


Large-Scale Synthesis of Iron Ore@Biomass Derived ESBC to Degrade Tetracycline Hydrochloride for Heterogeneous Persulfate Activation


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
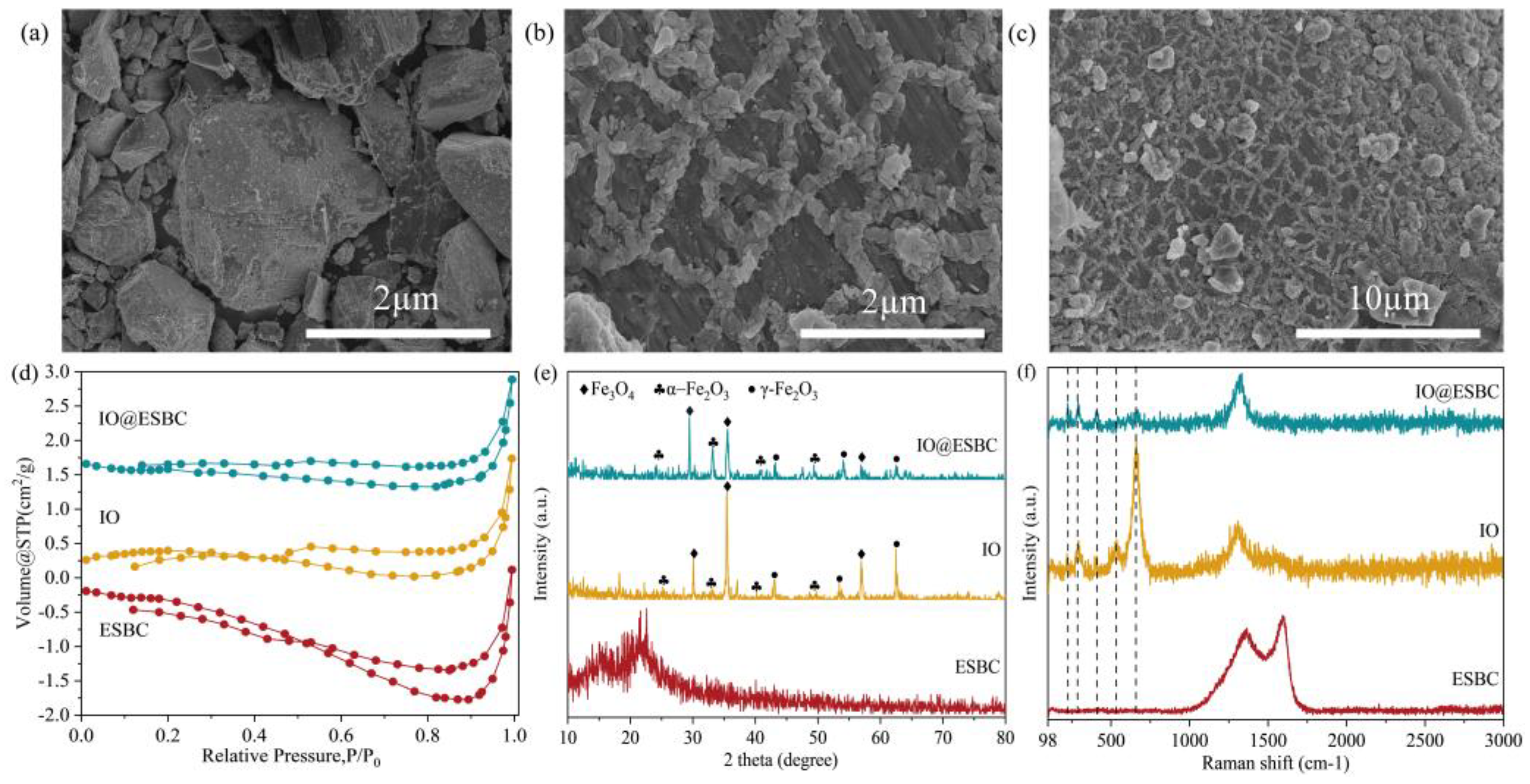
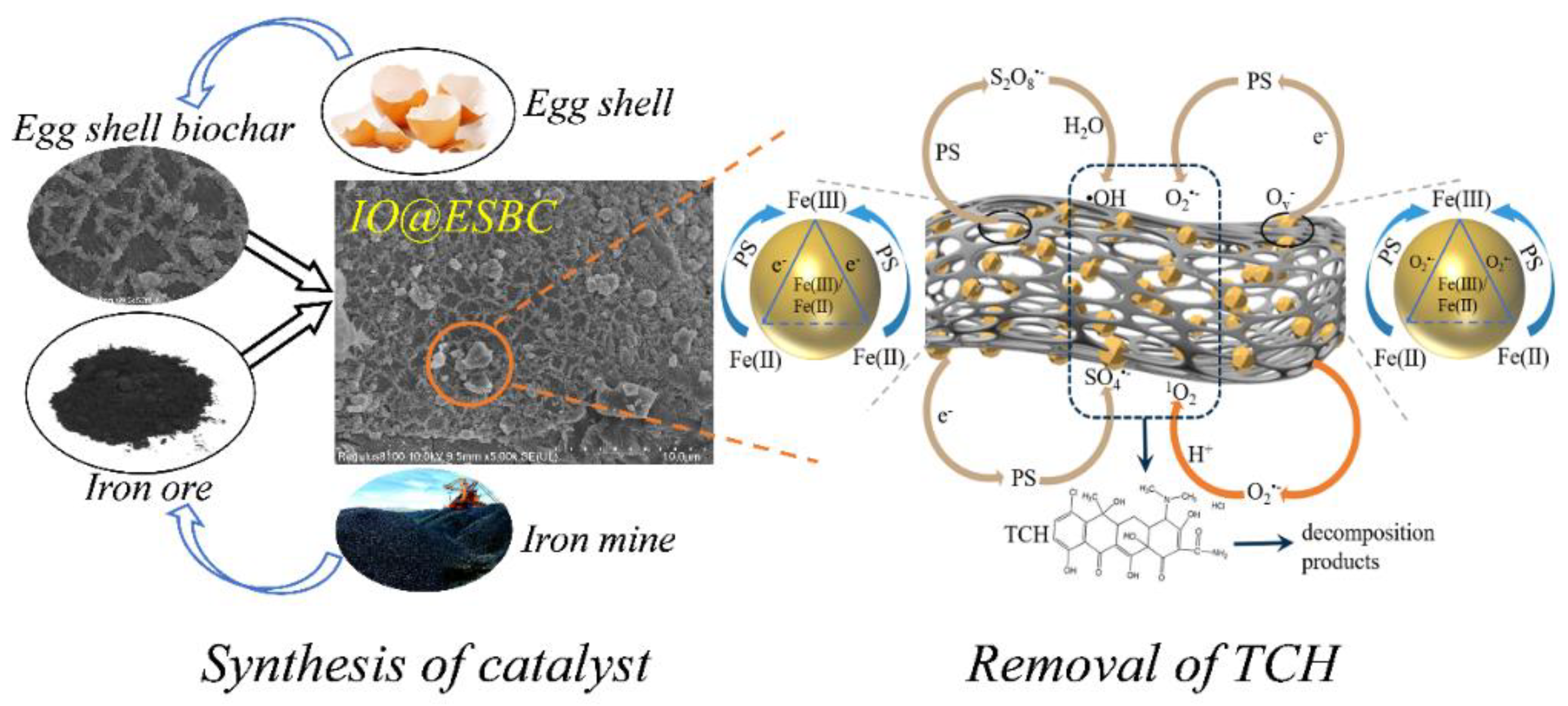
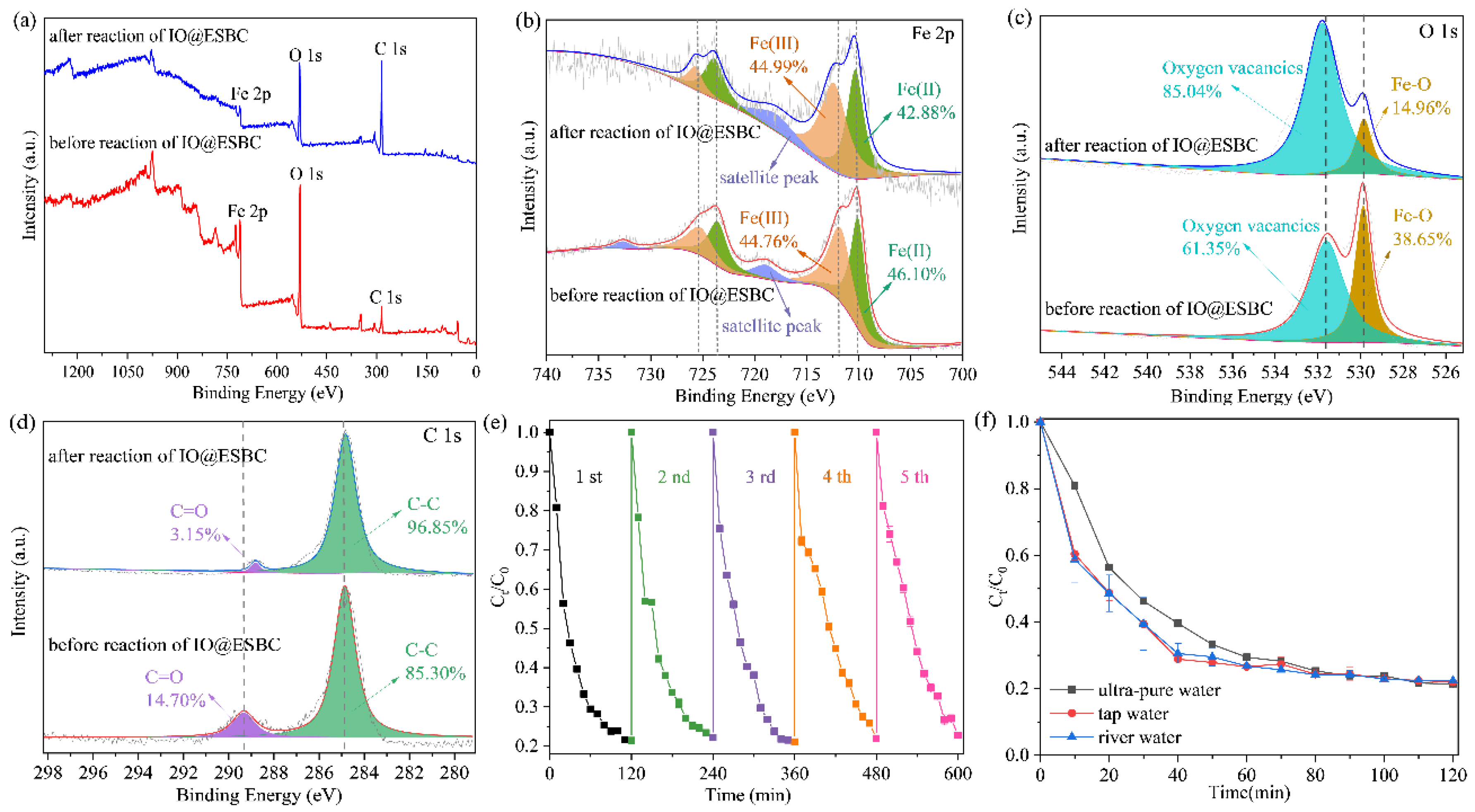
2.1. Characterization of the Green Synthesized IO@ESBC
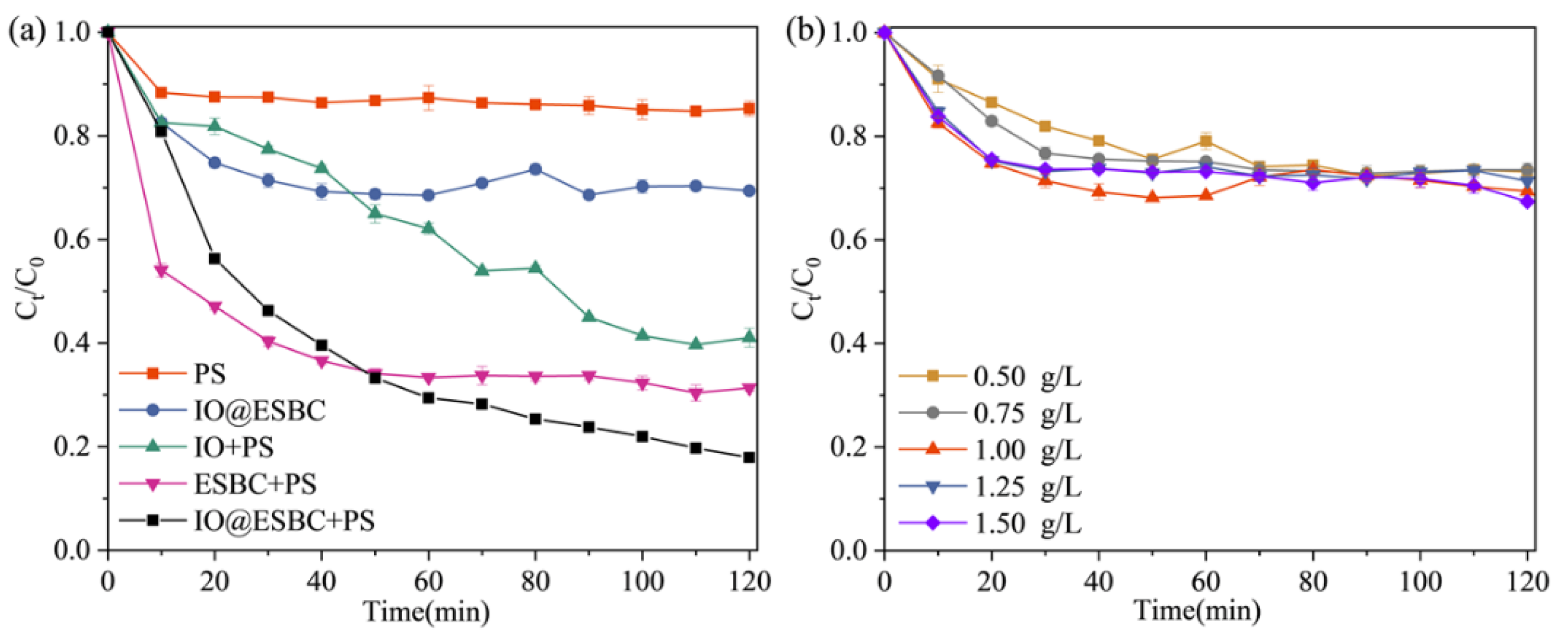
2.2. Catalytic Performance of IO@ESBC
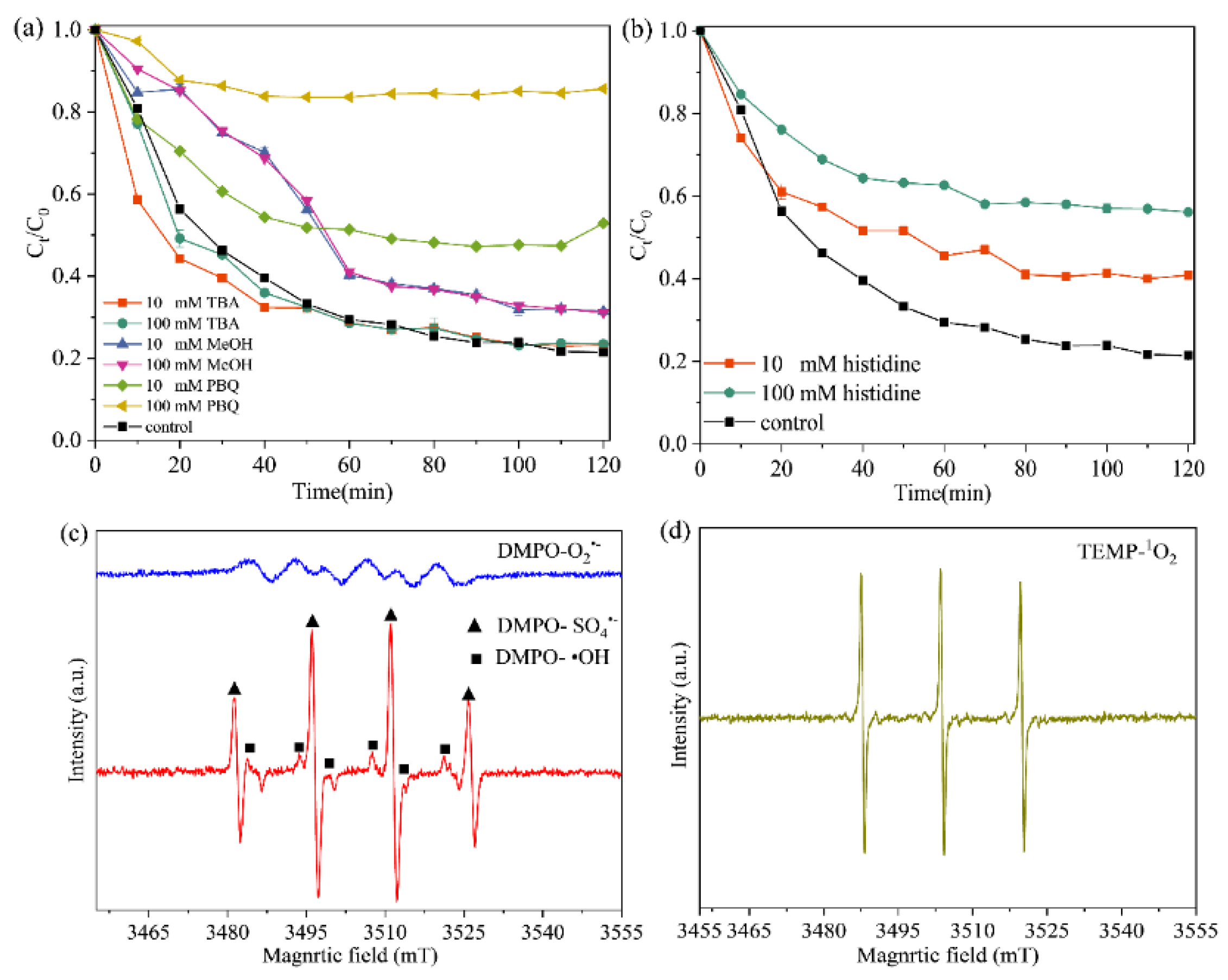
2.3. Possible Mechanism of IO@ESBC System for TCH Removal
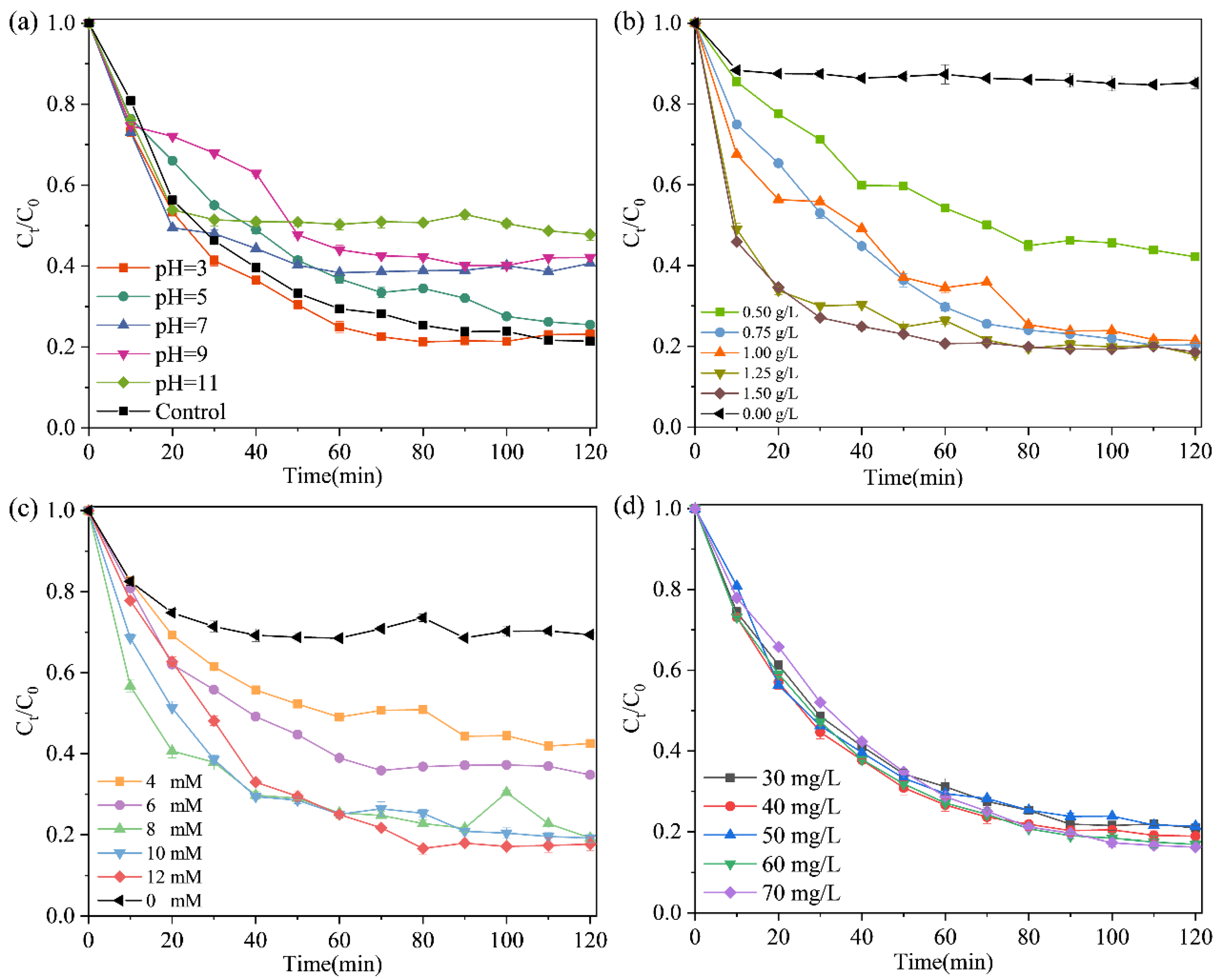
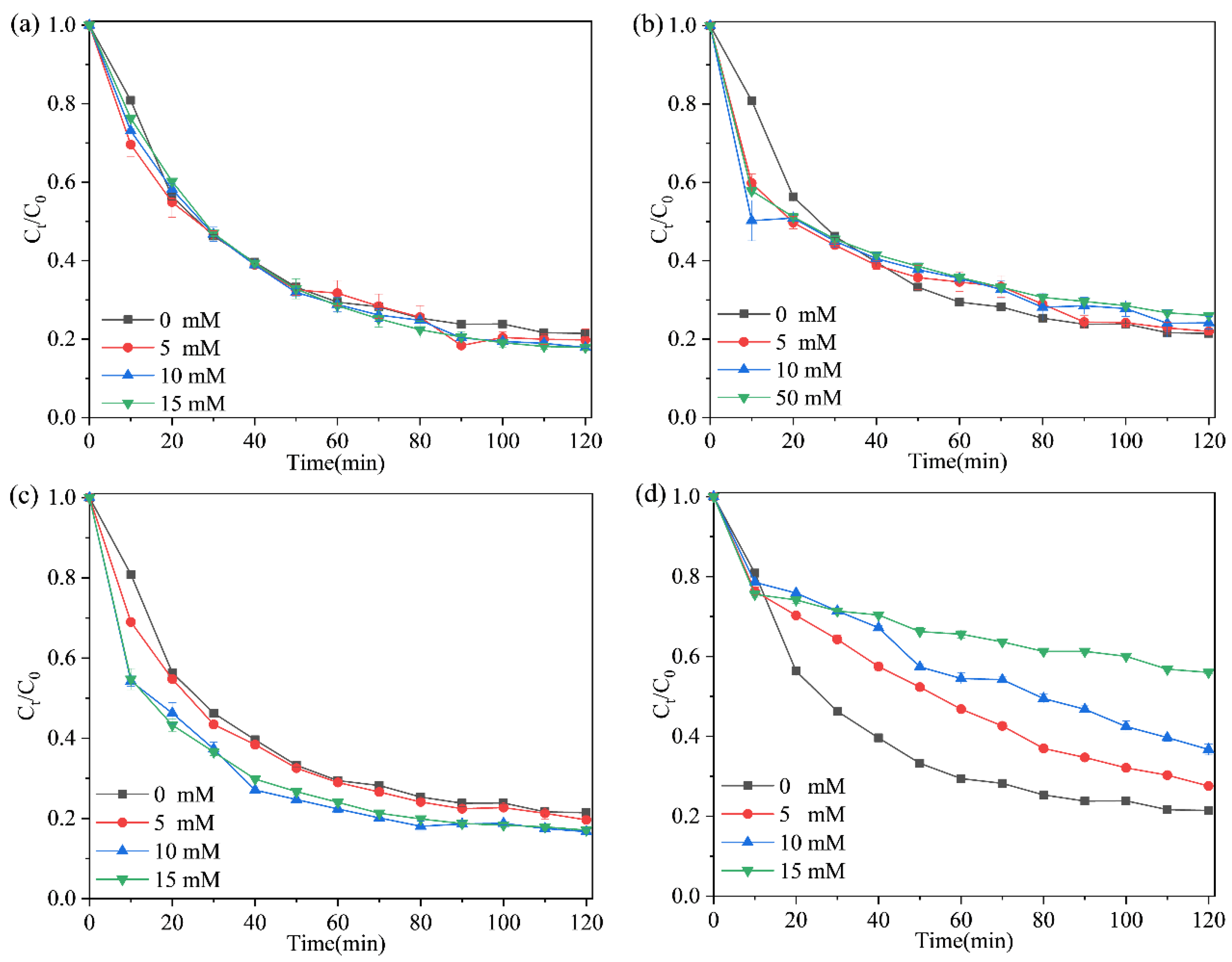
2.4. Effect of Different Conditions on TCH Removal
2.5. Reusability of IO@ESBC
3. Materials and Methods
3.1. Materials
3.2. IO@ESBC Synthesis
3.3. Characterization
3.4. Catalytic Activity Tests of IO@ESBC
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Barbosa, M.O.; Moreira, N.F.F.; Ribeiro, A.R.; Pereira, M.F.R.; Silva, A.M.T. Occurrence and removal of organic micropollutants: An overview of the watch list of EU Decision 2015/495. Water Res. 2016, 94, 257–279. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Zhang, G.; Xian, G.; Ren, Z.; Wei, T.; Li, Q.; Zhang, Y.; Zou, Z. Tetracycline degradation by persulfate activated with magnetic γ-Fe2O3/CeO2 catalyst: Performance, activation mechanism and degradation pathway. Sep. Purif. Technol. 2021, 259, 118156. [Google Scholar] [CrossRef]
- Habibi, M.; Habibi-Yangjeh, A.; Sabri, M.; Chand, H.; Krishnan, V.; Wang, C. Highly impressive activation of persulfate ions by novel ZnO/CuCo2O4 nanostructures for photocatalytic removal of tetracycline hydrochloride under visible light. Environ. Technol. Innov. 2021, 24, 102038. [Google Scholar] [CrossRef]
- Liu, X.; Huang, D.; Lai, C.; Zeng, G.; Qin, L.; Wang, H.; Yi, H.; Li, B.; Liu, S.; Zhang, M.; et al. Recent advances in covalent organic frameworks (COFs) as a smart sensing material. Chem. Soc. Rev. 2019, 48, 5266–5302. [Google Scholar] [CrossRef]
- Vellingiri, K.; Philip, L.; Kim, K.-H. Metal–organic frameworks as media for the catalytic degradation of chemical warfare agents. Coord. Chem. Rev. 2017, 353, 159–179. [Google Scholar] [CrossRef]
- Zhong, Q.; Lin, Q.; He, W.; Fu, H.; Huang, Z.; Wang, Y.; Wu, L. Study on the nonradical pathways of nitrogen-doped biochar activating persulfate for tetracycline degradation. Sep. Purif. Technol. 2021, 276, 119354. [Google Scholar] [CrossRef]
- Hu, L.; Ren, X.; Yang, M.; Guo, W. Facet-controlled activation of persulfate by magnetite nanoparticles for the degradation of tetracycline. Sep. Purif. Technol. 2021, 258, 118014. [Google Scholar] [CrossRef]
- Zhang, R.; Zheng, X.; Zhang, D.; Niu, X.; Ma, J.; Lin, Z.; Fu, M.; Zhou, S. Insight into the roles of endogenous minerals in the activation of persulfate by graphitized biochar for tetracycline removal. Sci. Total Environ. 2021, 768, 144281. [Google Scholar] [CrossRef]
- Shao, F.; Wang, Y.; Mao, Y.; Shao, T.; Shang, J. Degradation of tetracycline in water by biochar supported nanosized iron activated persulfate. Chemosphere 2020, 261, 127844. [Google Scholar] [CrossRef]
