
Bulk Co3O4 for Methane Oxidation: Effect of the Synthesis Route on Physico-Chemical Properties and Catalytic Performance



Abstract
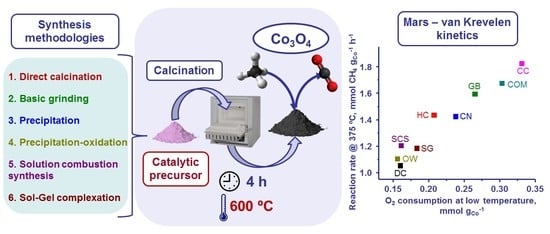
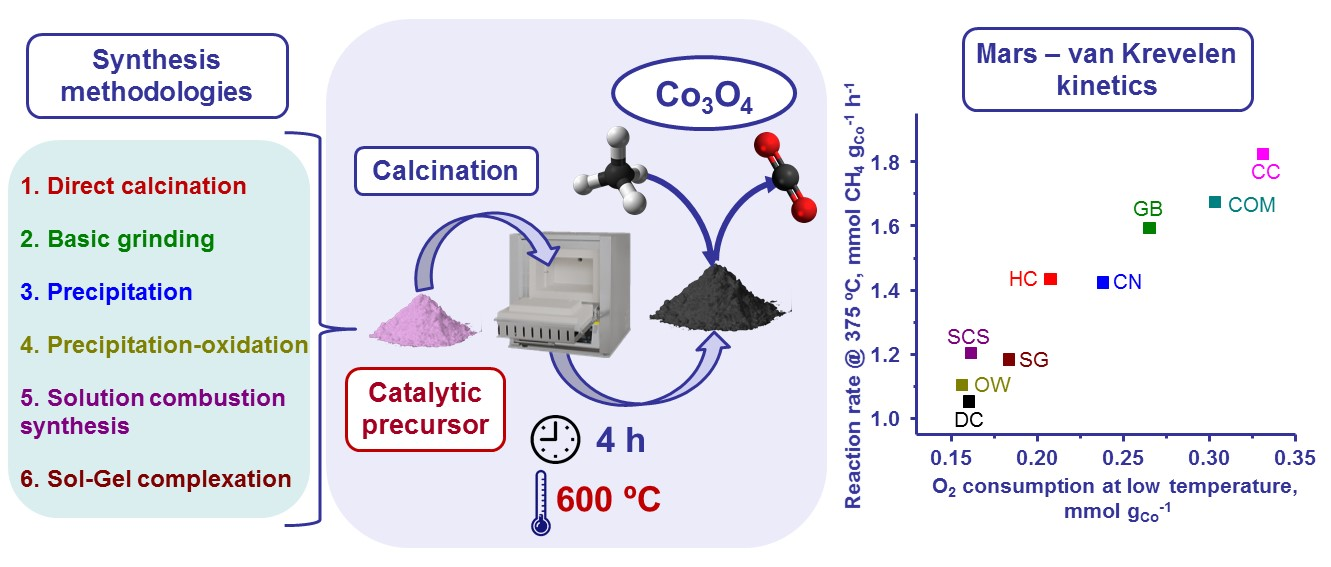
:
1. Introduction
2. Results and Discussion
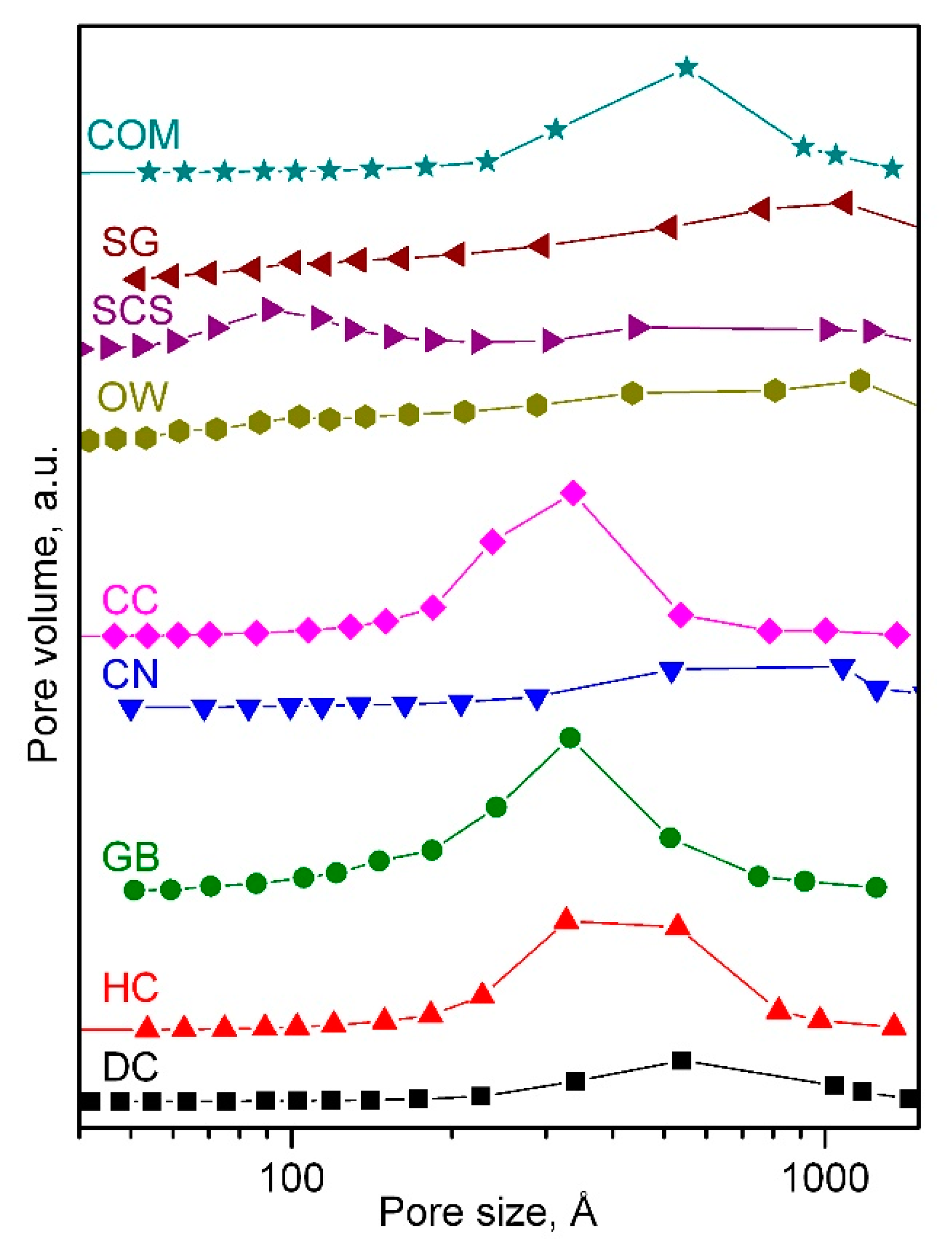
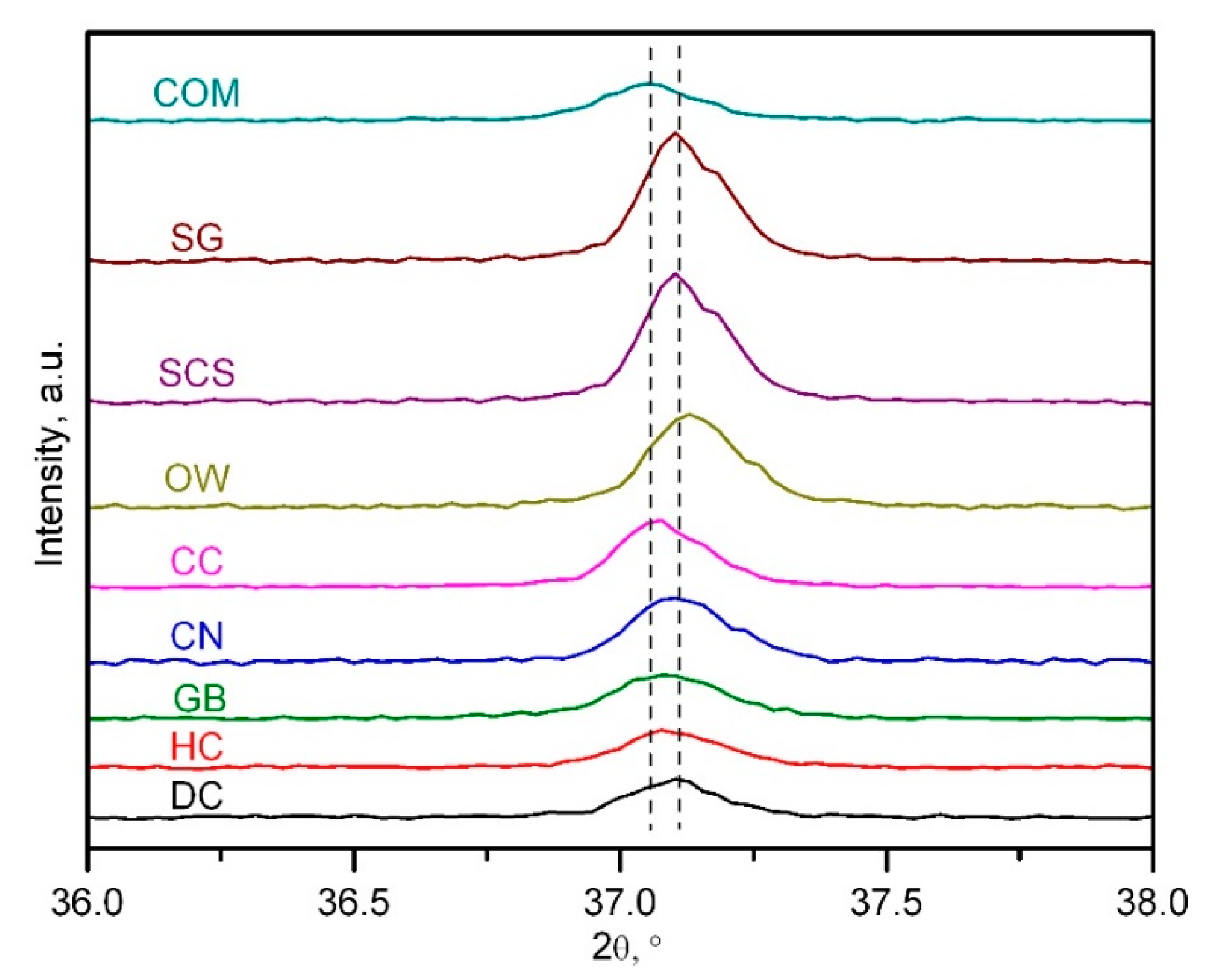
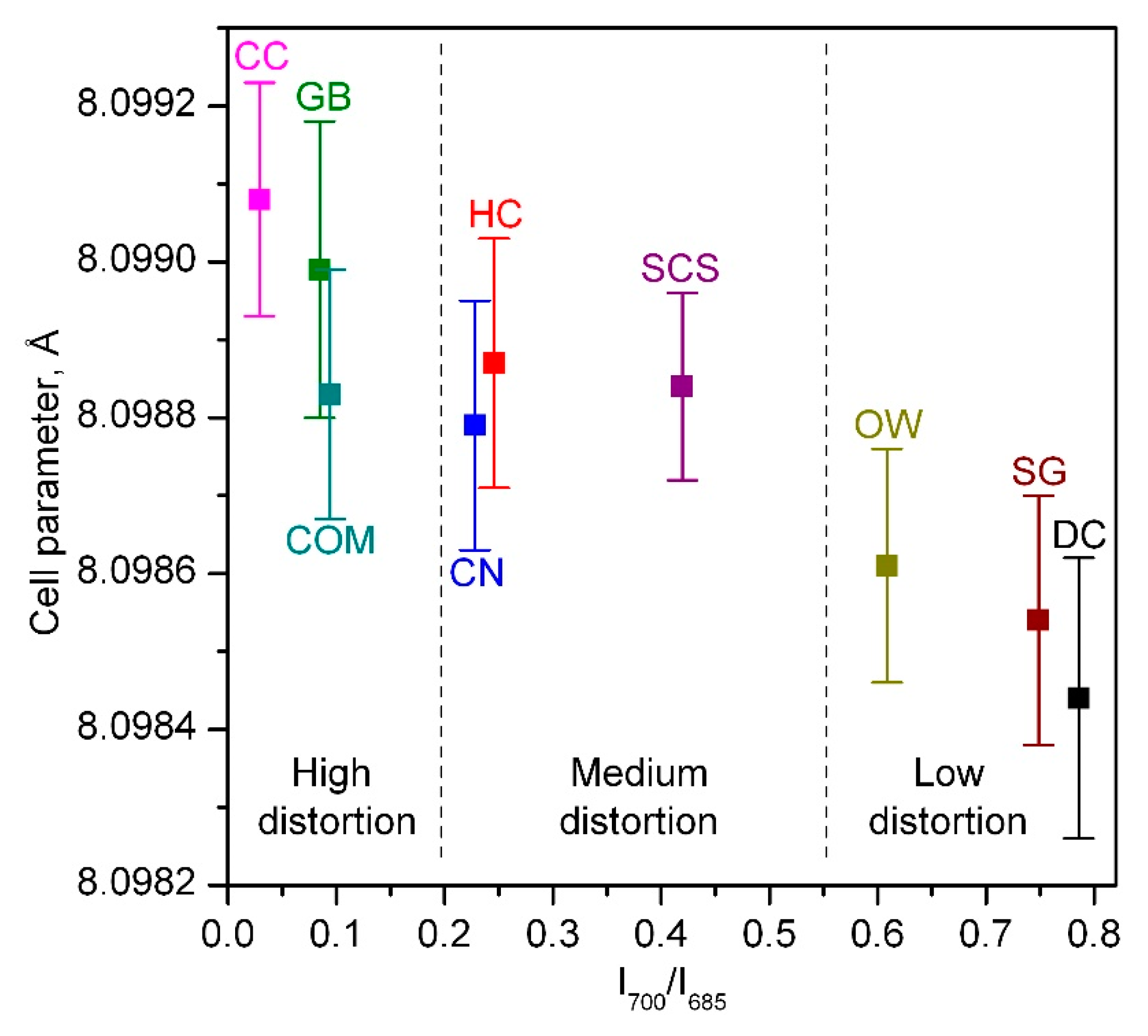
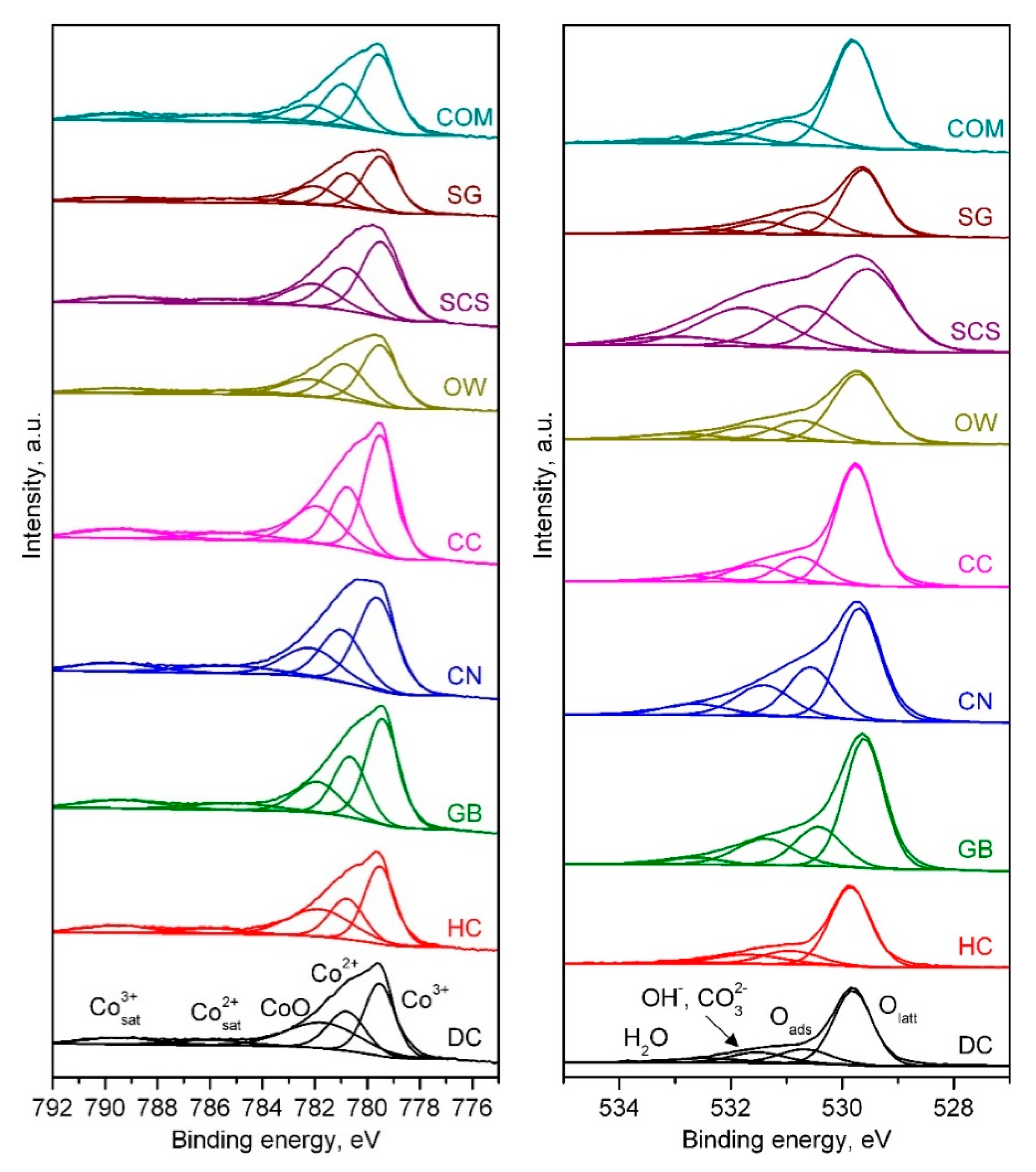
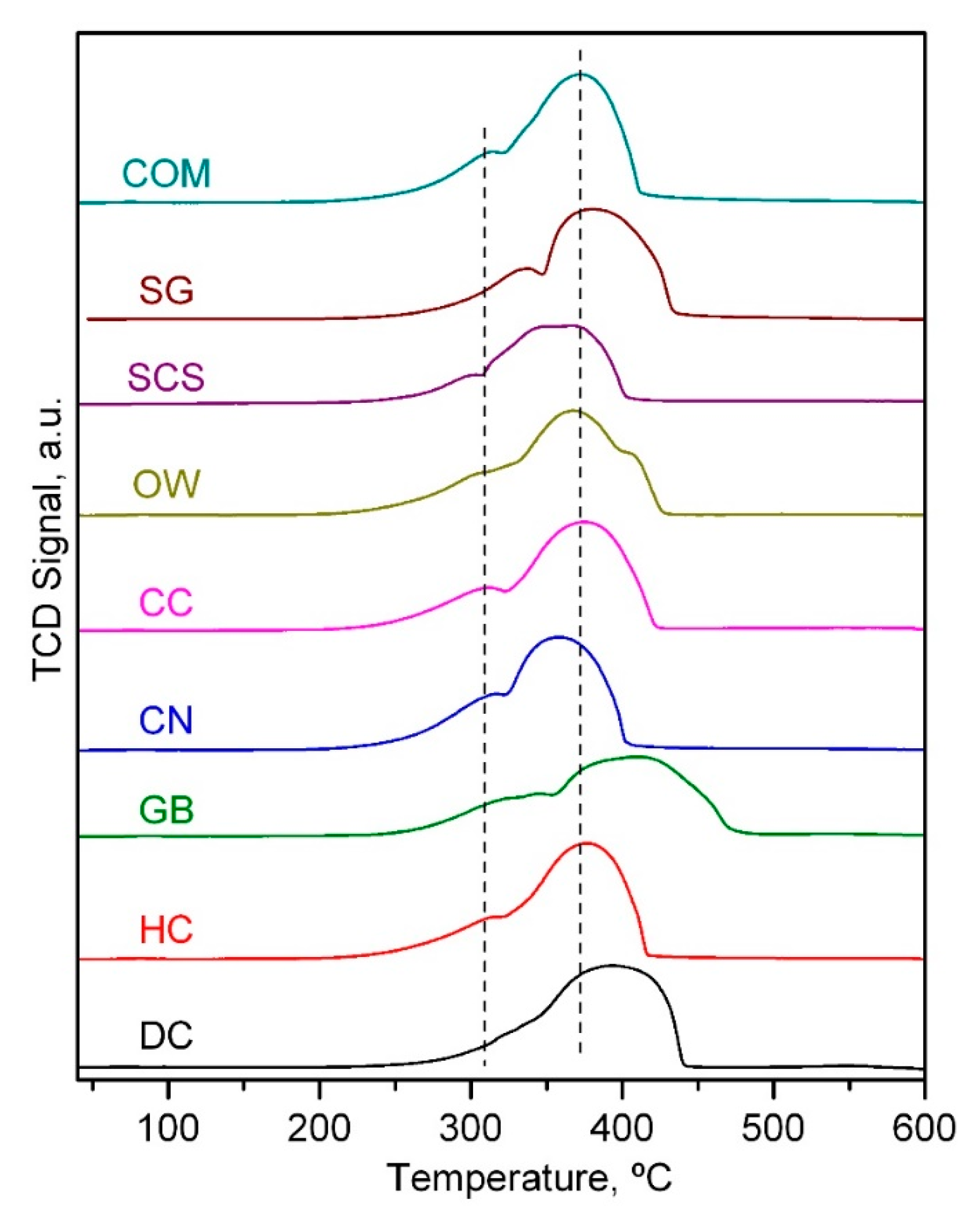
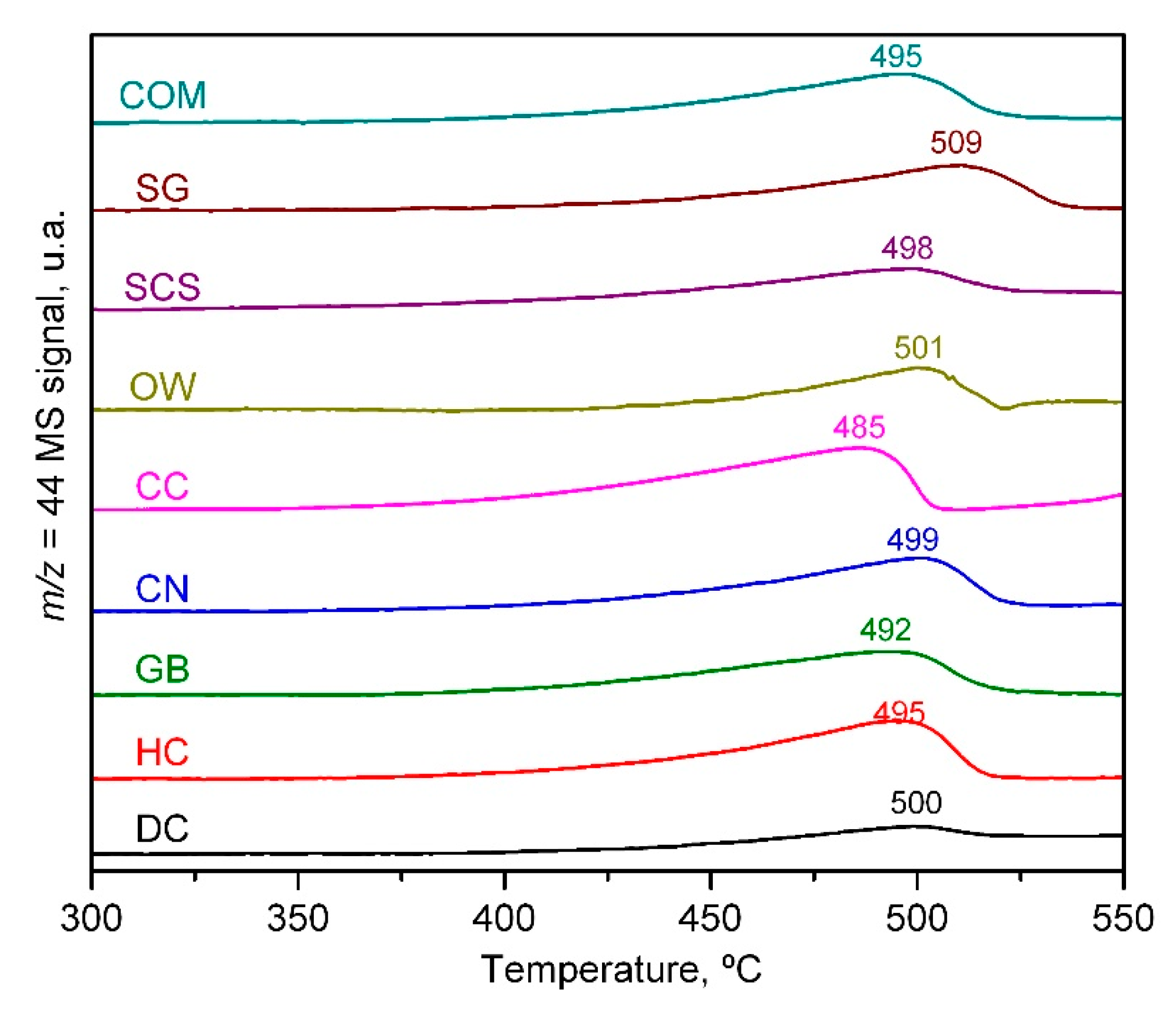
2.1. Physico-Chemical Characterisation of the Catalysts
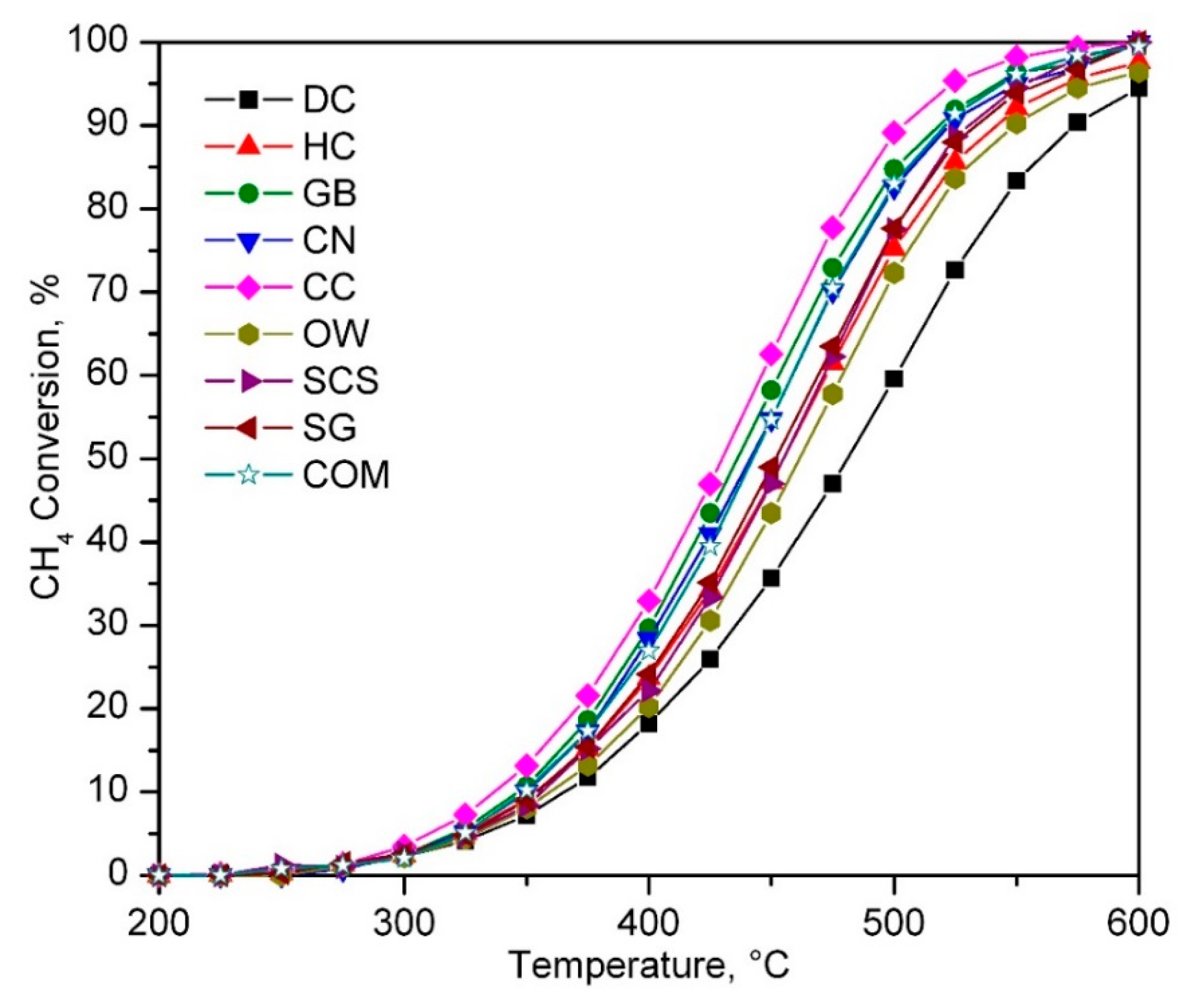
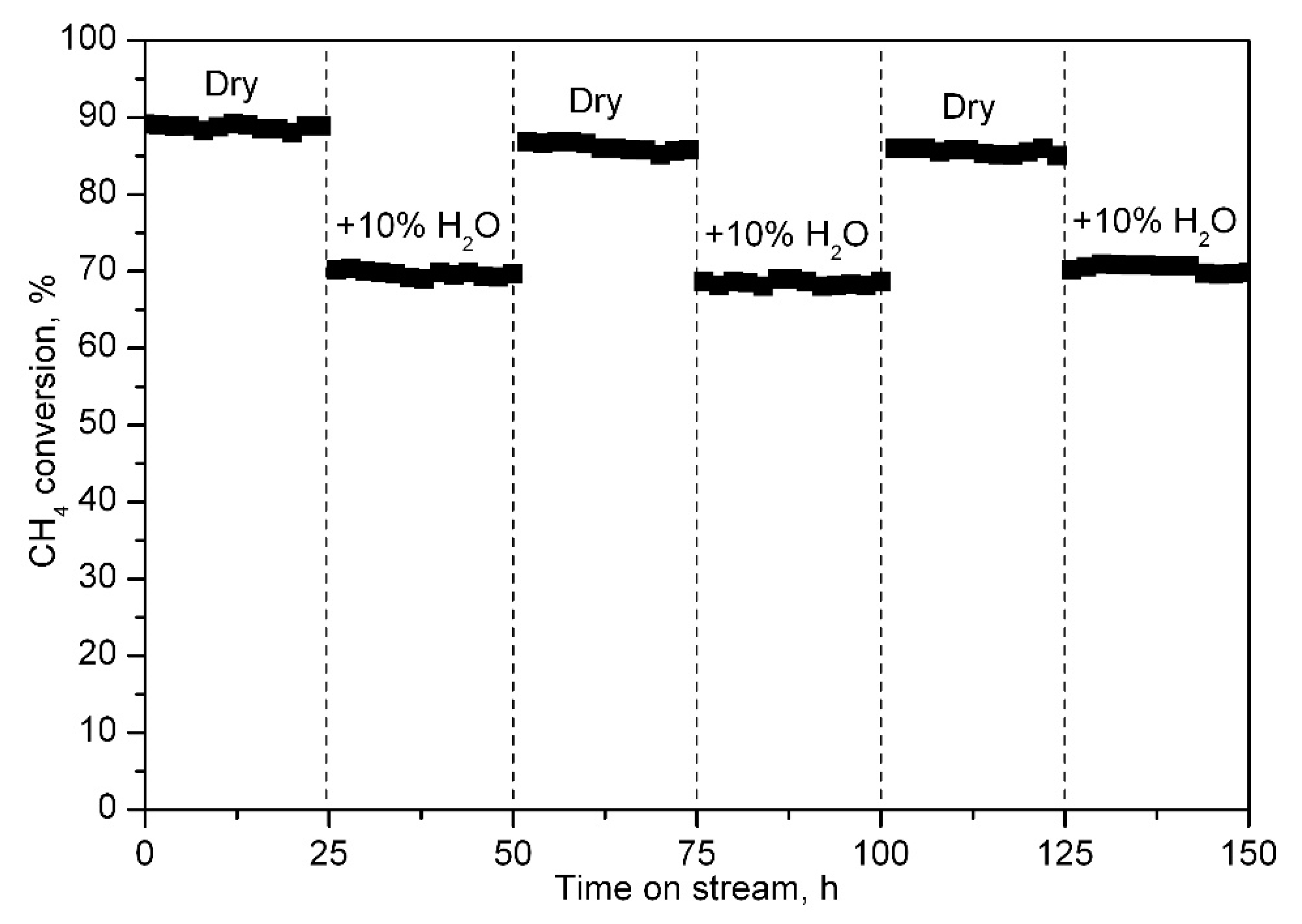
2.2. Catalytic Performance of the Synthesised Catalysts
3. Materials and Methods
3.1. Synthesis of the Bulk Catalysts
3.2. Characterisation Techniques
3.3. Evaluation of the Catalytic Activity and Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raj, A. Methane emission control. Johns. Matthey Technol. Rev. 2016, 60, 228–235. [Google Scholar] [CrossRef]
- Khan, M.I.; Yasmin, T.; Shakoor, A. Technical overview of compressed natural gas (CNG) as a transportation fuel. Renew. Sust. Energ. Rev. 2015, 51, 785–797. [Google Scholar] [CrossRef]
- Huang, S.; Li, T.; Wang, X.; Chen, R.; Yang, R.; Qian, Z. Effects of various discharge strategies on ignition and combustion of lean natural gas mixture under the static and turbulent conditions. Exp. Therm. Fluid Sci. 2022, 133, 110581. [Google Scholar] [CrossRef]
- Wang, J.; Gui, H.; Yang, Z.; Yu, T.; Zhang, X.; Liu, J. Real-world gaseous emission characteristics of natural gas heavy-duty sanitation trucks. J. Environ. Sci. 2022, 115, 319–329. [Google Scholar] [CrossRef]
