
Turning Carbon Dioxide and Ethane into Ethanol by Solar-Driven Heterogeneous Photocatalysis over RuO2- and NiO-co-Doped SrTiO3

 ,
,  ,
,  ,
, 
Abstract
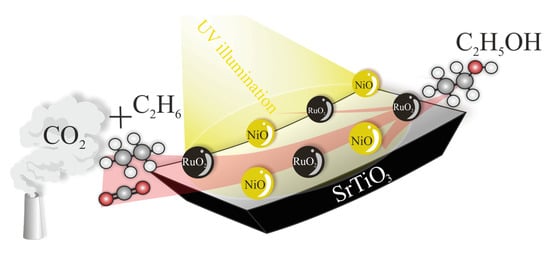
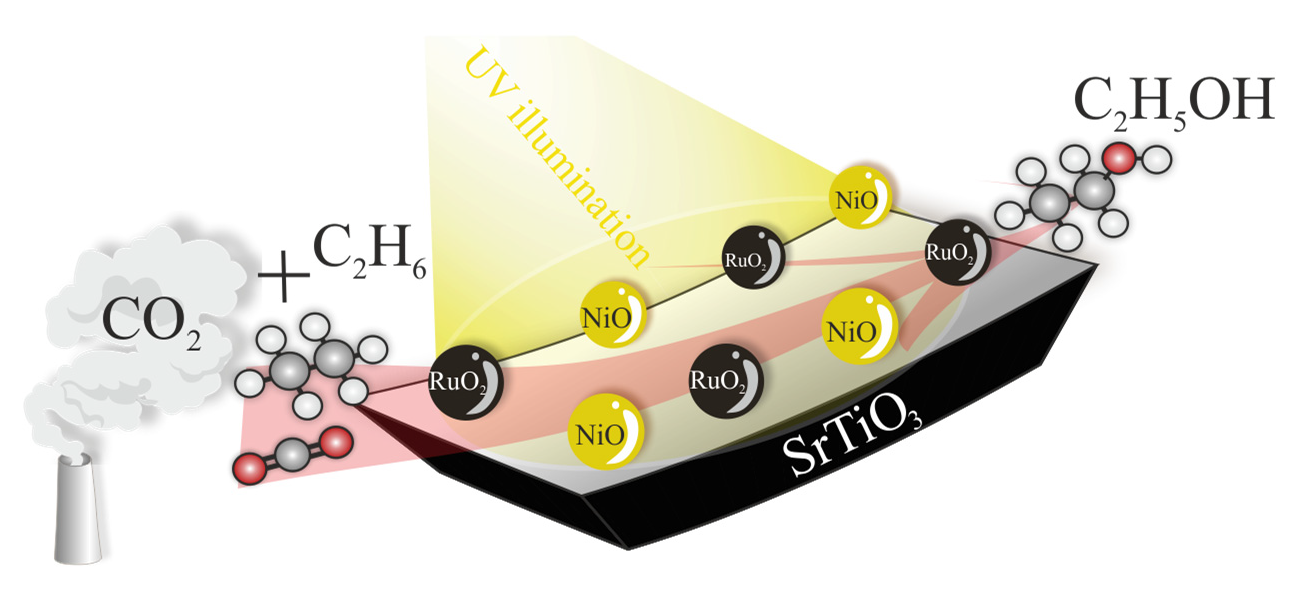
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
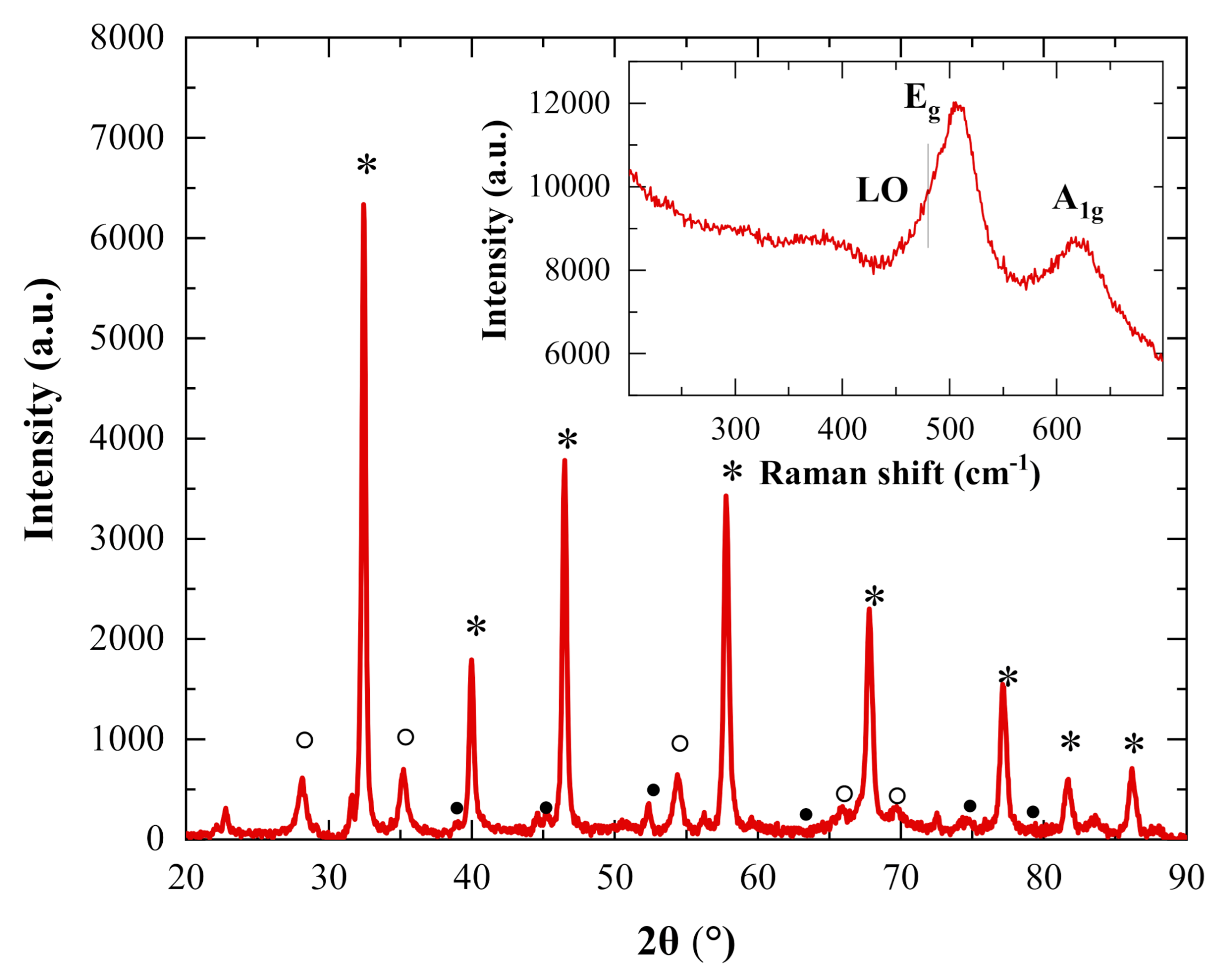
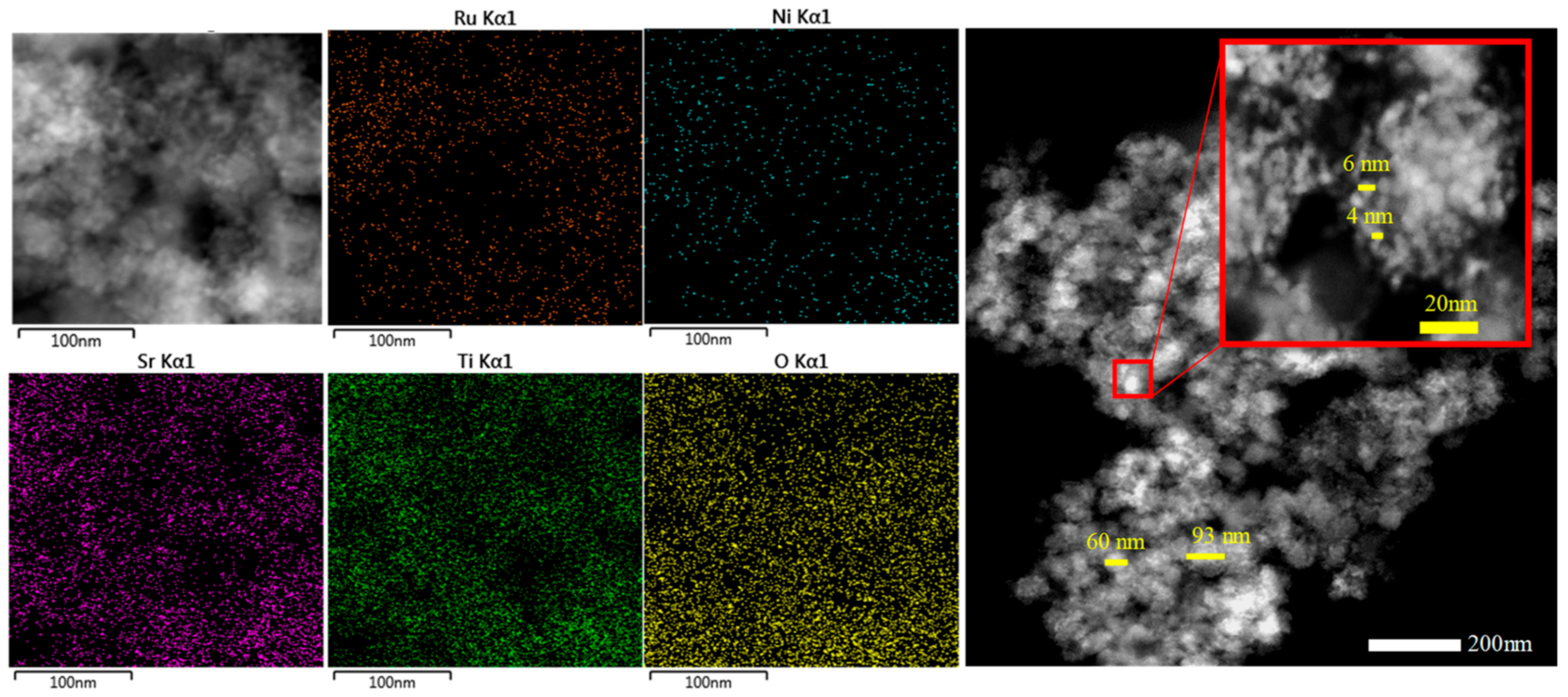
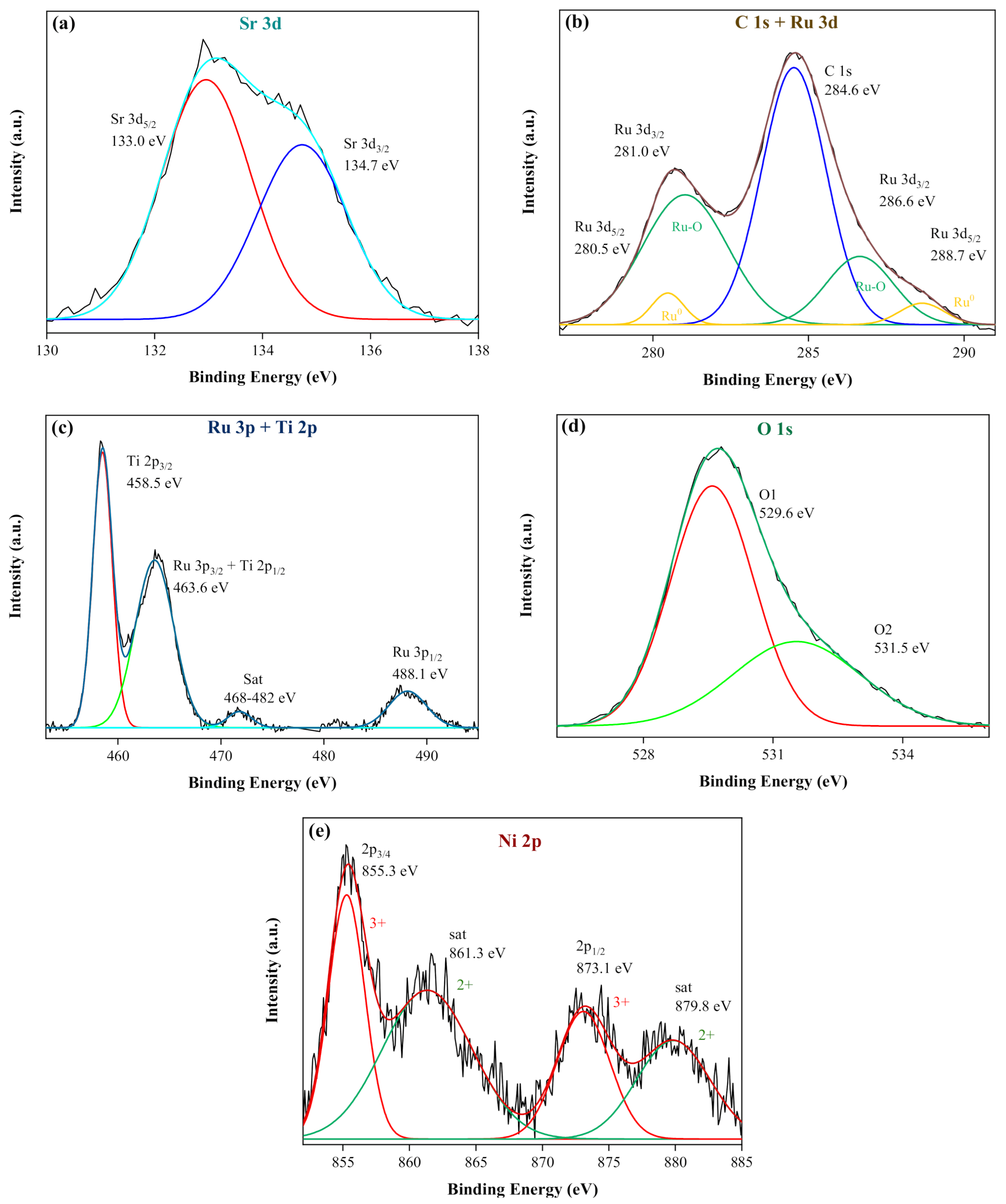
2.1. Characterization of the Photocatalysts
2.2. Assessment of the Photocatalytic Activity
2.2.1. Effect of the Co-Doping Metal Loading
2.2.2. Effect of the Reaction Temperature
2.2.3. Effect of the Optical Radiation Wavelength
2.2.4. Effect of the Photocatalyst Reuse
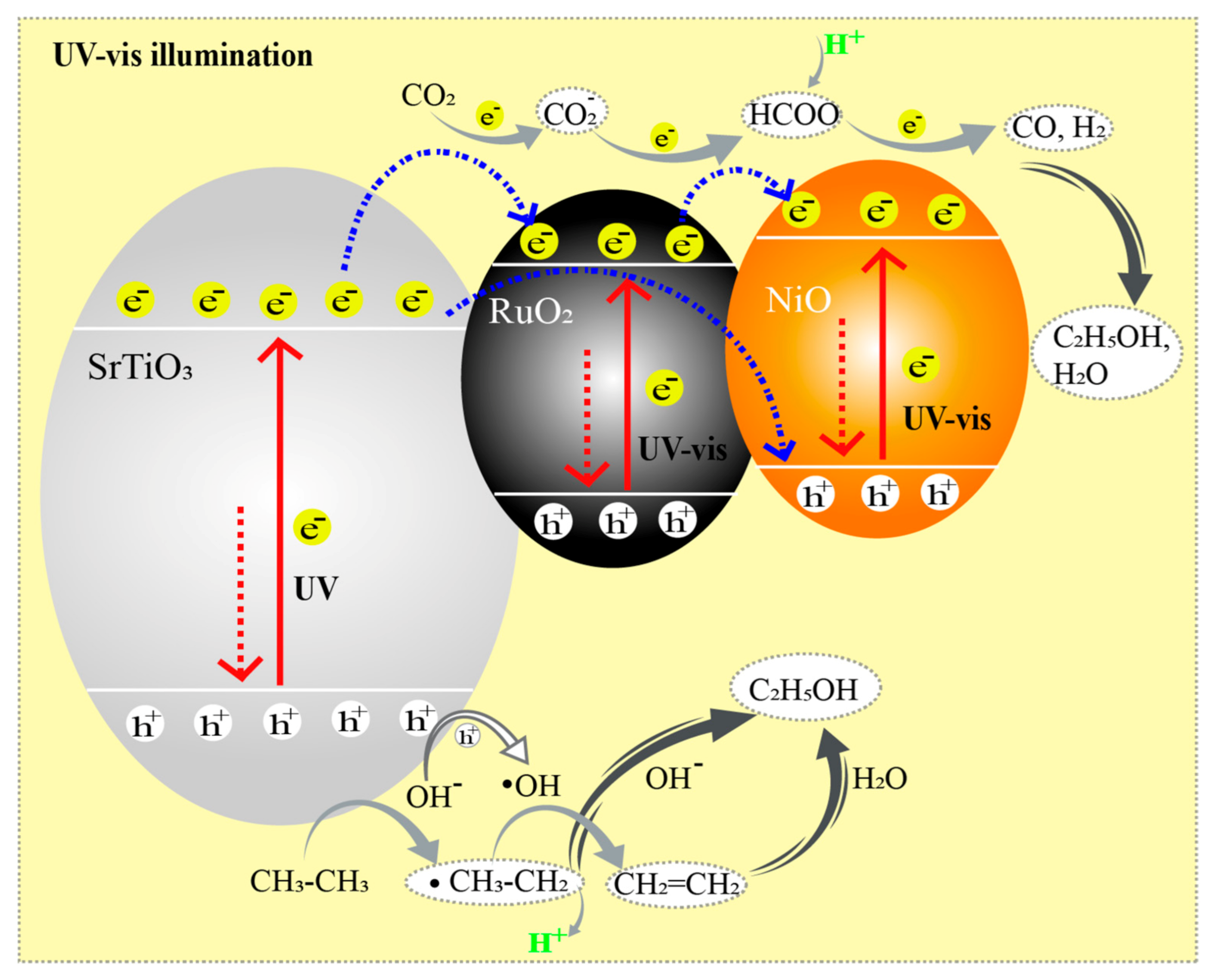
2.3. Reaction Mechanism Proposal
3. Materials and Methods
3.1. Chemicals
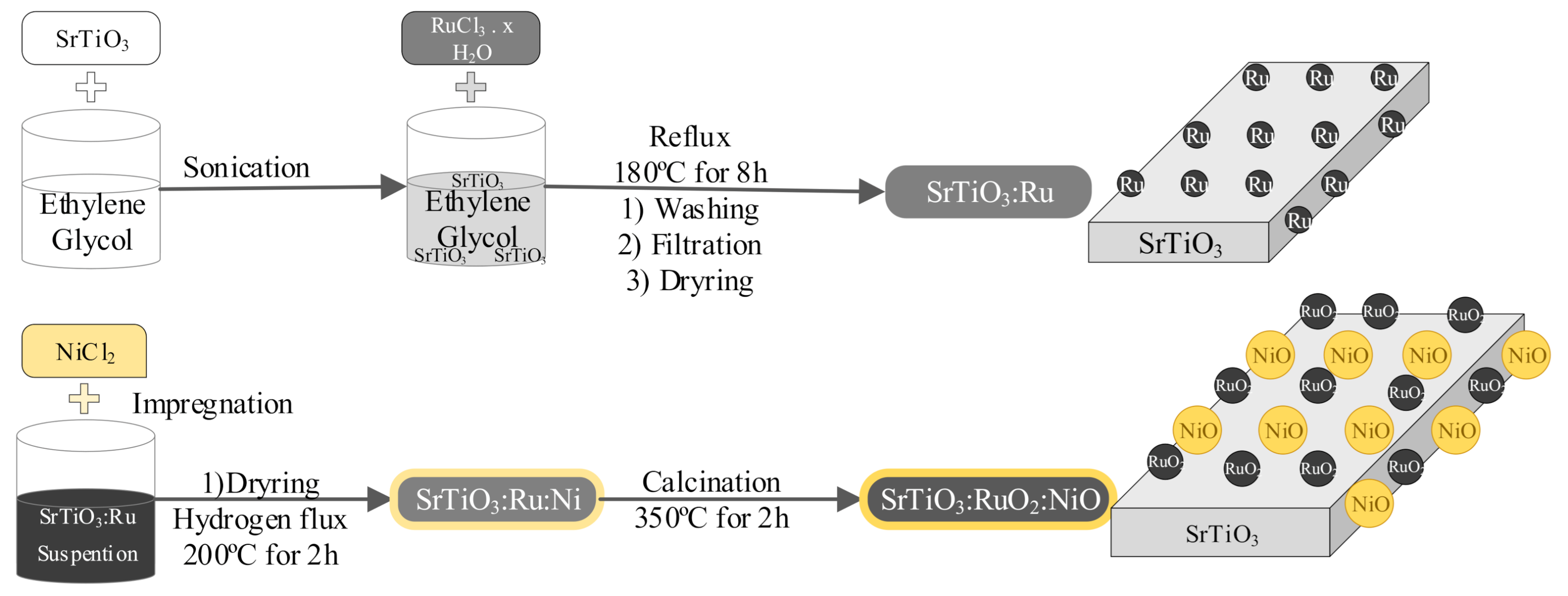
3.2. Preparation of the SrTiO3:RuO2:NiO Photocatalyst
3.3. Photocatalyst Characterization
3.4. Experimental Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chang, X.; Wang, T.; Gong, J. CO2 photo-reduction: Insights into CO2 activation and reaction on surfaces of photocatalysts. Energy Environ. Sci. 2016, 9, 2177–2196. [Google Scholar] [CrossRef]
- Li, M.; Li, P.; Chang, K.; Wang, T.; Liu, L.; Kang, Q.; Ouyang, S.; Ye, J. Highly efficient and stable photocatalytic reduction of CO2 to CH4 over Ru loaded NaTaO3. Chem. Commun. 2015, 51, 7645–7648. [Google Scholar] [CrossRef] [PubMed]
- Niu, F.; Shen, S.; Zhang, N.; Chen, J.; Guo, L. Cobaloxime coenzyme catalyzing artificial photosynthesis for hydrogen generation over CdS nanocrystals. Appl. Catal. B-Environ. 2016, 199, 134–141. [Google Scholar] [CrossRef]
- Noureldin, M.M.B.; Elbashir, N.O.; Gabriel, K.J.; El-Halwagi, M.M. A Process Integration Approach to the Assessment of CO2 Fixation through Dry Reforming. ACS Sustain. Chem. Eng. 2015, 3, 625–636. [Google Scholar] [CrossRef]
- Börner, A.; Franke, R. Hydroformylation: Fundamentals, Processes, and Applications in Organic Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Franke, R.; Selent, D.; Börner, A. Applied Hydroformylation. Chem. Rev. 2012, 112, 5675–5732. [Google Scholar] [CrossRef]
- Xie, Z.; Xu, Y.; Xie, M.; Chen, X.; Lee, J.H.; Stavitski, E.; Kattel, S.; Chen, J.G. Reactions of CO2 and ethane enable CO bond insertion for production of C3 oxygenates. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Myint, M.; Yan, B.; Wan, J.; Zhao, S.; Chen, J.G. Reforming and oxidative dehydrogenation of ethane with CO2 as a soft oxidant over bimetallic catalysts. J. Catal. 2016, 343, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Skoufa, Z.; Heracleous, E.; Lemonidou, A.A. On ethane ODH mechanism and nature of active sites over NiO-based catalysts via isotopic labeling and methanol sorption studies. J. Catal. 2015, 322, 118–129. [Google Scholar] [CrossRef]
