
Methane to Methanol through Heterogeneous Catalysis and Plasma Catalysis




Abstract
:1. Introduction
2. Heterogeneous Catalysis
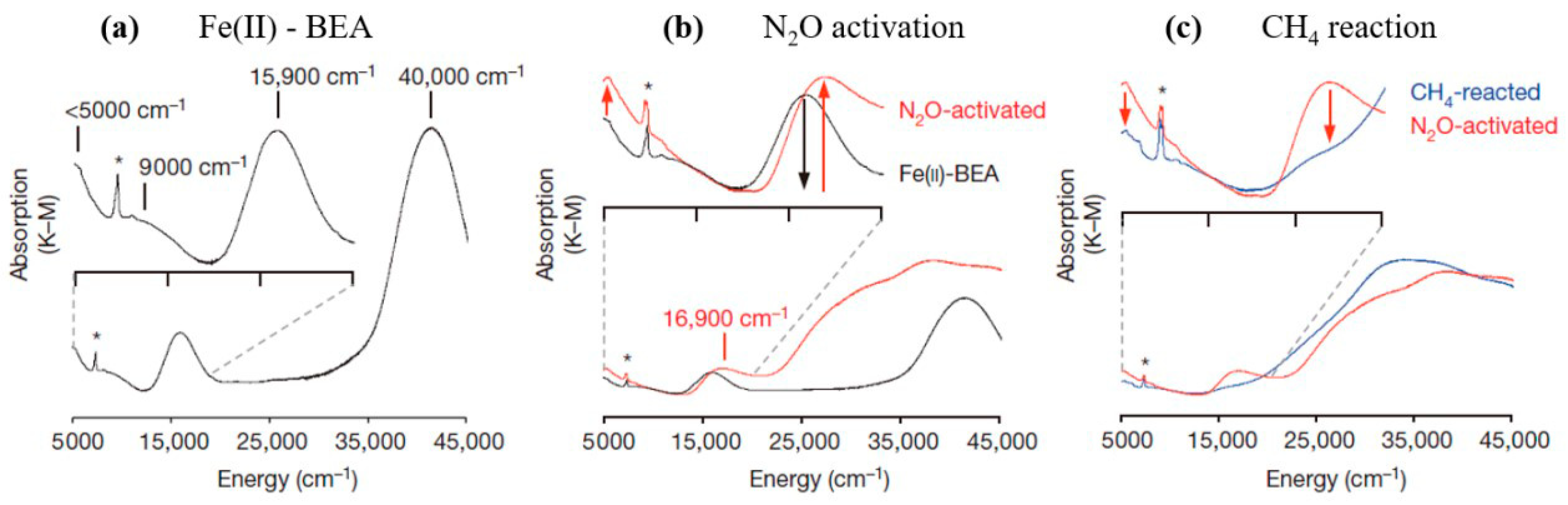
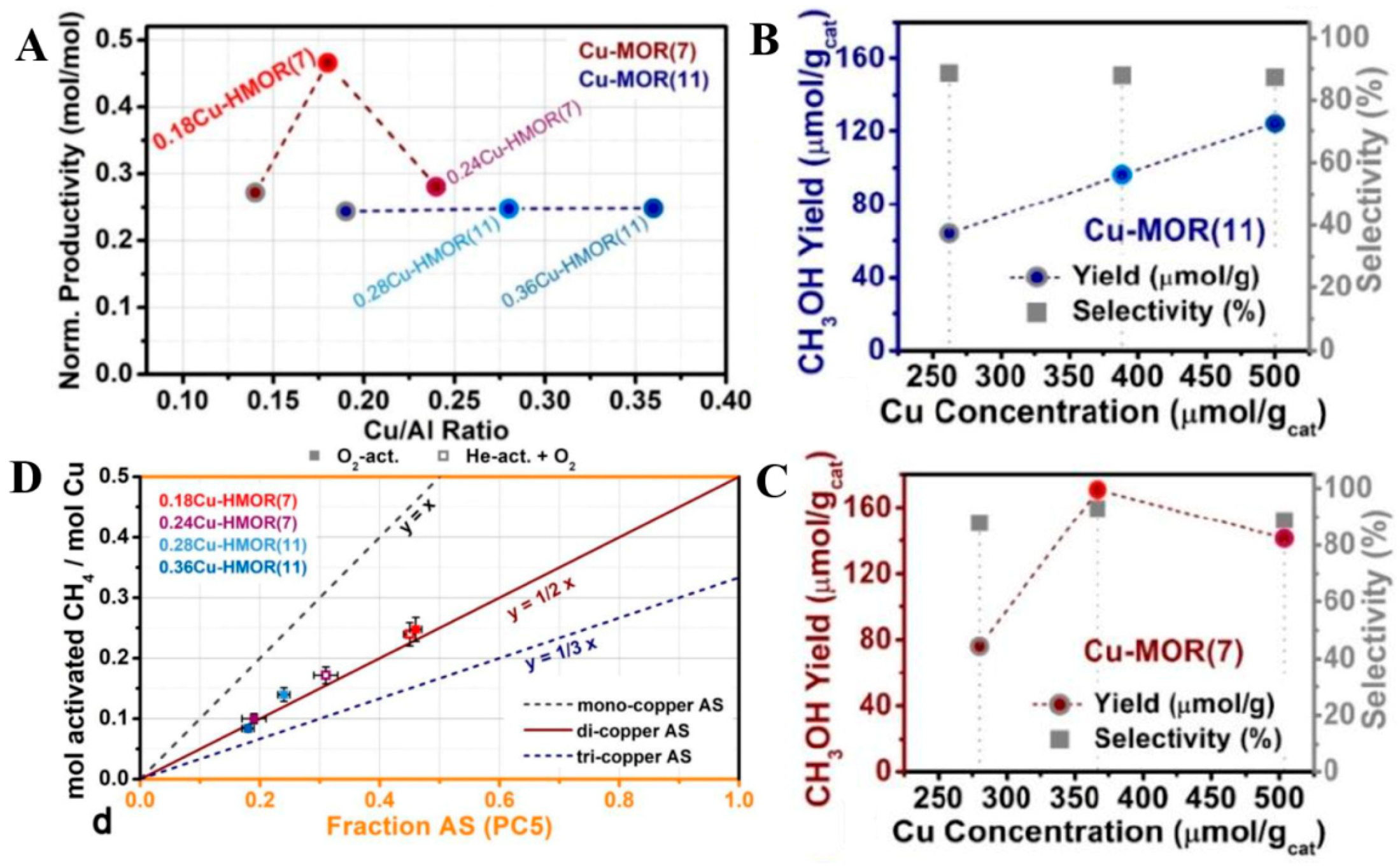
2.1. Nitrous Oxide as Oxidant
2.2. Hydrogen Peroxide as an Oxidant
2.3. Oxygen as Oxidants
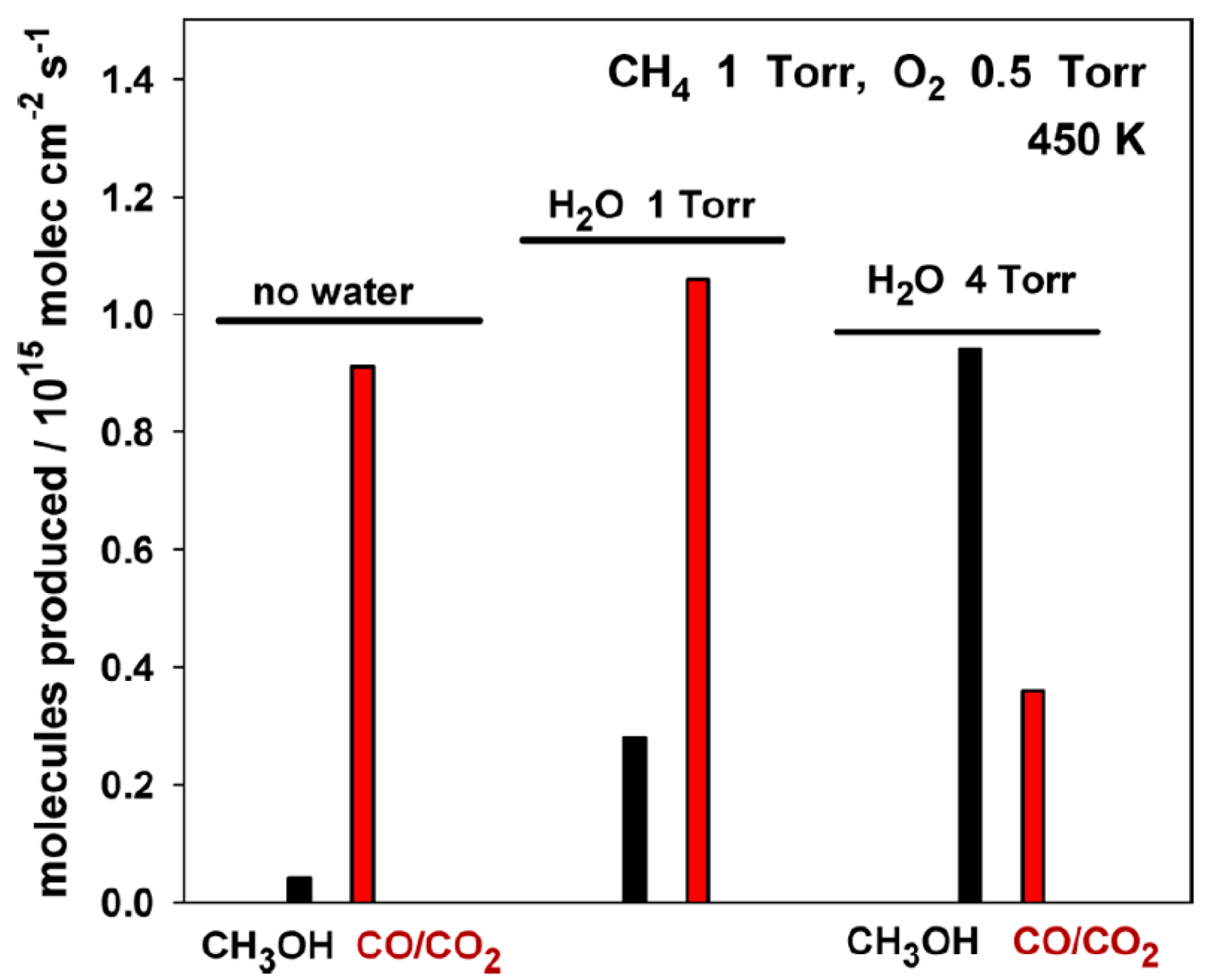
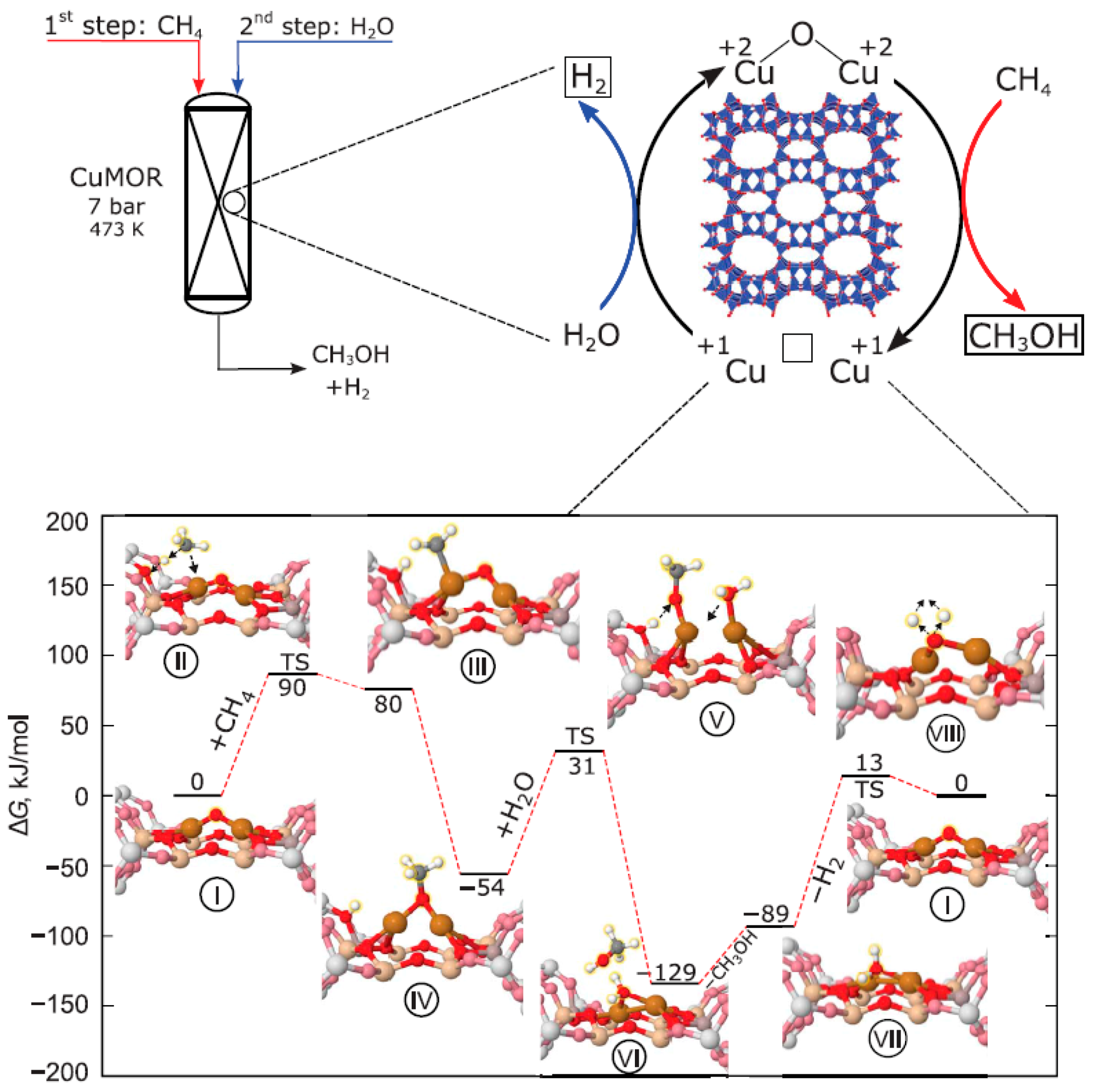
2.4. Water as Oxidant or Co-Oxidant
3. Plasma Catalysis
3.1. Nitrous Oxide as an Oxidant
3.2. Oxygen or Air as an Oxidant
3.3. Water as an Oxidant
3.4. Discussion
4. Outlook and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- IEA. World Energy-Outlook 2019; Part A: World Global Energy Trend, Outlook for Natural Gas; IEA: Paris, France, 2019; pp. 176–187. [Google Scholar]
- Schwach, P.; Pan, X.; Bao, X. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef]
- Ravi, M.; Ranocchiari, M.; Van Bokhoven, J.A. The Direct Catalytic Oxidation of Methane to Methanol A Critical Assessment. Angew. Chem. Int. Ed. 2017, 56, 16464–16483. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Yang, Y.; Xu, Y.; Etim, U.J.; Qiao, K.; Xu, B.; Yan, Z. A review of the direct oxidation of methane to methanol. Chin. J. Catal. 2016, 37, 1206–1215. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, B.; Zhang, Q.; Deng, W.; Wang, Y.; Yang, Y. Recent advances in heterogeneous selective oxidation catalysis for sustainable chemistry. Chem. Soc. Rev. 2014, 43, 3480–3524. [Google Scholar] [CrossRef] [PubMed]
- Caballero, A.; Perez, P.J. Methane as a raw material in synthetic chemistry the final frontier. Chem. Soc. Rev. 2013, 42, 8809–8820. [Google Scholar] [CrossRef]
- Zakaria, Z.; Kamarudin, S.K. Direct conversion technologies of methane to methanol: An overview. Renew. Sustain. Energy Rev. 2016, 65, 250–261. [Google Scholar] [CrossRef]
- Raynes, S.; Shah, M.A.; Taylor, R.A. Direct conversion of methane to methanol with zeolites: Towards understanding the role of extra-framework d-block metal and zeolite framework type. Dalton Trans. 2019, 48, 10364–10384. [Google Scholar] [CrossRef]
- Meng, X.; Cui, X.; Rajan, N.P.; Yu, L.; Deng, D.; Bao, X. Direct Methane Conversion under Mild Condition by Thermo-Electro or Photocatalysis. Chem 2019, 5, 2296–2325. [Google Scholar] [CrossRef]
- Cui, X.; Li, H.; Wang, Y.; Hu, Y.; Hua, L.; Li, H.; Han, X.; Liu, Q.; Yang, F.; He, L.; et al. Room-Temperature Methane Conversion by Graphene-Confined Single Iron Atoms. Chem 2018, 4, 1902–1910. [Google Scholar] [CrossRef] [Green Version]
- Periana, R.A.; Taube, D.J.; Evitt, E.R.; Loffler, D.G.; Wentrcek, P.R.; Voss, G.; Masuda, T. A Mercury Catalyzed High Yield System for the Oxidation of Methane to Methanol. Science 1993, 259, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Periana, R.A.; Tauba, D.J.; Gamble, S.; Taube, H.; Satoh, T.; Fujii, H. Platinum Catalysts for the High Yield Oxidation of Methane to a Methanol Derivative. Science 1998, 280, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Muehlhofer, M.; Strassner, T.; Herrmann, W.A. New Catalyst Systems for the Catalytic Conversion of Methane into Methanol. Angew. Chem. Int. Ed. 2002, 41, 1745–1747. [Google Scholar] [CrossRef]
- Smith, M.R.; Ozkan, U.S. The Partial Oxidation of Methane to Formaldehyde: Role of different Crystal Planes of MoO3. J. Catal. 1993, 141, 124–139. [Google Scholar] [CrossRef]
- Weng, T.; Wolf, E.E. Partial oxidation of methane on Mo/Sn/P silica supported catalysts. Appl. Catal. A Gen. 1993, 96, 383–396. [Google Scholar] [CrossRef]
- Millner, T.; Neugebauer, J. Volatility of the oxides of tungsten and molybdenum in the presence of water vapour. Nature 1949, 163, 601–602. [Google Scholar] [CrossRef]
- Barbero, J.A.; Alvarez, M.C.; Banares, M.A.; Pena, M.A.; Fierro, J.L.G. Breakthrough in the direct conversion of methane into C-1 Oxygenates. Chem. Commun. 2002, 11, 1184–1185. [Google Scholar] [CrossRef]
- Chen, S.Y.; Willcox, D. Effect of vanadium oxide loading on the selective oxidation of methane over vanadium oxide (V2O5)/silica. Ind. Eng. Chem. Res. 1993, 32, 584–587. [Google Scholar] [CrossRef]
- Marturano, P.; Drozdová, L.; Kogelbauer, A.; Prins, R. Fe/ZSM-5 Prepared by Sublimation of FeCl3 The Structure of the Fe Species as Determined by IR, 27Al MAS NMR, and EXAFS Spectroscopy. J. Catal. 2000, 192, 236–247. [Google Scholar] [CrossRef]
- Battiston, A.A.; Bitter, J.H.; De-Groot, F.M.F.; Overweg, A.R.; Stephan, O.; Van Bokhoven, J.A.; Kooyman, P.J.; van-der-Spek, C.; Vanko, G.; Koningsberger, D.C. Evolution of Fe species during the synthesis of over-exchanged Fe/ZSM5 obtained by chemical vapor deposition of FeCl3. J. Catal. 2003, 213, 251–271. [Google Scholar] [CrossRef] [Green Version]
- Groothaert, M.H.; Van Bokhoven, J.A.; Battiston, A.A.; Weckhuysen, B.M.; Schoonheydt, R.A. Bis(mu-oxo)dicopper in Cu-ZSM-5 and its role in the decomposition of NO: A combined in situ XAFS, UV-Vis-Near-IR, and kinetic study. J. Am. Chem. Soc. 2003, 125, 7629–7640. [Google Scholar] [CrossRef]