- Maunula, T.; Kallinen, K.; Kinnunen, N.; Keenan, M.; Wolff, T. Methane abatement and catalyst durability in heterogeneous lean-rich and dual-fuel conditions. Top. Catal. 2019, 62, 315–323. [Google Scholar] [CrossRef]
- Kinnunen, N.M.; Hirvi, J.T.; Kallinen, K.; Maunula, T.; Keenan, M.; Suvanto, M. Case study of a modern lean-burn methane combustion catalyst for automotive applications: What are the deactivation and regeneration mechanisms? Appl. Catal. B Environ. 2017, 207, 114–119. [Google Scholar] [CrossRef]
- Velin, P.; Ek, M.; Skoglundh, M.; Schaefer, A.; Raj, A.; Thompsett, D.; Smedler, G.; Carlsson, P.-A. Water inhibition in methane oxidation over alumina supported palladium catalysts. J. Phys. Chem. C 2019, 123, 25724–25737. [Google Scholar] [CrossRef]
- Mihai, O.; Smedler, G.; Nylén, U.; Olofsson, M.; Olsson, L. The effect of water on methane oxidation over Pd/Al2O3 under lean, stoichiometric and rich conditions. Catal. Sci. Technol. 2017, 7, 3084–3096. [Google Scholar] [CrossRef]
- Tang, W.; Weng, J.; Lu, X.; Wen, L.; Suburamanian, A.; Nam, C.-Y.; Gao, P.-X. Alkali-metal poisoning effect of total CO and propane oxidation over Co3O4 nanocatalysts. Appl. Catal. B Environ. 2019, 256, 117859. [Google Scholar] [CrossRef]
- González-Prior, J.; López-Fonseca, R.; Gutiérrez-Ortiz, J.I.; de Rivas, B. Catalytic removal of chlorinated compounds over ordered mesoporous cobalt oxides synthesised by hard-templating. Appl. Catal. B Environ. 2018, 222, 9–17. [Google Scholar] [CrossRef]
- Ma, Z. Cobalt oxide catalysts for environmental remediation. Curr. Catal. 2014, 3, 15–26. [Google Scholar] [CrossRef]