- Shen, Z.; Liu, J.; Xu, H.; Yue, Y.; Hua, W.; Shen, W. Dehydrogenation of ethane to ethylene over a highly efficient Ga2O3/HZSM-5 catalyst in the presence of CO2. Appl. Catal. A Gen. 2009, 356, 148–153. [Google Scholar] [CrossRef]
- Nair, V.; Muñoz-Batista, M.J.; Fernández-García, M.; Luque, R.; Colmenares, J.C. Thermo-Photocatalysis: Environmental and Energy Applications. ChemSusChem 2019, 12, 2098–2116. [Google Scholar] [CrossRef]
- Tu, W.; Zhou, Y.; Zou, Z. Photocatalytic Conversion of CO2 into Renewable Hydrocarbon Fuels: State-of-the-Art Accomplishment, Challenges, and Prospects. Adv. Mater. 2014, 26, 4607–4626. [Google Scholar] [CrossRef]
- Barber, J.; Tran, P.D. From natural to artificial photosynthesis. J. R. Soc. Interface R. Soc. 2013, 10. [Google Scholar] [CrossRef] [Green Version]
- Ola, O.; Maroto-Valer, M.M. Review of material design and reactor engineering on TiO2 photocatalysis for CO2 reduction. J. Photochem. Photobiol. C 2015, 24, 16–42. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Zhao, J.; Li, Y.; Zhao, W.; Zeng, Y.; Wang, C. Photocatalytic CO2 reduction over SrTiO3: Correlation between surface structure and activity. Appl. Surf. Sci. 2018, 447, 627–635. [Google Scholar] [CrossRef]
- Shao, K.; Wang, Y.; Iqbal, M.; Lin, L.; Wang, K.; Zhang, X.; He, M.; He, T. Modification of Ag nanoparticles on the surface of SrTiO3 particles and resultant influence on photoreduction of CO2. Appl. Surf. Sci. 2018, 434, 717–724. [Google Scholar] [CrossRef]
- Kang, H.W.; Lim, S.N.; Park, S.B. Co-doping schemes to enhance H2 evolution under visible light irradiation over SrTiO3:Ni/M (M = La or Ta) prepared by spray pyrolysis. Int. J Hydrogen Energy 2012, 37, 5540–5549. [Google Scholar] [CrossRef]
- Kang, H.W.; Park, S.B. H2 evolution under visible light irradiation from aqueous methanol solution on SrTiO3: Cr/Ta prepared by spray pyrolysis from polymeric precursor. Int. J Hydrogen Energy 2011, 36, 9496–9504. [Google Scholar] [CrossRef]
- Takata, T.; Domen, K. Defect engineering of photocatalysts by doping of aliovalent metal cations for efficient water splitting. J. Phys. Chem. C 2009, 113, 19386–19388. [Google Scholar] [CrossRef]
- Niishiro, R.; Kato, H.; Kudo, A. Nickel and either tantalum or niobium-codoped TiO2 and SrTiO3 photocatalysts with visible-light response for H2 or O2 evolution from aqueous solutions. Phys. Chem. Chem. Phys. 2005, 7, 2241–2245. [Google Scholar] [CrossRef]
- Mateo, D.; Albero, J.; García, H. Titanium-Perovskite-Supported RuO2 Nanoparticles for Photocatalytic CO2 Methanation. Joule 2019, 3, 1949–1962. [Google Scholar] [CrossRef]
- Ola, O.; Mercedes Maroto-Valer, M. Role of catalyst carriers in CO2 photoreduction over nanocrystalline nickel loaded TiO2-based photocatalysts. J. Catal. 2014, 309, 300–308. [Google Scholar] [CrossRef]