- Dang, V.C.; Tran, D.T.; Phan, A.T.; Pham, N.K.; Nguyen, V.N. Synergistic effect for the degradation of tetracycline by rGO-Co3O4 assisted persulfate activation. J. Phys. Chem. Solids 2021, 153, 110005. [Google Scholar] [CrossRef]
- Huang, H.; Guo, T.; Wang, K.; Li, Y.; Zhang, G. Efficient activation of persulfate by a magnetic recyclable rape straw biochar catalyst for the degradation of tetracycline hydrochloride in water. Sci. Total Environ. 2021, 758, 143957. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gao, Z.; Wu, Q. Activation of persulfate by magnetic zirconium-doped manganese ferrite for efficient degradation of tetracycline. Chem. Eng. J. 2021, 423, 130283. [Google Scholar] [CrossRef]
- Wu, Z.; Liang, Y.; Zou, D.; Yuan, X.; Xiao, Z.; Deng, Y.; Zhou, Y.; Jiang, L.; Qin, P. Enhanced heterogeneous activation of persulfate by NixCo3–xO4 for oxidative degradation of tetracycline and bisphenol A. J. Environ. Chem. Eng. 2020, 8, 104451. [Google Scholar] [CrossRef]
- Zhang, X.; Deng, H.; Zhang, G.; Yang, F.; Yuan, G.-E. Natural bornite as an efficient and cost-effective persulfate activator for degradation of tetracycline: Performance and mechanism. Chem. Eng. J. 2020, 381, 122717. [Google Scholar] [CrossRef]
- Feng, Q.; Zhou, J.; Luo, W.; Ding, L.; Cai, W. Photo-Fenton removal of tetracycline hydrochloride using LaFeO3 as a persulfate activator under visible light. Ecotoxicol. Environ. Saf. 2020, 198, 110661. [Google Scholar] [CrossRef]
- Tang, S.; Zhao, M.; Yuan, D.; Li, X.; Zhang, X.; Wang, Z.; Jiao, T.; Wang, K. MnFe2O4 nanoparticles promoted electrochemical oxidation coupling with persulfate activation for tetracycline degradation. Sep. Purif. Technol. 2021, 255, 117690. [Google Scholar] [CrossRef]
- Wang, G.; Ge, D.; Bai, L.; Dong, Y.; Bian, C.; Xu, J.; Zhu, N.; Yuan, H. Insight into the roles of electrolysis-activated persulfate oxidation in the waste activated sludge dewaterability: Effects and mechanism. J. Environ. Manag. 2021, 297, 113342. [Google Scholar] [CrossRef]
- Ge, D.; Zhu, Y.; Li, G.; Yuan, H.; Zhu, N. Identifying the key sludge properties characteristics in Fe2+-activated persulfate conditioning for dewaterability amelioration and engineering implementation. J. Environ. Manag. 2021, 296, 113204. [Google Scholar] [CrossRef]
- Syam Babu, D.; Nidheesh, P.V. Treatment of arsenite contaminated water by electrochemically activated persulfate oxidation process. Sep. Purif. Technol. 2022, 282, 119999. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, C.; Zhou, Z.; Wu, Y.; Xing, S. Degradation of ciprofloxacin by persulfate activation with CuO supported on Mg Al layered double hydroxide. J. Environ. Chem. Eng. 2021, 9, 106178. [Google Scholar] [CrossRef]
- Zheng, N.; He, X.; Hu, R.; Guo, W.; Hu, Z. Co-activation of persulfate by cation and anion from FeP for advanced oxidation processes. Appl. Catal. B Environ. 2021, 298, 120505. [Google Scholar] [CrossRef]
