
Synthesis of Novel Heteroleptic Oxothiolate Ni(II) Complexes and Evaluation of Their Catalytic Activity for Hydrogen Evolution


Abstract
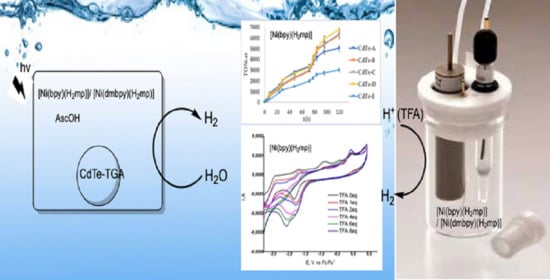
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization
2.2. Electrocatalytic Proton Reduction
2.3. Photocatalytic Hydrogen Evolution
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.3. Cyclic Voltammetry
3.4. X-ray Crystallography
3.5. Photocatalysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koshiba, K.; Yamauchi, K.; Sakai, K. Ligand-Based PCET Reduction in a Heteroleptic Ni(bpy)(dithiolene) Electrocatalyst Giving Rise to Higher Metal Basicity Required for Hydrogen Evolution. ChemElectroChem 2019, 6, 2273–2281. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous Photocatalyst Materials for Water Splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef]
- Reece, S.Y.; Hamel, J.A.; Sung, K.; Jarvi, T.D.; Esswein, A.J.; Pijpers, J.J.; Nocera, D.G. Wireless Solar Water Splitting Using Silicon-Based Semiconductors and Earth-Abundant Catalysts. Science 2011, 334, 645–648. [Google Scholar] [CrossRef] [Green Version]
- Hisatomi, T.; Kubota, J.; Domen, K. Recent advances in semiconductors for photocatalytic and photoelectrochemical water splitting. Chem. Soc. Rev. 2014, 43, 7520–7535. [Google Scholar] [CrossRef]
- Swierk, J.R.; Mallouk, T.E. Design and development of photoanodes for water-splitting dye-sensitized photoelectrochemical cells. Chem. Soc. Rev. 2013, 42, 2357–2387. [Google Scholar] [CrossRef]
- Esswein, A.J.; Nocera, D.G. Hydrogen Production by Molecular Photocatalysis. Chem. Rev. 2007, 107, 4022–4047. [Google Scholar] [CrossRef] [PubMed]
- Brudvig, G.W.; Reek, J.N.H.; Sakai, K.; Spiccia, L.; Sun, L. Catalytic Systems for Water Splitting. ChemPlusChem 2016, 81, 1017–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Chen, K.S.; Mishler, J.; Cho, S.C.; Adroher, X.C. A review of polymer electrolyte membrane fuel cells: Technology, applications, and needs on fundamental research. Appl. Energy 2011, 88, 981–1007. [Google Scholar] [CrossRef] [Green Version]
- Drosou, M.; Kamatsos, F.; Mitsopoulou, C.A. Recent advances in the mechanisms of the hydrogen evolution reaction by non-innocent sulfur-coordinating metal complexes. Inorg. Chem. Front. 2019, 7, 37–71. [Google Scholar] [CrossRef] [Green Version]
- Zarkadoulas, A.; Koutsouri, E.; Mitsopoulou, C.A. A perspective on solar energy conversion and water photosplitting by dithiolene complexes. Coord. Chem. Rev. 2012, 256, 2424–2434. [Google Scholar] [CrossRef]
- Frey, M. Hydrogenases: Hydrogen-Activating Enzymes. ChemBioChem 2002, 3, 153–160. [Google Scholar] [CrossRef]
- Vincent, K.A.; Parkin, A.; Armstrong, F.A. Investigating and Exploiting the Electrocatalytic Properties of Hydrogenases. Chem. Rev. 2007, 107, 4366–4413. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef]
- Kaiser, J.T.; Hu, Y.; Wiig, J.A.; Rees, D.C.; Ribbe, M.W. Structure of Precursor-Bound NifEN: A Nitrogenase FeMo Cofactor Maturase/Insertase. Science 2011, 331, 91–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spatzal, T.; Aksoyoglu, M.; Zhang, L.; Andrade, S.L.A.; Schleicher, E.; Weber, S.; Rees, D.C.; Einsle, O. Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor. Science 2011, 334, 940. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Koo, J. O2 sensitivity and H2 production activity of hydrogenases—A review. Biotechnol. Bioeng. 2019, 116, 3124–3135. [Google Scholar] [CrossRef]