- Li, Q.-S.; Domen, K.; Naito, S.; Onishi, T.; Tamaru, K. Photocatalytic synthesis and photodecomposition of ammonia over SrTiO3 and BaTiO3 based catalysts. Chem. Lett. 1983, 12, 321–324. [Google Scholar] [CrossRef]
- Kubelka, P.; Munk, F. A contribution to the optics of pigments. Z. Tech. Phys. 1931, 12, 193. [Google Scholar]
- Zhang, C.; Jiang, N.; Xu, S.; Li, Z.; Liu, X.; Cheng, T.; Han, A.; Lv, H.; Sun, W.; Hou, Y. Towards high visible light photocatalytic activity in rare earth and N co-doped SrTiO3: A first principles evaluation and prediction. RSC Adv. 2017, 7, 16282–16289. [Google Scholar] [CrossRef] [Green Version]
- Ramya, E.; Rao, M.V.; Jyothi, L.; Rao, D.N. Photoluminescence and nonlinear optical properties of transition metal (Ag, Ni, Mn) doped ZnO nanoparticles. J. Nanosci. Nanotechnol. 2018, 18, 7072–7077. [Google Scholar] [CrossRef]
- García-Ramírez, P.; Ramírez-Morales, E.; Solis Cortazar, J.C.; Sirés, I.; Silva-Martínez, S. Influence of ruthenium doping on UV- and visible-light photoelectrocatalytic color removal from dye solutions using a TiO2 nanotube array photoanode. Chemosphere 2021, 267, 128925. [Google Scholar] [CrossRef]
- Mondal, A.; Giri, N.; Sarkar, S.; Majumdar, S.; Ray, R. Tuning the photocatalytic activity of ZnO by TM (TM = Fe, Co, Ni) doping. Mat. Sci. Semicon. Proc. 2019, 91, 333–340. [Google Scholar] [CrossRef]
- Jia, A.; Su, Z.; Lou, L.-L.; Liu, S. Synthesis and characterization of highly-active nickel and lanthanum co-doped SrTiO3. Solid State Sci. 2010, 12, 1140–1145. [Google Scholar] [CrossRef]
- Mitsui, T.; Nouma, S.; Landolt, B. Numerical Data and Functional Relation in Science and Technology: Crystal and Solid State Physics; New Series, Group 2I; Springer: Berlin/Heidelberg, Germany, 1982. [Google Scholar]
- Glazer, A. Simple ways of determining perovskite structures. Acta Crystallogr. A 1975, 31, 756–762. [Google Scholar] [CrossRef] [Green Version]
- Al-Shomar, S. Investigation the effect of doping concentration in Ruthenium-doped TiO2 thin films for solar cells and sensors applications. Mater. Res. Express. 2020, 7, 036409. [Google Scholar] [CrossRef]
- Langford, J.I.; Wilson, A. Scherrer after sixty years: A survey and some new results in the determination of crystallite size. J. Appl. Crystallogr. 1978, 11, 102–113. [Google Scholar] [CrossRef]
- Scherrer, P. Bestimmung der inneren Struktur und der Größe von Kolloidteilchen mittels Röntgenstrahlen. Kolloidchem. Ein Lehrb. 1912, 387–409. [Google Scholar] [CrossRef]
- Chekuri, R.D.; Tirukkovalluri, S.R. Synthesis of cobalt doped titania nano material assisted by gemini surfactant: Characterization and application in degradation of Acid Red under visible light irradiation. Sajog S. Afr. J. Chem. Eng. 2017, 24, 183–195. [Google Scholar] [CrossRef]
