
The Power of Biocatalysts for Highly Selective and Efficient Phosphorylation Reactions


Abstract
:
{kind=link}
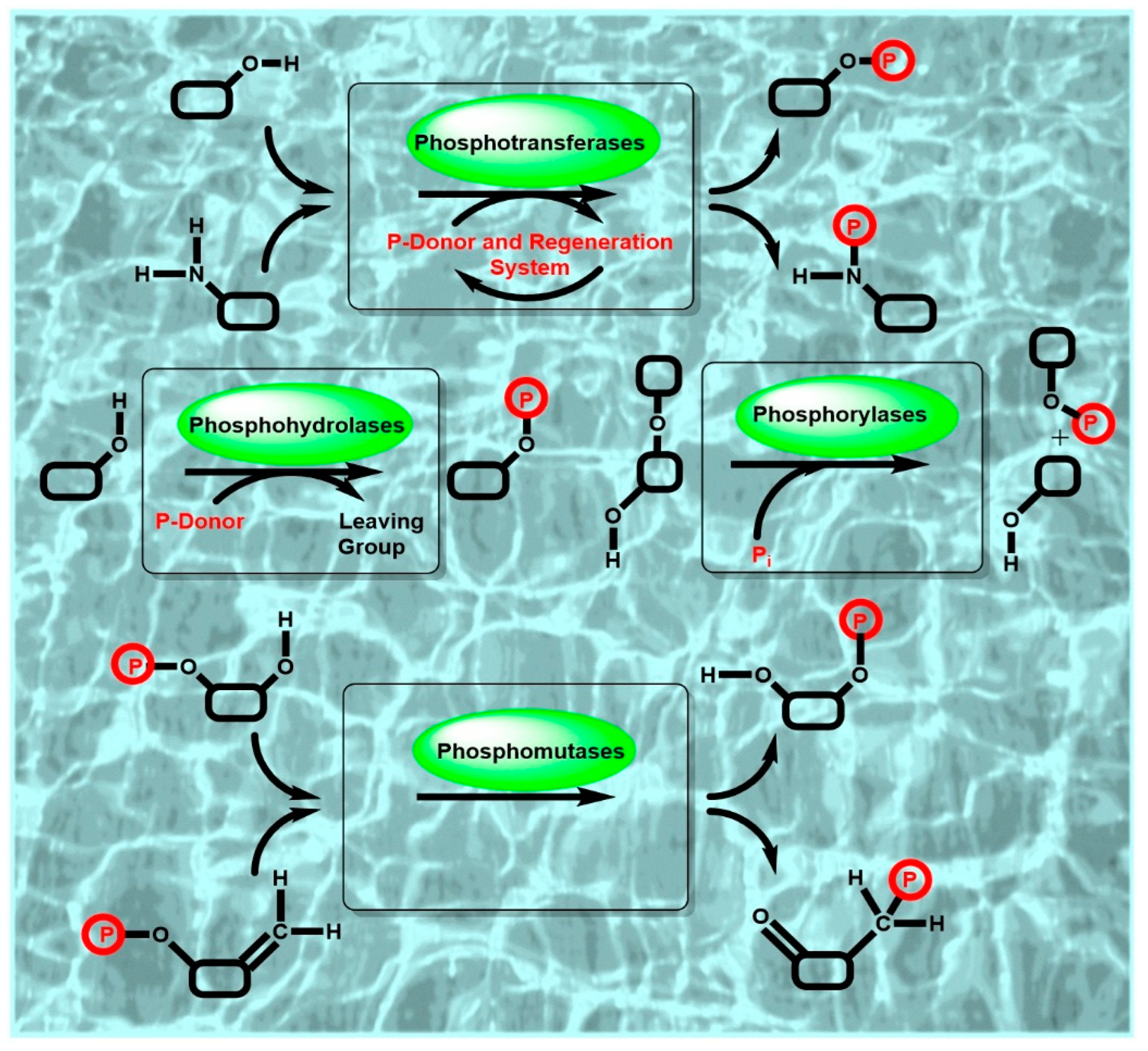