- Volbeda, A.; Garcin, E.; Piras, C.; De Lacey, A.L.; Fernandez, V.M.; Hatchikian, E.C.; Frey, M.; Fontecilla-Camps, J.C. Structure of the [NiFe] Hydrogenase Active Site: Evidence for Biologically Uncommon Fe Ligands. J. Am. Chem. Soc. 1996, 118, 12989–12996. [Google Scholar] [CrossRef]
- Volbeda, A.; Charon, M.-H.; Piras, C.; Hatchikian, E.C.; Frey, M.; Fontecilla-Camps, J.C. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 1995, 373, 580–587. [Google Scholar] [CrossRef]
- Higuchi, Y.; Yagi, T.; Yasuoka, N. Unusual ligand structure in Ni–Fe active center and an additional Mg site in hydrogenase revealed by high resolution X-ray structure analysis. Structure 1997, 5, 1671–1680. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, Y.; Ogata, H.; Miki, K.; Yasuoka, N.; Yagi, T. Removal of the bridging ligand atom at the Ni-Fe active site of [NiFe] hydrogenase upon reduction with H2, as revealed by X-ray structure analysis at 1.4 Å resolution. Structure 1999, 7, 549–556. [Google Scholar] [CrossRef]
- Rousset, M.; Montet, Y.; Guigliarelli, B.; Forget, N.; Asso, M.; Bertrand, P.; Fontecilla-Camps, J.C.; Hatchikian, E.C. [3Fe-4S] to [4Fe-4S] Cluster Conversion in Desulfovibrio Fructosovorans [NiFe] Hydrogenase by Site-Directed Mutagenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 11625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matias, P.M.; Soares, C.M.; Saraiva, L.M.; Coelho, R.; Morais, J.; Le Gall, J.; Carrondo, M.A. [NiFe] hydrogenase from Desulfovibrio desulfuricans ATCC 27774: Gene sequencing, three-dimensional structure determination and refinement at 1.8 Å and modelling studies of its interaction with the tetrahaem cytochrome C3. JBIC J. Biol. Inorg. Chem. 2001, 6, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Lamle, S.E.; Albracht, S.P.J.; Armstrong, F.A. The Mechanism of Activation of a [NiFe]-Hydrogenase by Electrons, Hydrogen, and Carbon Monoxide. J. Am. Chem. Soc. 2005, 127, 6595–6604. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-M.; Chiou, T.-W.; Chen, H.-H.; Chiang, C.-Y.; Kuo, T.-S.; Liaw, W.-F. Mononuclear Ni(II)-Thiolate Complexes with Pendant Thiol and Dinuclear Ni(III/II)-Thiolate Complexes with Ni···Ni Interaction Regulated by the Oxidation Levels of Nickels and the Coordinated Ligands. Inorg. Chem. 2007, 46, 8913–8923. [Google Scholar] [CrossRef]
- Peng, S.-M.; Goedken, V.L. Cofacial Dimer of a Dihydrooctaaza[14]Annulene Complex Containing a Nickel-Nickel Bond and Related Monomeric Complexes. J. Am. Chem. Soc. 1976, 98, 8500–8510. [Google Scholar] [CrossRef]
- Li, C.-H.; Chuang, H.-J.; Li, C.-Y.; Ko, B.-T.; Lin, C.-H. Bimetallic nickel and cobalt complexes as high-performance catalysts for copolymerization of carbon dioxide with cyclohexene oxide. Polym. Chem. 2014, 5, 4875–4878. [Google Scholar] [CrossRef] [Green Version]
- Barclay, T.M.; Hicks, R.G.; Lemaire, M.T.; Thompson, L.K. Synthesis, structure, and magnetism of bimetallic manganese or nickel complexes of a bridging verdazyl radical. Inorg. Chem. 2001, 40, 5581–5584. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.-M.; Chang, C.-H.; Chuang, H.-J.; Liu, C.-T.; Ko, B.-T.; Lin, C.-C. Bimetallic Nickel Complexes that Bear Diamine-Bis(Benzotriazole Phenolate) Derivatives as Efficient Catalysts for the Copolymerization of Carbon Dioxide with Epoxides. ChemCatChem 2016, 8, 984–991. [Google Scholar] [CrossRef]