- Yahmadi, B.; Kamoun, O.; Alhalaili, B.; Alleg, S.; Vidu, R.; Kamoun Turki, N. Physical Investigations of (Co, Mn) Co-Doped ZnO Nanocrystalline Films. Nanomaterials 2020, 10, 1507. [Google Scholar] [CrossRef]
- Álvarez, M.A.; Bobadilla, L.F.; Garcilaso, V.; Centeno, M.A.; Odriozola, J.A. CO2 reforming of methane over Ni-Ru supported catalysts: On the nature of active sites by operando DRIFTS study. J. CO2 Util. 2018, 24, 509–515. [Google Scholar] [CrossRef]
- Vasquez, R.P. SrTiO3 by XPS. Surf. Sci. Spectra 1992, 1, 129–135. [Google Scholar] [CrossRef]
- Guedes, E.; Abud, F.; Martins, H.; Abbate, M.; Jardim, R.; Mossanek, R. Role of Ti-Ru interaction in SrTi0.5Ru0.5O3: Physical properties, x-ray spectroscopy, and cluster model calculations. Phys. Rev. B 2019, 100. [Google Scholar] [CrossRef]
- Mahala, C.; Basu, M. Nanosheets of NiCo2O4/NiO as efficient and stable electrocatalyst for oxygen evolution reaction. ACS Omega 2017, 2, 7559–7567. [Google Scholar] [CrossRef] [Green Version]
- Mayer, B.; Neumann, M. XPS satellites in transition metal oxides. J. Electron. Spectrosc. 1996, 81, 63–67. [Google Scholar] [CrossRef]
- Shi, J.; Hui, F.; Yuan, J.; Yu, Q.; Mei, S.; Zhang, Q.; Li, J.; Wang, W.; Yang, J.; Lu, J. Ru-Ti oxide based catalysts for HCl oxidation: The favorable oxygen species and influence of Ce additive. Catalysts 2019, 9, 108. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.D.; Lee, C.W.; Jin, K.; Im, S.W.; Nam, K.T. Current status and bioinspired perspective of electrochemical conversion of CO2 to a long-chain hydrocarbon. J. Phys. Chem. Lett. 2017, 8, 538–545. [Google Scholar] [CrossRef]
- Zhang, M.; Yu, Y. Dehydration of ethanol to ethylene. Ind. Eng. Chem. 2013, 52, 9505–9514. [Google Scholar] [CrossRef]
- Oliveira, T.K.R.; Rosset, M.; Perez-Lopez, O.W. Ethanol dehydration to diethyl ether over Cu-Fe/ZSM-5 catalysts. Catal. Commun. 2018, 104, 32–36. [Google Scholar] [CrossRef]
- Schaaf, T.; Grünig, J.; Schuster, M.R.; Rothenfluh, T.; Orth, A. Methanation of CO2-storage of renewable energy in a gas distribution system. Energy Sustain. Soc. 2014, 4, 1–14. [Google Scholar] [CrossRef]
- Saladin, F.; Alxneit, I. Temperature dependence of the photochemical reduction of CO2 in the presence of H2O at the solid/gas interface of TiO2. J. Chem. Soc. Faraday Trans. 1997, 93, 4159–4163. [Google Scholar] [CrossRef]
- Moores, A.; Goettmann, F. The plasmon band in noble metal nanoparticles: An introduction to theory and applications. New J. Chem. 2006, 30, 1121–1132. [Google Scholar] [CrossRef]
- Devi, S.; Korake, P.; Achary, S.N.; Gupta, N.M. Genesis of enhanced photoactivity of CdS/Nix nanocomposites for visible-light-driven splitting of water. Int. J. Hydrogen Energy 2014, 39, 19424–19433. [Google Scholar] [CrossRef]