- Setiawan, A.; Kennedy, E.M.; Dlugogorski, B.Z.; Adesina, A.A.; Stockenhuber, M. The stability of Co3O4, Fe2O3, Au/Co3O4 and Au/Fe2O3 catalysts in the catalytic combustion of lean methane mixtures in the presence of water. Catal. Today 2015, 258, 276–283. [Google Scholar] [CrossRef]
- Zhang, W.; Descorme, C.; Valverde, J.L.; Giroir-Fendler, A. Cu-Co mixed oxide catalysts for the total oxidation of toluene and propane. Catal. Today 2022, 384–386, 238–245. [Google Scholar] [CrossRef]
- Karthick, S.N.; Hemalatha, K.V.; Justin Raj, C.; Kim, H.J.; Yi, M. Synthesis of nano-bound microsphere Co3O4 by simple polymer-assisted sol-gel technique. J. Nanopart. Res. 2013, 15, 1474. [Google Scholar] [CrossRef]
- Arango-Diaz, A.; Cecilia, J.A.; Marrero-Jerez, J.; Nuñez, P.; Jiménez-Jiménez, J.; Rodríguez-Castellón, E. Freeze-dried Co3O4-CeO2 catalysts for the preferential oxidation of CO with the presence of CO2 and H2O in the feed. Ceram. Int. 2016, 42, 7462–7474. [Google Scholar] [CrossRef]
- Ercolino, G.; Stelmachowski, P.; Specchia, S. Catalytic performance of Pd/Co3O4 on SiC and ZrO2 open cell foams for process intensification of methane combustion in lean conditions. Ind. Eng. Chem. Res. 2017, 56, 6625–6636. [Google Scholar] [CrossRef]
- Wang, Q.; Deng, W.; Lin, X.; Huang, X.; Wei, L.; Gong, L.; Liu, C.; Liu, G.; Liu, Q. Solid-state preparation of mesoporous Ce–Mn–Co ternary mixed oxide nanoparticles for catalytic degradation of methylene blue. J. Rare Earth. 2021, 39, 826–834. [Google Scholar] [CrossRef]