- Möller, F.; Merz, K.; Herrmann, C.; Apfel, U.-P. Bimetallic nickel complexes for selective CO2 carbon capture and sequestration. Dalton Trans. 2016, 45, 904–907. [Google Scholar] [CrossRef] [Green Version]
- Caris, R.; Peoples, B.C.; Valderrama, M.; Wu, G.; Rojas, R. Mono and bimetallic nickel bromide complexes bearing azolate-imine ligands: Synthesis, structural characterization and ethylene polymerization studies. J. Organomet. Chem. 2009, 694, 1795–1801. [Google Scholar] [CrossRef]
- Brazzolotto, D.; Gennari, M.; Queyriaux, N.; Simmons, T.R.; Pécaut, J.; Demeshko, S.; Meyer, F.; Orio, M.; Artero, V.; Duboc, C. Nickel-centred proton reduction catalysis in a model of [NiFe] hydrogenase. Nat. Chem. 2016, 8, 1054–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoi, V.S.; Sun, Y.; Long, J.R.; Chang, C.J. Complexes of earth-abundant metals for catalytic electrochemical hydrogen generation under aqueous conditions. Chem. Soc. Rev. 2013, 42, 2388–2400. [Google Scholar] [CrossRef] [PubMed]
- Artero, V.; Fontecave, M. Solar fuels generation and molecular systems: Is it homogeneous or heterogeneous catalysis? Chem. Soc. Rev. 2013, 42, 2338–2356. [Google Scholar] [CrossRef] [PubMed]
- McKone, J.R.; Marinescu, S.C.; Brunschwig, B.S.; Winkler, J.R.; Gray, H.B. Earth-abundant hydrogen evolution electrocatalysts. Chem. Sci. 2014, 5, 865–878. [Google Scholar] [CrossRef] [Green Version]
- Artero, V.; Saveant, J.-M. Toward the rational benchmarking of homogeneous H2-evolving catalysts. Energy Environ. Sci. 2014, 7, 3808–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutra, P.; Igau, A. Emerging Earth-abundant (Fe, Co, Ni, Cu) molecular complexes for solar fuel catalysis. Curr. Opin. Green Sustain. Chem. 2018, 10, 60–67. [Google Scholar] [CrossRef]
- Bhugun, I.; Lexa, D.; Savéant, J.-M. Homogeneous Catalysis of Electrochemical Hydrogen Evolution by Iron(0) Porphyrins. J. Am. Chem. Soc. 1996, 118, 3982–3983. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, R. Nickel-based cocatalysts for photocatalytic hydrogen production. Appl. Surf. Sci. 2015, 351, 779–793. [Google Scholar] [CrossRef]
- Zarkadoulas, A.; Field, M.J.; Papatriantafyllopoulou, C.; Fize, J.; Artero, V.; Mitsopoulou, C.A. Experimental and Theoretical Insight into Electrocatalytic Hydrogen Evolution with Nickel Bis(Aryldithiolene) Complexes as Catalysts. Inorg. Chem. 2016, 55, 432–444. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Wang, M.; Yang, Y.; Yao, T.; Sun, L. A Molecular Copper Catalyst for Electrochemical Water Reduction with a Large Hydrogen-Generation Rate Constant in Aqueous Solution. Angew. Chem. Int. Ed. 2014, 53, 13803–13807. [Google Scholar] [CrossRef]
- Lei, H.; Fang, H.; Han, Y.; Lai, W.; Fu, X.; Cao, R. Reactivity and Mechanism Studies of Hydrogen Evolution Catalyzed by Copper Corroles. ACS Catal. 2015, 5, 5145–5153. [Google Scholar] [CrossRef]
- Stubbert, B.D.; Peters, J.C.; Gray, H.B. Rapid Water Reduction to H2 Catalyzed by a Cobalt Bis (Iminopyridine) Complex. J. Am. Chem. Soc. 2011, 133, 18070–18073. [Google Scholar] [CrossRef] [PubMed]
- Aroua, S.; Todorova, T.K.; Mougel, V.; Hommes, P.; Reissig, H.-U.; Fontecave, M. New Cobalt-Bisterpyridyl Catalysts for Hydrogen Evolution Reaction. ChemCatChem 2017, 9, 2099–2105. [Google Scholar] [CrossRef]