- Dai, W.; Xu, H.; Yu, J.; Hu, X.; Luo, X.; Tu, X.; Yang, L. Photocatalytic reduction of CO2 into methanol and ethanol over conducting polymers modified Bi2WO6 microspheres under visible light. Appl. Surf. Sci 2015, 356, 173–180. [Google Scholar] [CrossRef]
- Cheng, M.; Yang, S.; Chen, R.; Zhu, X.; Liao, Q.; Huang, Y. Visible light responsive CdS sensitized TiO2 nanorod array films for efficient photocatalytic reduction of gas phase CO2. Mol. Catal. 2018, 448, 185–194. [Google Scholar] [CrossRef]
- Panayotov, D.; Kondratyuk, P.; Yates, J. Photooxidation of a mustard gas simulant over TiO2− SiO2 mixed-oxide photocatalyst: Site poisoning by oxidation products and reactivation. Langmuir 2004, 20, 3674–3678. [Google Scholar] [CrossRef]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous catalyst deactivation and regeneration: A review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef] [Green Version]
- Fatiadi, A.J. Addition and cycloaddition reactions of tetracyanoethylene (TCNE) in organic chemistry. Synthesis 1987, 1987, 749–789. [Google Scholar] [CrossRef]
- Wang, V.C.C.; Can, M.; Pierce, E.; Ragsdale, S.W.; Armstrong, F.A. A Unified Electrocatalytic Description of the Action of Inhibitors of Nickel Carbon Monoxide Dehydrogenase. J. Am. Chem. Soc. 2013, 135, 2198–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmeier, A.; Hall, S.; Ragsdale, S.W.; Armstrong, F.A. Selective Visible-Light-Driven CO2 Reduction on a p-Type Dye-Sensitised NiO Photocathode. J. Am. Chem. Soc. 2014, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Dey, G.R. Chemical Reduction of CO2 to Different Products during Photo Catalytic Reaction on TiO2 under Diverse Conditions: An Overview. J. Nat. Gas Chem. 2007, 16, 217–226. [Google Scholar] [CrossRef]
- Bergner, J.B.; Öberg, K.I.; Rajappan, M. Oxygen atom reactions with C2H6, C2H4, and C2H2 in ices. Astrophys. J. 2019, 874, 115. [Google Scholar] [CrossRef] [Green Version]
- DeMore, W.B. Reaction of O(1D) with hydrocarbons in liquid argon. J. Phys. Chem. 1969, 73, 391–395. [Google Scholar] [CrossRef]
- Zumdahl, S.S. Chemical Principles, 6th ed.; D. C. Heath: Lexington, MA, USA, 1992. [Google Scholar]
- Albero, J.; Peng, Y.; Garcia, H. Photocatalytic CO2 reduction to C2+ products. ACS Catal. 2020, 10, 5734–5749. [Google Scholar] [CrossRef]





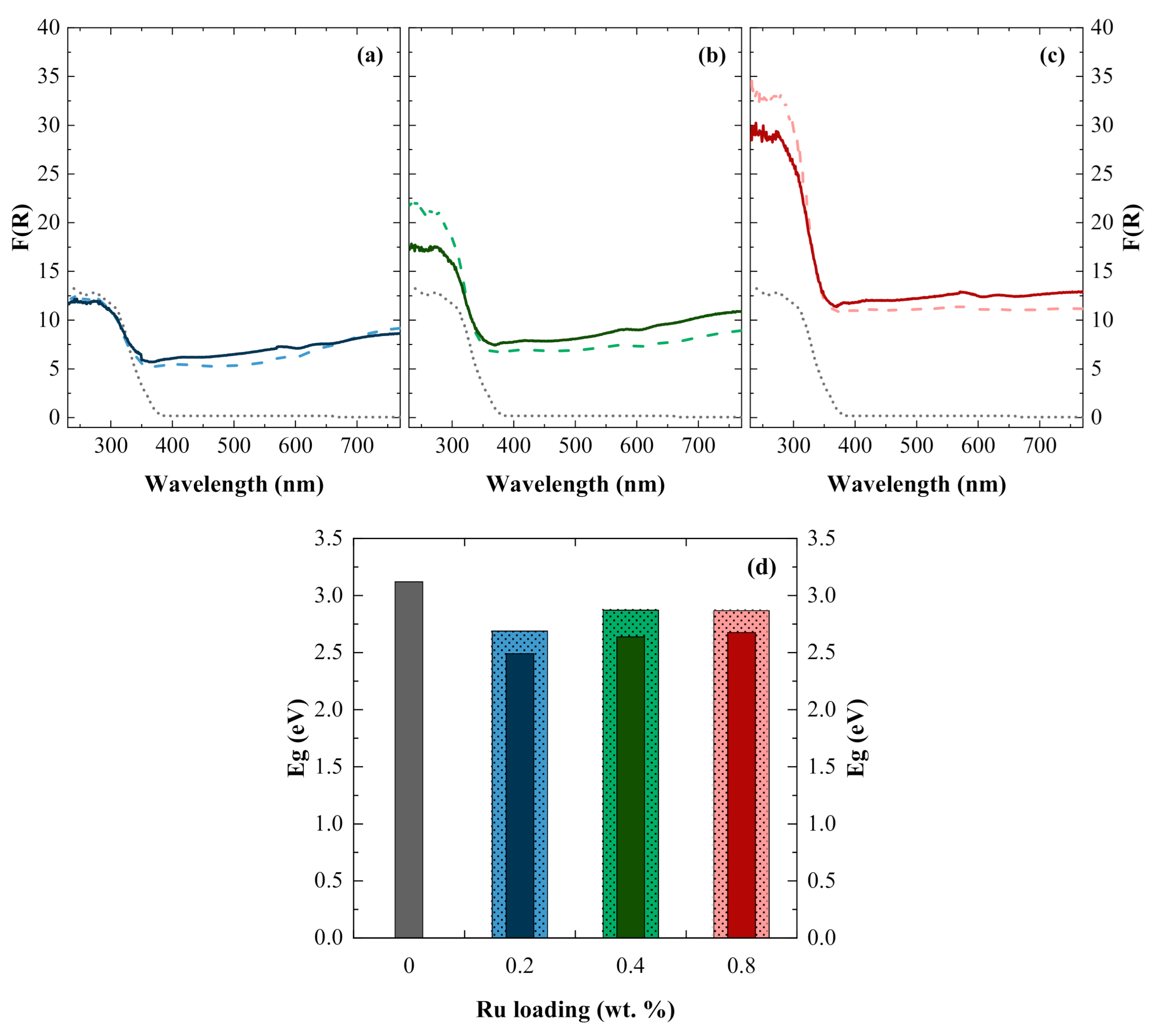
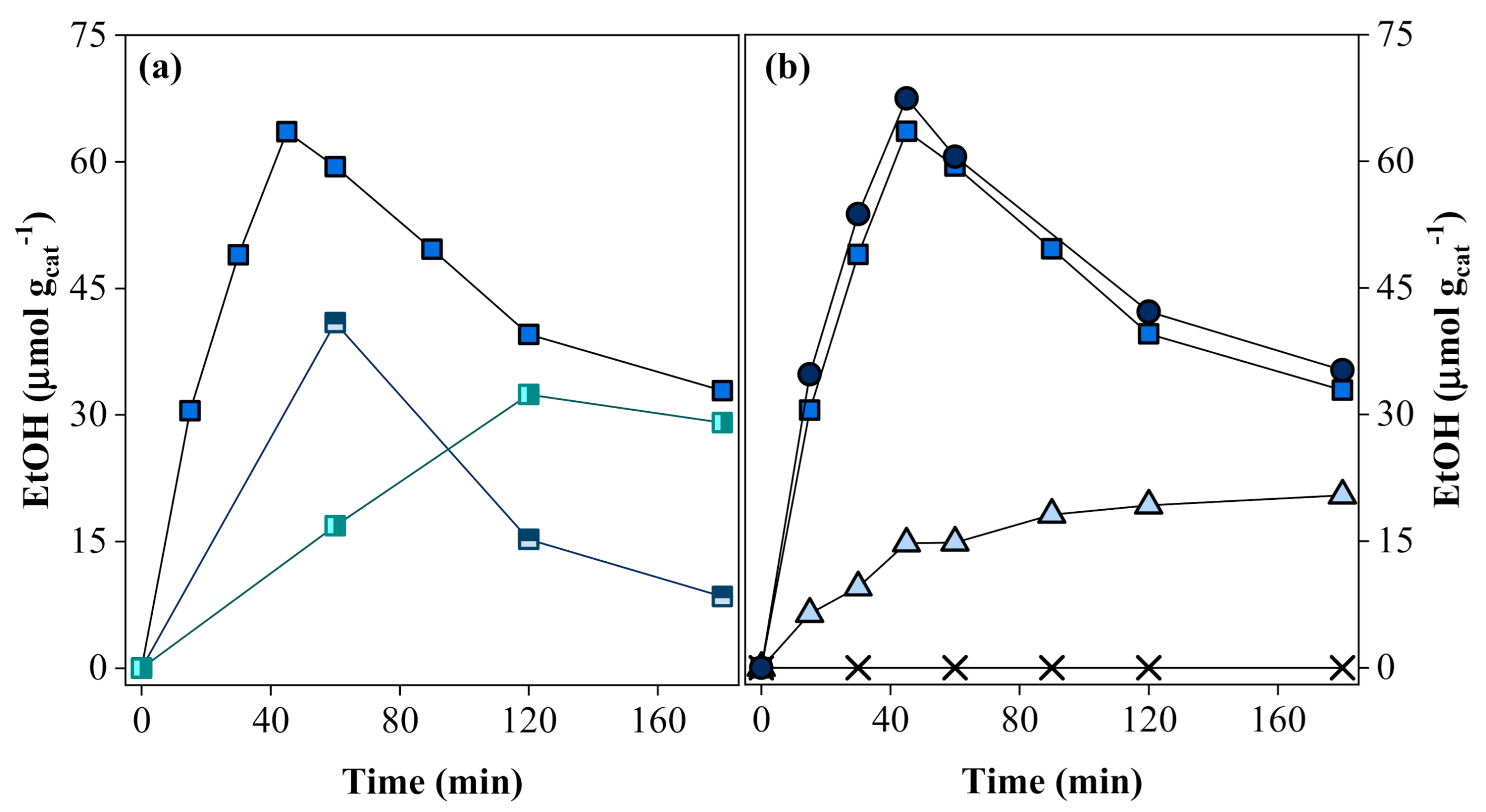
 ), 0.4 wt.% (
), 0.4 wt.% (  ), and 0.8 wt.% (
), and 0.8 wt.% (  ), keeping a [Ru]:[Ni] molar ratio of 1:1; (b) [Ru]:[Ni] molar ratios of 1:0 (
), keeping a [Ru]:[Ni] molar ratio of 1:1; (b) [Ru]:[Ni] molar ratios of 1:0 (  ), 1:0.3 (
), 1:0.3 (  ), 1:1 ( ), and 1:2 (
), 1:1 ( ), and 1:2 (  ), fixing the Ru content on 0.8 wt. %. Experimental conditions: PC2H6 = 1.01 bar; PCO2 = 0.35 bar; 20 mg of photocatalyst; T = 200 °C; and I = 1000 W m−2.
