2.1. Characteristics of CuO/CeO2 Catalysts
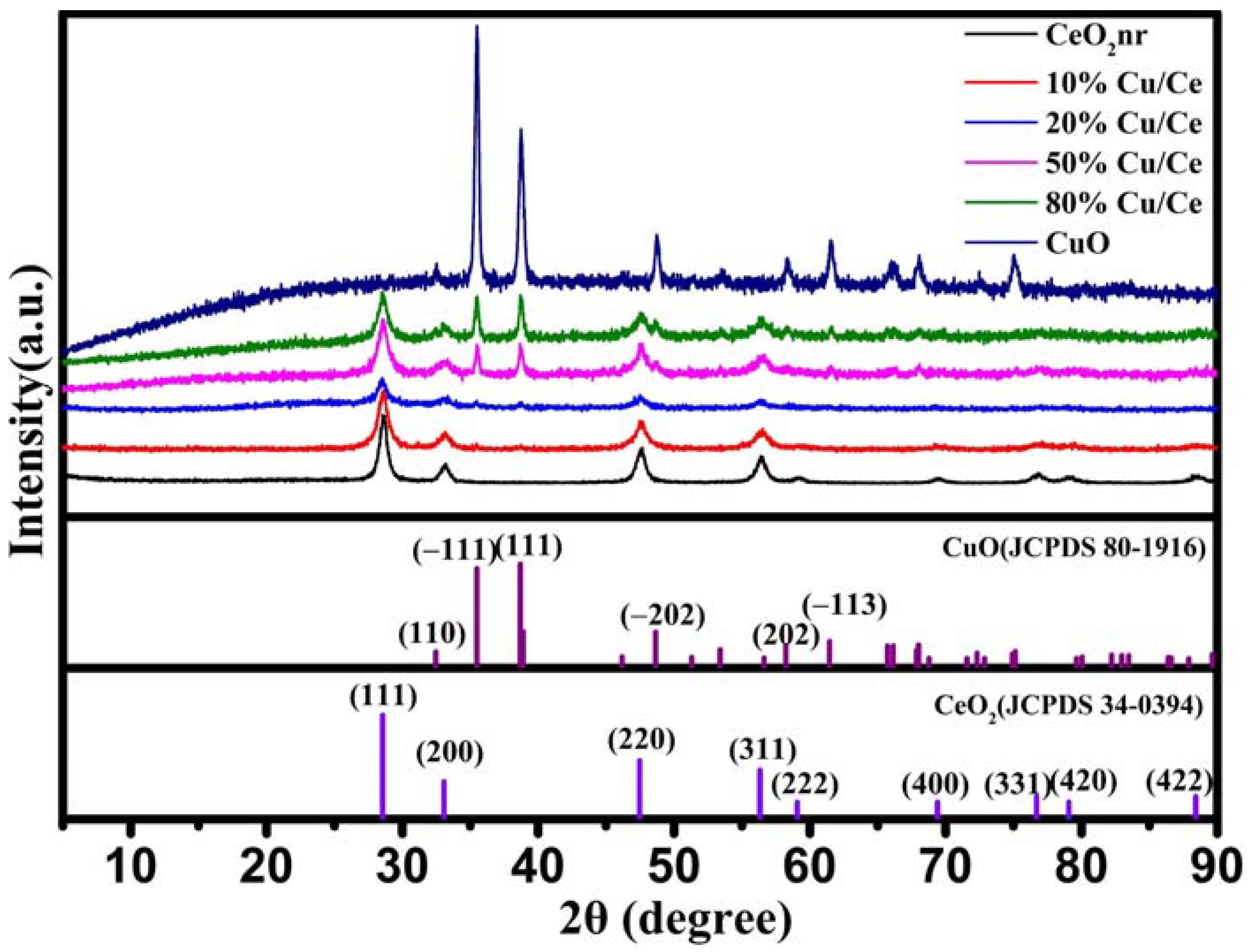
The as-prepared materials were characterized using XRD to identify crystal phases (
Figure 2). The
Figure 2 shows that the CeO
2 and CuO diffraction patterns match the characteristic peaks of the fluorite structure of CeO
2 (JCPDS 34-0394) and the monoclinic structure of CuO (JCPDS 80-1916), respectively. The intensity of the CuO diffraction peaks gradually increased with increased Cu/Ce ratio, and some CeO
2 diffraction peaks disappeared. When the Cu/Ce ratio was 10%, weak diffraction peaks appeared at 36.49° and 38.68°, corresponding to the (−111) and (111) crystal planes of the CuO diffraction peaks, respectively. This phenomenon may have been caused by the low loading of CuO. When the Cu/Ce ratio was 50%, the intensities of the diffraction peaks of the (−111) and (111) crystal planes increased, and new diffraction peaks appeared at 48.6° and 61.6°, corresponding to the (−202) and (−113) crystal planes of CuO, respectively. The crystalline size of the catalyst was calculated using the Scherrer equation, as shown in
Table 1. Owing to the smaller Cu diffraction peaks in 10%Cu/Ce and 20%Cu/Ce, the crystalline size of CuO could not be calculated.
Table 1 shows that the grain sizes of CeO
2 and CuO prepared using the secondary alkaline method did not change significantly.
The actual ratio of Cu/Ce determined by ICP-OES was higher than the theoretically calculated value (
Table 1), indicating that CeO
2 was partially dissolved in the secondary alkaline hydrothermal environment. The specific surface area (S
BET) of the samples was determined using the N
2 adsorption–desorption method. As shown in
Table 1, the S
BET of the CuO/CeO
2 catalyst decreased, mainly due to the formation of sheet-morphology CuO (
Figure 3f). The S
BET of sheet-morphology CuO is significantly lower than that of CeO
2 (
Table 1). Therefore, the S
BET of the CuO/CeO
2 catalyst decreased with increased in CuO doping. Moreover, the S
BET of 50% Cu/Ce and 80% Cu/Ce catalysts decrease more significantly compared with that of 10% Cu/Ce and 20%Cu/Ce catalysts as a result of the appearance of a large amount of sheet-morphology CuO (
Figure 3d,e) on CeO
2 nanorods.
Figure 3 shows a TEM image of the CuO/CeO
2 catalyst.
Figure 3a,f shows that the morphologies of pure CeO
2 and CuO were nanorods and nanoplates, respectively.
Figure 3b–e shows TEM images of the CuO/CeO
2 catalysts with Cu/Ce ratios of 10, 20, 50, and 80%, respectively. The morphology of the CeO
2 nanorods did not change after loading with CuO, and when the Cu/Ce ratio was 50 and 80%, CuO nanoplates began to appear (
Figure 3d,e).
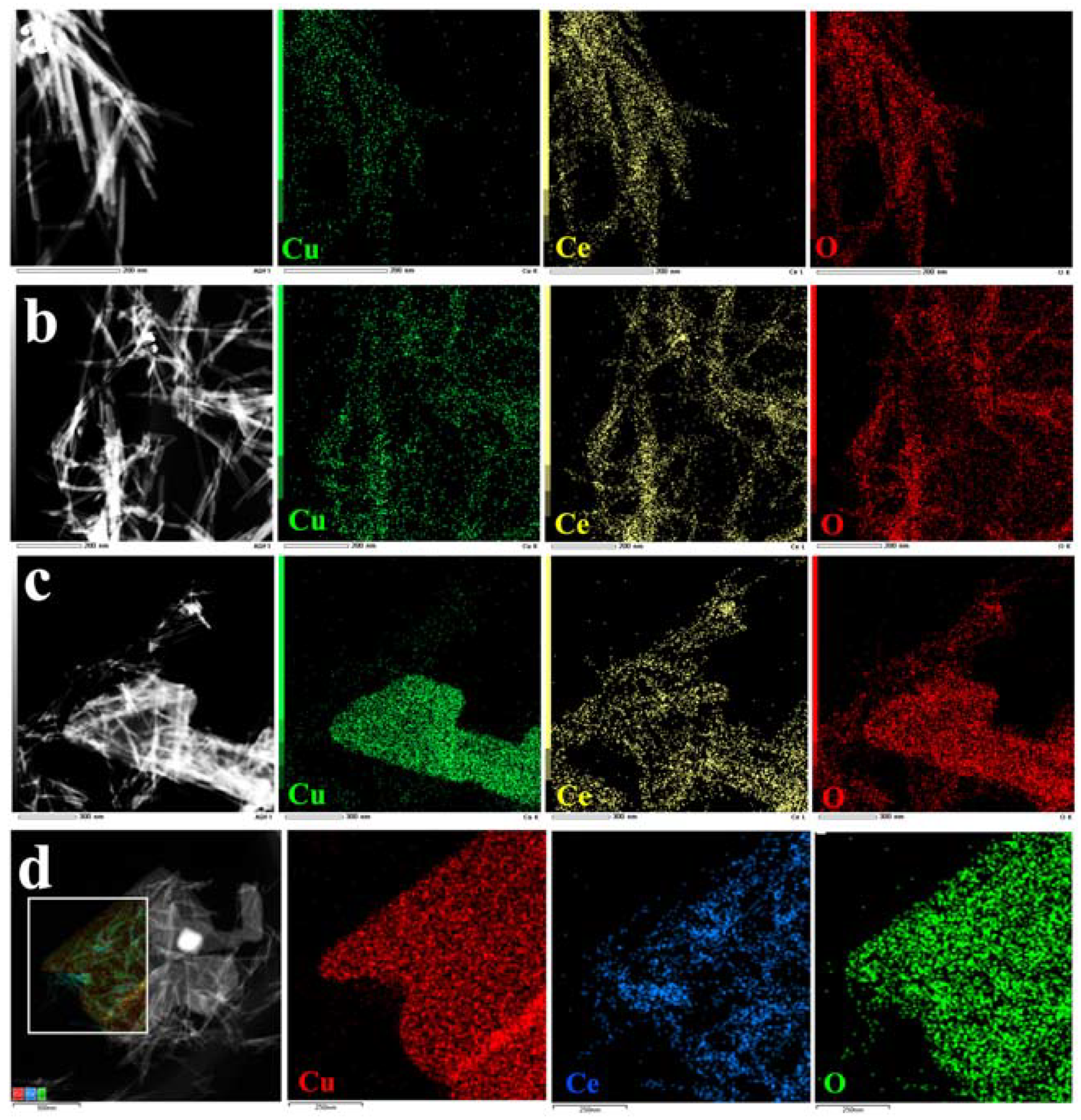
Figure 4 shows EDX images of the Cu/Ce catalysts with varying loading ratios, demonstrating that Cu was evenly distributed on the CeO nanorods (
Figure 4a,b).