{kind=link}
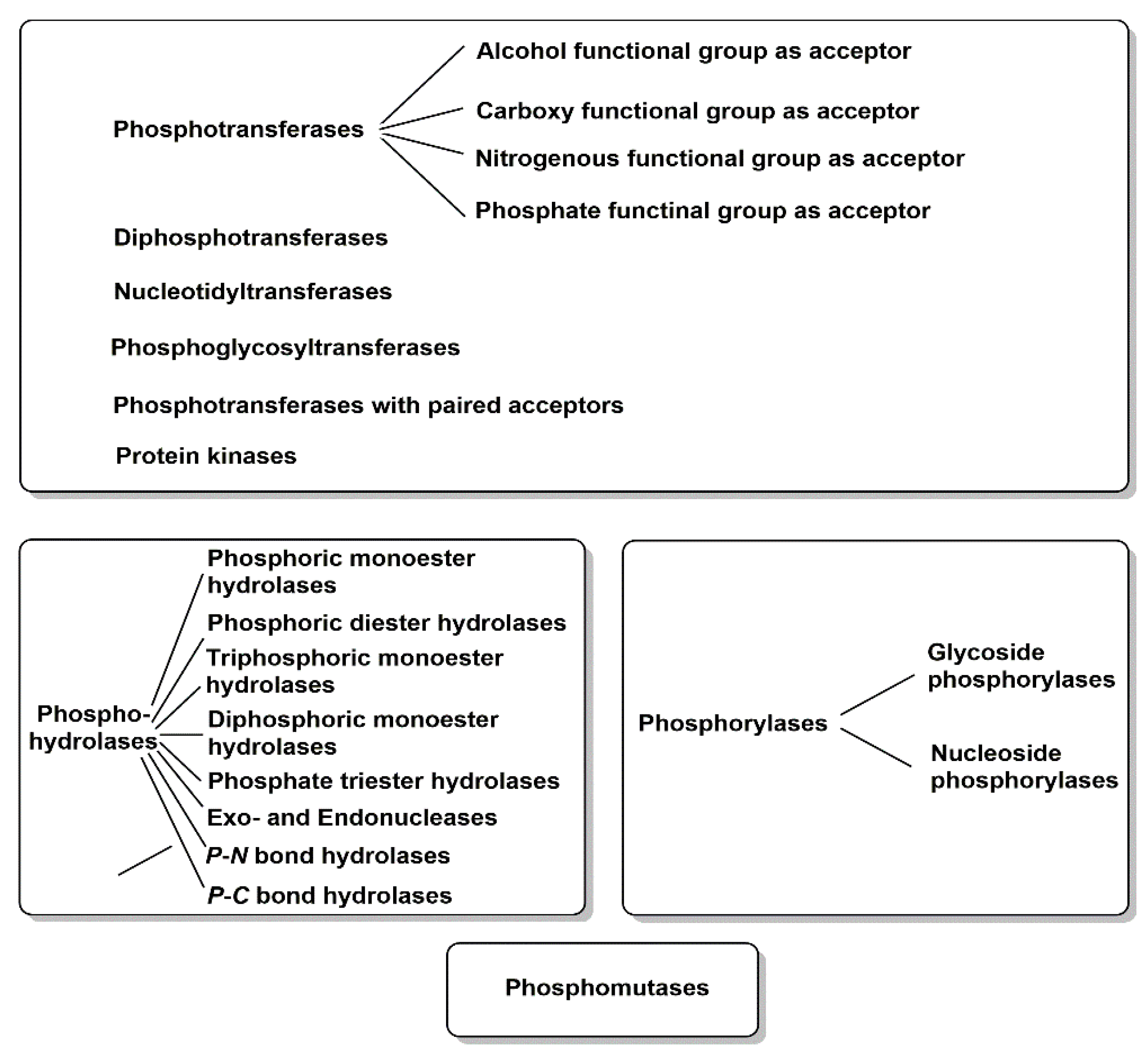