), 0.4 wt.% ( ), and 0.8 wt.% ( ), keeping a [Ru]:[Ni] molar ratio of 1:1; (b) [Ru]:[Ni] molar ratios of 1:0 ( ), 1:0.3 ( ), 1:1 ( ), and 1:2 ( ), fixing the Ru content on 0.8 wt. %. Experimental conditions: PC2H6 = 1.01 bar; PCO2 = 0.35 bar; 20 mg of photocatalyst; T = 200 °C; and I = 1000 W m−2.
), fixing the Ru content on 0.8 wt. %. Experimental conditions: PC2H6 = 1.01 bar; PCO2 = 0.35 bar; 20 mg of photocatalyst; T = 200 °C; and I = 1000 W m−2.
), 0.4 wt.% ( ), and 0.8 wt.% ( ), keeping a [Ru]:[Ni] molar ratio of 1:1; (b) [Ru]:[Ni] molar ratios of 1:0 ( ), 1:0.3 ( ), 1:1 ( ), and 1:2 ( ), fixing the Ru content on 0.8 wt. %. Experimental conditions: PC2H6 = 1.01 bar; PCO2 = 0.35 bar; 20 mg of photocatalyst; T = 200 °C; and I = 1000 W m−2.
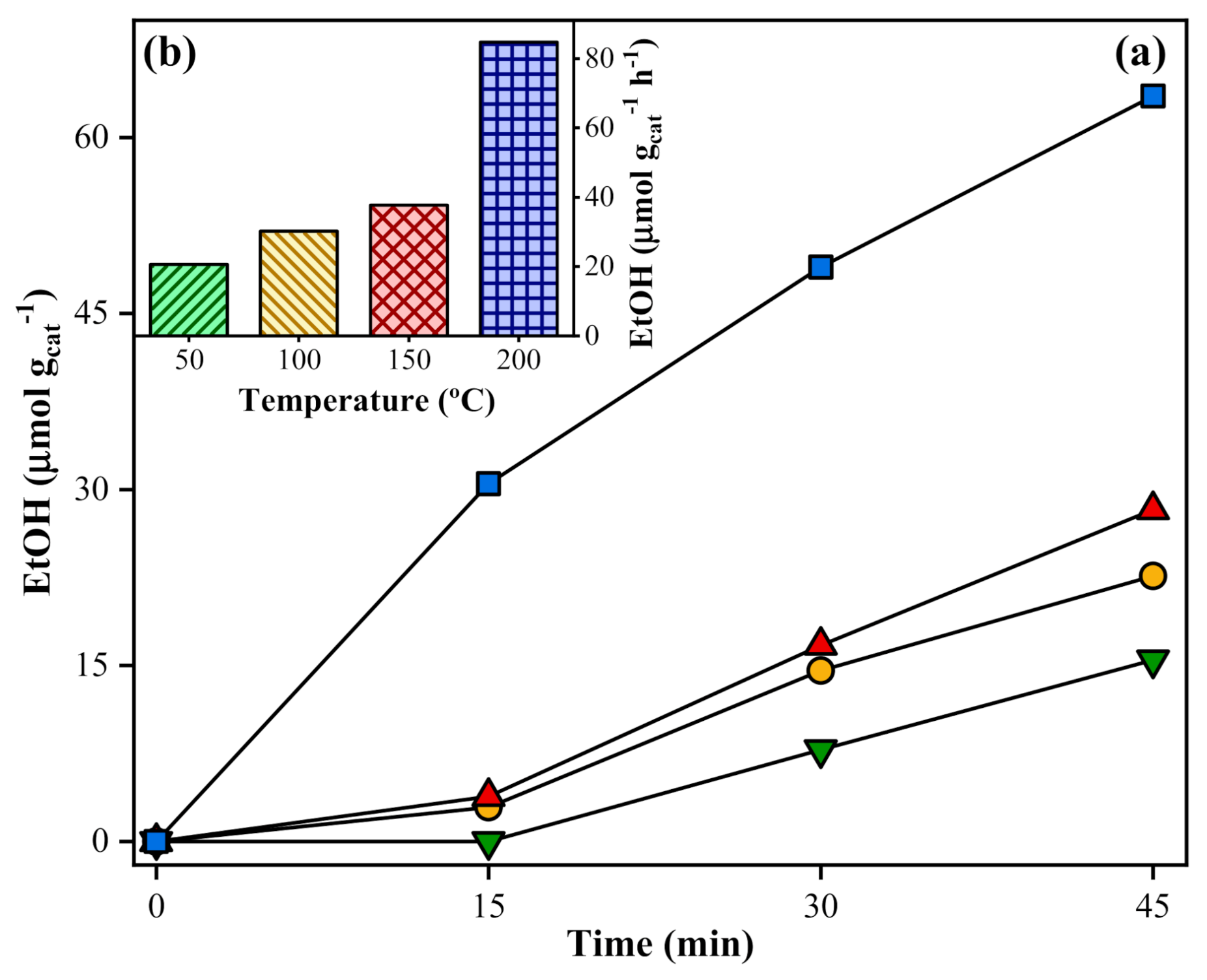
 ); 100 °C (
); 100 °C (  ); 150°C (
); 150°C (  ), and 200°C (
), and 200°C (  ). Experimental conditions: 20 mg of SrTiO3:RuO2:NiO; [Ru]:[Ni] = 1:1 (molar); Ru = 0.8 wt. %; PC2H6 = 1.01 bar; PCO2 = 0.35 bar; and I = 1000 W m−2.
); 100 °C ( ); 150°C ( ), and 200°C ( ). Experimental conditions: 20 mg of SrTiO3:RuO2:NiO; [Ru]:[Ni] = 1:1 (molar); Ru = 0.8 wt. %; PC2H6 = 1.01 bar; PCO2 = 0.35 bar; and I = 1000 W m−2.
). Experimental conditions: 20 mg of SrTiO3:RuO2:NiO; [Ru]:[Ni] = 1:1 (molar); Ru = 0.8 wt. %; PC2H6 = 1.01 bar; PCO2 = 0.35 bar; and I = 1000 W m−2.
); 100 °C ( ); 150°C ( ), and 200°C ( ). Experimental conditions: 20 mg of SrTiO3:RuO2:NiO; [Ru]:[Ni] = 1:1 (molar); Ru = 0.8 wt. %; PC2H6 = 1.01 bar; PCO2 = 0.35 bar; and I = 1000 W m−2.