- Yue, X.; Guo, W.; Li, X.; Zhou, H.; Wang, R. Core-shell Fe3O4@MIL-101(Fe) composites as heterogeneous catalysts of persulfate activation for the removal of Acid Orange 7. Environ. Sci. Pollut. Res. 2016, 23, 15218–15226. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Chen, C.; Zhao, X.; Bu, X.; Liao, X.; Fan, H.; Gao, W.; Hu, H.; Zhang, Y.; Huang, Z. Malachite green degradation by persulfate activation with CuFe2O4@biochar composite: Efficiency, stability and mechanism. J. Environ. Chem. Eng. 2021, 9, 105800. [Google Scholar] [CrossRef]
- Gao, Y.; Yang, F.; Jian, H.; Zhen, K.; Zhang, P.; Tang, X.; Fu, Z.; Xu, W.; Wang, C.; Sun, H. Pyrene degradation in an aqueous system using ferrous citrate complex activated persulfate over a wide pH range. J. Environ. Chem. Eng. 2021, 9, 106733. [Google Scholar] [CrossRef]
- Guo, J.; Gao, Q.; Yang, S.; Zheng, F.; Du, B.; Wen, S.; Wang, D. Degradation of pyrene in contaminated water and soil by Fe2+-activated persulfate oxidation: Performance, kinetics, and background electrolytes (Cl-, HCO3- and humic acid) effects. Process Saf. Environ. Prot. 2021, 146, 686–693. [Google Scholar] [CrossRef]
- Andrew Lin, K.-Y.; Hsu, F.-K. Magnetic iron/carbon nanorods derived from a metal organic framework as an efficient heterogeneous catalyst for the chemical oxidation process in water. RSC Adv. 2015, 5, 50790–50800. [Google Scholar] [CrossRef]
- Li, X.; Liao, F.; Ye, L.; Yeh, L. Controlled pyrolysis of MIL-88A to prepare iron/carbon composites for synergistic persulfate oxidation of phenol: Catalytic performance and mechanism. J. Hazard. Mater. 2020, 398, 122938. [Google Scholar] [CrossRef]
- Liu, C.; Wang, Y.; Zhang, Y.; Li, R.; Meng, W.; Song, Z.; Qi, F.; Xu, B.; Chu, W.; Yuan, D.; et al. Enhancement of Fe@porous carbon to be an efficient mediator for peroxymonosulfate activation for oxidation of organic contaminants: Incorporation NH2-group into structure of its MOF precursor. Chem. Eng. J. 2018, 354, 835–848. [Google Scholar] [CrossRef]
- Pu, M.; Wan, J.; Zhang, F.; Brusseau, M.L.; Ye, D.; Niu, J. Insight into degradation mechanism of sulfamethoxazole by metal-organic framework derived novel magnetic Fe@C composite activated persulfate. J. Hazard. Mater. 2021, 414, 125598. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Pan, Y.; Xiong, Z.; Yao, G.; Xie, R.; Lai, B. Peroxymonosulfate activation on FeCo2S4 modified g-C3N4 (FeCo2S4-CN): Mechanism of singlet oxygen evolution for nonradical efficient degradation of sulfamethoxazole. Chem. Eng. J. 2020, 384, 123361. [Google Scholar] [CrossRef]
- He, J.; Tang, J.; Zhang, Z.; Wang, L.; Liu, Q.; Liu, X. Magnetic ball-milled FeS@biochar as persulfate activator for degradation of tetracycline. Chem. Eng. J. 2021, 404, 126997. [Google Scholar]
- Escobedo, E.; Cho, K.; Chang, Y.-S. Electrochemical activation of hydrogen peroxide, persulfate, and free chlorine using sacrificial iron anodes for decentralized wastewater treatment. J. Hazard. Mater. 2022, 423, 127068. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-T.; Li, D.; Lai, L.-J.; Li, Y.-H. Remediation of petroleum hydrocarbon contaminated soil by using activated persulfate with ultrasound and ultrasound/Fe. Chemosphere 2020, 238, 124657. [Google Scholar] [CrossRef] [PubMed]
- Nashat, M.; Mossad, M.; El-Etriby, H.K.; Gar Alalm, M. Optimization of electrochemical activation of persulfate by BDD electrodes for rapid removal of sulfamethazine. Chemosphere 2022, 286, 131579. [Google Scholar] [CrossRef] [PubMed]