{kind=link}
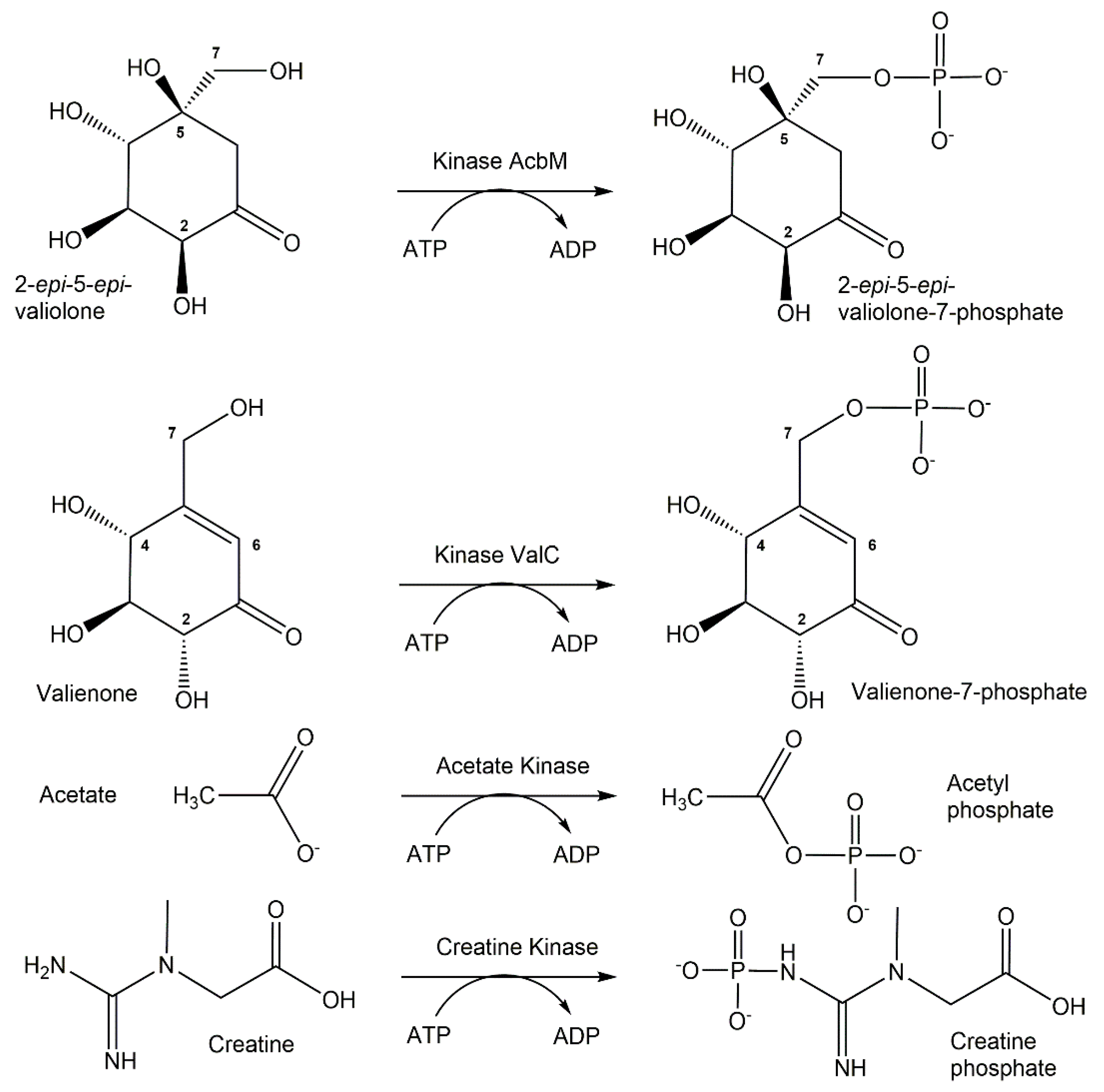{kind=link}