- Vanelderen, P.; Hadt, R.G.; Smeets, P.J.; Solomon, E.I.; Schoonheydt, R.A.; Sels, B.F. Cu-ZSM-5: A biomimetic inorganic model for methane oxidation. J. Catal. 2011, 284, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, N.; Freakley, S.J.; McVicker, R.U.; Althahban, S.M.; Dimitratos, N.; He, Q.; Morgan, D.J.; Jenkins, R.L.; Willock, D.J.; Taylor, S.H.; et al. Aqueous Au-Pd colloids catalyze selective CH4 oxidation to CH3OH with O2 under mild conditions. Science 2017, 358, 223–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, J.; Li, M.; Allard, L.F.; Lee, S.; Flytzani-Stephanopoulos, M. Mild oxidation of methane to methanol or acetic acid on supported isolated rhodium catalysts. Nature 2017, 551, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Starokon, E.V.; Parfenov, M.V.; Arzumanov, S.S.; Pirutko, L.V.; Stepanov, A.G.; Panov, G.I. Oxidation of methane to methanol on the surface of FeZSM-5 zeolite. J. Catal. 2013, 300, 47–54. [Google Scholar] [CrossRef]
- Bols, M.L.; Hallaert, S.D.; Snyder, B.E.R.; Devos, J.; Plessers, D.; Rhoda, H.M.; Dusselier, M.; Schoonheydt, R.A.; Pierloot, K.; Solomon, E.I.; et al. Spectroscopic Identification of the alpha-Fe/alpha-O Active Site in Fe-CHA Zeolite for the Low-Temperature Activation of the Methane C–H Bond. J. Am. Chem. Soc. 2018, 140, 12021–12032. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, T.Y.; Lee, H.; Yi, J. Distinct activation of Cu-MOR for direct oxidation of methane to methanol. Chem. Commun. 2017, 53, 4116–4119. [Google Scholar] [CrossRef] [PubMed]
- Ab Rahim, M.H.; Forde, M.M.; Jenkins, R.L.; Hammond, C.; He, Q.; Dimitratos, N.; Lopez-Sanchez, J.A.; Carley, A.F.; Taylor, S.H.; Willock, D.J.; et al. Oxidation of Methane to Methanol with Hydrogen Peroxide Using Supported Gold–Palladium Alloy Nanoparticles. Angew. Chem. Int. Ed. 2013, 52, 1280–1284. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Wang, L.; Zuidema, E.; Mondal, K.; Zhang, M.; Zhang, J.; Wang, C.; Meng, X.; Yang, H.; Mesters, C.; et al. Hydrophobic zeolite modification for in situ peroxide formation in methane oxidation to methanol. Science 2020, 367, 193–197. [Google Scholar] [CrossRef]
- Kwon, Y.; Kim, T.Y.; Kwon, G.; Yi, J.; Lee, H. Selective Activation of Methane on Single-Atom Catalyst of Rhodium Dispersed on Zirconia for Direct Conversion. J. Am. Chem. Soc. 2017, 139, 17694–17699. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.; Forde, M.M.; Ab Rahim, M.H.; Thetford, A.; He, Q.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Dummer, N.F.; Murphy, D.M.; et al. Direct catalytic conversion of methane to methanol in an aqueous medium by using copper-promoted Fe-ZSM-5. Angew. Chem. Int. Ed. 2012, 51, 5129–5133. [Google Scholar] [CrossRef]
- Groothaert, M.H.; Smeets, P.J.; Sels, B.F.; Jacobs, P.A.; Schoonheydt, R.A. Selective Oxidation of Methane by the Bis(µ-oxo)dicopper Core Stabilized on ZSM-5 and Mordenite Zeolites. J. Am. Chem. Soc. 2005, 127, 1394–1395. [Google Scholar] [CrossRef]
- Pappas, D.K.; Martini, A.; Dyballa, M.; Kvande, K.; Teketel, S.; Lomachenko, K.A.; Baran, R.; Glatzel, P.; Arstad, B.; Berlier, G.; et al. The Nuclearity of the Active Site for Methane to Methanol Conversion in Cu-Mordenite: A Quantitative Assessment. J. Am. Chem. Soc. 2018, 140, 15270–15278. [Google Scholar] [CrossRef] [PubMed]
- Pappas, D.K.; Borfecchia, E.; Dyballa, M.; Pankin, I.; Lomachenko, K.A.; Martini, A.; Signorile, M.; Teketel, S.; Arstad, B.; Berlier, G.; et al. Methane to Methanol: Structure-Activity Relationships for Cu-CHA. J. Am. Chem. Soc. 2017, 139, 14961–14975. [Google Scholar] [CrossRef] [Green Version]
- Knorpp, A.J.; Pinar, A.B.; Newton, M.; Sushkevich, V.L.; Van Bokhoven, J.A. Copper-Exchanged Omega (MAZ) Zeolite: Copper-concentration Dependent Active Sites and its Unprecedented Methane to Methanol Conversion. ChemCatChem 2018, 10, 5593–5596. [Google Scholar] [CrossRef]
- Beznis, N.V.; Weckhuysen, B.M.; Bitter, J.H. Partial Oxidation of Methane Over Co-ZSM-5: Tuning the Oxygenate Selectivity by Altering the Preparation Route. Catal. Lett. 2010, 136, 52–56. [Google Scholar] [CrossRef] [Green Version]
- Shan, J.; Huang, W.; Nguyen, L.; Yu, Y.; Zhang, S.; Li, Y.; Frenkel, A.I.; Tao, F.F. Conversion of methane to methanol with a bent mono(mu-oxo)dinickel anchored on the internal surfaces of micropores. Langmuir 2014, 30, 8558–8569. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Huang, E.; Orozco, I.; Liao, W.; Palomino, R.M.; Rui, N.; Duchon, T.; Nemsak, S.; Grinter, D.C.; Mahapatra, M.; et al. Water-promoted interfacial pathways in methane oxidation to methanol on a CeO2-Cu2O catalyst. Science 2020, 368, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Lustemberg, P.G.; Palomino, R.M.; Gutiérrez, R.A.; Grinter, D.C.; Vorokhta, M.; Liu, Z.; Ramírez, P.J.; Matolín, V.; Ganduglia-Pirovano, M.V.; Senanayake, S.D.; et al. Direct Conversion of Methane to Methanol on Ni-Ceria Surfaces: Metal–Support Interactions and Water-Enabled Catalytic Conversion by Site Blocking. J. Am. Chem. Soc. 2018, 140, 7681–7687. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Yao, Q.; Xu, Y.; Cao, K.; Huang, X. Strong synergy in a lichen-like RuCu nanosheet boosts the direct methane oxidation to methanol. Nano Energy 2020, 71, 104566. [Google Scholar] [CrossRef]