- Manbeck, G.F.; Canterbury, T.; Zhou, R.; King, S.; Nam, G.; Brewer, K.J. Electrocatalytic H2 Evolution by Supramolecular RuII–RhIII–RuII Complexes: Importance of Ligands as Electron Reservoirs and Speciation upon Reduction. Inorg. Chem. 2015, 54, 8148–8157. [Google Scholar] [CrossRef]
- Bindra, G.S.; Schulz, M.; Paul, A.; Groarke, R.; Soman, S.; Inglis, J.L.; Browne, W.R.; Pfeffer, M.G.; Rau, S.; MacLean, B.J.; et al. The role of bridging ligand in hydrogen generation by photocatalytic Ru/Pd assemblies. Dalton Trans. 2012, 41, 13050–13059. [Google Scholar] [CrossRef] [Green Version]
- Kowacs, T.; O’Reilly, L.; Pan, Q.; Huijser, A.; Lang, P.; Rau, S.; Browne, W.R.; Pryce, M.T.; Vos, J.G. Subtle Changes to Peripheral Ligands Enable High Turnover Numbers for Photocatalytic Hydrogen Generation with Supramolecular Photocatalysts. Inorg. Chem. 2016, 55, 2685–2690. [Google Scholar] [CrossRef] [PubMed]
- Karnahl, M.; Kuhnt, C.; Ma, F.; Yartsev, A.; Schmitt, M.; Dietzek, B.; Rau, S.; Popp, J. Tuning of Photocatalytic Hydrogen Production and Photoinduced Intramolecular Electron Transfer Rates by Regioselective Bridging Ligand Substitution. ChemPhysChem 2011, 12, 2101–2109. [Google Scholar] [CrossRef]
- Das, A.; Han, Z.; Brennessel, W.W.; Holland, P.L.; Eisenberg, R. Nickel Complexes for Robust Light-Driven and Electrocatalytic Hydrogen Production from Water. ACS Catal. 2015, 5, 1397–1406. [Google Scholar] [CrossRef]
- Jain, R.; Al Mamun, A.; Buchanan, R.M.; Kozlowski, P.M.; Grapperhaus, C.A. Ligand-Assisted Metal-Centered Electrocatalytic Hydrogen Evolution upon Reduction of a Bis(thiosemicarbazonato) Ni(II) Complex. Inorg. Chem. 2018, 57, 13486–13493. [Google Scholar] [CrossRef]
- Abul-Futouh, H.; Zagranyarski, Y.; Müller, C.; Schulz, M.; Kupfer, S.; Görls, H.; El-Khateeb, M.; Gräfe, S.; Dietzek, B.; Peneva, K. [FeFe]-Hydrogenase H-Cluster Mimics Mediated by Naphthalene Monoimide Derivatives of Peri-Substituted Dichalcogenides. Dalton Trans. 2017, 46, 11180–11191. [Google Scholar] [CrossRef]
- Gao, W.; Liu, J.; Jiang, W.; Wang, M.; Weng, L.; Åkermark, B.; Sun, L. An Azadithiolate Bridged Fe2S2 Complex as Active Site Model of FeFe-Hydrogenase Covalently Linked to a Re (CO)3 (Bpy)(Py) Photosensitizer Aiming for Light-Driven Hydrogen Production. Comptes Rendus Chim. 2008, 11, 915–921. [Google Scholar] [CrossRef]
- Felton, G.A.N.; Vannucci, A.K.; Chen, J.; Lockett, L.T.; Okumura, N.; Petro, B.J.; Zakai, U.I.; Evans, D.H.; Glass, R.S.; Lichtenberger, D.L. Hydrogen Generation from Weak Acids: Electrochemical and Computational Studies of a Diiron Hydrogenase Mimic. J. Am. Chem. Soc. 2007, 129, 12521–12530. [Google Scholar] [CrossRef]
- Kamatsos, F.; Drosou, M.; Mitsopoulou, C.A. Heteroleptic thiolate diamine nickel complexes: Noble-free-metal catalysts in electrocatalytic and light-driven hydrogen evolution reaction. Int. J. Hydrogen Energy 2021, in press. [Google Scholar] [CrossRef]
- Drosou, M.; Kamatsos, F.; Ioannidis, G.; Zarkadoulas, A.; Mitsopoulou, C.A.; Papatriantafyllopoulou, C.; Tzeli, D. Reactivity and Mechanism of Photo- and Electrocatalytic Hydrogen Evolution by a Diimine Copper(I) Complex. Catalysts 2020, 10, 1302. [Google Scholar] [CrossRef]