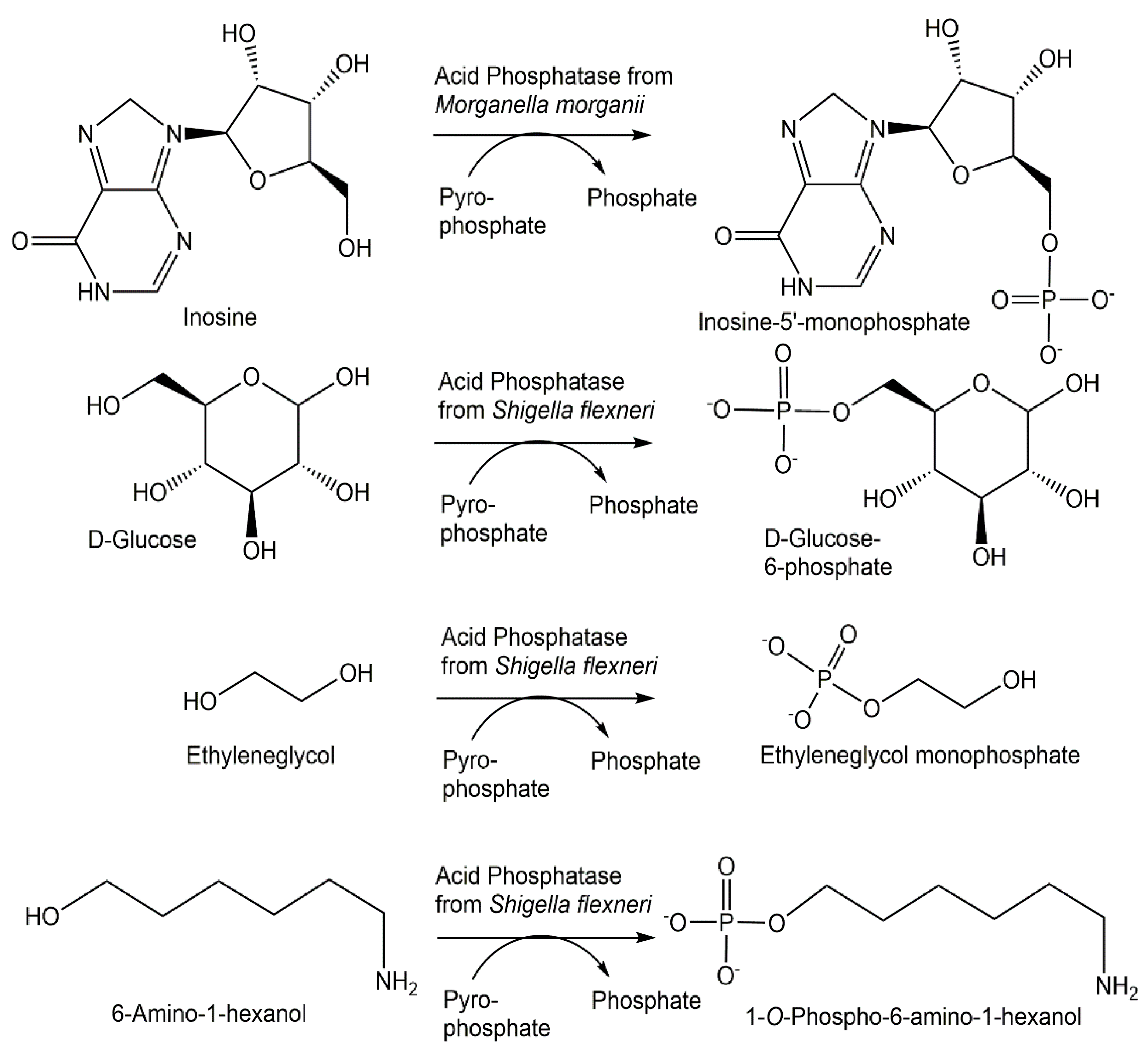{kind=link}