- Liu, R.S.; Iwamoto, M.; Lunsford, J.H. Partial Oxidation of Methane by Nitrous Oxide over Molybdenum Oxide supported on Silica. J. Chem. Soc. Chem. 1982, 1, 78–79. [Google Scholar] [CrossRef]
- Rosenzweig, A.; Frederick, C.; Lippard, S.; Nordlund, P. Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature 1993, 366, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Panov, G.I.; Soboley, V.I.; Kharitonov, A.S. The role of iron in N2O decomposition on ZSM-5 zeolite and reactivity of the surface oxygen formed. J. Mol. Catal. 1990, 61, 85–97. [Google Scholar] [CrossRef]
- Panov, G.I.; Sheveleva, G.A.; Kharitonov, A.S.; Romannikov, V.N.; Vostrikova, L.A. Oxidation of benzene to phenol by nitrous oxide over Fe-ZSM-5 zeolites. Appl. Catal. A Gen. 1992, 82, 31–36. [Google Scholar] [CrossRef]
- Panov, G.I.; Soboley, V.I.; Dubkov, K.A.; Parmon, V.N.; Ovanesyan, N.S.; Shilov, A.E.; Shteinman, A.A. Iron complexes in zeolites as a new model of methane monooxygenase. React. Kinet. Catal. Lett. 1997, 61, 251–258. [Google Scholar] [CrossRef]
- Snyder, B.E.R.; Vanelderen, P.; Bols, M.L.; Hallaert, S.D.; Bottger, L.H.; Ungur, L.; Pierloot, K.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. The active site of low-temperature methane hydroxylation in iron-containing zeolites. Nature 2016, 536, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Michalkiewicz, B. Partial oxidation of methane to formaldehyde and methanol using molecular oxygen over Fe-ZSM-5. Appl. Catal. A Gen. 2004, 277, 147–153. [Google Scholar] [CrossRef]
- Goltl, F.; Michel, C.; Andrikopoulos, P.C.; Love, A.M.; Hafner, J.; Hermans, I.; Sautet, P. Computationally Exploring Confinement Effects in the Methane-to-Methanol Conversion Over Iron Oxo Centers in Zeolites. ACS. Catal. 2016, 6, 8404–8409. [Google Scholar] [CrossRef]
- Andrikopoulos, P.C.; Sobalik, Z.; Novakova, J.; Sazama, P.; Sklenak, S. Mechanism of framework oxygen exchange in Fe-zeolites: A combined DFT and mass spectrometry study. ChemPhysChem 2013, 14, 520–531. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Shiota, Y.; Yoshizawa, K. Methane selective oxidation to methanol by metal-exchanged zeolites: A review of active sites and their reactivity. Catal. Sci. Techol. 2019, 9, 1744–1768. [Google Scholar] [CrossRef]
- Zhao, G.; Adesina, A.A.; Kennedy, E.M.; Stockenhuber, M. Formation of Surface Oxygen Species and the Conversion of Methane to Value-Added Products with N2O as Oxidant over Fe-Ferrierite Catalysts. ACS Catal. 2020, 10, 1406–1416. [Google Scholar] [CrossRef]
- Park, K.S.; Kim, J.H.; Park, S.H.; Moon, D.J.; Roh, H.-S.; Chung, C.-H.; Um, S.H.; Choi, J.-H.; Bae, J.W. Direct activation of CH4 to oxygenates and unsaturated hydrocarbons using N2O on Fe-modified zeolites. J. Mol. Catal. A Chem. 2017, 426, 130–140. [Google Scholar] [CrossRef]
- Starokon, E.V.; Parfenov, M.V.; Pirutko, L.V.; Abornev, S.I.; Panov, G.I. Room Temperature Oxidation of Methane by R Oxygen and Extraction of Products from the FeZSM-5 Surface. J. Phys. Chem. 2011, 115, 2155–2161. [Google Scholar] [CrossRef]
- Hammond, C.; Dimitratos, N.; Jenkins, R.; Lopez-Sanchez, T.; Kondrat, S.; Ab-Rahim, M.H.; Forde, M.; Thetford, A.; Taylor, S.H.; Hagen, H.; et al. Elucidation and Evolution of the Active Component within Cu/Fe/ZSM-5 for Catalytic Methane Oxidation: From Synthesis to Catalysis. ACS Catal. 2013, 3, 689–699. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Staykov, A.; Shiota, Y.; Yoshizawa, K. Direct Conversion of Methane to Methanol by Metal-Exchanged ZSM-5 Zeolite (Metal=Fe, Co, Ni, Cu). ACS Catal. 2016, 6, 8321–8331. [Google Scholar] [CrossRef]
- Li, S.; Fan, L.; Song, L.; Cheng, D.; Chen, F. Influence of extra-framework Al in Fe-MOR catalysts for the direct conversion of methane to oxygenates by nitrous oxide. Chin. J. Chem. Eng. 2020. [Google Scholar] [CrossRef]
- Parfenov, M.V.; Starokon, E.V.; Pirutko, L.V.; Panov, G.I. Quasicatalytic and catalytic oxidation of methane to methanol by nitrous oxide over FeZSM-5 zeolite. J. Catal. 2014, 318, 14–21. [Google Scholar] [CrossRef]
- Liu, H.F.; Liu, R.S.; Liew, K.Y.; Johnson, R.E.; Lunsford, J.H. Partial Oxidation of Methane by Nitrous Oxide over molybdenum on silica. J. Am. Chem. Soc. 1984, 106, 4117–4121. [Google Scholar] [CrossRef]
- Ipek, B.; Lobo, R.F. Catalytic conversion of methane to methanol on Cu-SSZ-13 using N2O as oxidant. Chem. Commun. 2016, 52, 13401–13404. [Google Scholar] [CrossRef] [PubMed]
- Nematollahi, P.; Neyts, E.C. Direct oxidation of methane to methanol on Co embedded N-doped graphene: Comparing the role of N2O and O2 as oxidants. Appl. Catal. A Gen. 2020, 602, 117716. [Google Scholar] [CrossRef]
- Montzka, S.A.; Dlugokencky, E.J.; Butler, J.H. Non-CO2 greenhouse gases and climate change. Nature 2011, 476, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Goor, G.; Glenneberg, J.; Jacobi, S.; Dadabhoy, J.; Candido, E. Hydrogen Peroxide. Ullmann’s Encycl. Ind. Chem. 2019. [Google Scholar] [CrossRef]
- Guo, X.; Fang, G.; Li, G.; Ma, H.; Fan, H.; Yu, L.; Ma, C.; Wu, X.; Deng, D.; Wei, M.; et al. Direct nonoxidative conversion of methane to ethylene, aromatics, and hydrogen. Science 2014, 344, 616–619. [Google Scholar] [CrossRef]