- Makedonas, C.; Mitsopoulou, C.A.; Lahoz, F.J.; Balana, A.I. Synthesis, Characterization, and Crystal Structure of the Pd(phen)(bdt) Complex. A DFT and TDDFT Study of Its Ground Electronic and Excited States Compared to Those of Analogous Complexes. Inorg. Chem. 2003, 42, 8853–8865. [Google Scholar] [CrossRef]
- Baker-Hawkes, M.J.; Billig, E.; Gray, H.B. Characterization and Electronic Structures of Metal Complexes Containing Benzene-1,2-dithiolate and Related Ligands. J. Am. Chem. Soc. 1966, 88, 4870–4875. [Google Scholar] [CrossRef]
- Balch, A.L. Electron-transfer series of the [M-O2S2] type. Complexes derived from o-mercaptophenol, 1-mercapto-2-naphthol, and 1-hydroxy-2-pyridinethione. J. Am. Chem. Soc. 1969, 91, 1948–1953. [Google Scholar] [CrossRef]
- Robinson, F.V.; Topp, N.E. The extraction of rare earth nitrates with tri-n-butyl phosphate (TBP). J. Inorg. Nucl. Chem. 1964, 26, 473–476. [Google Scholar] [CrossRef]
- Holm, R.H.; Balch, A.L.; Davison, A.; Maki, A.H.; Berry, T.E. Electron-transfer complexes of the [M-N2S2] type. The existence of cation-stabilized free-radical complexes. J. Am. Chem. Soc. 1967, 89, 2866–2874. [Google Scholar] [CrossRef]
- Makedonas, C.; Mitsopoulou, C.A. A vibrational and DFT study of M(diimine)(dithiolate) complexes and their complexation route. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2006, 64, 918–930. [Google Scholar] [CrossRef]
- De Mello, M.T.S.; Ribeiro, M.C.; Santos, P.S. Resonance Raman Spectroscopy of Benzenedithiolate Complexes: Evidences of Extensive Delocalization of the Chromophore. J. Mol. Struct. 1995, 372, 1–7. [Google Scholar]
- Gerasimova, T.P.; Katsyuba, S.A. Bipyridine and phenanthroline IR-spectral bands as indicators of metal spin state in hexacoordinated complexes of Fe(ii), Ni(ii) and Co(ii). Dalton Trans. 2013, 42, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Makedonas, C.; Mitsopoulou, C.A. Tuning the properties of M(diimine)(dithiolate) complexes—The role of the metal and solvent effect. A combined experimental, DFT and TDDFT study. Inorg. Chim. Acta 2007, 360, 3997–4009. [Google Scholar] [CrossRef]
- Makedonas, C.; Mitsopoulou, C.A. An Investigation of the Reactivity of [(diimine)(dithiolato)M] Complexes Using the Fukui Functions Concept. Eur. J. Inorg. Chem. 2006, 2006, 590–598. [Google Scholar] [CrossRef]
- Zarkadoulas, A.; Koutsouri, E.; Semidalas, E.; Psycharis, V.; Raptopoulou, C.P.; Mitsopoulou, C.A. Photocatalytic hydrogen production with alkylated nickel bis-dithiolene complexes. Polyhedron 2018, 152, 138–146. [Google Scholar] [CrossRef]
- Eckenhoff, W.T. Molecular catalysts of Co, Ni, Fe, and Mo for hydrogen generation in artificial photosynthetic systems. Coord. Chem. Rev. 2018, 373, 295–316. [Google Scholar] [CrossRef]
- Herebian, D.; Bothe, E.; Bill, E.; Weyhermüller, T.; Wieghardt, K. Experimental Evidence for the Noninnocence of O-Aminothiophenolates: Coordination Chemistry of o-Iminothionebenzosemiquinonate(1-) π-Radicals with Ni(II), Pd(II), Pt(II). J. Am. Chem. Soc. 2001, 123, 10012–10023. [Google Scholar] [CrossRef] [PubMed]
- Bachler, V.; Olbrich, G.; Neese, F.; Wieghardt, K. Theoretical Evidence for the Singlet Diradical Character of Square Planar Nickel Complexes Containing Two O-Semiquinonato Type Ligands. Inorg. Chem. 2002, 41, 4179–4193. [Google Scholar] [CrossRef]