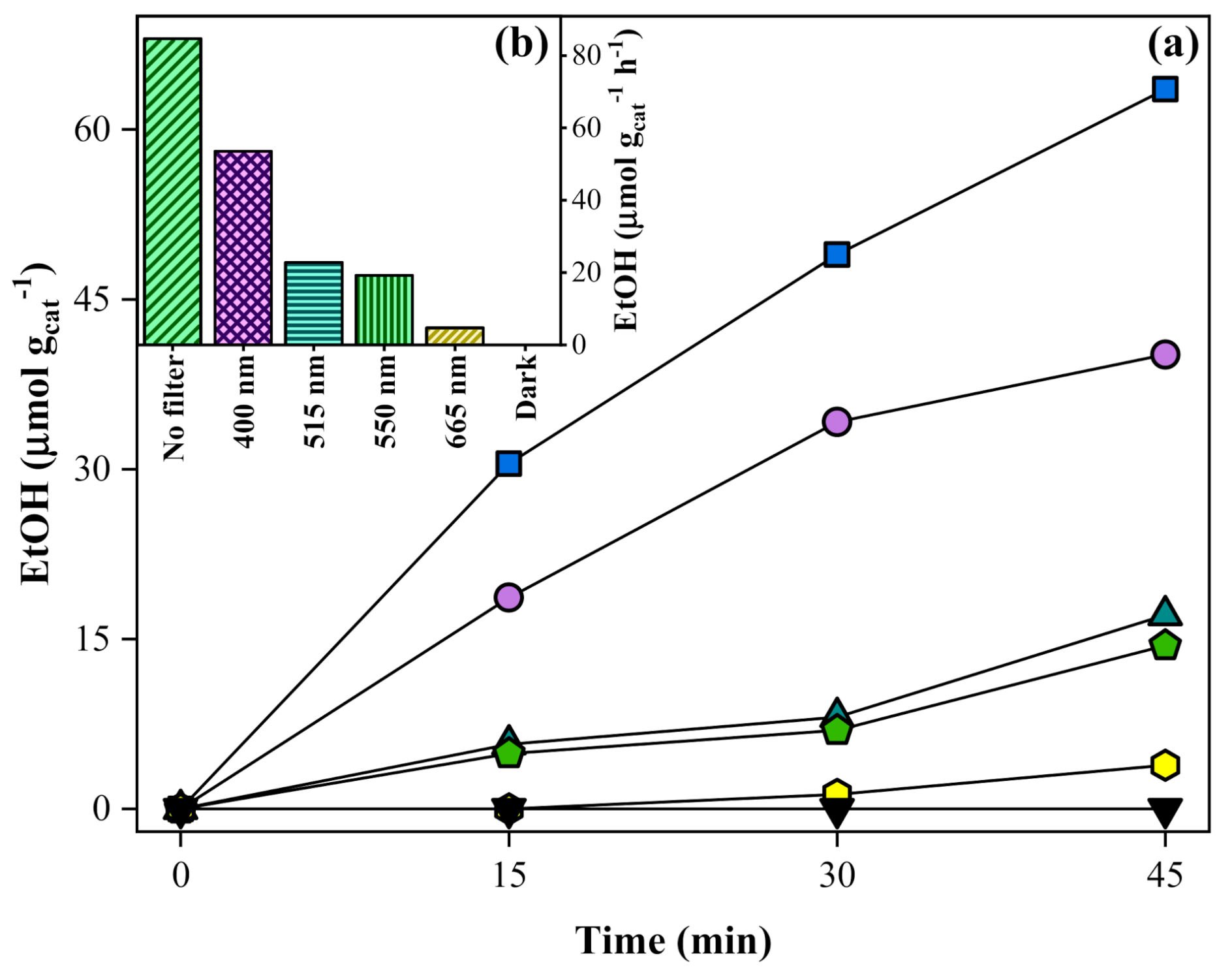
 ), 400 nm (
), 400 nm (  ), 515 nm (
), 515 nm (  ), 550 nm (
), 550 nm (  ), 665 nm (
), 665 nm (  ), and dark (
), and dark (  , covering the photoreactor with aluminum foil). Experimental conditions: 20 mg of SrTiO3:RuO2:NiO; [Ru]:[Ni] = 1:1 (molar); Ru = 0.8 wt. %; PC2H6 = 1.01 bar; PCO2 = 0.35 bar; and T = 200 °C.
), 400 nm ( ), 515 nm ( ), 550 nm ( ), 665 nm ( ), and dark ( , covering the photoreactor with aluminum foil). Experimental conditions: 20 mg of SrTiO3:RuO2:NiO; [Ru]:[Ni] = 1:1 (molar); Ru = 0.8 wt. %; PC2H6 = 1.01 bar; PCO2 = 0.35 bar; and T = 200 °C.
, covering the photoreactor with aluminum foil). Experimental conditions: 20 mg of SrTiO3:RuO2:NiO; [Ru]:[Ni] = 1:1 (molar); Ru = 0.8 wt. %; PC2H6 = 1.01 bar; PCO2 = 0.35 bar; and T = 200 °C.
), 400 nm ( ), 515 nm ( ), 550 nm ( ), 665 nm ( ), and dark ( , covering the photoreactor with aluminum foil). Experimental conditions: 20 mg of SrTiO3:RuO2:NiO; [Ru]:[Ni] = 1:1 (molar); Ru = 0.8 wt. %; PC2H6 = 1.01 bar; PCO2 = 0.35 bar; and T = 200 °C.
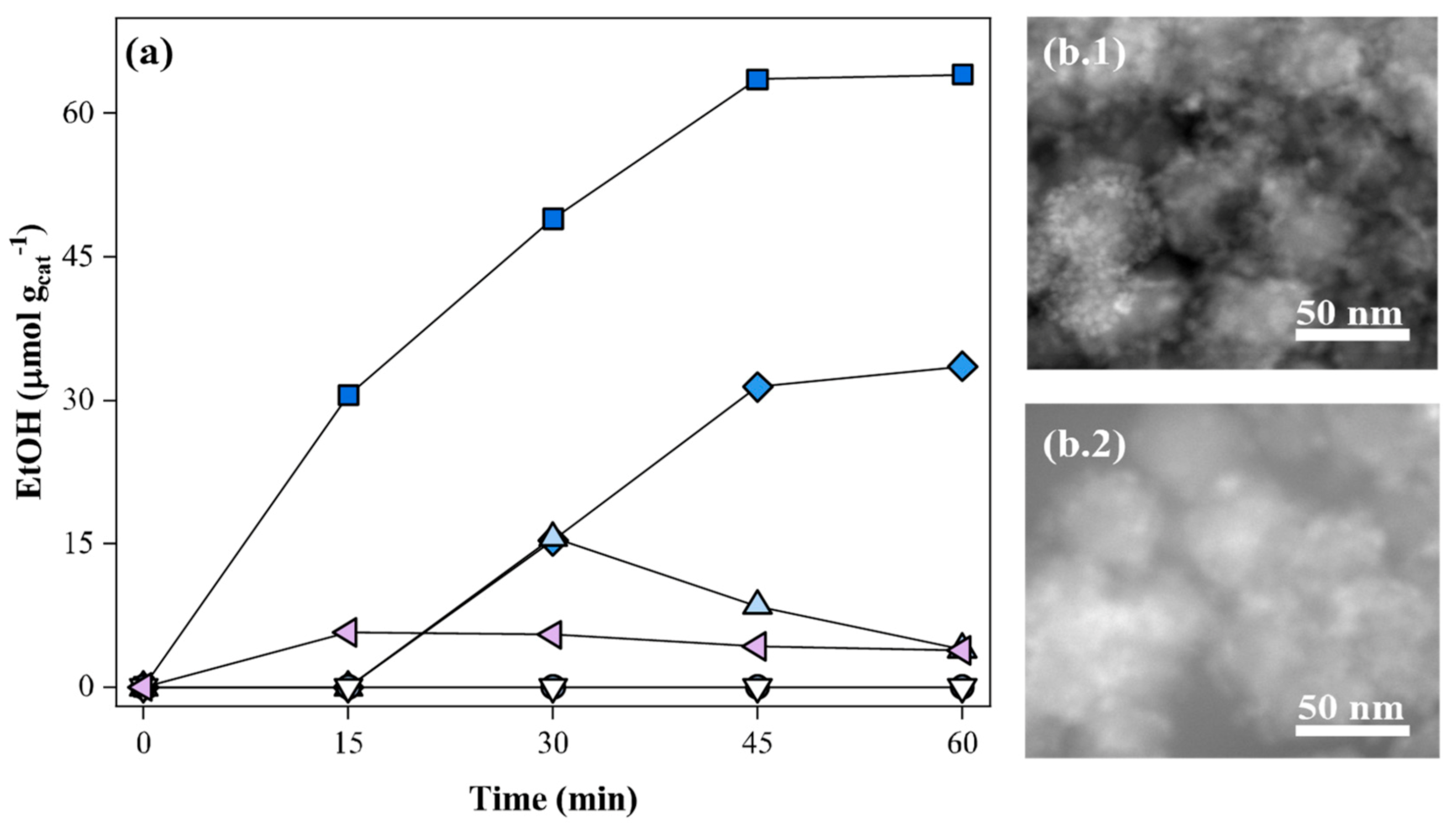
 ), 2 cycles (
), 2 cycles (  ), 3 cycles (
), 3 cycles (  ) and 4 cycles (
) and 4 cycles (  ) of the photocatalyst use, and (ii) reactivation of the photocatalyst, resulting from the 4th cycle, with H2 flow (
) of the photocatalyst use, and (ii) reactivation of the photocatalyst, resulting from the 4th cycle, with H2 flow (  , at 200 °C for 2 h) or air-driven calcination (
, at 200 °C for 2 h) or air-driven calcination (  , at 350 °C for 2 h). Experimental conditions: 20 mg of SrTiO3:RuO2:NiO; [Ru]:[Ni] = 1:1 (molar); Ru = 0.8 wt. %; PC2H6 = 1.01 bar; PCO2 = 0.35 bar; T = 200 °C and I = 1000 W m−2. (b) TEM images of the (.1) fresh and (.2) inactivated photocatalyst (after the 4th use).