- Song, S.; Wang, X.; Zhang, H. CeO2-encapsulated noble metal nanocatalysts: Enhanced activity and stability for catalytic application. NPG Asia Mater. 2015, 7, e179. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Zhang, S.; Tang, Y.; Li, Y.; Nguyen, L.; Li, Y.; Shan, J.; Xiao, D.; Gagne, R.; Frenkel, A.I.; et al. Low-Temperature Transformation of Methane to Methanol on Pd1 O4 Single Sites Anchored on the Internal Surface of Microporous Silicate. Angew. Chem. Int. Ed. 2016, 55, 13441–13445. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, Y.; Fung, V.; Jiang, D.E.; Huang, W.; Zhang, S.; Iwasawa, Y.; Sakata, T.; Nguyen, L.; Zhang, X.; et al. Single rhodium atoms anchored in micropores for efficient transformation of methane under mild conditions. Nat. Commun. 2018, 9, 1231. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.K.; Solsona, B.E.; Landon, P.; Carley, A.F.; Herzing, A.; Kiely, C.J.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 using TiO2-supported Au–Pd catalysts. J. Catal. 2005, 236, 69–79. [Google Scholar] [CrossRef]
- Kesavan, L.; Tiruvalam, R.; Ab Rahim, M.H.; Bin Saiman, M.I.; Enache, D.I.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Taylor, S.H.; Knight, D.W.; et al. Solvent-free oxidation of primary carbon-hydrogen bonds in toluene using Au-Pd alloy nanoparticles. Science 2011, 331, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.; Piccinini, M.; Tiruvalam, R.; He, Q.; Dimitratos, N.; Lopez-Sanchez, J.A.; Morgan, D.J.; Carley, A.F.; Edwards, J.K.; Kiely, C.J.; et al. Effect of heat treatment on Au–Pd catalysts synthesized by sol immobilisation for the direct synthesis of hydrogen peroxide and benzyl alcohol oxidation. Catal. Sci. Technol. 2013, 3, 308–317. [Google Scholar] [CrossRef]
- Thetford, A.; Hutchings, G.J.; Taylor, S.H.; Willock, D.J. The decomposition of H2O2 over the components of Au/TiO2 catalysts. Proc. Math. Phys. Eng. Sci. 2011, 467, 1885–1899. [Google Scholar]
- Ab-Rahim, M.H.; Armstrong, R.D.; Hammond, C.; Dimitratos, N.; Freakley, S.J.; Forde, M.M.; Morgan, D.J.; Lalev, G.; Jenkins, R.L.; Lopez-Sanchez, J.A.; et al. Low temperature selective oxidation of methane to methanol using titania supported gold palladium copper catalysts. Catal. Sci. Technol. 2016, 6, 3410–3418. [Google Scholar] [CrossRef] [Green Version]
- Serra-Maia, R.; Michel, F.M.; Kang, Y.; Stach, E.A. Decomposition of Hydrogen Peroxide Catalyzed by AuPd Nanocatalysts during Methane Oxidation to Methanol. ACS Catal. 2020, 10, 5115–5123. [Google Scholar] [CrossRef]
- Xie, J.; Jin, R.; Li, A.; Bi, Y.; Ruan, Q.; Deng, Y.; Zhang, Y.; Yao, S.; Sankar, G.; Ma, D.; et al. Highly selective oxidation of methane to methanol at ambient conditions by titanium dioxide-supported iron species. Nat. Catal. 2018, 1, 889–896. [Google Scholar] [CrossRef]
- Xu, J.; Armstrong, R.D.; Shaw, G.; Dummer, N.F.; Freakley, S.J.; Taylor, S.H.; Hutchings, G.J. Continuous selective oxidation of methane to methanol over Cu- and Fe-modified ZSM-5 catalysts in a flow reactor. Catal. Today 2016, 270, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Park, K.H.; Cho, S.J.; Park, E.D. Partial oxidation of methane with hydrogen peroxide over Fe-ZSM-5 catalyst. Catal. Today 2020. [Google Scholar] [CrossRef]
- Kalamaras, C.; Palomas, D.; Bos, R.; Horton, A.; Crimmin, M.; Hellgardt, K. Selective Oxidation of Methane to Methanol Over Cu- and Fe-Exchanged Zeolites: The Effect of Si/Al Molar Ratio. Catal. Lett. 2016, 146, 483–492. [Google Scholar] [CrossRef]
- Shahami, M.; Shantz, D.F. Zeolite acidity strongly influences hydrogen peroxide activation and oxygenate selectivity in the partial oxidation of methane over M,Fe-MFI (M: Ga, Al, B) zeolites. Catal. Sci. Technol. 2019, 9, 2945–2951. [Google Scholar] [CrossRef]
- Forde, M.M.; Armstrong, R.D.; Hammond, C.; He, Q.; Jenkins, R.L.; Kondrat, S.A.; Dimitratos, N.; Lopez-Sanchez, J.A.; Taylor, S.H.; Willock, D.; et al. Partial oxidation of ethane to oxygenates using Fe- and Cu-containing ZSM-5. J. Am. Chem. Soc. 2013, 135, 11087–11099. [Google Scholar] [CrossRef] [PubMed]
- Tomkins, P.; Ranocchiari, M.; Van Bokhoven, J.A. Direct Conversion of Methane to Methanol under Mild Conditions over Cu-Zeolites and beyond. Acc. Chem. Res. 2017, 50, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Markovits, M.A.C.; Jentys, A.; Tromp, M.; Sanchez-Sanchez, M.; Lercher, J.A. Effect of Location and Distribution of Al Sites in ZSM-5 on the Formation of Cu-Oxo Clusters Active for Direct Conversion of Methane to Methanol. Top. Catal. 2016, 59, 1554–1563. [Google Scholar] [CrossRef]
- Smeets, P.J.; Groothaert, M.H.; Schoonheydt, R.A. Cu based zeolites: A UV–vis study of the active site in the selective methane oxidation at low temperatures. Catal. Today 2005, 110, 303–309. [Google Scholar] [CrossRef]
- Woertink, J.S.; Smeets, P.J.; Groothaert, M.H.; Vance, M.A.; Sels, B.F.; Schoonheydt, R.A.; Solomon, E.I. A [Cu2O]2+ core in Cu-ZSM-5, the active site in the oxidation of methane to methanol. Proc. Natl. Acad. Sci. USA 2009, 106, 18908–18913. [Google Scholar] [CrossRef] [Green Version]