- Fagan, W.P.; Zhao, J.; Villamena, F.A.; Zweier, J.L.; Weavers, L.K. Synergistic, aqueous PAH degradation by ultrasonically-activated persulfate depends on bulk temperature and physicochemical parameters. Ultrason. Sonochem. 2020, 67, 105172. [Google Scholar] [CrossRef]
- Zhou, X.; Lai, C.; Liu, S.; Li, B.; Qin, L.; Liu, X.; Yi, H.; Fu, Y.; Li, L.; Zhang, M.; et al. Activation of persulfate by swine bone derived biochar: Insight into the specific role of different active sites and the toxicity of acetaminophen degradation pathways. Sci. Total Environ. 2021, 807, 151059. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, H.; Leng, H.; You, H.; Jia, Y.; Wang, S. Ultrasonic role to activate persulfate/chlorite with foamed zero-valent-iron: Sonochemical applications and induced mechanisms. Ultrason. Sonochem. 2021, 78, 105750. [Google Scholar] [CrossRef]
- Song, H.; Li, Q.; Ye, Y.; Pan, F.; Zhang, D.; Xia, D. Degradation of cephalexin by persulfate activated with magnetic loofah biochar: Performance and mechanism. Sep. Purif. Technol. 2021, 272, 118971. [Google Scholar] [CrossRef]
- Liu, X.; Yuan, B.; Zou, J.; Wu, L.; Dai, L.; Ma, H.; Li, K.; Ma, J. Cu(II)-enhanced degradation of acid orange 7 by Fe(II)-activated persulfate with hydroxylamine over a wide pH range. Chemosphere 2020, 238, 124533. [Google Scholar] [CrossRef]
- Kang, Y.-G.; Chi Vu, H.; Chang, Y.-Y.; Chang, Y.-S. Fe(III) adsorption on graphene oxide: A low-cost and simple modification method for persulfate activation. Chem. Eng. J. 2020, 387, 124012. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, L.; Fan, J.; Song, Y.; Chu, G.; Shao, L. Degradation of indigo carmine by coupling Fe(II)-activated sodium persulfate and ozone in a rotor-stator reactor. Chem. Eng. Process. Process Intensif. 2020, 148, 107791. [Google Scholar] [CrossRef]
- Wang, H.; Deng, J.; Lu, X.; Wan, L.; Huang, J.; Liu, Y. Rapid and continuous degradation of diclofenac by Fe(II)-activated persulfate combined with bisulfite. Sep. Purif. Technol. 2021, 262, 118335. [Google Scholar] [CrossRef]
- Ali, J.; Wenli, L.; Shahzad, A.; Ifthikar, J.; Aregay, G.G.; Shahib, I.I.; Elkhlifi, Z.; Chen, Z.; Chen, Z. Regulating the redox centers of Fe through the enrichment of Mo moiety for persulfate activation: A new strategy to achieve maximum persulfate utilization efficiency. Water Res. 2020, 181, 115862. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-G.; Vu, H.C.; Le, T.T.; Chang, Y.-S. Activation of persulfate by a novel Fe(II)-immobilized chitosan/alginate composite for bisphenol A degradation. Chem. Eng. J. 2018, 353, 736–745. [Google Scholar] [CrossRef]
- Zhu, K.; Bin, Q.; Shen, Y.; Huang, J.; He, D.; Chen, W. In-situ formed N-doped bamboo-like carbon nanotubes encapsulated with Fe nanoparticles supported by biochar as highly efficient catalyst for activation of persulfate (PS) toward degradation of organic pollutants. Chem. Eng. J. 2020, 402, 126090. [Google Scholar] [CrossRef]