Figure 5 shows HRTEM images of the catalysts. The exposed (111) and (200) crystal planes of the CeO
2 nanorods correspond to the lattice fringes at 0.31 and 0.28 nm, respectively (
Figure 5a); the exposed (020) crystal planes of the CuO nanoplates correspond to the lattice fringes at 0.17 nm (
Figure 5f).
Figure 4c–e shows that a lattice spacing of 0.29 nm appeared with increasing CuO load due to the change in lattice spacing caused by the doping of Cu atoms into the CeO
2 lattice.
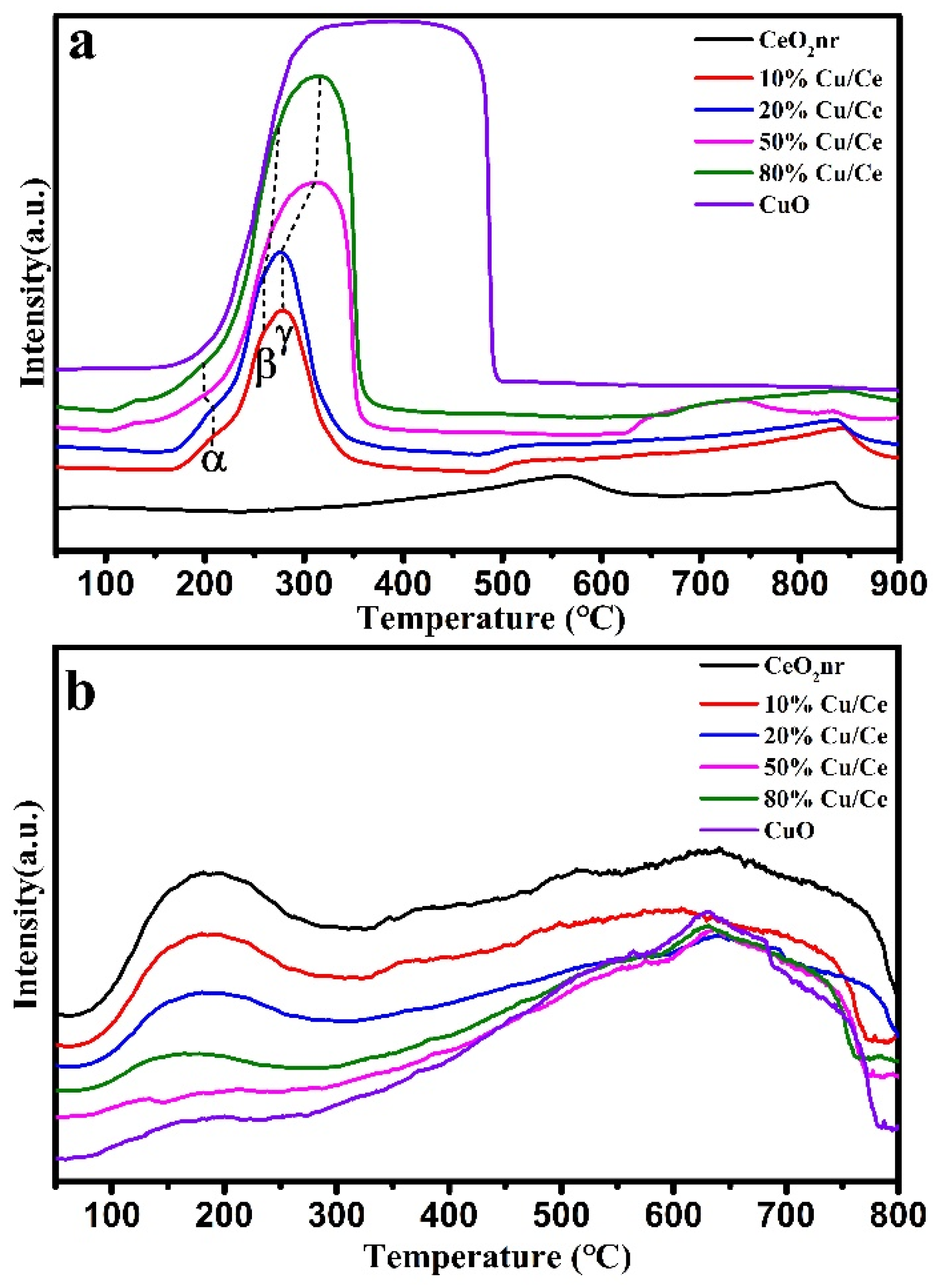
The oxidation activity of the catalyst material was characterized using H
2-TPR (
Figure 6a and
Table 2). There are usually two reduction peaks of CeO
2: a peak at approximately 675 °C corresponding to the surface oxygen reduction peak of CeO
2 and a peak at approximately 835 °C corresponding to the bulk oxygen reduction peak of CeO
2 [
36]. A broad peak appeared in CuO at 170–500 °C, which may be due to the closeness of the surface oxygen and bulk oxygen reduction peaks of CuO, such that the two peaks cannot be completely separated. CuO/CeO
2 has three main peaks at 170–202 °C, 250 °C, and 275 °C, which are denoted by α, β, and γ, respectively. The α peak corresponds to CuO
x, which interacts strongly with CeO
2; the β peak corresponds to CuO
x, which interacts weakly with CeO
2; and the γ peak is attributed to bulk CuO [
37]. The H
2-TPR diagram (
Figure 6) clearly shows that after CuO was loaded onto CeO
2, the reduction temperature of the CuO/CeO
2 catalyst was significantly lower than that of the CeO
2, indicating that the loaded catalyst had more active oxygen. H
2 consumption amounts of as-prepared catalyst were calculated using H
2-TPR (
Table 2). As shown in
Table 2, the theoretical and the calculated value of H
2 consumption increased with an increase in the Cu/Ce loading ratio. The calculated hydrogen consumption amount of 10%Cu/Ce catalyst exceeded theoretical values; however, with an increase in the Cu/Ce ratio, the theoretical calculated value exceeded the calculated value, implying a interaction between CeO
2 and CuO species [
38]. Evidently, the addition of CuO improved the CeO
2 oxidation activity.
O
2-TPD tests were performed on the CuO/CeO
2 catalysts, CeO
2, and CuO to investigate the desorption behavior of oxygen (
Figure 6b and
Table 3). Two types of oxygen exist in the catalyst, namely surface-adsorbed oxygen and lattice oxygen [
39]. The peak at 180 °C is related to chemisorbed oxygen around the oxygen vacancies, and the peak at 640 °C is related to oxygen escaping from the lattice. As shown in
Table 3, with an increase in the Cu/Ce ratio, the amount of oxygen adsorbed at low temperatures decreased, and the lattice oxygen at high temperatures increased.
XPS characterizations were used to obtain the composition information and determine the chemical state on the surface of CeO
2, CuO, and CuO/CeO
2.
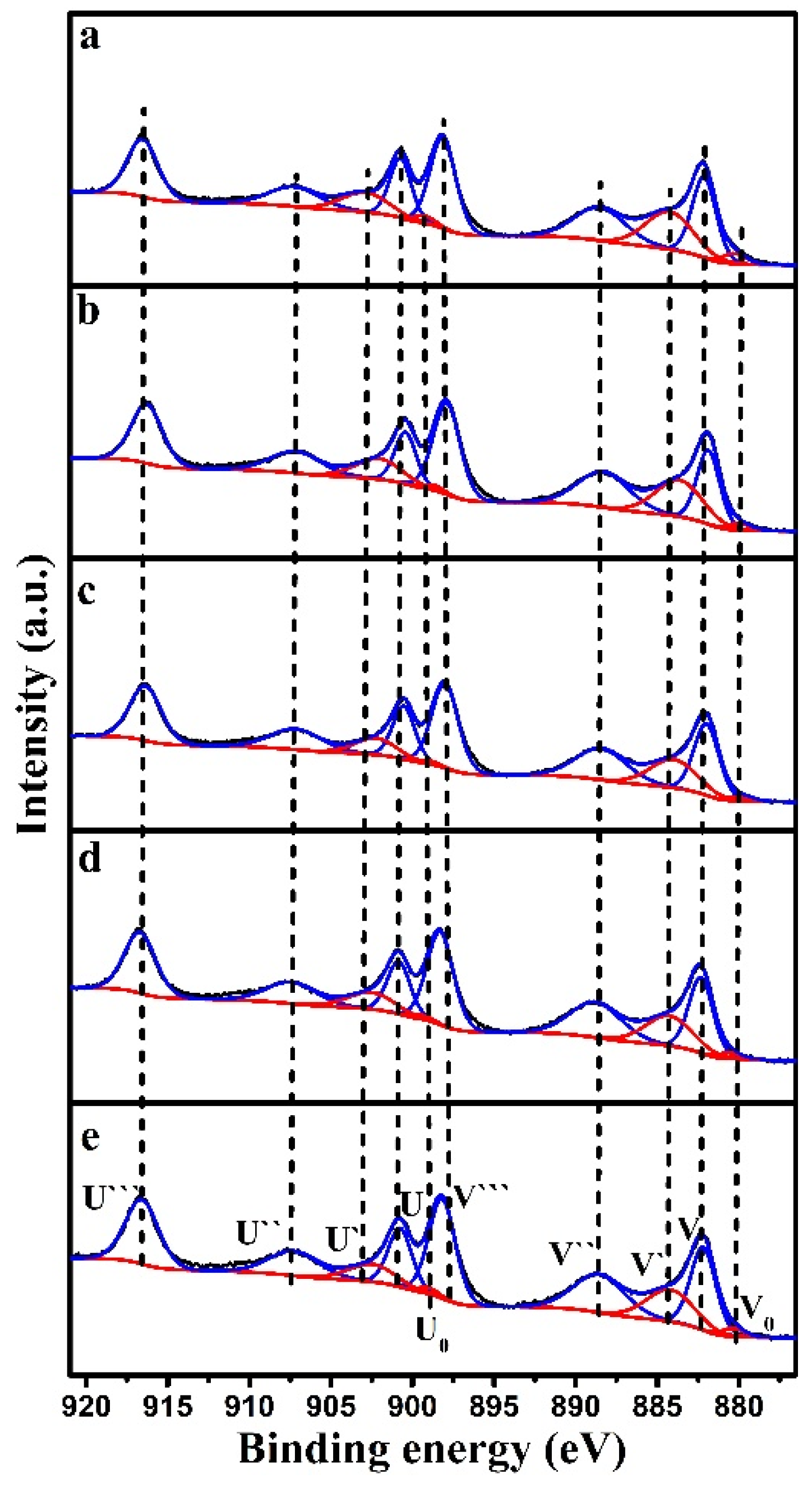
Figure 7 shows the Ce 3d XPS spectra of the CeO
2 and CuO/CeO
2 catalysts, which are deconvoluted into ten peaks [
36,
40,
41]. The Ce 3d spectra are composed of two-group spin orbitals of the overlapping peaks, labeled as U
0–U``` for 3d
3/2 and V
0–V``` for 3d
5/2. The binding energy peaks of V (882.14 eV), V`` (888.51 eV), V``` (898.05 eV), U (900.63 eV), U`` (907.32 eV), and U``` (916.45 eV) are related to Ce
4+, whereas the binding energy peaks of V
0 (880.2 eV), V` (884.7 eV), U
0 (898.65 eV), and U` (903.09 eV) are associated with Ce
3+. The Ce
3+ content was calculated using the peak area ratio of Ce
3+/(Ce
3+ + Ce
4+), and the results are listed in
Table 4. The Ce
3+ content decreased after loading CuO. It has been speculated that a reversible reaction of Ce
3+ + Cu
2+ ↔ Ce
4+ + Cu
+ and 2Ce
3+ + Cu
2+ ↔ 2Ce
4+ + Cu
0 occurs on the surface of the CuO/CeO
2 catalyst [
42,
43,
44].