- Smeets, P.J.; Hadt, R.G.; Woertink, J.S.; Vanelderen, P.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Oxygen Precursor to the Reactive Intermediate in Methanol Synthesis by Cu-ZSM-5. J. Am. Chem. Soc. 2010, 132, 14736–14738. [Google Scholar] [CrossRef] [Green Version]
- Groothaert, M.H.; Lievens, K.; Leeman, H.; Weckhuysen, B.M.; Schoonheydt, R.A. An operando optical fiber UV–vis spectroscopic study of the catalytic decomposition of NO and N2O over Cu-ZSM-5. J. Catal. 2003, 220, 500–512. [Google Scholar] [CrossRef] [Green Version]
- Grundner, S.; Markovits, M.A.C.; Li, G.; Tromp, M.; Pidko, E.A.; Hensen, E.J.M.; Jentys, A.; Sanchez-Sanchez, M.; Lercher, J.A. Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol. Nat. Commun. 2015, 6, 7546. [Google Scholar] [CrossRef] [PubMed]
- Sushkevich, V.L.; Palagin, D.; Van Bokhoven, J.A. The Effect of the Active-Site Structure on the Activity of Copper Mordenite in the Aerobic and Anaerobic Conversion of Methane into Methanol. Angew. Chem. Int. Ed. 2018, 57, 8906–8910. [Google Scholar] [CrossRef] [PubMed]
- Park, M.B.; Ahn, S.H.; Ranocchiari, M.; Van Bokhoven, J.A. Comparative Study of Diverse Cu-Zeolites for Conversion of Methane-to-Methanol. ChemCatChem 2017, 9, 3705–3713. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Tanaka, T.; Shiota, Y.; Staykov, A.; Yoshizawa, K. Methane Partial Oxidation over [Cu2(μ-O)]2+ and [Cu3(μ-O)3]2+ Active Species in Large-Pore Zeolites. ACS Catal. 2018, 8, 1500–1509. [Google Scholar] [CrossRef]
- Vanelderen, P.; Snyder, B.E.R.; Tsai, M.L.; Hadt, R.G.; Vancauwenbergh, J.; Coussens, O.; Schoonheydt, R.A.; Sels, B.F.; Solomon, E.I. Spectroscopic definition of the copper active sites in mordenite: Selective methane oxidation. J. Am. Chem. Soc. 2015, 137, 6383–6392. [Google Scholar] [CrossRef] [Green Version]
- Wulfers, M.J.; Teketel, S.; Ipek, B.; Lobo, R.F. Conversion of methane to methanol on copper-containing small-pore zeolites and zeotypes. Chem. Commun. 2015, 51, 4447–4450. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, T.; Hamill, C.D.; Goguet, A.; Rooney, D.W.; Thompson, J.M. A low temperature, isothermal gas-phase system for conversion of methane to methanol over Cu-ZSM-5. Chem. Commun. 2014, 50, 11053–11055. [Google Scholar] [CrossRef]
- Tomkins, P.; Mansouri, A.; Bozbag, S.E.; Krumeich, F.; Park, M.B.; Alayon, E.M.C.; Ranocchiari, M.; Van Bokhoven, J.A. Isothermal Cyclic Conversion of Methane into Methanol over Copper-Exchanged Zeolite at Low Temperature. Angew. Chem. Int. Ed. 2016, 55, 5467–5471. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Van Bokhoven, J.A. Methane-to-Methanol: Activity Descriptors in Copper-Exchanged Zeolites for the Rational Design of Materials. ACS Catal. 2019, 9, 6293–6304. [Google Scholar] [CrossRef]
- Krisnandi, Y.K.; Putra, B.A.P.; Bahtiar, M.; Zahara; Abdullah, I.; Howe, R.F. Partial Oxidation of Methane to Methanol over Heterogeneous Catalyst Co/ZSM-5. Procedia Chem. 2015, 14, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Meesub, C.; Tannititam, P.; Kruemek, P.; Triamnak, N.; Chotigkrai, N. Direct stepwise oxidation of methane to methanol via zinc promoted copper containing mordenite. In Proceedings of the 8th International Thai Institute of Chemical Engineering and Applied Chemistry Conference, Pattaya, Thailand, 8–9 November 2018; pp. 45–50. [Google Scholar]
- Zuo, Z.; Ramirez, P.J.; Senanayake, S.; Liu, P.; Rodriguez, J.A. Low-Temperature Conversion of Methane to Methanol on CeOx/Cu2O Catalysts: Water Controlled Activation of the C–H Bond. J. Am. Chem. Soc. 2016, 138, 13810–13813. [Google Scholar] [CrossRef]
- Lange, J.P.; Sushkevich, V.L.; Knorpp, M.J.; Van Bokhoven, J.A. Methane to Methanol via Chemical Looping Economic Potential and Guidance for Future Research. Ind. Eng. Chem. Res. 2019, 58, 8674–8680. [Google Scholar] [CrossRef]
- Sushkevich, V.L.; Palagin, D.; Ranocchiari, M.; Van Bokhoven, J.A. Selective anaerobic oxidation of methane enables direct synthesis of methanol. Science 2017, 356, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Palagin, D.; Sushkevich, V.L.; van Bokhoven, J.A. Water Molecules Facilitate Hydrogen Release in Anaerobic Oxidation of Methane to Methanol over Cu/Mordenite. ACS Catal. 2019, 9, 10365–10374. [Google Scholar] [CrossRef]
- Jeong, Y.R.; Jung, H.; Kang, J.; Han, J.W.; Park, E.D. Continuous Synthesis of Methanol from Methane and Steam over Copper-Mordenite. ACS Catal. 2021, 11, 1065–1070. [Google Scholar] [CrossRef]
- Koishybay, A.; Shantz, D.F. Water Is the Oxygen Source for Methanol Produced in Partial Oxidation of Methane in a Flow Reactor over Cu-SSZ-13. J. Am. Chem. Soc. 2020, 142, 11962–11966. [Google Scholar] [CrossRef] [PubMed]
- Periana, R.A. Comment on “Selective anaerobic oxidation of methane enables direct synthesis of methanol”. Science 2017, 358, eaan5970. [Google Scholar] [CrossRef] [Green Version]