- Huang, L.-Z.; Zhou, C.; Shen, M.; Gao, E.; Zhang, C.; Hu, X.-M.; Chen, Y.; Xue, Y.; Liu, Z. Persulfate activation by two-dimensional MoS2 confining single Fe atoms: Performance, mechanism and DFT calculations. J. Hazard. Mater. 2020, 389, 122137. [Google Scholar] [CrossRef]
- Shang, W.; Dong, Z.; Li, M.; Song, X.; Zhang, M.; Jiang, C.; Feiyun, S. Degradation of diatrizoate in water by Fe(II)-activated persulfate oxidation. Chem. Eng. J. 2019, 361, 1333–1344. [Google Scholar] [CrossRef]
- Xu, X.; Yang, Y.; Jia, Y.; Lian, X.; Zhang, Y.; Feng, F.; Liu, Q.; Xi, B.; Jiang, Y. Heterogeneous catalytic degradation of 2,4-dinitrotoluene by the combined persulfate and hydrogen peroxide activated by the as-synthesized Fe-Mn binary oxides. Chem. Eng. J. 2019, 374, 776–786. [Google Scholar] [CrossRef]
- Huang, S.; Li, Z.; Chen, C.; Tang, S.; Cheng, X.; Guo, X. Synergetic activation of persulfate by heat and Fe(II)-complexes for hydrolyzed polyacrylamide degradation at high pH condition: Kinetics, mechanism, and application potential for filter cake removal during cementing in CO2 storage wells. Sci. Total Environ. 2020, 713, 136561. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, B.; Ma, Y.; Xing, S. Enhanced superoxide radical production for ofloxacin removal via persulfate activation with Cu-Fe oxide. Chem. Eng. J. 2018, 354, 473–480. [Google Scholar] [CrossRef]
- Zhu, J.-P.; Lin, Y.-L.; Zhang, T.-Y.; Cao, T.-C.; Xu, B.; Pan, Y.; Zhang, X.-T.; Gao, N.-Y. Modelling of iohexol degradation in a Fe(II)-activated persulfate system. Chem. Eng. J. 2019, 367, 86–93. [Google Scholar] [CrossRef]
- Wang, H.; Guo, W.; Yin, R.; Du, J.; Wu, Q.; Luo, H.; Liu, B.; Sseguya, F.; Ren, N. Biochar-induced Fe(III) reduction for persulfate activation in sulfamethoxazole degradation: Insight into the electron transfer, radical oxidation and degradation pathways. Chem. Eng. J. 2019, 362, 561–569. [Google Scholar] [CrossRef]
- Xu, L.; Li, J.; Zeng, W.; Liu, K.; Ma, Y.; Fang, L.; Shi, C. Surfactant-assisted removal of 2,4-dichlorophenol from soil by zero-valent Fe/Cu activated persulfate. Chin. J. Chem. Eng. 2021, 44, 447–455. [Google Scholar] [CrossRef]
- Zhang, W.; Li, X.; Yang, Q.; Wang, D.; Wu, Y.; Zhu, X.; Wei, J.; Liu, Y.; Hou, L.; Chen, C. Pretreatment of landfill leachate in near-neutral pH condition by persulfate activated Fe-C micro-electrolysis system. Chemosphere 2019, 216, 749–756. [Google Scholar] [CrossRef]
- Dong, Y.; Wang, P.; Li, B. Fe complex immobilized on waste polypropylene fibers for fast degradation of Reactive Red 195 via enhanced activation of persulfate under LED visible irradiation. J. Clean. Prod. 2019, 208, 1347–1356. [Google Scholar] [CrossRef]
- Li, X.; Zhou, M.; Pan, Y. Enhanced degradation of 2,4-dichlorophenoxyacetic acid by pre-magnetization Fe-C activated persulfate: Influential factors, mechanism and degradation pathway. J. Hazard. Mater. 2018, 353, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Prulho, R.; Brigante, M.; Dong, W.; Hanna, K.; Mailhot, G. Activation of persulfate by Fe(III) species: Implications for 4-tert-butylphenol degradation. J. Hazard. Mater. 2017, 322, 380–386. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Wang, B.; Wang, S.; Liu, M.; Wu, Y.; Lu, L.; Ren, H.; Li, H.; Dong, W.; et al. Degradation of ibuprofen in soil systems by persulfate activated with pyrophosphate chelated Fe(II). Chem. Eng. J. 2020, 379, 122145. [Google Scholar] [CrossRef]