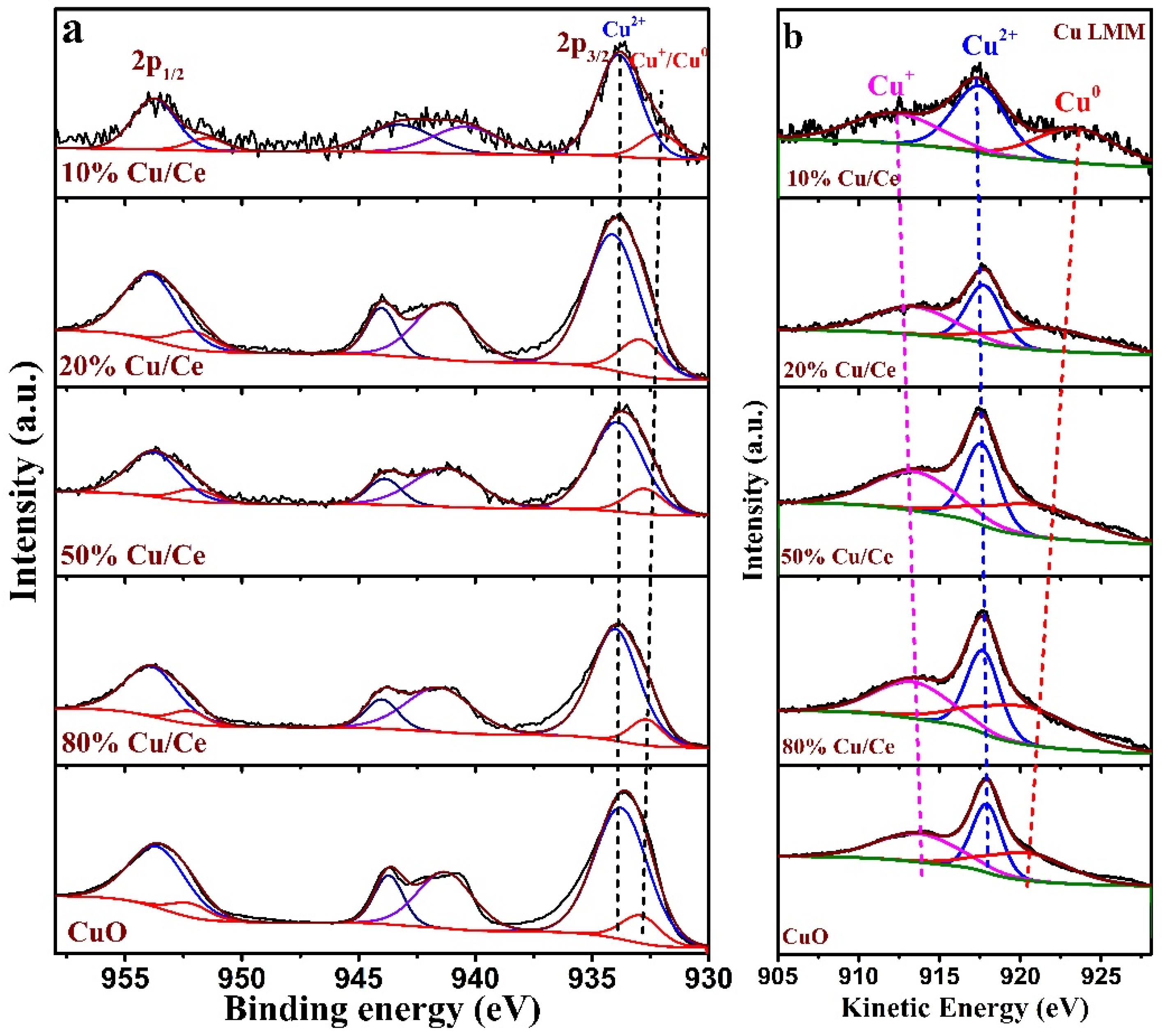
Figure 8 shows the Cu 2p spectra and Cu LMM Auger spectra of the CuO/CeO
2 catalysts, where two characteristic peaks are visible, namely Cu 2p
3/2 and Cu 2p
1/2, along with a broad satellite peak in the Cu 2p spectra. The peaks centered at 934 and 954 eV correspond to Cu 2p
3/2 and Cu 2p
1/2, respectively, and the broad satellite peak is in the range of 937–947 eV. The peaks located at 934 eV and the corresponding satellite peaks are associated with Cu
2+, and the peaks located at 932–933 eV are associated with Cu
+ or/and Cu
0 [
38,
45]. The two reduced states (Cu
+ and Cu
0) are difficult to distinguish in the Cu 2p spectra [
46,
47,
48,
49]. In order to distinguish Cu
+ and Cu
0, Auger Cu LMM spectra were used. The broad feature in the kinetic energy spectra consists of three peaks at 917.5 eV, 913.2–916.0 eV, and 918.5 eV–919 eV, which are assigned to Cu
2+, Cu
+, and Cu
0 species, respectively [
45,
50,
51,
52]. Although the Cu LMM Auger spectra show that both Cu
0 and Cu
+ occur simultaneously in the prepared catalysts, the presence of Cu
0 species possible due to the photoreduction in the XPS test [
37,
49].
Table 4 presents the calculated Cu
0 and Cu
+ to Cu
2+ ratios; it is clear that (Cu
0 + Cu
+)/Cu
2+ is related to the change in Ce
3+ content (ΔCe
3+), which further indicates that reversible reactions of Ce
3+ + Cu
2+ ↔ Ce
4+ + Cu
+ and 2Ce
3+ + Cu
2+ ↔ 2Ce
4+ + Cu
0 occurred on the surface of the CuO/CeO
2 catalyst. The (Cu
0 + Cu
+)/Cu
2+ of 20%Cu/Ce, 50%Cu/Ce, and 80%Cu/Ce were nearly the same and correspond to a minimal change in ΔCe
3+.