- Holyer, R.H.; Hubbard, C.D.; Kettle, S.F.A.; Wilkins, R.G. The Kinetics of Replacement Reactions of Complexes of the Transition Metals with 1,10-Phenanthroline and 2,2′-Bipyridine. Inorg. Chem. 1965, 4, 929–935. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Gupta, P.; Basuli, F.; Pierpont, C.G. Structural Systematics for O-C6H4XY Ligands with X, Y = O, NH, and S Donor Atoms. o-Iminoquinone and o-Iminothioquinone Complexes of Ruthenium and Osmium. Inorg. Chem. 2002, 41, 5810–5816. [Google Scholar] [CrossRef]
- Köckerling, M.; Henkel, G. Einkernige Nickel-Thiolato-Komplexe mit Nickel-Zentren in unterschiedlichen Oxidationszuständen: Molekularer Aufbau von [Ni(SC6H4O)2]2− und [Ni(SC6H4O)2]. Chem. Ber. 1993, 126, 951–953. [Google Scholar] [CrossRef]
- Ghiasi, Z.; Amani, V.; Mirzaei, P.; Safari, N.; Abedi, A. Trichloridothallium(III) Complexes with Bipyridine Derivatives: From Structure to Luminescence Properties. Aust. J. Chem. 2013, 66, 676–684. [Google Scholar] [CrossRef]
- Goss, C.A.; Abruna, H.D. Spectral, electrochemical and electrocatalytic properties of 1,10-phenanthroline-5,6-dione complexes of transition metals. Inorg. Chem. 1985, 24, 4263–4267. [Google Scholar] [CrossRef]
- Jacques, P.-A.; Artero, V.; Pecaut, J.; Fontecave, M. Cobalt and Nickel Diimine-Dioxime Complexes as Molecular Electrocatalysts for Hydrogen Evolution with Low Overvoltages. Proc. Natl. Acad. Sci. USA 2009, 106, 20627–20632. [Google Scholar] [CrossRef] [Green Version]
- Hawley, M.; Tatawawadi, S.; Piekarski, S.; Adams, R.N. Electrochemical Studies of the Oxidation Pathways of Catecholamines. J. Am. Chem. Soc. 1967, 89, 447–450. [Google Scholar] [CrossRef]
- Engstrom, R.C. Electrochemical pretreatment of glassy carbon electrodes. Anal. Chem. 1982, 54, 2310–2314. [Google Scholar] [CrossRef]
- Fortier, S.; Le Roy, J.J.; Chen, C.-H.; Vieru, V.; Murugesu, M.; Chibotaru, L.F.; Mindiola, D.J.; Caulton, K.G. A Dinuclear Cobalt Complex Featuring Unprecedented Anodic and Cathodic Redox Switches for Single-Molecule Magnet Activity. J. Am. Chem. Soc. 2013, 135, 14670–14678. [Google Scholar] [CrossRef]
- Banerjee, S.; Sheet, D.; Sarkar, S.; Halder, P.; Paine, T.K. Nickel complexes of ligands derived from (o-hydroxyphenyl) dichalcogenide: Delocalised redox states of nickel and o-chalcogenophenolate ligands. Dalton Trans. 2019, 48, 17355–17363. [Google Scholar] [CrossRef]
- Fourmond, V.; Canaguier, S.; Golly, B.; Field, M.J.; Fontecave, M.; Artero, V. A nickel–manganese catalyst as a biomimic of the active site of NiFe hydrogenases: A combined electrocatalytical and DFT mechanistic study. Energy Environ. Sci. 2011, 4, 2417–2427. [Google Scholar] [CrossRef]
- Elgrishi, N.; McCarthy, B.D.; Rountree, E.S.; Dempsey, J.L. Reaction Pathways of Hydrogen-Evolving Electrocatalysts: Electrochemical and Spectroscopic Studies of Proton-Coupled Electron Transfer Processes. ACS Catal. 2016, 6, 3644–3659. [Google Scholar] [CrossRef]
- Costentin, C.; Passard, G.; Robert, M.; Savéant, J.-M. Concertedness in proton-coupled electron transfer cleavages of carbon-metal bonds illustrated by the reduction of an alkyl cobalt porphyrin. Chem. Sci. 2013, 4, 819–823. [Google Scholar] [CrossRef]
- Hu, X.; Brunschwig, B.S.; Peters, J.C. Electrocatalytic Hydrogen Evolution at Low Overpotentials by Cobalt Macrocyclic Glyoxime and Tetraimine Complexes. J. Am. Chem. Soc. 2007, 129, 8988–8998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letko, C.S.; Panetier, J.A.; Head-Gordon, M.; Tilley, T.D. Mechanism of the Electrocatalytic Reduction of Protons with Diaryldithiolene Cobalt Complexes. J. Am. Chem. Soc. 2014, 136, 9364–9376. [Google Scholar] [CrossRef]