- Sushkevich, V.L.; Palagin, D.; Ranocchiari, M.; Van Bokhoven, J.A. Response to Comment on “Selective anaerobic oxidation ofmethane enables direct synthesis ofmethanol”. Science 2017, 358, eaan6083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labinger, J.A. Comment on “Selective anaerobic oxidation of methane enables direct synthesis of methanol”. Science 2018, 359, eaar4968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinh, K.T.; Sullivan, M.M.; Narsimhan, K.; Serna, P.; Meyer, R.J.; Dinca, M.; Roman-Leshkov, Y. Continuous Partial Oxidation of Methane to Methanol Catalyzed by Diffusion-Paired Copper Dimers in Copper-Exchanged Zeolites. J. Am. Chem. Soc. 2019, 141, 11641–11650. [Google Scholar] [CrossRef] [PubMed]
- Le, H.V.; Parishan, S.; Sagaltchik, A.; Ahi, H.; Trunschke, A.; Schomacker, R.; Thomas, A. Stepwise Methane-to-Methanol Conversion on CuO/SBA-15. Chemistry 2018, 24, 12592–12599. [Google Scholar] [CrossRef] [PubMed]
- Yumura, T.; Hirose, Y.; Wakasugi, T.; Kuroda, Y.; Kobayashi, H. Roles of Water Molecules in Modulating the Reactivity of Dioxygen-Bound Cu-ZSM-5 toward Methane: A Theoretical Prediction. ACS Catal. 2016, 6, 2487–2495. [Google Scholar] [CrossRef]
- Snoeckx, R.; Bogaerts, A. Plasma technology—A novel solution for CO2 conversion? Chem. Soc. Rev. 2017, 46, 5805–5863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehead, J.C. Plasma catalysis the known knowns, the known unknowns and the unknown unknowns. J. Phys. D Appl. Phys. 2016, 49, 243001. [Google Scholar] [CrossRef]
- Bogaerts, A.; Tu, X.; Whitehead, J.C.; Centi, G.; Lefferts, L.; Guaitella, O.; Azzolina-Jury, F.; Kim, H.-H.; Murphy, A.B.; Schneider, W.F.; et al. The 2020 plasma catalysis roadmap. J. Phys. D Appl. Phys. 2020, 53, 443001. [Google Scholar] [CrossRef]
- Neyts, E.C.; Ostrikov, K.K.; Sunkara, M.K.; Bogaerts, A. Plasma Catalysis: Synergistic Effects at the Nanoscale. Chem. Rev. 2015, 115, 13408–13446. [Google Scholar] [CrossRef]
- Kim, H.H.; Teramoto, Y.; Ogata, A.; Takagi, H.; Nanba, T. Plasma Catalysis for Environmental Treatment and Energy Applications. Plasma Chem. Plasma Process. 2015, 36, 45–72. [Google Scholar] [CrossRef]
- Liu, M.; Yi, Y.; Wang, L.; Guo, H.; Bogaerts, A. Hydrogenation of Carbon Dioxide to Value-Added Chemicals by Heterogeneous Catalysis and Plasma Catalysis. Catalysts 2019, 9, 275. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Yi, Y.; Wang, L.; Yan, J.; Guo, H. Pt/TS-1 Catalyst Promoted C–N Coupling Reaction in CH4–NH3 Plasma for HCN Synthesis at Low Temperature. ACS Catal. 2018, 8, 10219–10224. [Google Scholar] [CrossRef]
- Wang, L.; Yi, Y.; Wu, C.; Guo, H.; Tu, X. One-Step Reforming of CO2 and CH4 into High-Value Liquid Chemicals and Fuels at Room Temperature by Plasma-Driven Catalysis. Angew. Chem. Int. Ed. 2017, 56, 13679–13683. [Google Scholar] [CrossRef] [Green Version]
- Tu, X.; Whitehead, J.C. Plasma-catalytic dry reforming of methane in an atmospheric dielectric barrier discharge: Understanding the synergistic effect at low temperature. Appl. Catal. B Environ. 2012, 125, 439–448. [Google Scholar] [CrossRef]
- Wang, L.; Yi, Y.; Guo, H.; Tu, X. Atmospheric Pressure and Room Temperature Synthesis of Methanol through Plasma-Catalytic Hydrogenation of CO2. ACS Catal. 2017, 8, 90–100. [Google Scholar] [CrossRef]
- Chawdhury, P.; Wang, Y.; Ray, D.; Mathieu, S.; Wang, N.; Harding, J.; Bin, F.; Tu, X.; Subrahmanyam, C. A promising plasma-catalytic approach towards single-step methane conversion to oxygenates at room temperature. Appl. Catal. B Environ. 2021, 284, 119735. [Google Scholar] [CrossRef]
- Puliyalil, H.; Jurkovic, D.L.; Dasireddy, V.D.B.C.; Likozar, B. A review of plasma-assisted catalytic conversion of gaseous carbon dioxide and methane into value-added platform chemicals and fuels. RSC Adv. 2018, 8, 27481–27508. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Winter, L.R.; Chen, J.G. Review of plasma-assisted catalysis for selective generation of oxygenates from CO2 and CH4. ACS Catal. 2020, 10, 2855–2871. [Google Scholar] [CrossRef]
- Matsumoto, H.; Tanabe, S.; Okitsu, K.; Hayashi, Y.; Suib, S.L. Selective Oxidation of Methane to Methanol and Formaldehyde with Nitrous Oxide in a Dielectric-Barrier Discharge-Plasma Reactor. J. Phys. Chem. A 2001, 105, 5304–5308. [Google Scholar] [CrossRef]
- Mahammadunnisa, S.; Krushnamurty, K.; Subrahmanyam, C. Catalytic nonthermal plasma assisted co-processing of methane and nitrous oxide for methanol production. Catal. Today 2015, 256, 102–107. [Google Scholar] [CrossRef]
- Mahammadunnisa, S.; Reddy, P.M.K.; Subrahmanyam, C. Nonthermal plasma assisted co-processing of CH4 and N2O for methanol production. RSC Adv. 2014, 4, 4034–4036. [Google Scholar] [CrossRef]
- Yao, S.L.; Takemoto, T.; Ouyang, F.; Nakayama, A.; Suzuki, E.; Mizuno, A.; Okumoto, M. Selective Oxidation of Methane Using a Non-Thermal Pulsed Plasma. Energy Fuels 2000, 14, 459–463. [Google Scholar] [CrossRef]
- Chawdhury, P.; Bhargavi, K.V.S.S.; Subrahmanyam, C. Enhanced synergy by plasma reduced Pd nanoparticles on in-plasma catalytic methane conversion to liquid oxygenates. Catal. Commun. 2020, 147, 106139. [Google Scholar] [CrossRef]
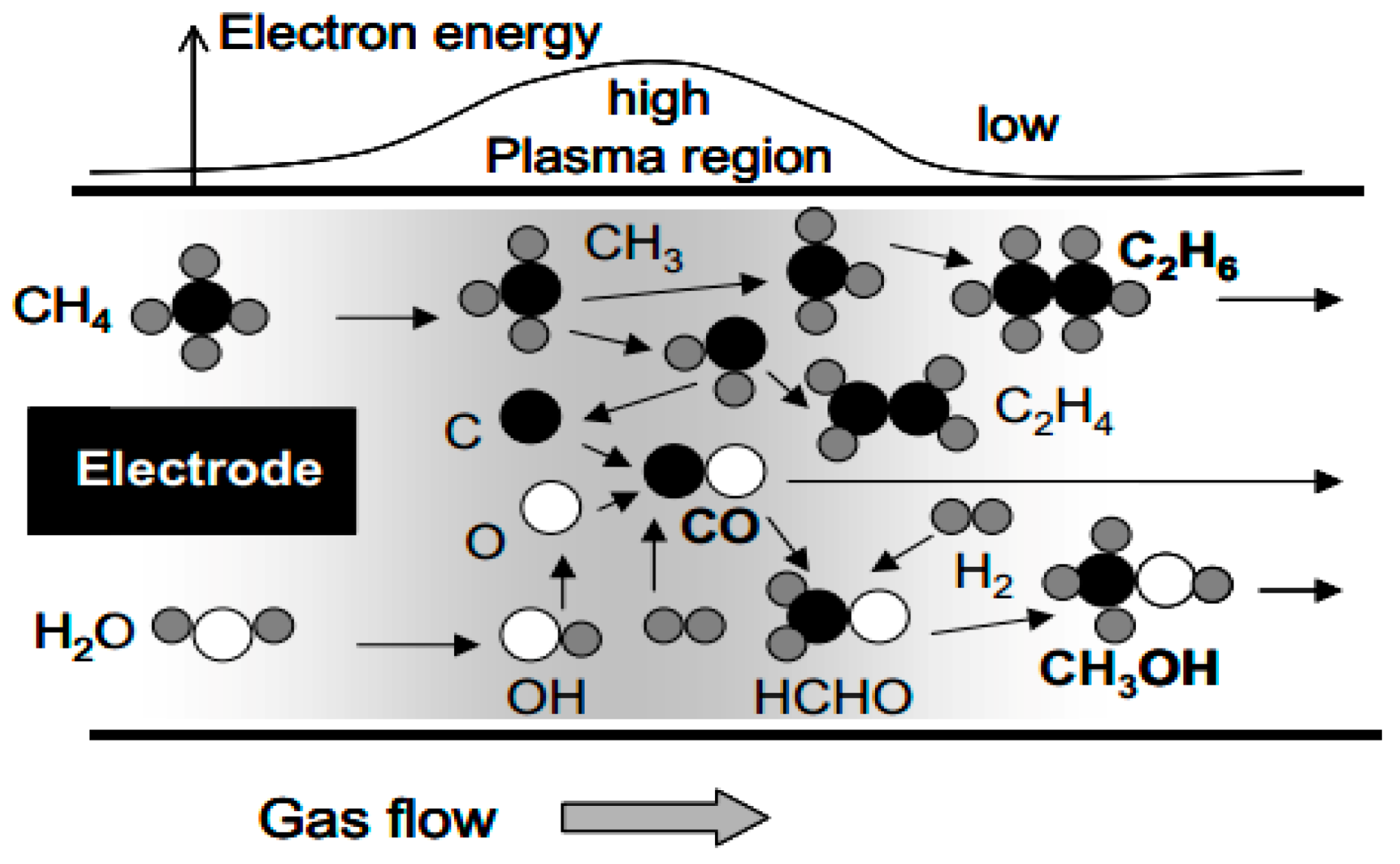