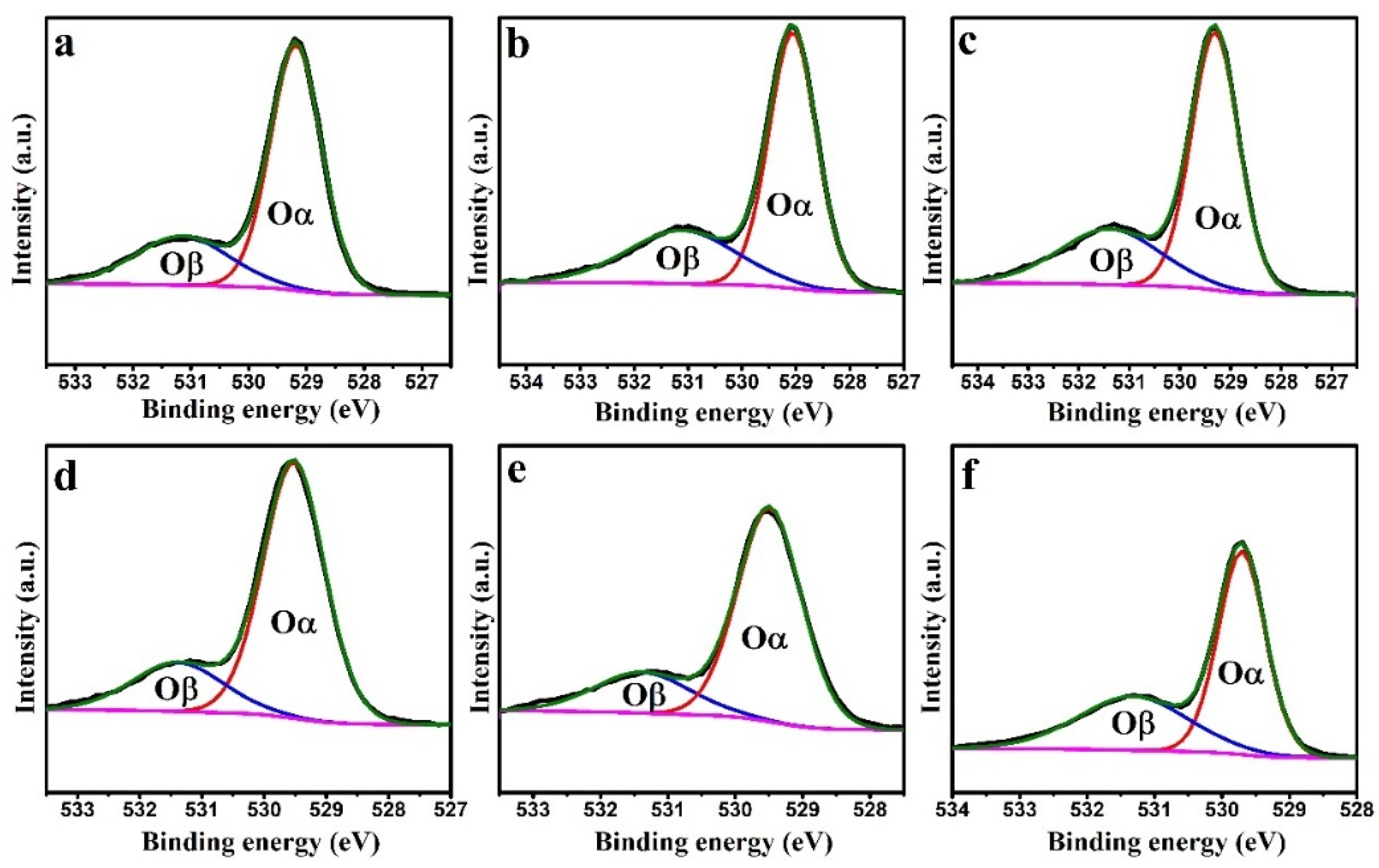
Figure 9 shows that the O1s spectra of the CeO
2, CuO, and CuO/CeO
2 catalysts were deconvoluted into two distinct peaks [
36]. The low-binding-energy peak located at 528.5–529.5 eV (denoted as Oα) was attributed to surface lattice oxygen. The higher-binding-energy peak located at 530–531.5 eV (denoted as Oβ) was attributed to oxygen vacancies, surface-adsorbed O
2−/O
−, surface-adsorbed O
2, hydroxyl groups, and carbonates [
39,
53]. The Oα and Oβ contents were calculated based on the areas of the deconvoluted peaks (
Table 4). With an increase in the Cu/Ce ratio, the Oβ content decreased, whereas the Oα content increased; this trend is consistent with the O
2-TPD results.
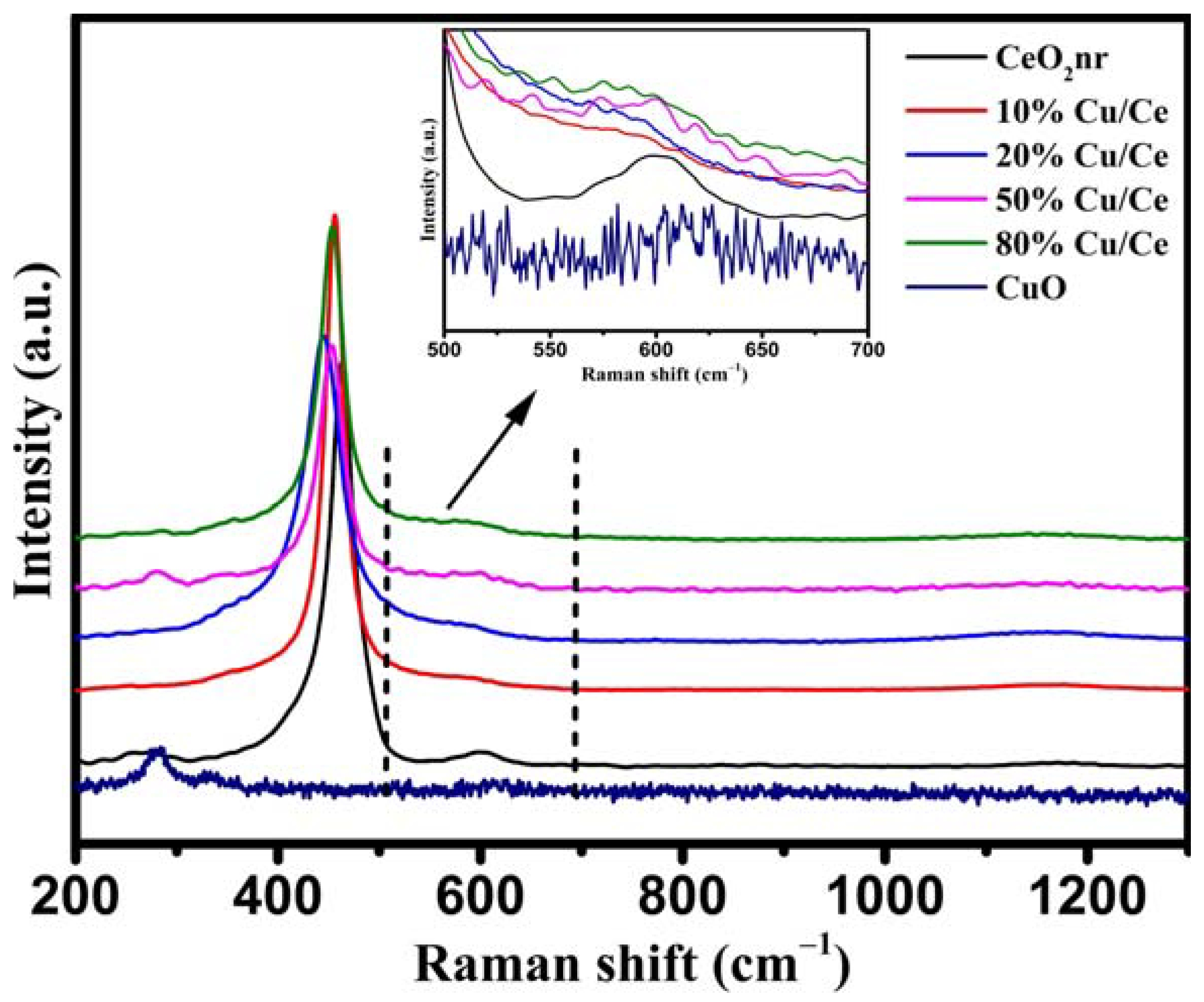
Visible Raman spectroscopy provides additional information about the lattice vibrations and internal defects of catalysts.
Figure 10 shows that a strong band was detected at 450–460 cm
−1, which was ascribed to the Raman-active vibrational F
2g mode of CeO
2 with a fluorite-type structure. Another broad peak located at approximately 598 cm
−1 was also detected (
Figure 10 inset), which corresponds to the defect-induced (D) mode associated with O vacancies caused by the presence of Ce
3+ [
36]. The F
2g mode of CeO
2 shifted from 463 to 445 cm
−1 for the CuO/CeO
2 catalysts, which is generally attributed to the shrinkage and distortion of the CeO
2 lattice caused by the electronic redistribution of the Cu-V-Ce structure. The ratio of the two peak intensities between the defect-induced (D) mode and the F
2g mode (I
D/I
F2g) represents the relative concentration of O vacancies (
Table 4) [
54,
55]. The I
D/I
F2g first increased and then decreased with an increase in the Cu/Ce ratio, and 50%Cu/Ce exhibited the highest O vacancy concentration of 0.083.