- Derien, S.; Dunach, E.; Perichon, J. From stoichiometry to catalysis: Electroreductive coupling of alkynes and carbon dioxide with nickel-bipyridine complexes. Magnesium ions as the key for catalysis. J. Am. Chem. Soc. 1991, 113, 8447–8454. [Google Scholar] [CrossRef]
- Appel, A.M.; Helm, M.L. Determining the Overpotential for a Molecular Electrocatalyst. ACS Catal. 2014, 4, 630–633. [Google Scholar] [CrossRef]
- Cavell, A.C.; Hartley, C.L.; Liu, D.; Tribble, C.S.; McNamara, W.R. Sulfinato Iron(III) Complex for Electrocatalytic Proton Reduction. Inorg. Chem. 2015, 54, 3325–3330. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Mingot, M.; Porcher, J.-P.; Todorova, T.K.; Fogeron, T.; Mellot-Draznieks, C.; Li, Y.; Fontecave, M. Bioinspired Tungsten Dithiolene Catalysts for Hydrogen Evolution: A Combined Electrochemical, Photochemical, and Computational Study. J. Phys. Chem. B 2015, 119, 13524–13533. [Google Scholar] [CrossRef] [PubMed]
- Rountree, E.S.; McCarthy, B.D.; Eisenhart, T.T.; Dempsey, J.L. Evaluation of Homogeneous Electrocatalysts by Cyclic Voltammetry. Inorg. Chem. 2014, 53, 9983–10002. [Google Scholar] [CrossRef]
- Zhao, P.-H.; Li, J.-R.; Gu, X.-L.; Jing, X.-B.; Liu, X.-F. Diiron and trinuclear NiFe2 dithiolate complexes chelating by PCNCP ligands: Synthetic models of [FeFe]- and [NiFe]-hydrogenases. J. Inorg. Biochem. 2020, 210, 111126. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Engelhard, M.H.; Zhu, Z.; Helm, M.L.; Roberts, J.A.S. Electrodeposition from Acidic Solutions of Nickel Bis(benzenedithiolate) Produces a Hydrogen-Evolving Ni–S Film on Glassy Carbon. ACS Catal. 2014, 4, 90–98. [Google Scholar] [CrossRef]
- Wu, S.; Dou, J.; Zhang, J.; Zhang, S. A simple and economical one-pot method to synthesize high-quality water soluble CdTe QDs. J. Mater. Chem. 2012, 22, 14573–14578. [Google Scholar] [CrossRef]
- Yin, J.; Cogan, N.M.B.; Burke, R.; Hou, Z.; Sowers, K.L.; Krauss, T.D. Size dependence of photocatalytic hydrogen generation for CdTe quantum dots. J. Chem. Phys. 2019, 151, 174707. [Google Scholar] [CrossRef] [PubMed]
- Benazzi, E.; Coni, V.C.; Boni, M.; Mazzaro, R.; Morandi, V.; Natali, M. The role of the capping agent and nanocrystal size in photoinduced hydrogen evolution using CdTe/CdS quantum dot sensitizers. Dalton Trans. 2020, 49, 10212–10223. [Google Scholar] [CrossRef] [PubMed]
- Huo, P.; Uyeda, C.; Goodpaster, J.D.; Peters, J.C.; Miller, T.F. Breaking the Correlation between Energy Costs and Kinetic Barriers in Hydrogen Evolution via a Cobalt Pyridine-Diimine-Dioxime Catalyst. ACS Catal. 2016, 6, 6114–6123. [Google Scholar] [CrossRef]
- Koutsouri, E.; Mitsopoulou, C.A. Photocatalytic Hydrogen Evolution by tris-dithiolene tungsten complexes. Open Chem. 2016, 14, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Armarego, W.L. Purification of Laboratory Chemicals; Butterworth-Heinemann: Oxford, UK, 2017; ISBN 0-12-805456-5. [Google Scholar]
- Arounaguiri, S.; Maiya, B.G. Dipyridophenazine Complexes of Cobalt(III) and Nickel(II): DNA-Binding and Photocleavage Studies. Inorg. Chem. 1996, 35, 4267–4270. [Google Scholar] [CrossRef] [PubMed]
- Bruker, S. V8. 32A; Bruker AXS, Inc.: Madison, WI, USA, 2013. [Google Scholar]