- Liu, B.; Peng, J.; Zhang, L.; Wan, R.; Guo, S.; Zhou, L. Optimization of preparation for Co3O4 by calcination from cobalt oxalate using response surface methodology. Chem. Eng. Res. Des. 2010, 88, 971–976. [Google Scholar] [CrossRef]
- Biabani-Ravandi, A.; Rezaei, M. Low temperature CO oxidation over Fe-Co mixed oxide nanocatalysts. Chem. Eng. J. 2012, 184, 141–146. [Google Scholar] [CrossRef]
- Shi, C.; Wang, Y.; Zhu, A.; Chen, B.; Au, C. MnxCo3-xO4 solid solution as high-efficient catalysts for low-temperature oxidation of formaldehyde. Catal. Commun. 2012, 28, 18–22. [Google Scholar] [CrossRef]
- Zou, G.; Xu, Y.; Wang, S.; Chen, M.; Shangguan, W. The synergistic effect in Co-Ce oxides for catalytic oxidation of diesel soot. Catal. Sci. Technol. 2015, 5, 1084–1092. [Google Scholar] [CrossRef]
- Li, D.; Wang, L.; Koike, M.; Nakagawa, Y.; Tomishige, K. Steam reforming of tar from pyrolysis of biomass over Ni/Mg/Al catalysts prepared from hydrotalcite-like precursors. Appl. Catal. B Environ. 2011, 102, 528–538. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, M.; Song, Z.; Zhao, H.; Liu, W.; Zhao, J.; Ma, Z.; Xing, Y. The effect of different metal oxides on the catalytic activity of a Co3O4 catalyst for toluene combustion: Importance of the structure-property relationship and surface active species. New J. Chem. 2019, 43, 10868–10877. [Google Scholar] [CrossRef]
- Zhang, Q.; Mo, S.; Chen, B.; Zhang, W.; Huang, C.; Ye, D. Hierarchical Co3O4 nanostructures in-situ grown on 3D nickel foam towards toluene oxidation. Mol. Catal. 2018, 454, 12–20. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Cañón, J.; Teplyakov, A.V. XPS characterization of cobalt impregnated SiO2 and γ-Al2O3. Surf. Interface Anal. 2021, 53, 475–481. [Google Scholar] [CrossRef]