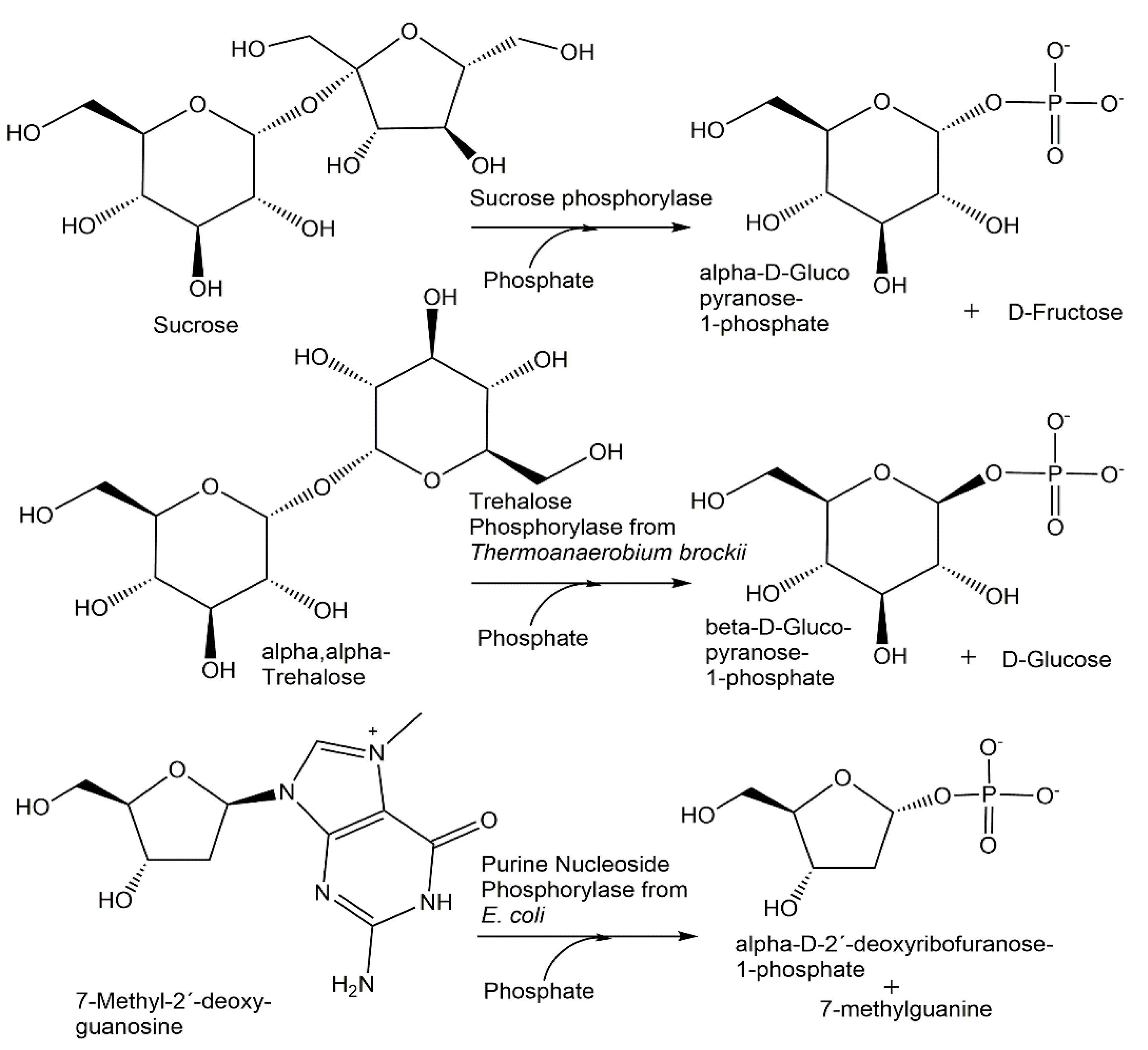{kind=link}
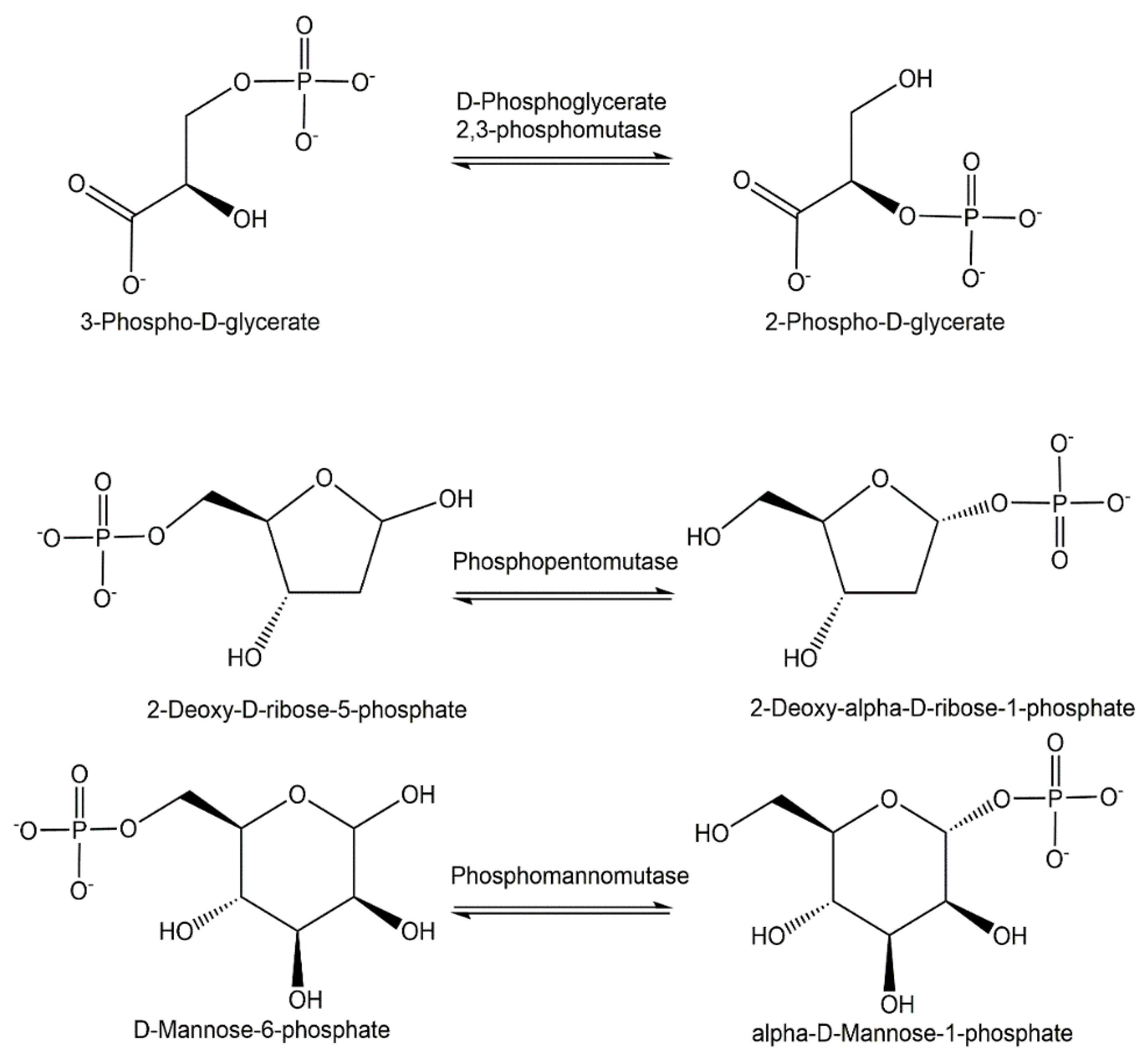