2.3. Reaction Mechanism for the Thermocatalytic Decomposition of DMMP
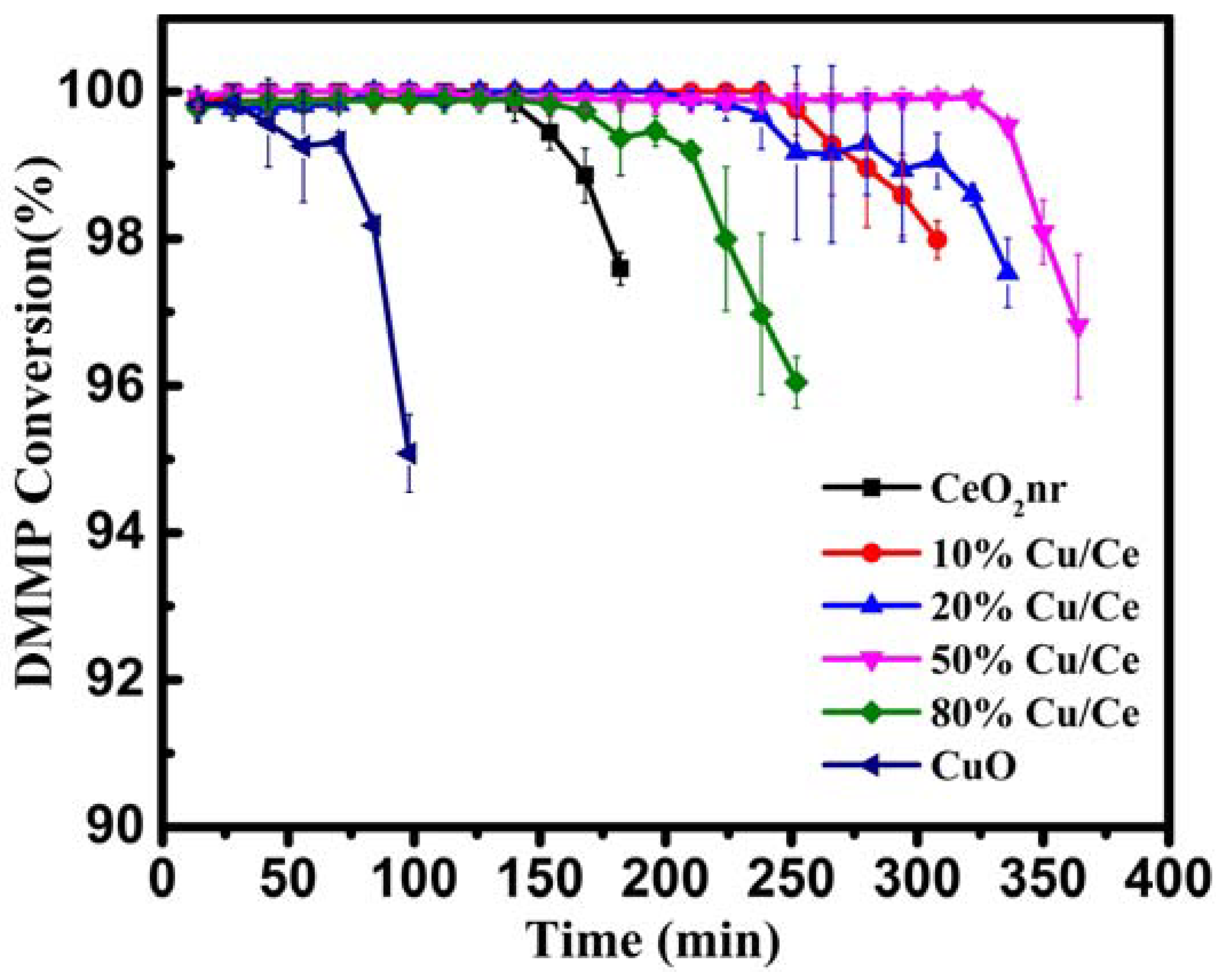
50%Cu/Ce catalyst with the longest protection time, CeO
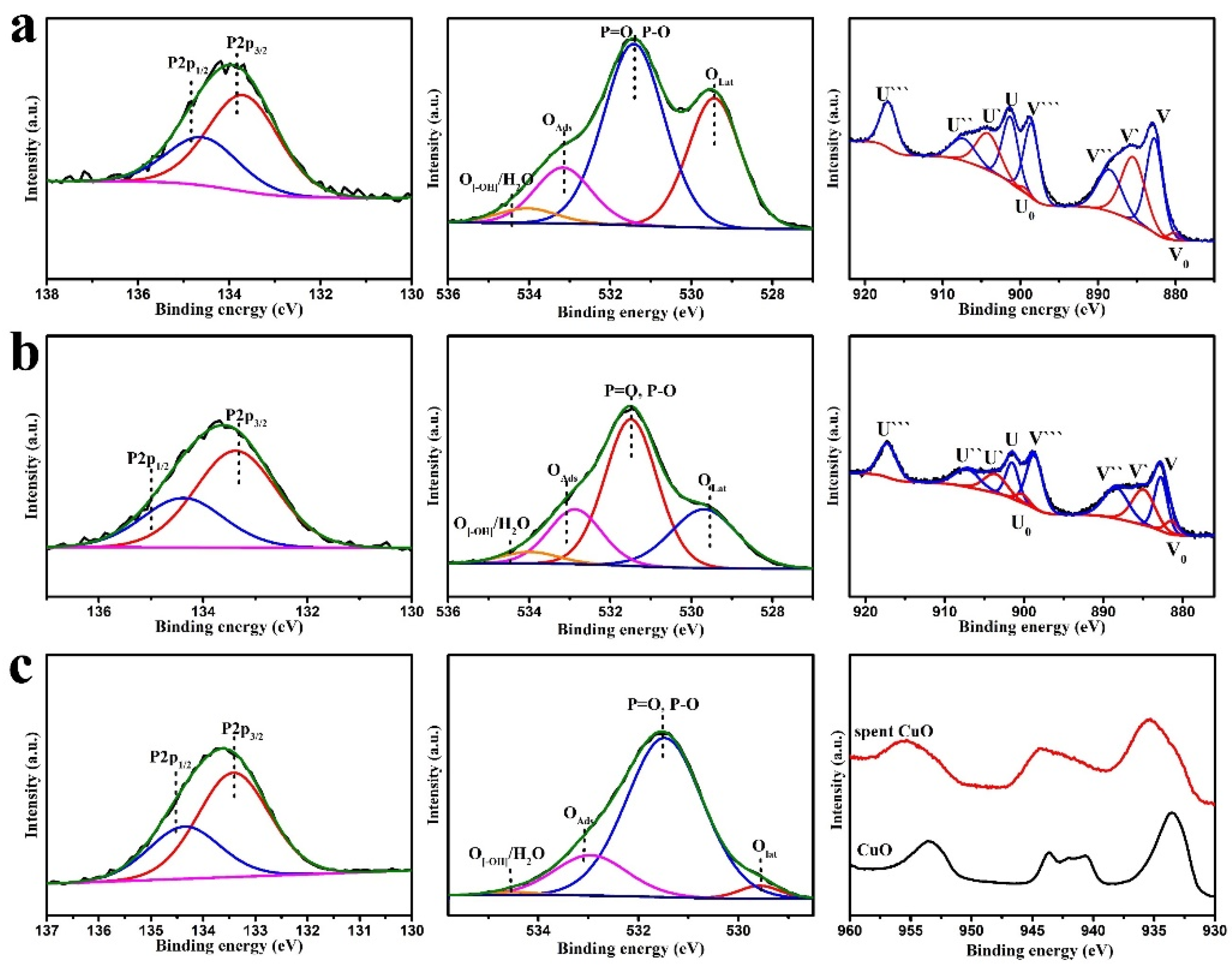
2, and CuO were selected to investigate the DMMP thermocatalytic decomposition mechanism. After the protection-time test, XPS analyses were performed on the deactivated catalyst; the results are shown in
Figure 12. A P2p peak was obvious on the spent catalysts, indicating that the deactivated catalyst surface was covered with P species. The O1s spectrum of the deactivated catalyst was fitted to four peaks, namely surface lattice oxygen (Oα), P–O, adsorbed oxygen (O
Ads), and adsorbed water/hydroxyl oxygen [
19]. The appearance of the P–O peak indicates the formation of P–O byproducts on the catalyst surface. Oα contents before and after the catalytic decomposition of DMMP were calculated (
Table 6). It is obvious that the proportion of Oα in the deactivated catalyst was significantly lower that of the fresh catalyst, which indicates that lattice oxygen participates in the reaction and may play an important role in the catalytic oxidative decomposition of DMMP.
Figure 12c shows the Cu2p spectra of CuO before and after the reaction. The Cu2p
3/2 peak changed from 534 to 535.5 eV, with a broad satellite peak at 935–945 eV. The Cu2p satellite peak of CuO in the deactivated catalyst was similar to that of Cu2p
3/2 when comparing the Cu2p of copper carbonate and copper phosphate, and the peak position was consistent with that of Cu2p
3/2. Therefore, copper phosphate and copper carbonate products were speculated to exist on the surface of the deactivated catalyst.
The phosphorus-containing byproducts on the surface of the deactivated catalysts were qualitatively analyzed using ion chromatography, which showed the presence of PO
43− and methyl phosphonate species, as shown in
Table 7. DMMP decomposition amounts and the corresponding P element amounts are presented in
Table 7. Mass spectrometry was used to qualitatively analyze the tail gas produced by the catalytic decomposition of DMMP on CeO
2, 50%Cu/Ce, and CuO to determine its composition.
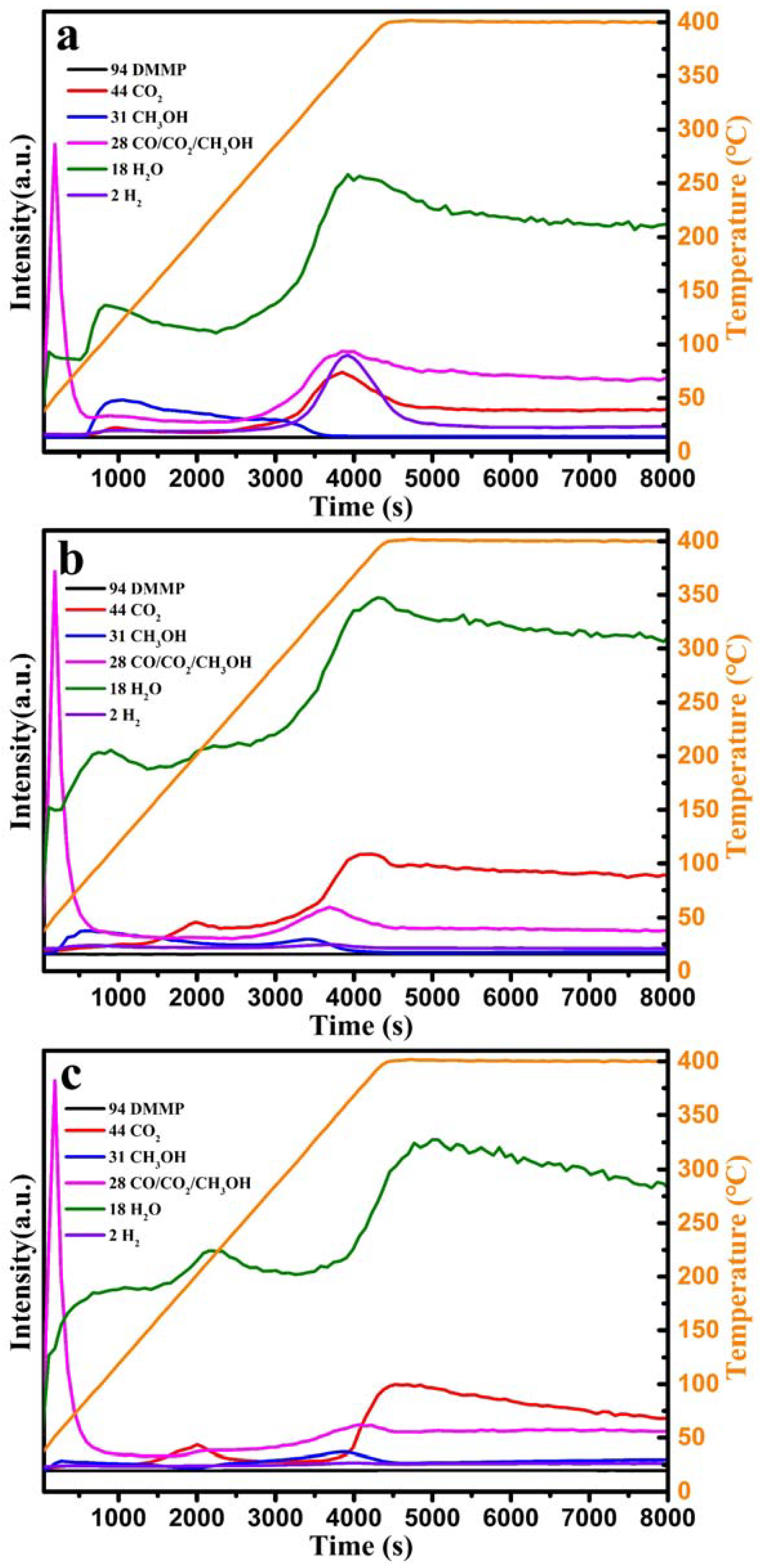
Figure 13 shows the mass spectra of the tail gas generated by CeO
2, 50%Cu/Ce, and CuO, demonstrating that methanol, CO
2, H
2O, and H
2 were detected in the reaction tail gas. Additionally, except for the fragment mass-to-charge ratio of methanol and CO
2 (28), the mass-to-charge ratio of CO was 28. Therefore, gas chromatography was used to determine the presence of CO in the generated exhaust gas. CO was detected in the reaction tail gas of CeO
2 and CuO; however, no CO was detected for 50%Cu/Ce, indicating that the oxidative activity of the 50%Cu/Ce catalyst was higher than that of CeO
2 and CuO.