- Fan, J.; Gu, L.; Wu, D.; Liu, Z. Mackinawite (FeS) activation of persulfate for the degradation of p-chloroaniline: Surface reaction mechanism and sulfur-mediated cycling of iron species. Chem. Eng. J. 2018, 333, 657–664. [Google Scholar] [CrossRef]
- Liang, C.; Liang, C.-P.; Chen, C.-C. pH dependence of persulfate activation by EDTA/Fe(III) for degradation of trichloroethylene. J. Contam. Hydrol. 2009, 106, 173–182. [Google Scholar] [CrossRef]
- Li, X.; Guo, W.; Liu, Z.; Wang, R.; Liu, H. Fe-based MOFs for efficient adsorption and degradation of acid orange 7 in aqueous solution via persulfate activation. Appl. Surf. Sci. 2016, 369, 130–136. [Google Scholar] [CrossRef]
- Zhu, L.; Ai, Z.; Ho, W.; Zhang, L. Core–shell Fe–Fe2O3 nanostructures as effective persulfate activator for degradation of methyl orange. Sep. Purif. Technol. 2013, 108, 159–165. [Google Scholar] [CrossRef]
- Bu, L.; Shi, Z.; Zhou, S. Modeling of Fe(II)-activated persulfate oxidation using atrazine as a target contaminant. Sep. Purif. Technol. 2016, 169, 59–65. [Google Scholar] [CrossRef]
- Yu, S.; Gu, X.; Lu, S.; Xue, Y.; Zhang, X.; Xu, M.; Qiu, Z.; Sui, Q. Degradation of phenanthrene in aqueous solution by a persulfate/percarbonate system activated with CA chelated-Fe(II). Chem. Eng. J. 2018, 333, 122–131. [Google Scholar] [CrossRef]
- Zhen, G.; Wang, J.; Lu, X.; Su, L.; Zhu, X.; Zhou, T.; Zhao, Y. Effective gel-like floc matrix destruction and water seepage for enhancing waste activated sludge dewaterability under hybrid microwave-initiated Fe(II)-persulfate oxidation process. Chemosphere 2019, 221, 141–153. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, Z.; Feng, M.; Liu, W.; Wang, W.; Yang, Q.; Hu, Y. Degradation of 2,4-dichlorophenoxyacetic acid in water by persulfate activated with FeS (mackinawite). Chem. Eng. J. 2017, 313, 498–507. [Google Scholar] [CrossRef]
- Zhen, G.; Lu, X.; Zhao, Y.; Chai, X.; Niu, D. Enhanced dewaterability of sewage sludge in the presence of Fe(II)-activated persulfate oxidation. Bioresour. Technol. 2012, 116, 259–265. [Google Scholar] [CrossRef]
- Rao, Y.F.; Qu, L.; Yang, H.; Chu, W. Degradation of carbamazepine by Fe(II)-activated persulfate process. J. Hazard. Mater. 2014, 268, 23–32. [Google Scholar] [CrossRef]
- Idrees, A.; Shan, A.; Danish, M.; Zaman, W.Q.; Mohsin, A.; Abbas, Z.; Huang, J.; Shahzad, T.; Sun, Y.; Xu, Z.; et al. Influence of preparation method on copper ferrite characteristics for the efficient degradation of trichloroethylene in persulfate activated system. J. Environ. Chem. Eng. 2021, 9, 106044. [Google Scholar] [CrossRef]
- Han, D.; Wan, J.; Ma, Y.; Wang, Y.; Li, Y.; Li, D.; Guan, Z. New insights into the role of organic chelating agents in Fe(II) activated persulfate processes. Chem. Eng. J. 2015, 269, 425–433. [Google Scholar] [CrossRef]