- Larkin, D.W.; Lobban, L.L.; Mallinson, R.G. The direct partial oxidation of methane to organic oxygenates using a dielectric barrier discharge reactor as a catalytic reactor analog. Catal. Today 2001, 71, 199–210. [Google Scholar] [CrossRef]
- Zhou, L.M.; Xue, B.; Kogelschatz, U.; Eliasson, B. Partial Oxidation of Methane to Methanol with Oxygen or Air in a Nonequilibrium Discharge Plasma. Plasma Chem. Plasma Process. 1998, 18, 375–393. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, X.-W.; Huang, L.; Lei, L.-C. Partial oxidation of methane with air for methanol production in a post-plasma catalytic system. Chem. Eng. Process. 2009, 48, 1333–1340. [Google Scholar] [CrossRef]
- Chawdhury, P.; Ray, D.; Subrahmanyam, C. Single step conversion of methane to methanol assisted by nonthermal plasma. Fuel Process. Technol. 2018, 179, 32–41. [Google Scholar] [CrossRef]
- Chawdhury, P.; Ray, D.; Vinodkumar, T.; Subrahmanyam, C. Catalytic DBD plasma approach for methane partial oxidation to methanol under ambient conditions. Catal. Today 2019, 337, 117–125. [Google Scholar] [CrossRef]
- Indarto, A. Methanol synthesis from methane and oxygen with [Ga Cr]/Cu–Zn–Al catalyst in a dielectric barrier discharge. Ionics 2014, 20, 445–449. [Google Scholar] [CrossRef]
- Indarto, A.; Lee, H.; Choi, J.W.; Song, H.K. Partial Oxidation of Methane with Yttria-stabilized Zirconia Catalyst in a Dielectric Barrier Discharge. Energy Sources Part A 2008, 30, 1628–1636. [Google Scholar] [CrossRef]
- Chawdhury, P.; Kumar, D.; Subrahmanyam, C. NTP reactor for a single stage methane conversion to methanol: Influence of catalyst addition and effect of promoters. Chem. Eng. J. 2019, 372, 638–647. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, X.; Huang, L.; Lei, L. Post-Plasma Catalysis for Methane Partial Oxidation to Methanol: Role of the Copper-Promoted Iron Oxide Catalyst. Chem. Eng. Technol. 2010, 33, 2073–2081. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, X.; Huang, L.; Lei, L. Application of in-plasma catalysis and post-plasma catalysis for methane partial oxidation to methanol over a Fe2O3-CuO/γ-Al2O3 catalyst. J. Nat. Gas Chem. 2010, 19, 628–637. [Google Scholar] [CrossRef]
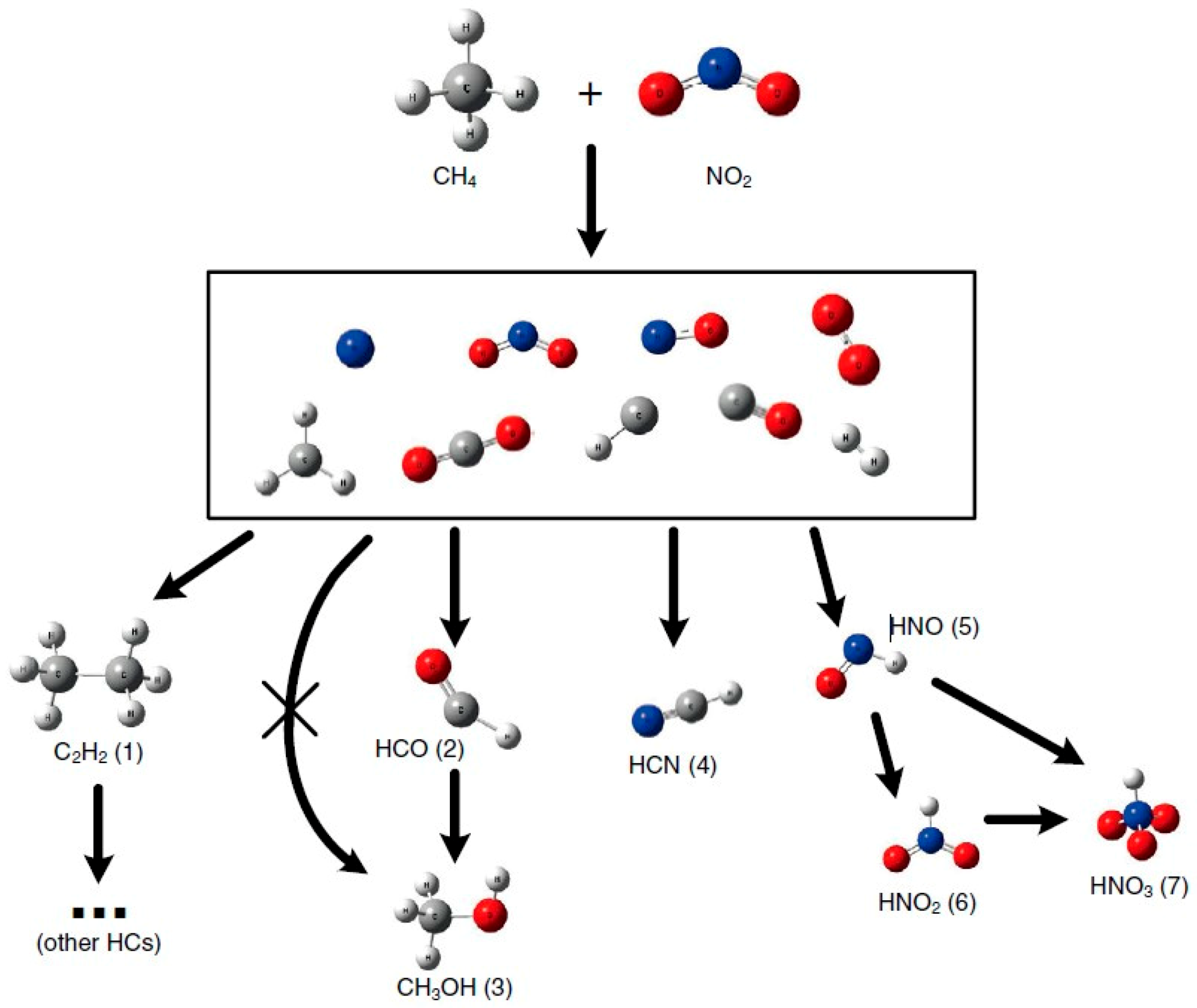
- Indarto, A. Partial oxidation of methane to methanol with nitrogen dioxide in dielectric barrier discharge plasma: Experimental and molecular modeling. Plasma Sources Sci. Technol. 2016, 25, 025002. [Google Scholar] [CrossRef]
- Aghamir, F.M.; Matin, N.S.; Jalili, A.H.; Esfarayeni, M.H.; Khodagholi, M.A.; Ahmadi, R. Conversion of methane to methanol in an ac dielectric barrier discharge. Plasma Sources Sci. Technol. 2004, 13, 707–711. [Google Scholar] [CrossRef]
- Larkin, D.W.; Lobban, L.L.; Mallinson, R.G. Production of Organic Oxygenates in the Partial Oxidation of Methane in a Silent Electric Discharge Reactor. Ind. Eng. Chem. Res. 2001, 40, 1594–1601. [Google Scholar] [CrossRef]
- Indarto, A. A review of direct methane conversion to methanol by dielectric barrier discharge. IEEE Trans. Dielectr. Electr. Insul. 2008, 15, 1038–1043. [Google Scholar] [CrossRef]
- Nozaki, T.; Ağıral, A.; Yuzawa, S.; Han Gardeniers, J.G.E.; Okazaki, K. A single step methane conversion into synthetic fuels using microplasma reactor. Chem. Eng. J. 2011, 166, 288–293. [Google Scholar] [CrossRef]
- Nozaki, T.; Goujard, V.; Yuzawa, S.; Moriyama, S.; Agıral, A.; Okazaki, K. Selective conversion of methane to synthetic fuels using dielectric barrier discharge contacting liquid film. J. Phys. D Appl. Phys. 2011, 44, 274010. [Google Scholar] [CrossRef]
- Nozaki, T.; Hattori, A.; Okazaki, K. Partial oxidation of methane using a microscale non-equilibrium plasma reactor. Catal. Today 2004, 98, 607–616. [Google Scholar] [CrossRef]
- Indarto, A.; Jae Wook, C.; Hwaung, L.; Hyung Keun, S. Methanol Synthesis over Cu and Cu-Oxide-Containing ZnO/Al2O3 Using Dielectric Barrier Discharge. IEEE Trans. Plasma Sci. 2008, 36, 516–518. [Google Scholar] [CrossRef]
- Zhang, Q.Z.; Bogaerts, A. Propagation of a plasma streamer in catalyst pores. Plasma Sources Sci. Technol. 2018, 27, 035009. [Google Scholar] [CrossRef]
- Zhang, Q.Z.; Wang, W.; Bogaerts, A. Importance of surface charging during plasma streamer propagation in catalyst pores. Plasma Sources Sci. Technol. 2018, 27, 065009. [Google Scholar] [CrossRef] [Green Version]
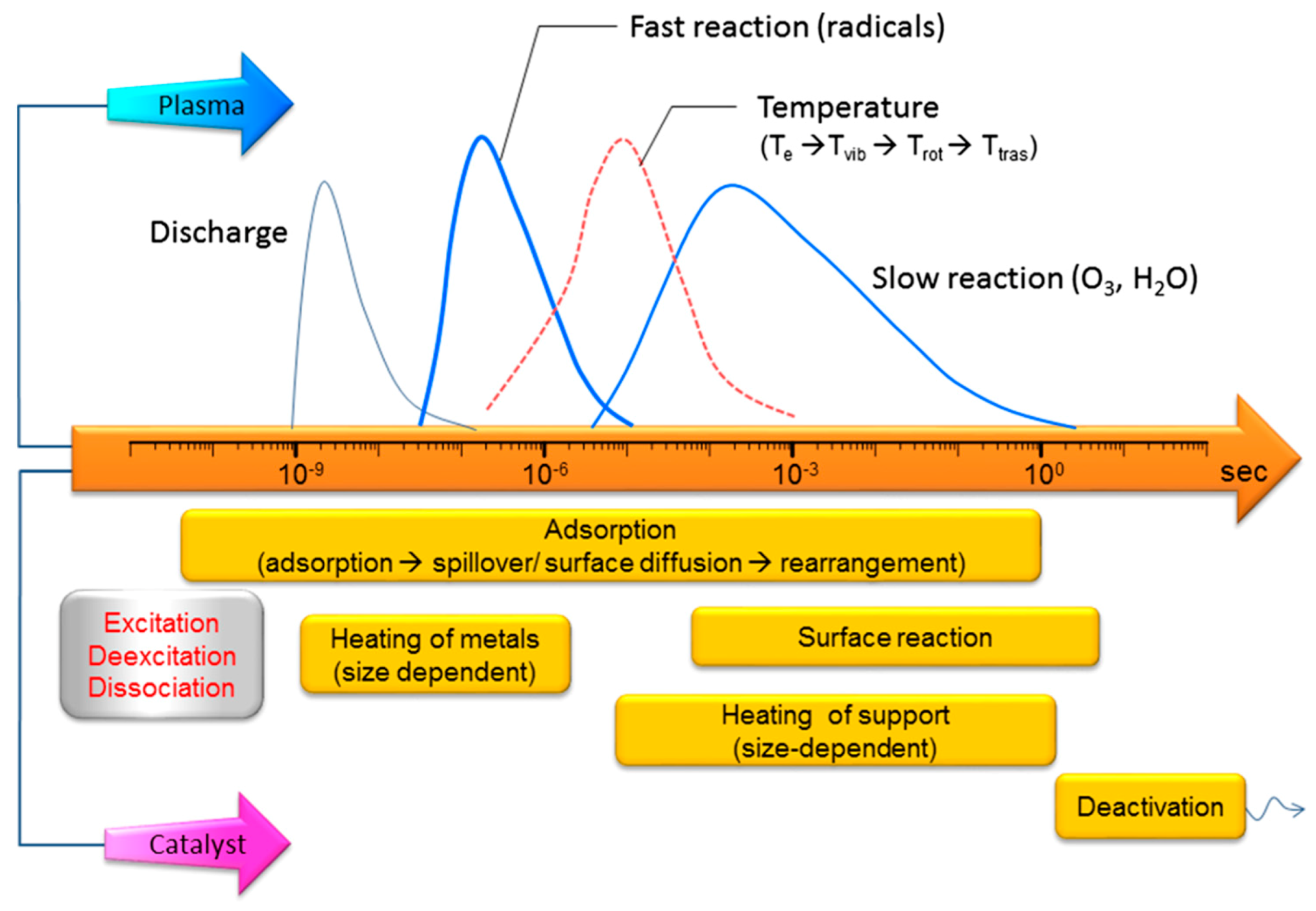
- Bogaerts, A.; Zhang, Q.Z.; Zhang, Y.-R.; Van Laer, K.; Wang, W. Burning questions of plasma catalysis: Answers by modeling. Catal. Today 2019, 337, 3–14. [Google Scholar] [CrossRef]
- Van Laer, K.; Bogaerts, A. How bead size and dielectric constant affect the plasma behaviour in a packed bed plasma reactor: A modelling study. Plasma Sources Sci. Technol. 2017, 26, 085007. [Google Scholar] [CrossRef]
- Wang, Y.; Craven, M.; Yu, X.; Ding, J.; Bryant, P.; Huang, J.; Tu, X. Plasma-Enhanced Catalytic Synthesis of Ammonia over a Ni/Al2O3 Catalyst at Near-Room Temperature: Insights into the Importance of the Catalyst Surface on the Reaction Mechanism. ACS Catal. 2019, 9, 10780–10793. [Google Scholar]