- Cole, K.M.; Kirk, D.W.; Thorpe, S.J. Co3O4 nanoparticles characterized by XPS and UPS. Surf. Sci. Spectra 2021, 28, 014001. [Google Scholar] [CrossRef]
- Ilieva, L.; Petrova, P.; Venezia, A.M.; Anghel, E.M.; State, R.; Avdeev, G.; Tabakova, T. Mechanochemically Prepared Co3O4-CeO2 Catalysts for Complete Benzene Oxidation. Catalysts 2021, 11, 1316. [Google Scholar] [CrossRef]
- Zasada, F.; Janas, J.; Piskorz, W.; Gorczynska, M.; Sojka, Z. Total oxidation of lean methane over cobalt spinel nanocubes controlled by the self-adjusted redox state of the catalyst: Experimental and theoretical account for interplay between the Langmuir-Hinshelwood and Mars-Van Krevelen mechanisms. ACS Catal. 2017, 7, 2853–2867. [Google Scholar] [CrossRef]
- Rosen, J.; Hutchings, G.S.; Jiao, F. Ordered mesoporous cobalt oxide as highly efficient oxygen evolution catalyst. J. Am. Chem. Soc. 2013, 135, 4516–4521. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Li, Y.; Mo, Y.; Lan, J.; Jiang, Y.; Feng, S. Rod-like and mushroom-like Co3O4–CeO2 catalysts derived from Ce-1,3,5-benzene tricarboxylic acid for CO preferential oxidation: Effects of compositions and morphology. React. Kinet. Mech. Catal. 2020, 129, 135–151. [Google Scholar] [CrossRef]
- Zheng, Y.; Yu, Y.; Zhou, H.; Huang, W.; Pu, Z. Combustion of lean methane over Co3O4 catalysts prepared with different cobalt precursors. RSC Adv. 2020, 10, 4490–4498. [Google Scholar] [CrossRef] [Green Version]