The in situ DRIFTS of DMMP on CeO
2, 50%Cu/Ce, and CuO at varying temperatures were determined to ascertain the species change of DMMP on the surface of the three catalysts. As shown in
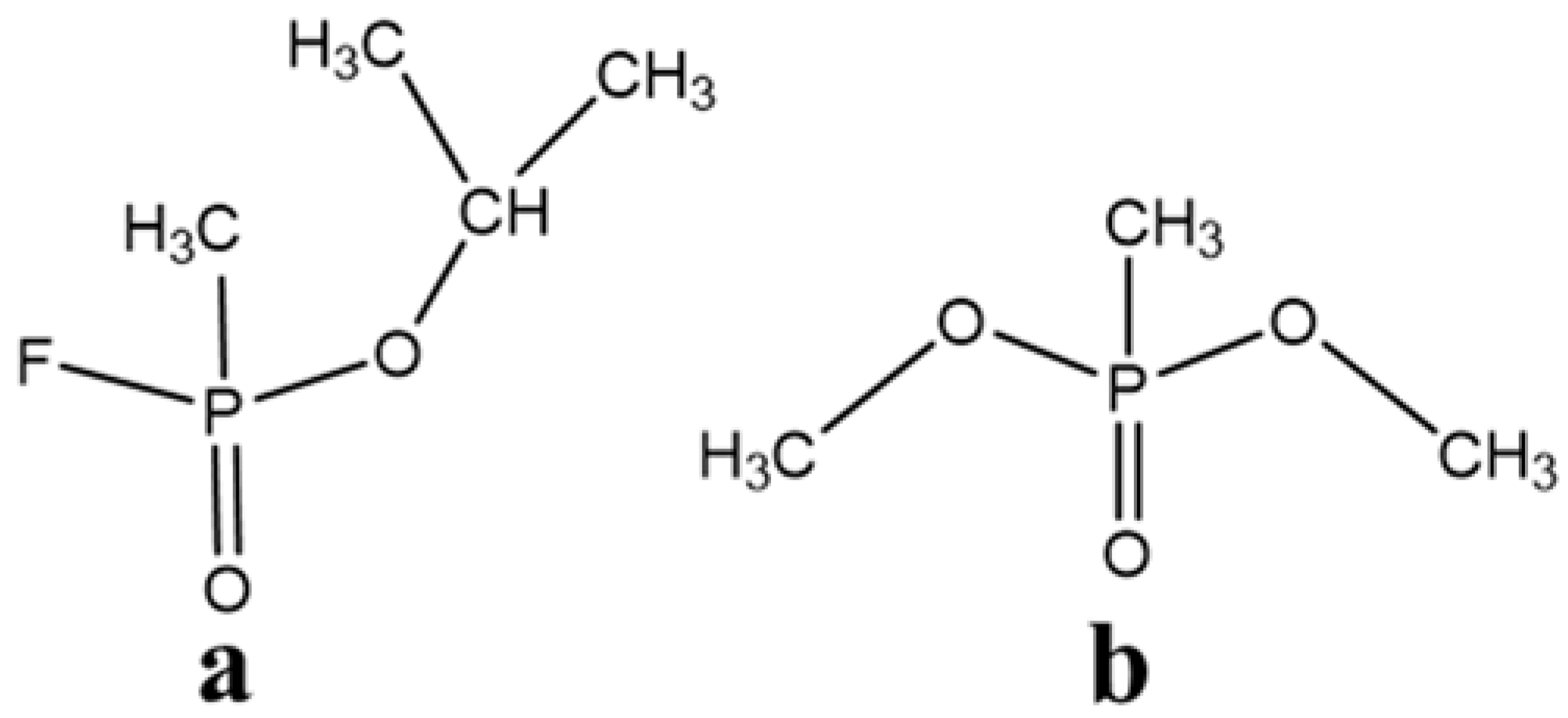
Figure 1, the molecular structure of DMMP contains P–CH
3, O–CH
3, P=O, P–O, P–C, C–O, and C–H bonds. The vibration peaks of chemical bonds in DMMP are mainly distributed in two regions. C–H bond-stretching vibrations of P–CH
3 and O–CH
3 are located in the high-frequency region from approximately 2800 cm
−1 to 3200 cm
−1, whereas stretching vibrations of P=O, C–O, and P–O, as well as deformation vibrations and rocking vibrations of PCH
3 and OCH
3 are located in the low-frequency region from 800 cm
−1 to 1500 cm
−1. The corresponding functional groups of the DMMP molecules were consistent with those reported in the literature [
58,
59], as shown in
Table 8. The corresponding functional groups of surface species on CeO
2, 50%Cu/Ce, and CuO are shown in
Table 9. The characteristic peak located at 3500–3800 cm
−1 was attributed to the stretching vibration of the surface hydroxyl groups [
60,
61]. As the temperature increased, hydroxyl vibration peaks of the negative-going OH bands were observed. The depletion of the surface hydroxyl groups may have originated from the formation of H bonds between the DMMP molecules and surface hydroxyl groups of CeO
2, CuO, and Cu/Ce or from the surface hydroxyl groups participating in the DMMP decomposition reaction [
59,
62] (
Figure 14,
Figure 15 and
Figure 16).
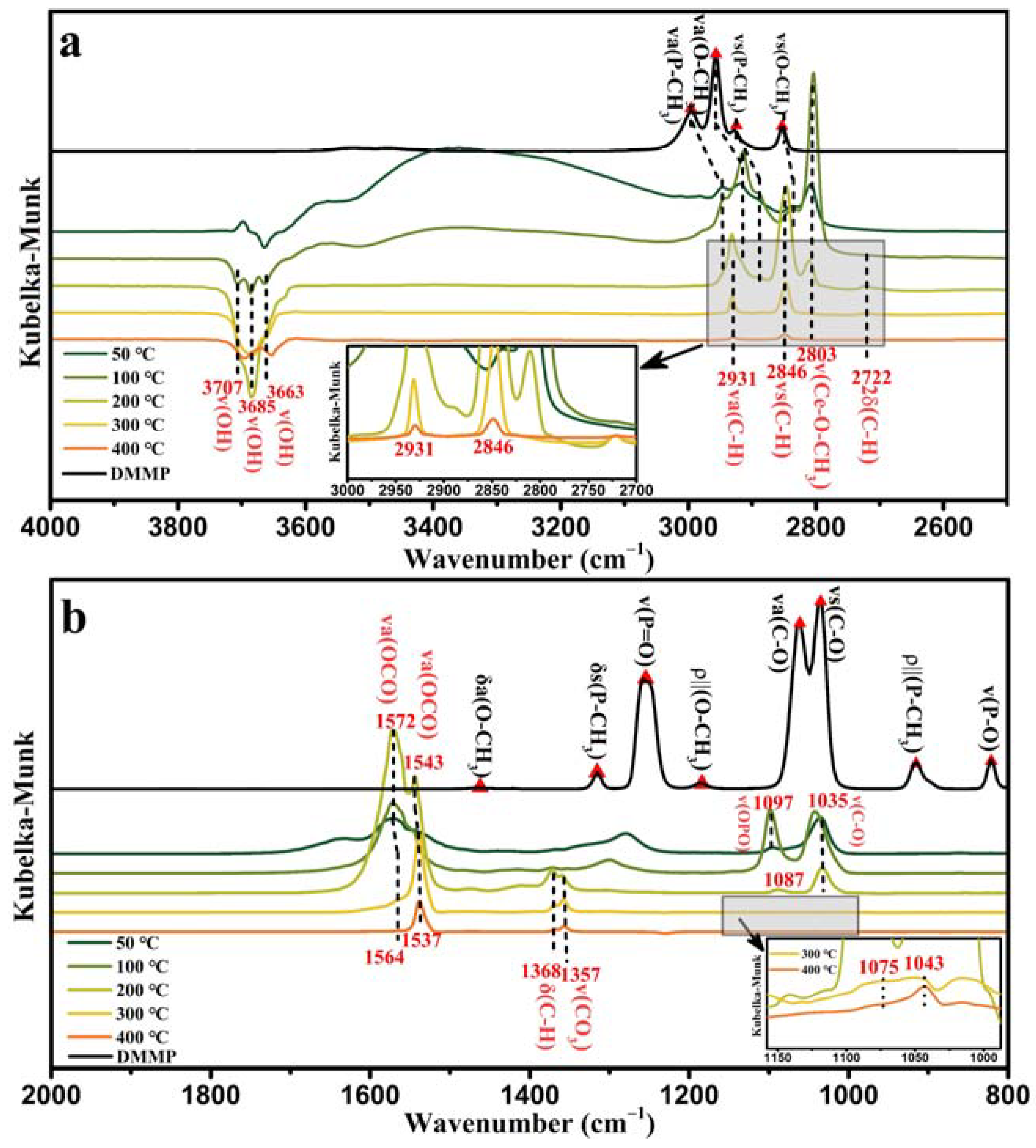
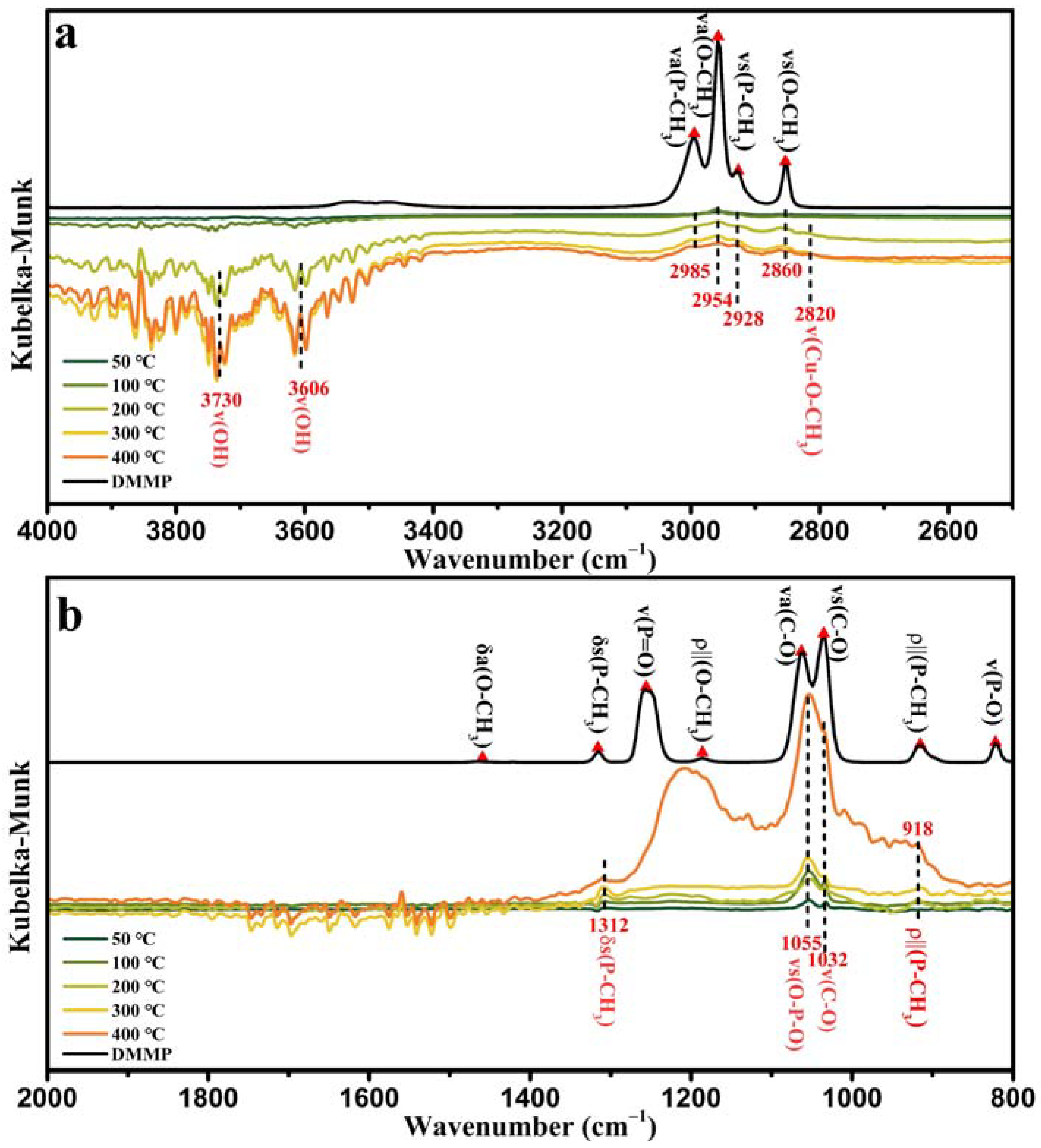
Figure 14a shows the in situ DRIFTS of CeO
2 in the high-frequency region.
The characteristic infrared peaks of ν
a(P–CH
3), ν
a(O–CH
3), ν
s(P–CH
3), and ν
s(O–CH
3) in the DMMP molecules gradually weakened with increasing temperature and disappeared at 400 °C. New characteristic peaks simultaneously appeared at 2931 cm
−1 and 2846 cm
−1, and the peak intensity weakened with increasing temperature. Therefore, we speculated that these two peaks correspond to the ν(C–H) vibrational modes of the intermediates generated during DMMP decomposition. Morris et al. reported that cleavage of P–OCH
3 led to the formation of methoxy groups, which combined with Ti ions to form surface methoxy groups. The vibrational peak of the surface methoxy shifts to a lower frequency than that of DMMP [
63]. A similar phenomenon was observed in the in situ DRIFTS of CeO
2. Therefore, the peak at 2803 cm
−1 was attributed to ν(Ce–O–CH
3).
Figure 14a shows a partially enlarged view of the 2600–2900 cm
−1 band in the high-frequency region, which clearly demonstrates that the peak disappeared when the temperature reached 400 °C. This indicates that the methoxy group in Ce–O–CH