- De Bie, C.; Verheyde, B.; Martens, T.; Dijk, J.V.; Paulussen, S.; Bogaerts, A. Fluid Modeling of the Conversion of Methane into Higher Hydrocarbons in an Atmospheric Pressure Dielectric Barrier Discharge. Plasma Process. Polym. 2011, 8, 1033–1058. [Google Scholar] [CrossRef]
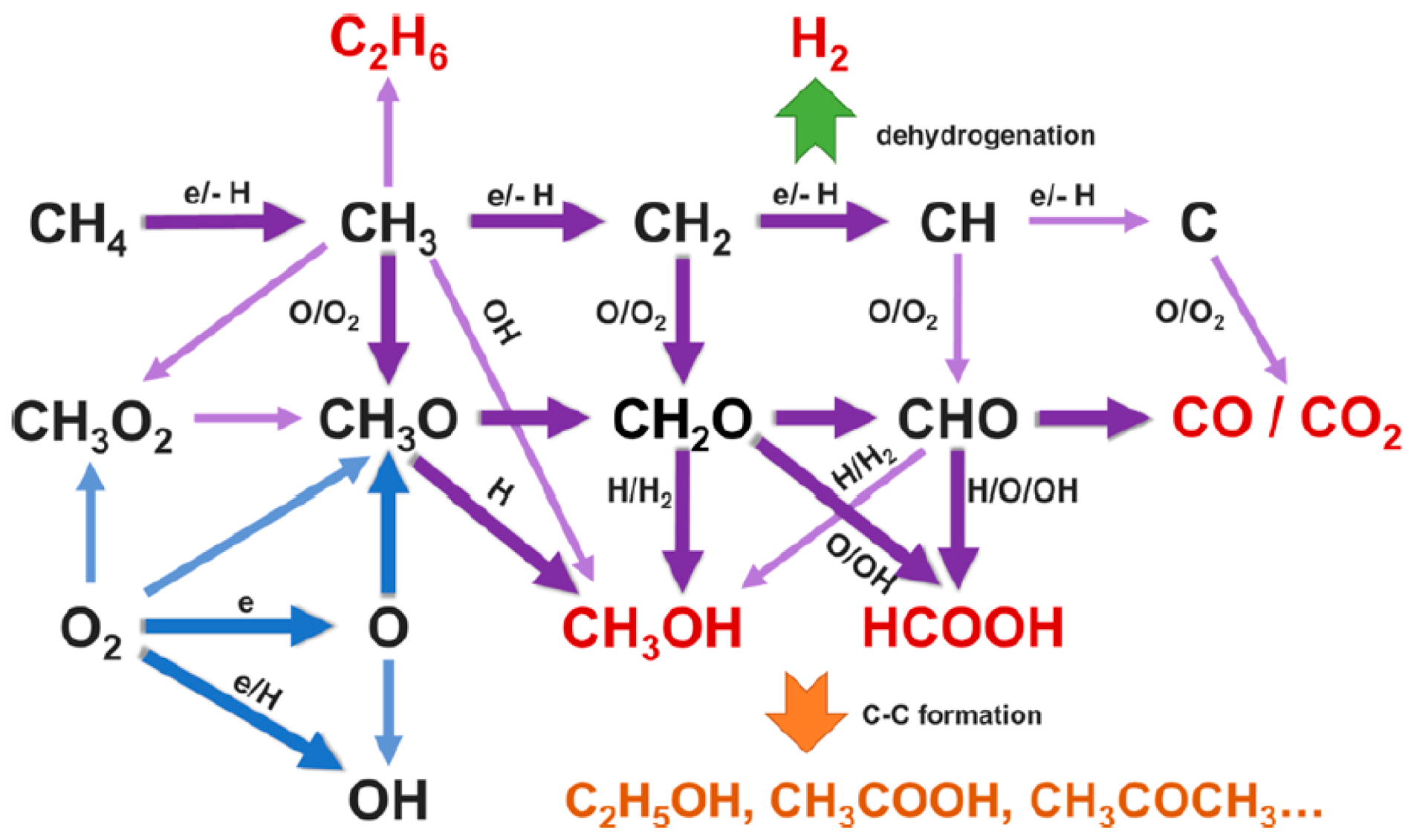
- De-Bie, C.; Dijk, J.V.; Bogaerts, A. The Dominant Pathways for the Conversion of Methane into Oxygenates and Syngas in an Atmospheric Pressure Dielectric Barrier Discharge. J. Phys. Chem. C 2015, 119, 22331–22350. [Google Scholar] [CrossRef]
- Loenders, B.; Engelmann, Y.; Bogaerts, A. Plasma-catalytic partial oxidation of methane on Pt(111): A microkinetic study of the role of different plasma species. J. Phys. Chem. C 2021, 125, 2966–2983. [Google Scholar] [CrossRef]
- Takayuki Tsuchiya, S.I. Conversion of Methane to Methanol by a Low-Pressure Steam Plasma. J. Environ. Eng. Technol. 2013, 2, 35–39. [Google Scholar]
- Pearce, M.P.; Bussemaker, M.J.; Cooper, P.D.; Lapere, K.M.; Wild, D.A.; McKinley, A.J. Formation of methanol from methane and water in an electrical discharge. Phys. Chem. Chem. Phys. 2012, 14, 3444–3449. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, Y.; Mehta, P.; Neyts, E.C.; Schneider, W.F.; Bogaerts, A. Predicted Influence of Plasma Activation on Non-Oxidative Coupling of Methane on Transition Metal Catalysts. ACS Sustain. Chem. Eng. 2020, 8, 6043–6054. [Google Scholar] [CrossRef]
- Nozaki, T.; Okazaki, K. Non-thermal plasma catalysis of methane: Principles, energy efficiency, and applications. Catal. Today 2013, 211, 29–38. [Google Scholar] [CrossRef]
- Yi, Y.; Wang, X.; Jafarzadeh, A.; Wang, L.; Liu, P.; He, B.; Yan, J.; Zhang, R.; Zhang, H.; Liu, X.; et al. Plasma-Catalytic Ammonia Reforming of Methane over Cu-Based Catalysts for the Production of HCN and H2 at Reduced Temperature. ACS Catal. 2021, 11, 1765–1773. [Google Scholar] [CrossRef]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxidant | Catalysts | PCH4 (bar) | Temp (°C) a | CH3OH Yield (μmol·gcat−1) | Productivity (mmol /molmetal) | CH3OH Sel. (%) | Ref. |
|---|---|---|---|---|---|---|---|
| N2O | Fe/ZSM-5 | 6.6 × 10−4 | 160 | 160 | / | 76 | [25] |
| Fe/CHA | / | RT | 26.8 | 681 | / | [26] | |
| Cu/MOR | / | 150 | 97 | >300 | / | [27] | |
| Cu/SSZ-13 | 0.3 | 260 | 19 | / | 27 | [27] | |
| H2O2 | Au-Pd/TiO2 | 30.5 | 2 | 54.6 | 13.1 | 45.2 | [28] |
| Au-Pd colloids | 30 | ≤50 | 2.8–7.6 | / | 14–28.3 | [23] | |
| AuPd@ZSM-5 | 0.48 | 70 | / | 91.6 | 92 | [29] | |
| FeN4/Graphene | 18 | 25 | / | / | / | [10] | |
| Rh/ZrO2 | 28.5 | 70 | / | 1.25 | / | [30] | |
| Cu-Fe/ZSM-5 | 30.8 | 50 | 5.2 × 103 | / | 88 | [31] | |
| O2 | Cu/ZSM-5 | / | 175 | 8.2 | / | 98 | [32] |
| Cu/MOR | / | 200 | 170 | 0.47 | / | [33] | |
| Cu/SSZ-13 | 0.1 | 200 | 125 | 0.2 | >90 | [34] | |
| Cu/Omega | 30 | 200 | 200 | 265 | / | [35] | |
| Co/ZSM-5 | 1 | 150 | 0.3–0.4 | / | 40–100 | [36] | |
| Ni/ZSM-5 | 1 | 175 | 14.9 | / | / | [37] | |
| O2+H2O | CeOx/Cu2O/Cu(111) | 2.7 × 10−5 | 25 | / | / | 70 | [38] |
| Ni/CeO2 | 1.3 × 10−3 | 177 | / | / | <40 | [39] | |
| O2+CO | Rh/ZSM-5 | 20 | 150 | 1224 | / | 6.2 | [24] |
| O2+H2O2 | RuCu/NL b | 25 | 50 | ~1.5 × 103 | / | / | [40] |
| N2O+H2O | Mo/SiO2 | / | 300 | / | 16 | 60 | [41] |
| System | Oxidant | Packed Material | Power (W) | SEI (kJ/L) a | Temp. (K) | CH4 Conv. (%) | CH3OH Sel. (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Plasma only | N2O | / | 0.27–7.7 | - | - | 5 | 43 | [121] |
| N2O | / | 6 | 6 | - | 15 | 31 | [122] | |
| N2O | / | 6 | 6 | - | <15 | 28 | [123] | |
| O2 | / | 5.7 | 3.4 | RT b | 1.9 | 47 | [124] | |
| O2 | / | 1.7 | 3.4 | RT | 7 | 20 | [125] | |
| O2 | / | 118 | 35.4 | 288 | 6 | 19 | [126] | |
| O2 | / | 200 | 4.0 | 353 | 3 | 30 | [127] | |
| Air | / | 400 | 11.9 | 353 | 15 | 13.3 | [127] | |
| Air | / | 140 | 28 | 523 | ~25 | ~8 | [128] | |
| Plasma catalysis | N2O | Cu-Ni/CeO2 | 6 | 6 | - | 23 | 36 | [122] |
| O2 | Fe/γ-Al2O3 | 1.8 | 3.6 | RT | 13 | 36 | [118] | |
| O2 | Glass Beads | 1.7 | 5.1 | - | 15.4 | 35.4 | [129] | |
| O2 | Cu/γ-Al2O3 | 1.9 | 3.6 | - | 9 | 37 | [130] | |
| O2 | Ga/CZA | 50 | / | - | 54.5 | 22.2 | [131] | |
| O2 | Ni/YSZ | 80 | 160 | RT | 35.3 | 23.5 | [132] | |
| Air | CuZn/Al2O3 | 1.7 | 3.4 | - | 11 | 28 | [133] | |
| Air | Fe2O3-CuO /CP | 140 | 28 | 473 | ~26 | 11 | [134] | |
| Air | Fe2O3/CuO/Al2O3 | 120 | 24 | 473 | 43 | 3.7 | [135] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Ahmed, R.; Yi, Y.; Bogaerts, A. Methane to Methanol through Heterogeneous Catalysis and Plasma Catalysis. Catalysts 2021, 11, 590. https://doi.org/10.3390/catal11050590
Li S, Ahmed R, Yi Y, Bogaerts A. Methane to Methanol through Heterogeneous Catalysis and Plasma Catalysis. Catalysts. 2021; 11(5):590. https://doi.org/10.3390/catal11050590
Chicago/Turabian StyleLi, Shangkun, Rizwan Ahmed, Yanhui Yi, and Annemie Bogaerts. 2021. "Methane to Methanol through Heterogeneous Catalysis and Plasma Catalysis" Catalysts 11, no. 5: 590. https://doi.org/10.3390/catal11050590
APA StyleLi, S., Ahmed, R., Yi, Y., & Bogaerts, A. (2021). Methane to Methanol through Heterogeneous Catalysis and Plasma Catalysis. Catalysts, 11(5), 590. https://doi.org/10.3390/catal11050590