- Zasada, F.; Grybos, J.; Budiyanto, E.; Janas, J.; Sojka, Z. Oxygen species stabilized on the cobalt spinel nano-octahedra at various reaction conditions and their role in catalytic CO and CH4 oxidation, N2O decomposition and oxygen isotopic exchange. J. Catal. 2019, 371, 224–235. [Google Scholar] [CrossRef]
- Xu, X.; Han, H.; Liu, J.; Liu, W.; Li, W.; Wang, X. Promotional effects of samarium on Co3O4 spinel for CO and CH4 oxidation. J. Rare Earth. 2014, 32, 159–169. [Google Scholar] [CrossRef]
- Li, G.; Li, N.; Sun, Y.; Qu, Y.; Jiang, Z.; Zhao, Z.; Zhang, Z.; Cheng, J.; Hao, Z. Efficient defect engineering in Co-Mn binary oxides for low-temperature propane oxidation. Appl. Catal. B Environ. 2021, 282, 119512. [Google Scholar] [CrossRef]
- Choya, A.; de Rivas, B.; González-Velasco, J.R.; Gutiérrez-Ortiz, J.I.; López-Fonseca, R. Optimisation of bimetallic Co-Ni supported catalysts for oxidation of methane in natural gas vehicles. Appl. Catal. B Environ. 2021, 284, 119712. [Google Scholar] [CrossRef]
- Rodrigues, J.M.; Ribeiro, M.F.; Fernandes, E.C. Catalytic activity of electrodeposited cobalt oxide films for methane combustion in a micro-channel reactor. Fuel 2018, 232, 51–59. [Google Scholar] [CrossRef]
- Stefanov, P.; Todorova, S.; Naydenov, A.; Tzaneva, B.; Kolev, H.; Atanasova, G.; Stoyanova, D.; Karakirova, Y.; Aleksieva, K. On the development of active and stable Pd-Co/γ-Al2O3 catalyst for complete oxidation of methane. Chem. Eng. J. 2015, 266, 329–338. [Google Scholar] [CrossRef]
- Nasr, S.; Hayes, R.E.; Semagina, N. Stability of kinetic parameters of Co3O4/CeO2 catalyzed methane combustion. Can. J. Chem. Eng. 2021, 99, 2670–2676. [Google Scholar] [CrossRef]
