs/GDE | 0.1 M KOH/CO | MWCNTs | Ag | n.d. | 95% at −0.77 V vs. RHE | 338 at −0.77 V vs. RHE | −0.1 V vs. RHE | n.d. | Electrochemicalflow reactor | [216] |
| Ni@NC-900/Carbon fiber paper | 0.1 M KHCO3/CO | Carbon shells | Ni (~0.203 nm) | ~3 | 96% at −1 V vs. RHE | 17.5 at −1 V vs. RHE | 0.89 V | 1 h at −0.9 V | Two compartment gas-tight H-cell (Nafion membrane) | [217] |
| 0.1 M KHCO3/CO | 85% | ~30 at 2.6 V | n.d. | 18 h at 2.6 V | GDE system (Sustainion X37-50 membrane) | |||||
| 5nm Ag/C/Glassy carbon plate | 0.5 M KHCO3/CO | Carbon black | Ag (5 nm) | n.d. | 84.4% at −0.75 V RHE | ~4 at −0.75 V RHE | 0.64 V | n.d. | Two-compartmentelectrochemical cell, Nafion 117 membrane | [183] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez-Valencia, L.D.; Bailón-García, E.; Carrasco-Marín, F.; Pérez-Cadenas, A.F. From CO2 to Value-Added Products: A Review about Carbon-Based Materials for Electro-Chemical CO2 Conversion. Catalysts 2021, 11, 351. https://doi.org/10.3390/catal11030351
Ramírez-Valencia LD, Bailón-García E, Carrasco-Marín F, Pérez-Cadenas AF. From CO2 to Value-Added Products: A Review about Carbon-Based Materials for Electro-Chemical CO2 Conversion. Catalysts. 2021; 11(3):351. https://doi.org/10.3390/catal11030351
Chicago/Turabian StyleRamírez-Valencia, Lilian D., Esther Bailón-García, Francisco Carrasco-Marín, and Agustín F. Pérez-Cadenas. 2021. "From CO2 to Value-Added Products: A Review about Carbon-Based Materials for Electro-Chemical CO2 Conversion" Catalysts 11, no. 3: 351. https://doi.org/10.3390/catal11030351
APA StyleRamírez-Valencia, L. D., Bailón-García, E., Carrasco-Marín, F., & Pérez-Cadenas, A. F. (2021). From CO2 to Value-Added Products: A Review about Carbon-Based Materials for Electro-Chemical CO2 Conversion. Catalysts, 11(3), 351. https://doi.org/10.3390/catal11030351