), 2 cycles ( ), 3 cycles ( ) and 4 cycles ( ) of the photocatalyst use, and (ii) reactivation of the photocatalyst, resulting from the 4th cycle, with H2 flow ( , at 200 °C for 2 h) or air-driven calcination ( , at 350 °C for 2 h). Experimental conditions: 20 mg of SrTiO3:RuO2:NiO; [Ru]:[Ni] = 1:1 (molar); Ru = 0.8 wt. %; PC2H6 = 1.01 bar; PCO2 = 0.35 bar; T = 200 °C and I = 1000 W m−2. (b) TEM images of the (.1) fresh and (.2) inactivated photocatalyst (after the 4th use).
, at 350 °C for 2 h). Experimental conditions: 20 mg of SrTiO3:RuO2:NiO; [Ru]:[Ni] = 1:1 (molar); Ru = 0.8 wt. %; PC2H6 = 1.01 bar; PCO2 = 0.35 bar; T = 200 °C and I = 1000 W m−2. (b) TEM images of the (.1) fresh and (.2) inactivated photocatalyst (after the 4th use).
), 2 cycles ( ), 3 cycles ( ) and 4 cycles ( ) of the photocatalyst use, and (ii) reactivation of the photocatalyst, resulting from the 4th cycle, with H2 flow ( , at 200 °C for 2 h) or air-driven calcination ( , at 350 °C for 2 h). Experimental conditions: 20 mg of SrTiO3:RuO2:NiO; [Ru]:[Ni] = 1:1 (molar); Ru = 0.8 wt. %; PC2H6 = 1.01 bar; PCO2 = 0.35 bar; T = 200 °C and I = 1000 W m−2. (b) TEM images of the (.1) fresh and (.2) inactivated photocatalyst (after the 4th use).
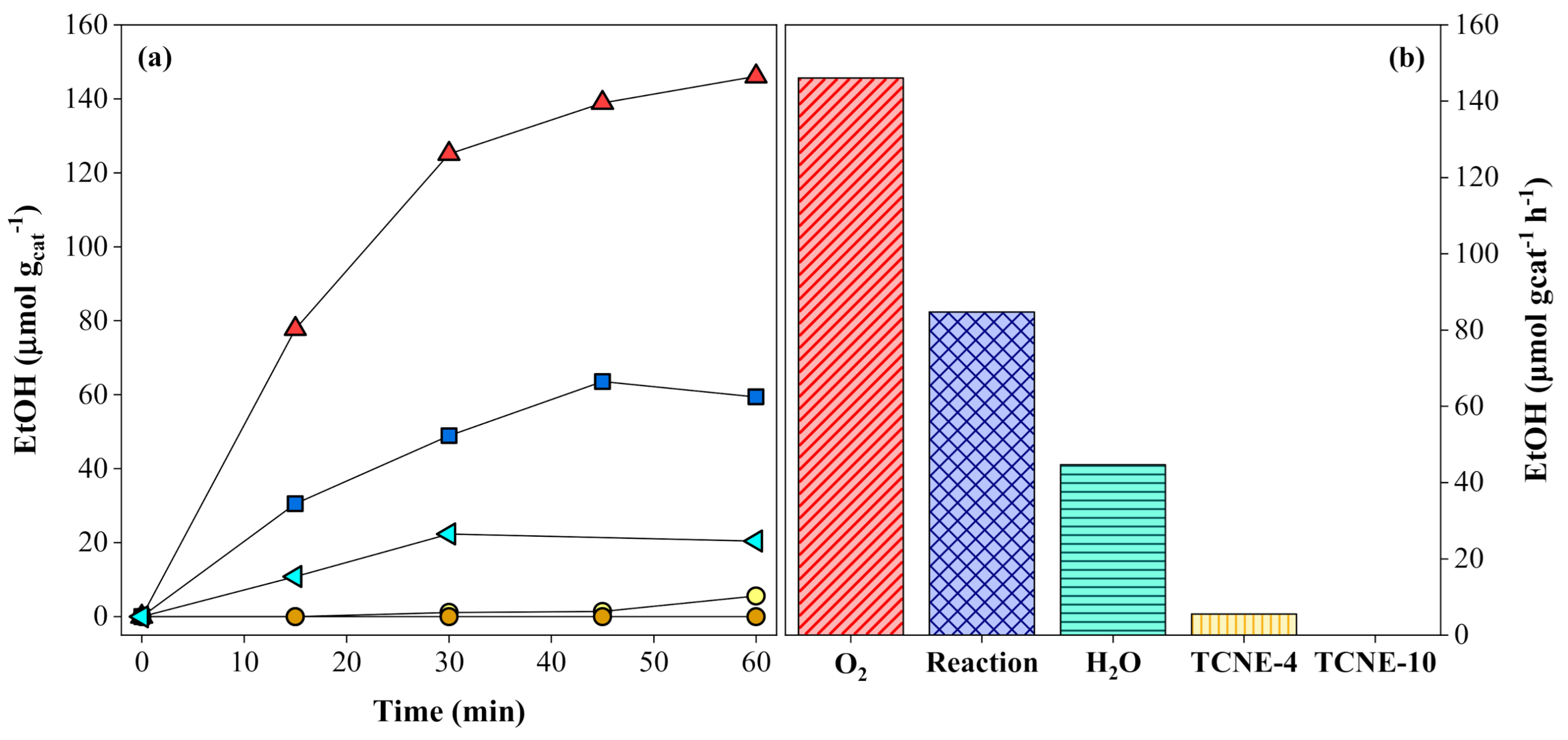
 ) and the presence of electron scavengers: 4 (
) and the presence of electron scavengers: 4 (  ) and 10 (
) and 10 (  ) µmol of TCNE; 0.2 mmol of O2 (
) µmol of TCNE; 0.2 mmol of O2 (  ); or 0.3 µmol of H2O (
); or 0.3 µmol of H2O (  ). Experimental conditions: 20 mg of SrTiO3:RuO2:NiO; [Ru]:[Ni] = 1:1 (molar); Ru = 0.8 wt. %; PC2H6 = 1.01 bar; PCO2 = 0.35 bar; T = 200 °C and I = 1000 W m-2.
) and the presence of electron scavengers: 4 ( ) and 10 ( ) µmol of TCNE; 0.2 mmol of O2 ( ); or 0.3 µmol of H2O ( ). Experimental conditions: 20 mg of SrTiO3:RuO2:NiO; [Ru]:[Ni] = 1:1 (molar); Ru = 0.8 wt. %; PC2H6 = 1.01 bar; PCO2 = 0.35 bar; T = 200 °C and I = 1000 W m-2.
). Experimental conditions: 20 mg of SrTiO3:RuO2:NiO; [Ru]:[Ni] = 1:1 (molar); Ru = 0.8 wt. %; PC2H6 = 1.01 bar; PCO2 = 0.35 bar; T = 200 °C and I = 1000 W m-2.
) and the presence of electron scavengers: 4 ( ) and 10 ( ) µmol of TCNE; 0.2 mmol of O2 ( ); or 0.3 µmol of H2O ( ). Experimental conditions: 20 mg of SrTiO3:RuO2:NiO; [Ru]:[Ni] = 1:1 (molar); Ru = 0.8 wt. %; PC2H6 = 1.01 bar; PCO2 = 0.35 bar; T = 200 °C and I = 1000 W m-2.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paulista, L.O.; Albero, J.; Martins, R.J.E.; Boaventura, R.A.R.; Vilar, V.J.P.; Silva, T.F.C.V.; García, H. Turning Carbon Dioxide and Ethane into Ethanol by Solar-Driven Heterogeneous Photocatalysis over RuO2- and NiO-co-Doped SrTiO3. Catalysts 2021, 11, 461. https://doi.org/10.3390/catal11040461
Paulista LO, Albero J, Martins RJE, Boaventura RAR, Vilar VJP, Silva TFCV, García H. Turning Carbon Dioxide and Ethane into Ethanol by Solar-Driven Heterogeneous Photocatalysis over RuO2- and NiO-co-Doped SrTiO3. Catalysts. 2021; 11(4):461. https://doi.org/10.3390/catal11040461
Chicago/Turabian StylePaulista, Larissa O., Josep Albero, Ramiro J. E. Martins, Rui A. R. Boaventura, Vítor J. P. Vilar, Tânia F. C. V. Silva, and Hermenegildo García. 2021. "Turning Carbon Dioxide and Ethane into Ethanol by Solar-Driven Heterogeneous Photocatalysis over RuO2- and NiO-co-Doped SrTiO3" Catalysts 11, no. 4: 461. https://doi.org/10.3390/catal11040461
APA StylePaulista, L. O., Albero, J., Martins, R. J. E., Boaventura, R. A. R., Vilar, V. J. P., Silva, T. F. C. V., & García, H. (2021). Turning Carbon Dioxide and Ethane into Ethanol by Solar-Driven Heterogeneous Photocatalysis over RuO2- and NiO-co-Doped SrTiO3. Catalysts, 11(4), 461. https://doi.org/10.3390/catal11040461