- Zasada, F.; Piskorz, W.; Janas, J.; Grybos, J.; Indyka, P.; Sojka, Z. Reactive oxygen species on the (100) facet of cobalt spinel nanocatalyst and their relevance in 16O2/18O2 isotopic exchange, deN2O, and deCH4 processes–A theoretical and experimental account. ACS Catal. 2015, 5, 6879–6892. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | BET Surface, m2 g−1 | Pore Volume, cm3 g−1 | Mean Pore Diameter, Å | Crystallite Size, nm | Cell Parameter, Å |
|---|---|---|---|---|---|
| DC | 5 | 0.02 | 355 | 84 | 8.09844 ± 0.00018 |
| HC | 12 | 0.06 | 350 | 64 | 8.09887 ± 0.00016 |
| GB | 16 | 0.10 | 275 | 58 | 8.09899 ± 0.00019 |
| CN | 6 | 0.02 | 460 | 92 | 8.09879 ± 0.00016 |
| CC | 14 | 0.09 | 255 | 63 | 8.09908 ± 0.00015 |
| OW | 9 | 0.02 | 170 | 86 | 8.09861 ± 0.00015 |
| SCS | 14 | 0.04 | 170 | 89 | 8.09884 ± 0.00012 |
| SG | 5 | 0.01 | 370 | 102 | 8.09854 ± 0.00016 |
| COM | 8 | 0.03 | 410 | 75 | 8.09883 ± 0.00016 |
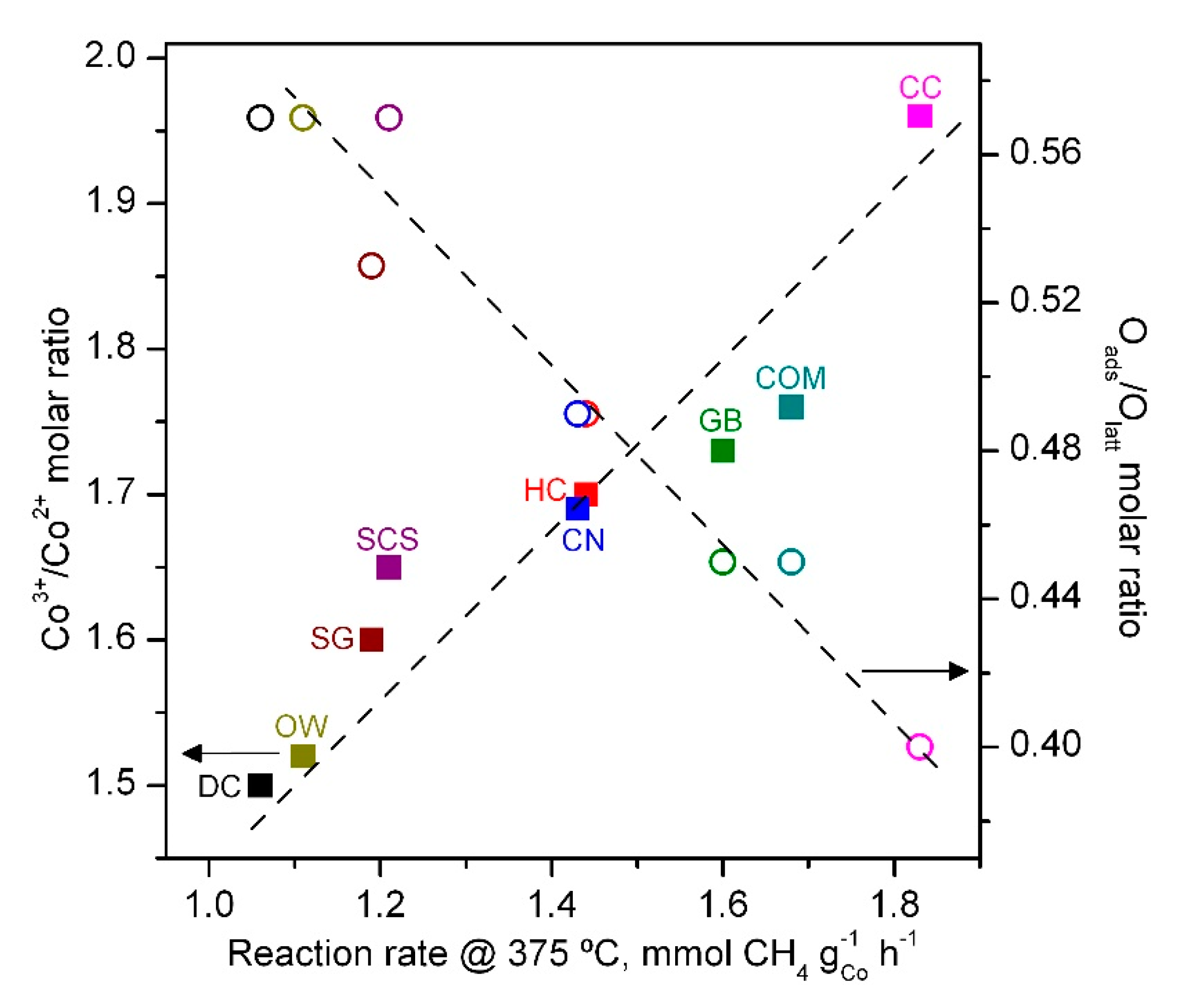
| Catalyst | Co3+/Co2+ Molar Ratio | Oads/Olatt Molar Ratio |
|---|---|---|
| DC | 1.50 | 0.57 |
| HC | 1.70 | 0.49 |
| GB | 1.73 | 0.45 |
| CN | 1.69 | 0.49 |
| CC | 1.96 | 0.40 |
| OW | 1.52 | 0.57 |
| SCS | 1.65 | 0.57 |
| SG | 1.60 | 0.53 |
| COM | 1.76 | 0.45 |
| Catalyst | Low-Temperature H2 Uptake, mmol g−1 | High-Temperature H2 Uptake, mmol g−1 | Relative H2 Uptake at Low and High Temperature | Onset Reduction Temperature, °C |
|---|---|---|---|---|
| DC | 2.6 | 13.9 | 0.19 | 300 |
| HC | 3.3 | 13.4 | 0.25 | 270 |
| GB | 4.3 | 12.4 | 0.34 | 285 |
| CN | 4.1 | 12.6 | 0.32 | 270 |
| CC | 4.6 | 12.0 | 0.38 | 265 |
| OW | 3.4 | 12.3 | 0.25 | 270 |