3 is an unstable intermediate product that can be completely decomposed by the CeO
2 catalyst at 400 °C. The vibrational peak at 2722 cm
−1 shown in
Figure 14a was assigned to the 2δ(C–H) vibrational mode of the formate product [
61]. The characteristic infrared peaks of DMMP molecules in the mid–low frequency range of CeO
2 in situ DRIFTS (
Figure 14b), δ
a(O–CH
3), δ
a(P–CH
3), ν(P=O), ρ‖(O–CH
3), ν
a(C–O), ν
s(C–O), ρ‖(P–CH
3), and ν(P–O), disappeared at 400 °C. The vibrational peaks at 1572–1564 cm
−1 and 1537 cm
−1 belong to the vibrational mode of bidentate (bdt) formate ν
a(OCO), and the δ(CH) vibrational mode of the formate appeared at 1368 cm
−1 [
61]. Additionally, the vibrational mode ν(CO
3) of the carbonate appeared at 1357 cm
−1 [
60].
Figure 15 shows the in situ DRIFTS spectra of CuO at varying temperatures. CuO exhibited a multimodal overlap at 3100–2800 cm
−1 in the high-frequency region of the in situ DRIFTS. Moreover, the in situ DRIFTS spectral peak signal of CuO in the high-frequency region was relatively weak, and it was impossible to directly distinguish the peak attribution in this region. The ν(C–H) characteristic peak of the intermediate product appeared in this region with reference to the in situ infrared spectrum of CeO
2 (
Figure 15a). Therefore, we speculated that the CuO peaks in this region might be the ν(C–H) characteristic peaks of the undecomposed DMMP itself and the ν(C–H) characteristic peaks of the partially decomposed DMMP products. This is similar to the in situ DRIFTS spectrum of CeO
2; a ν(Cu–O–CH
3) vibrational peak was also generated on the CuO surface at 2820 cm
−1. The in situ DRIFTS (
Figure 15b) in the mid–low frequency region clearly shows that the ν(P=O) vibration mode in DMMP disappeared, and a new bridging P species ν
s(O–P–O) appeared at 1055 cm
−1, implying an interaction between the phosphoryl oxide and catalyst surface [
21,
64]. The disappearance of the ν(P–O) vibration mode in DMMP indicated that the methoxy group was separated from the DMMP molecules, and the disappearance of the δ
a(O–CH
3), ρ‖(O–CH
3), ν
a(C–O), and ν
s(C–O) vibration modes in DMMP indicated that the methoxy group completely decomposed. Although the characteristic peaks of ρ‖(P–CH
3) and δ
s(P–CH
3) in DMMP were still visible, these characteristic peaks were speculated to be characteristic peaks of methyl-containing products. At 400 °C, the peak intensities of the two bands at 1290–1150 and 1100–1000 cm
−1 significantly increased, indicating that a large number of phosphorus and carbon oxygen products were produced on the surface of CuO at 400 °C [
21,
65].