- Sheldrick, G. SADABS—Area Detector Scaling and Adsorption Correction; Bruker AXS: Madison, WI, USA, 2012. [Google Scholar]
- Bruker, A.; Saint, B. AXS, Inc.: Madison, WI, USA, 2004; Search PubMed;(b) Sheldrick, G.M. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2015, 71, 3–8. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Thorn, A.; Dittrich, B.; Sheldrick, G.M. Enhanced rigid-bond restraints. Acta Crystallogr. Sect. A Found. Crystallogr. 2012, 68, 448–451. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | Å | Angle | Deg | Angle | Deg |
|---|---|---|---|---|---|
| Ni1-O1 | 1.834(4) | O1-Ni1-S1 | 90.09(12) | Ni1-O1-C14 | 119.2(3) |
| Ni1-S1 | 2.1588(16) | O1-Ni1-N1 | 172.69(19) | Ni1-S1-C13 | 96.19(18) |
| Ni1-N1 | 1.904(5) | O1-Ni1-N2 | 90.13(18) | Ni1-N1-C1 | 114.8(4) |
| Ni1-N2 | 1.908(5) | S1-Ni1-N1 | 96.38(15) | Ni1-N1-C15 | 127.4(4) |
| O1-C14 | 1.339(6) | S1-Ni1-N2 | 176.02(14) | Ni1-N2-C7 | 115.4(4) |
| S1-C13 | 1.767(5) | N1-Ni1-N2 | 83.6(2) | Ni1-N2-C8 | 126.3(4) |
| N1-C1 | 1.364(7) | ||||
| N2-C7 | 1.358(7) | ||||
| C13-C14 | 1.410 (7) |
| Complex | Overpotential (mV) | Faradaic Efficiency | TON/3 h | TOF (h−1) |
|---|---|---|---|---|
| 1 | 580 | 58% | 4.63 | 1.54 |
| 2 | 720 | 73% | 39.17 | 13.1 |
| [Ni(mp2)]− | 690 | 59% | 14.3 | 4.76 |
| Empirical Formula | C19 H18 Cl2 N2 Ni O S |
|---|---|
| Formula weight | 452.02 |
| Temperature | 120(2) Κ |
| Wavelength | 1.54178 Å |
| Crystal system | Monoclinic |
| Space Group | P21/c |
| Unit cell dimensions | a = 8.2538(18) Å b = 18.097(4) Å, β = 96.238(7)° c = 12.863(3) Å |
| Volume | 1909.9(7) Å3 |
| Z | 4 |
| Density (calculated) | 1.572 Mg/m3 |
| Absorption coefficient | 5.148 mm−1 |
| F(000) | 928 |
| Crystal size | 0.320 × 0.200 × 0.100 mm3 |
| Theta range for data collection | 4.233 to 66.592°. |
| Index ranges | −9 < =h < = 9, −21 < = k < = 21, −15 < = l < = 15 |
| Reflections collected | 22,231 |
| Independent reflections | 3313 [R(int) = 0.1037] |
| Completeness to theta = 66.592o | 98.3% |
| Refinement method | Full-matrix least-squares on F2 |
| Data/Restraints/Parameters | 3313/180/291 |
| Goodness-of-fit on F2 | 1.061 |
| Final R indices [I >2σ(I)] | R1 = 0.0852, wR2 = 0.2529 |
| R indices (all data) | R1 = 0.0994, wR2 = 0.2701 |
| Largest diff. peak and hole | 0.593 and −0.773 e·Å−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamatsos, F.; Bethanis, K.; Mitsopoulou, C.A. Synthesis of Novel Heteroleptic Oxothiolate Ni(II) Complexes and Evaluation of Their Catalytic Activity for Hydrogen Evolution. Catalysts 2021, 11, 401. https://doi.org/10.3390/catal11030401
Kamatsos F, Bethanis K, Mitsopoulou CA. Synthesis of Novel Heteroleptic Oxothiolate Ni(II) Complexes and Evaluation of Their Catalytic Activity for Hydrogen Evolution. Catalysts. 2021; 11(3):401. https://doi.org/10.3390/catal11030401
Chicago/Turabian StyleKamatsos, Fotios, Kostas Bethanis, and Christiana A. Mitsopoulou. 2021. "Synthesis of Novel Heteroleptic Oxothiolate Ni(II) Complexes and Evaluation of Their Catalytic Activity for Hydrogen Evolution" Catalysts 11, no. 3: 401. https://doi.org/10.3390/catal11030401
APA StyleKamatsos, F., Bethanis, K., & Mitsopoulou, C. A. (2021). Synthesis of Novel Heteroleptic Oxothiolate Ni(II) Complexes and Evaluation of Their Catalytic Activity for Hydrogen Evolution. Catalysts, 11(3), 401. https://doi.org/10.3390/catal11030401