| SCS | 3.9 | 12.7 | 0.30 | 280 |
| SG | 4.0 | 12.7 | 0.32 | 290 |
| COM | 4.2 | 12.4 | 0.34 | 275 |
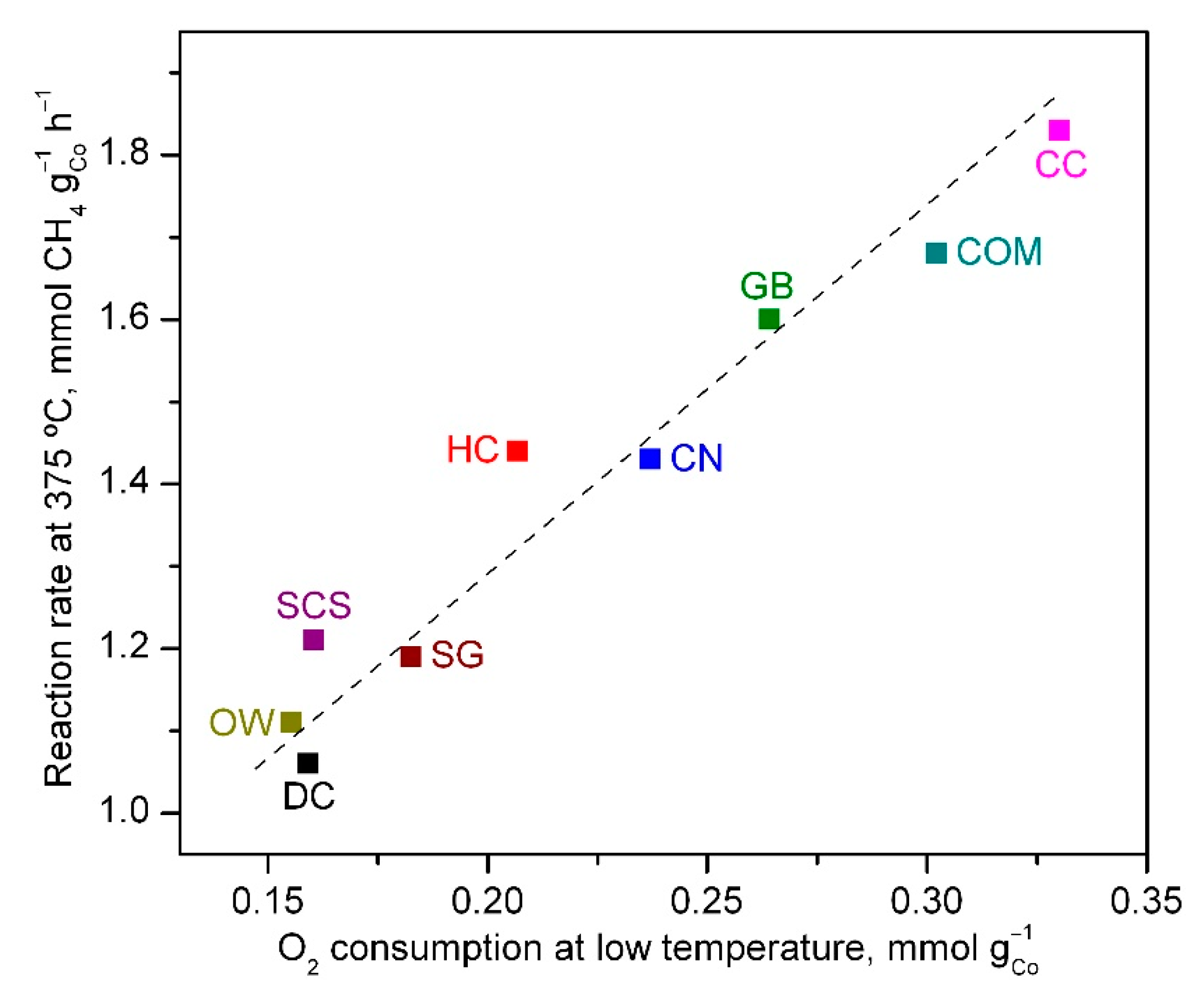
| Catalyst | T50, °C | Reaction Rate at 375 °C, mmol CH4 gCo−1 h−1 | Ea, kJ mol−1 |
|---|---|---|---|
| DC | 480 | 1.06 | 71 ± 2 |
| HC | 455 | 1.44 | 74 ± 2 |
| GB | 435 | 1.60 | 74 ± 1 |
| CN | 440 | 1.43 | 74 ± 2 |
| CC | 430 | 1.83 | 74 ± 2 |
| OW | 460 | 1.11 | 73 ± 2 |
| SCS | 455 | 1.21 | 75 ± 1 |
| SG | 450 | 1.19 | 73 ± 2 |
| COM | 440 | 1.68 | 74 ± 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choya, A.; de Rivas, B.; Gutiérrez-Ortiz, J.I.; López-Fonseca, R. Bulk Co3O4 for Methane Oxidation: Effect of the Synthesis Route on Physico-Chemical Properties and Catalytic Performance. Catalysts 2022, 12, 87. https://doi.org/10.3390/catal12010087
Choya A, de Rivas B, Gutiérrez-Ortiz JI, López-Fonseca R. Bulk Co3O4 for Methane Oxidation: Effect of the Synthesis Route on Physico-Chemical Properties and Catalytic Performance. Catalysts. 2022; 12(1):87. https://doi.org/10.3390/catal12010087
Chicago/Turabian StyleChoya, Andoni, Beatriz de Rivas, Jose Ignacio Gutiérrez-Ortiz, and Rubén López-Fonseca. 2022. "Bulk Co3O4 for Methane Oxidation: Effect of the Synthesis Route on Physico-Chemical Properties and Catalytic Performance" Catalysts 12, no. 1: 87. https://doi.org/10.3390/catal12010087
APA StyleChoya, A., de Rivas, B., Gutiérrez-Ortiz, J. I., & López-Fonseca, R. (2022). Bulk Co3O4 for Methane Oxidation: Effect of the Synthesis Route on Physico-Chemical Properties and Catalytic Performance. Catalysts, 12(1), 87. https://doi.org/10.3390/catal12010087