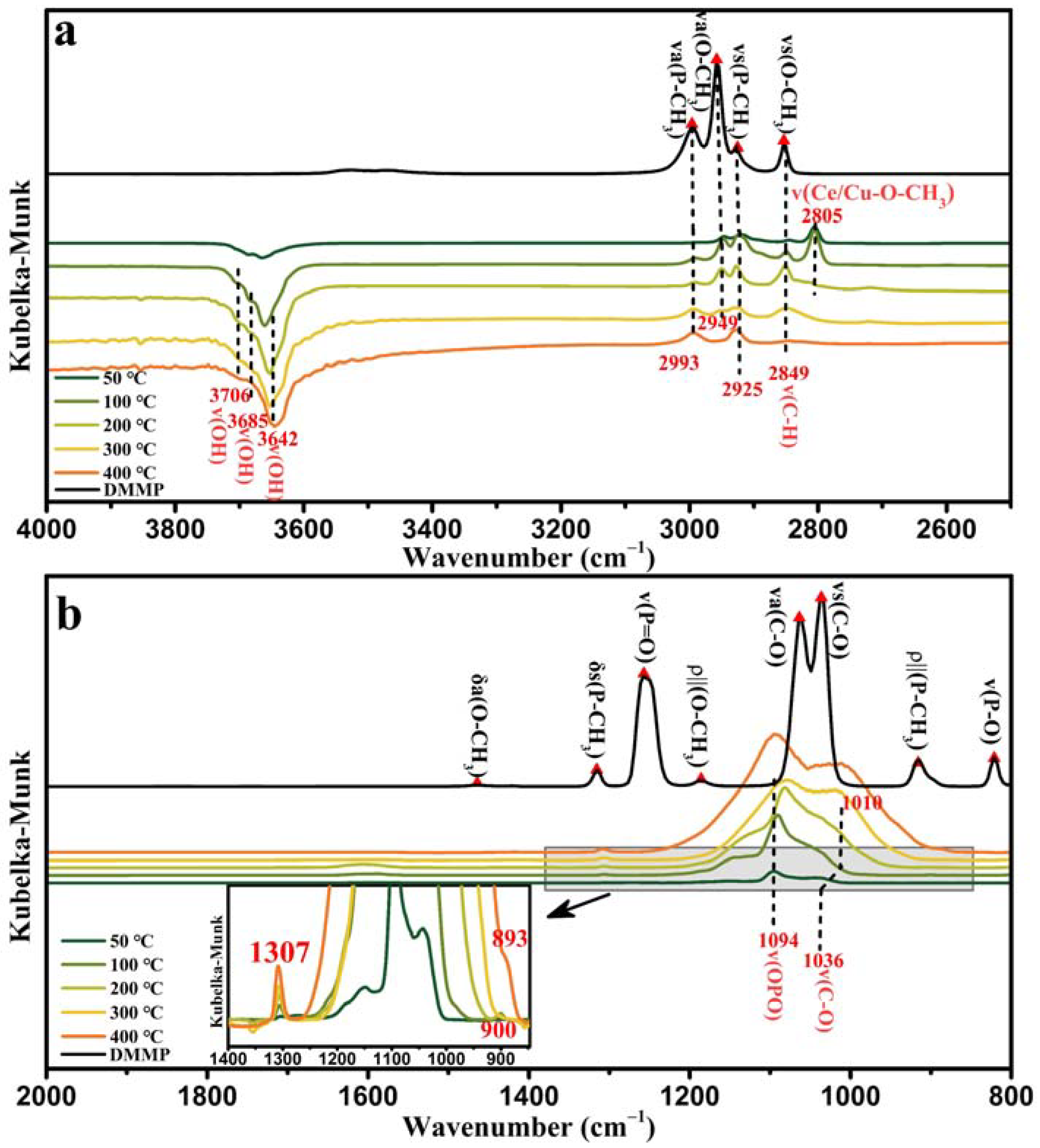
Figure 16 shows the in situ DRIFTS spectra of 50%Cu/Ce at varying temperatures. In the in situ DRIFTS (
Figure 16a) in the high-frequency region of 50%Cu/Ce, the stretching vibration mode ν
a(P–CH
3) of DMMP molecules shifted from 2996 to 2993 cm
−1; ν
s(P–CH
3) shifted from 2927 to 2925 cm
−1, and the peak did not decay with increasing temperature. Therefore, we speculated that the vibration peaks of ν(P–CH
3) at 2993 cm
−1 and 2925 cm
−1 were the DMMP decomposition products. The stretching vibration mode ν
a(O–CH
3) in DMMP shifted from 2958 to 2949 cm
−1, and the stretching vibration of ν
a(O–CH
3) weakened with increasing temperature, disappearing at 400 °C. The vibrational mode of ν
s(O–CH
3) in DMMP was similar to that of CeO
2; therefore, we speculated that the peak at 2849 cm
−1 is the ν(C–H) vibrational mode of the intermediate product generated during the decomposition of DMMP. Similar to CeO
2 and CuO, the ν(Ce/Cu–O–CH
3) vibrational mode of the unstable intermediate appeared near 2805 cm
−1 in the 50%Cu/Ce in situ DRIFTS and disappeared at 400 °C. The spectral characteristic peaks in the low-frequency region of the in situ DRIFTS (
Figure 16b) of 50%Cu/Ce at 400 °C were similar to those of CuO, i.e., the methoxy group in DMMP completely decomposed. A methyl-containing product peak was present at 1307 cm
−1. Large quantities of phosphorus and carbon oxygen products were produced in the 1250–900 cm
−1 band, and the magnified image of the 1350–850 cm
−1 band shows that the 1307 cm
−1 and 900–893 cm
−1 bands are characteristic peaks of methyl-containing products.
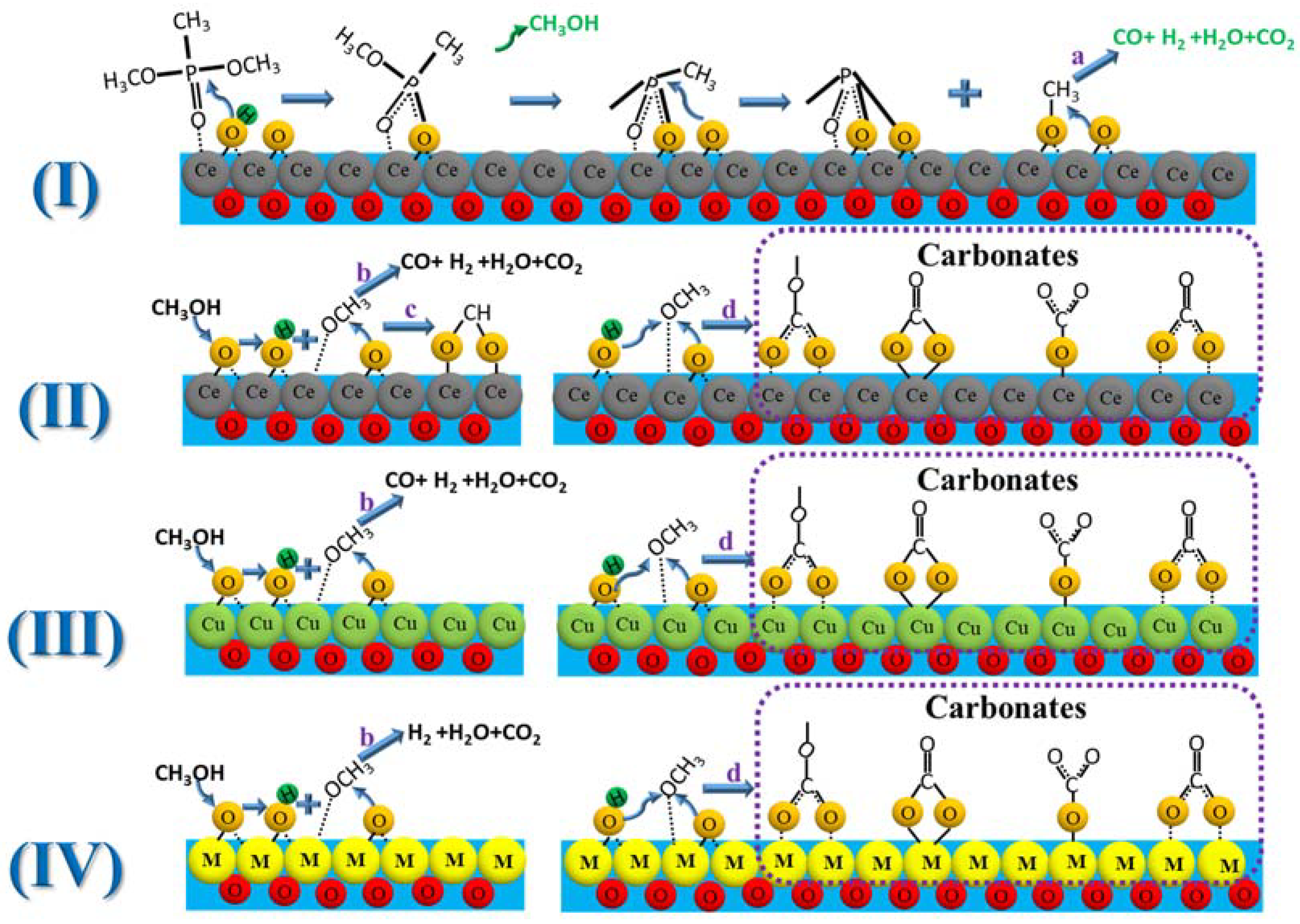
The reaction pathways for the catalytic decomposition of DMMP on CeO
2, CuO, and CuO/CeO
2 were speculated using the combined ion chromatography, mass spectrometry, and in situ DRIFTS data (
Scheme 1).
Scheme 1(I) shows the presumed reaction pathway for the decomposition of DMMP on the surface of CeO
2. First, the electron-rich O in DMMP combines with Ce or surface hydroxyl groups on the surface of CeO
2. The surface hydroxyl groups nucleophilically attack P, breaking the P–OCH
3 bond, and combine the methoxy group with H to generate gaseous methanol. Simultaneously, methyl phosphates with bridged O–P–O structures formed on the surface [
4,
21,
59] The surface lattice O in CeO
2 nucleophilically attacks the methyl group in methyl phosphate, forming a PO
x species on the CeO
2 surface, and the methyl group is further dehydrogenated to CO, H
2, CO
2, and H
2O (
Scheme 1a). According to Wu et al., methanol can induce a series of side reactions on the CeO
2 surface [
61]. First, in
Scheme 1(II) methanol reacts with the CeO
2 surface lattice O to form surface methoxy Ce–O–CH
3. The methoxy groups continue to react with the surface lattice O or hydroxyl groups, fully dehydrogenating to form CO, H
2, CO
2, and H
2O (
Scheme 1b) and partially dehydrogenating to form formate (
Scheme 1c) [
66], which reacts with the surface lattice O and completely dehydrogenates to form carbonate (
Scheme 1d) [
67]. Based on the in situ DRIFTS data, we speculated that the carbonates may contain one or more species, including polydentate, bidentate, bridged, and monodentate carbonates, due to the broadband nature of their vibrational modes. The catalytic decomposition pathways of DMMP on CuO and CuO/CeO
2 are essentially the same as those on CeO
2 (
Scheme 1(I)). The difference is that there is no formate in the decomposition products of the CuO and CuO/CeO
2 catalytic decomposition of DMMP (
Scheme 1c), and no CO is generated in the decomposition products of the CuO/CeO
2 catalytic decomposition of DMMP. This indicates that the synergistic effect of the bimetals enhances the selectivity for the fully catalytic oxidation product of DMMP, CO
2.
Various mechanisms have been proposed over ceria-based catalysts in the literature [
68,
69,
70], which can be generally broken down into three main categories: the Eley-Rideal mechanism [
71], the Langmuir-Hinshelwood mechanism [
72], and the Mars-van-Krevelen mechanism [
73,
74]. In catalytic reactions at higher temperatures, the mechanism of CeO
2-based materials is mainly the Mars-van-Krevelen mechanism. The Mars-van-Krevelen mechanism consists of a redox cycle. First, reactant molecules interact with the lattice oxygen on the catalyst surface, and oxygen vacancies are formed on the catalyst surface. Then, the oxygen vacancies are refilled by gaseous oxygen or oxygen atoms from the bulk. Kinetic experimentation is needed in future studies to verify the thermal catalytic DMMP decomposition mechanism of as-prepared materials reported in the present study.

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}