- Li, M.; Yang, X.; Wang, D.; Yuan, J. Enhanced oxidation of erythromycin by persulfate activated iron powder–H2O2 system: Role of the surface Fe species and synergistic effect of hydroxyl and sulfate radicals. Chem. Eng. J. 2017, 317, 103–111. [Google Scholar] [CrossRef]
- Zhu, J.; Song, Y.; Wang, L.; Zhang, Z.; Gao, J.; Tsang, D.C.W.; Ok, Y.S.; Hou, D. Green remediation of benzene contaminated groundwater using persulfate activated by biochar composite loaded with iron sulfide minerals. Chem. Eng. J. 2022, 429, 132292. [Google Scholar] [CrossRef]
- Sun, C.; Chen, T.; Huang, Q.; Zhan, M.; Li, X.; Yan, J. Activation of persulfate by CO2-activated biochar for improved phenolic pollutant degradation: Performance and mechanism. Chem. Eng. J. 2020, 380, 122519. [Google Scholar] [CrossRef]
- Han, D.; Wan, J.; Ma, Y.; Wang, Y.; Huang, M.; Chen, Y.; Li, D.; Guan, Z.; Li, Y. Enhanced decolorization of Orange G in a Fe(II)-EDDS activated persulfate process by accelerating the regeneration of ferrous iron with hydroxylamine. Chem. Eng. J. 2014, 256, 316–323. [Google Scholar] [CrossRef]
- Xu, J.; Wang, S.; Yan, C.; Adeel Sharif, H.M.; Yang, B. Activation of sodium persulfate by TiO2@MIL-101(Fe): Boosting the Fenton-like process by interfacial charge transfer. Chemosphere 2022, 288, 132666. [Google Scholar] [CrossRef]
- Li, Y.; Yang, F.; Miao, S.; Wang, D.; Li, Z.; Yuan, X.; Yuan, L.; Liu, Q. Achieved deep-dewatering of dredged sediments by Fe(II) activating persulfate pretreatment: Filtrating performance and mechanistic insights. Chem. Eng. J. 2021, 405, 126847. [Google Scholar] [CrossRef]
- Li, Y.; Wang, D.; Xu, Q.; Liu, X.; Wang, Y.; Wu, Y.; Yang, G.; Yuan, X.; Wu, Z.; Guan, R.; et al. New insight into modification of extracellular polymeric substances extracted from waste activated sludge by homogeneous Fe(II)/persulfate process. Chemosphere 2020, 247, 125804. [Google Scholar] [CrossRef]
- Zeng, G.; Yang, R.; Fu, X.; Zhou, Z.; Xu, Z.; Zhou, Z.; Qiu, Z.; Sui, Q.; Lyu, S. Naphthalene degradation in aqueous solution by Fe(II) activated persulfate coupled with citric acid. Sep. Purif. Technol. 2021, 264, 118441. [Google Scholar] [CrossRef]
- Rodríguez, S.; Lorenzo, D.; Santos, A.; Romero, A. Comparison of real wastewater oxidation with Fenton/Fenton-like and persulfate activated by NaOH and Fe(II). J. Environ. Manag. 2020, 255, 109926. [Google Scholar] [CrossRef]
- Sühnholz, S.; Kopinke, F.-D.; Mackenzie, K. Reagent or catalyst?—FeS as activator for persulfate in water. Chem. Eng. J. 2020, 387, 123804. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, J.; Chen, X.; Wang, Z.; Ji, H.; Chen, L.; Liu, W.; Wang, C.-C. Bifunctional Bi12O17Cl2/MIL-100(Fe) composites toward photocatalytic Cr(VI) sequestration and activation of persulfate for bisphenol A degradation. Sci. Total Environ. 2021, 752, 141901. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, P.; Wang, C.; Jia, H.; Shang, X.; Tang, J.; Sun, H. Metal-rich hyperaccumulator-derived biochar as an efficient persulfate activator: Role of intrinsic metals (Fe, Mn and Zn) in regulating characteristics, performance and reaction mechanisms. J. Hazard. Mater. 2022, 424, 127225. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Jiang, Y.; Armutlulu, A.; Shen, Z.; Lai, B.; Wang, H. One-step fabrication of oxygen vacancy-enriched Fe@Ti/C composite for highly efficient degradation of organic pollutants through persulfate activation. J. Colloid Interface Sci. 2021, 583, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Rykov, A.I.; Zhang, B.; Zhang, Y.; Wang, J. Graphene encapsulated FexCoy nanocages derived from metal–organic frameworks as efficient activators for peroxymonosulfate. Catal. Sci. Technol. 2016, 6, 7486–7494. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Jiang, X.; Xie, R.; Zhang, Y.; Jin, Y.; Jiang, W. A novel porous biochar-supported Fe-Mn composite as a persulfate activator for the removal of acid red 88. Sep. Purif. Technol. 2020, 250, 117232. [Google Scholar] [CrossRef]
- Hao, H.; Zhang, Q.; Qiu, Y.; Meng, L.; Wei, X.; Sang, W.; Tao, J. Insight into the degradation of Orange G by persulfate activated with biochar modified by iron and manganese oxides: Synergism between Fe and Mn. J. Water Process Eng. 2020, 37, 101470. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, B.-T.; Teng, Y.; Zhao, J.; Sun, X. Heterogeneous activation of persulfate by carbon nanofiber supported Fe3O4@carbon composites for efficient ibuprofen degradation. J. Hazard. Mater. 2021, 401, 123428. [Google Scholar] [CrossRef]
- Shan, A.; Idrees, A.; Zaman, W.Q.; Abbas, Z.; Ali, M.; Rehman, M.S.U.; Hussain, S.; Danish, M.; Gu, X.; Lyu, S. Synthesis of nZVI-Ni@BC composite as a stable catalyst to activate persulfate: Trichloroethylene degradation and insight mechanism. J. Environ. Chem. Eng. 2021, 9, 104808. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, H.; Wang, Z.; Huang, D.; Qin, H.; He, Y.; Chen, M.; Zeng, G.; Xu, P. Ferrocene modified g-C3N4 as a heterogeneous catalyst for photo-assisted activation of persulfate for the degradation of tetracycline. Colloids Surf. A Physicochem. Eng. Asp. 2021, 626, 127024. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, T.; Zhu, X.; Song, Z.; Li, X.; Zhang, J.; Mao, Y.; Wu, J.; Zhang, W.; Wang, C. Large-Scale Synthesis of Iron Ore@Biomass Derived ESBC to Degrade Tetracycline Hydrochloride for Heterogeneous Persulfate Activation. Catalysts 2022, 12, 1345. https://doi.org/10.3390/catal12111345
Tian T, Zhu X, Song Z, Li X, Zhang J, Mao Y, Wu J, Zhang W, Wang C. Large-Scale Synthesis of Iron Ore@Biomass Derived ESBC to Degrade Tetracycline Hydrochloride for Heterogeneous Persulfate Activation. Catalysts. 2022; 12(11):1345. https://doi.org/10.3390/catal12111345
Chicago/Turabian StyleTian, Tingting, Xinfeng Zhu, Zhongxian Song, Xindong Li, Jinhui Zhang, Yanli Mao, Junfeng Wu, Wei Zhang, and Chaohai Wang. 2022. "Large-Scale Synthesis of Iron Ore@Biomass Derived ESBC to Degrade Tetracycline Hydrochloride for Heterogeneous Persulfate Activation" Catalysts 12, no. 11: 1345. https://doi.org/10.3390/catal12111345
APA StyleTian, T., Zhu, X., Song, Z., Li, X., Zhang, J., Mao, Y., Wu, J., Zhang, W., & Wang, C. (2022). Large-Scale Synthesis of Iron Ore@Biomass Derived ESBC to Degrade Tetracycline Hydrochloride for Heterogeneous Persulfate Activation. Catalysts, 12(11), 1345. https://doi.org/10.3390/catal12111345