{kind=link}
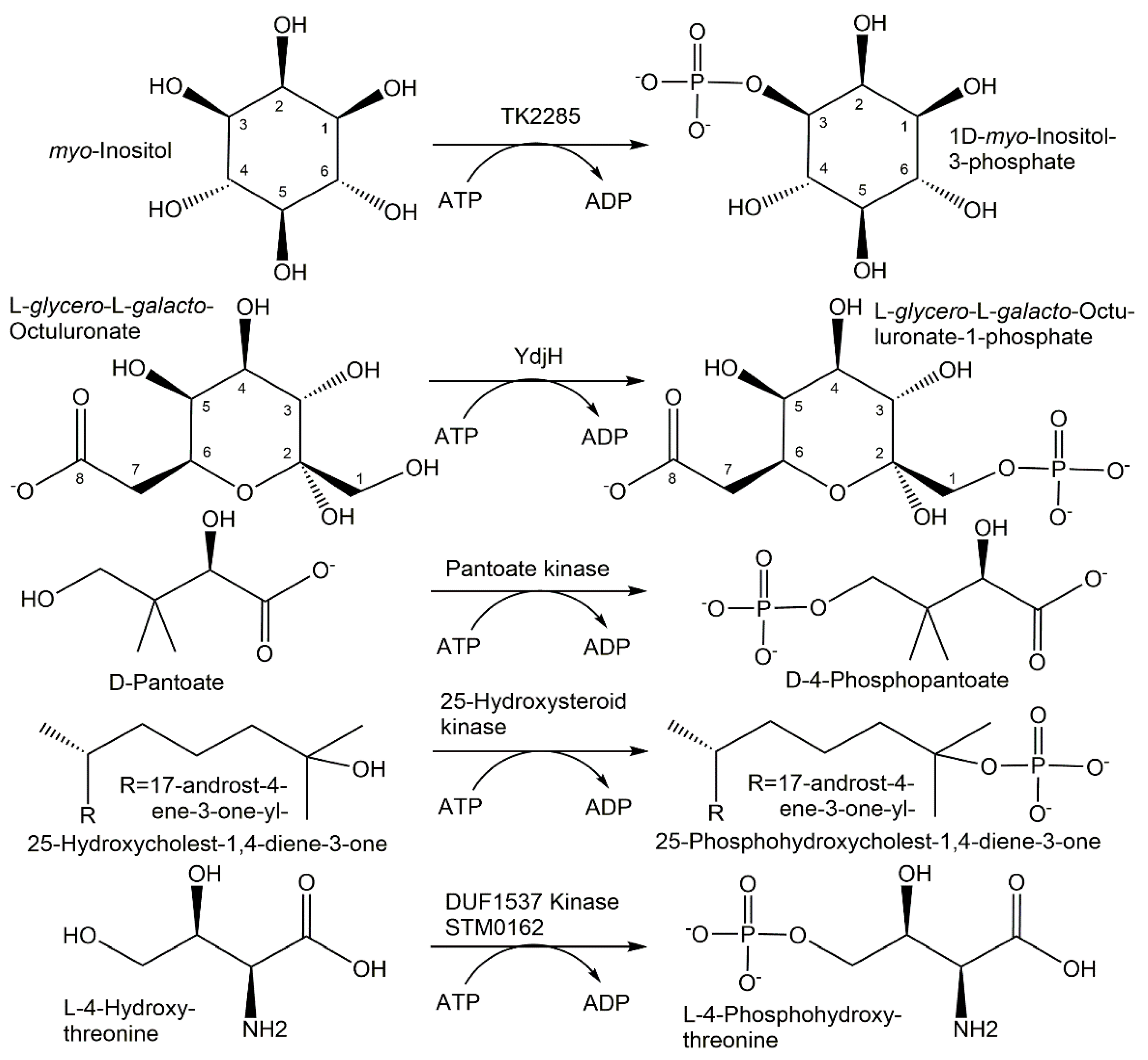{kind=link}
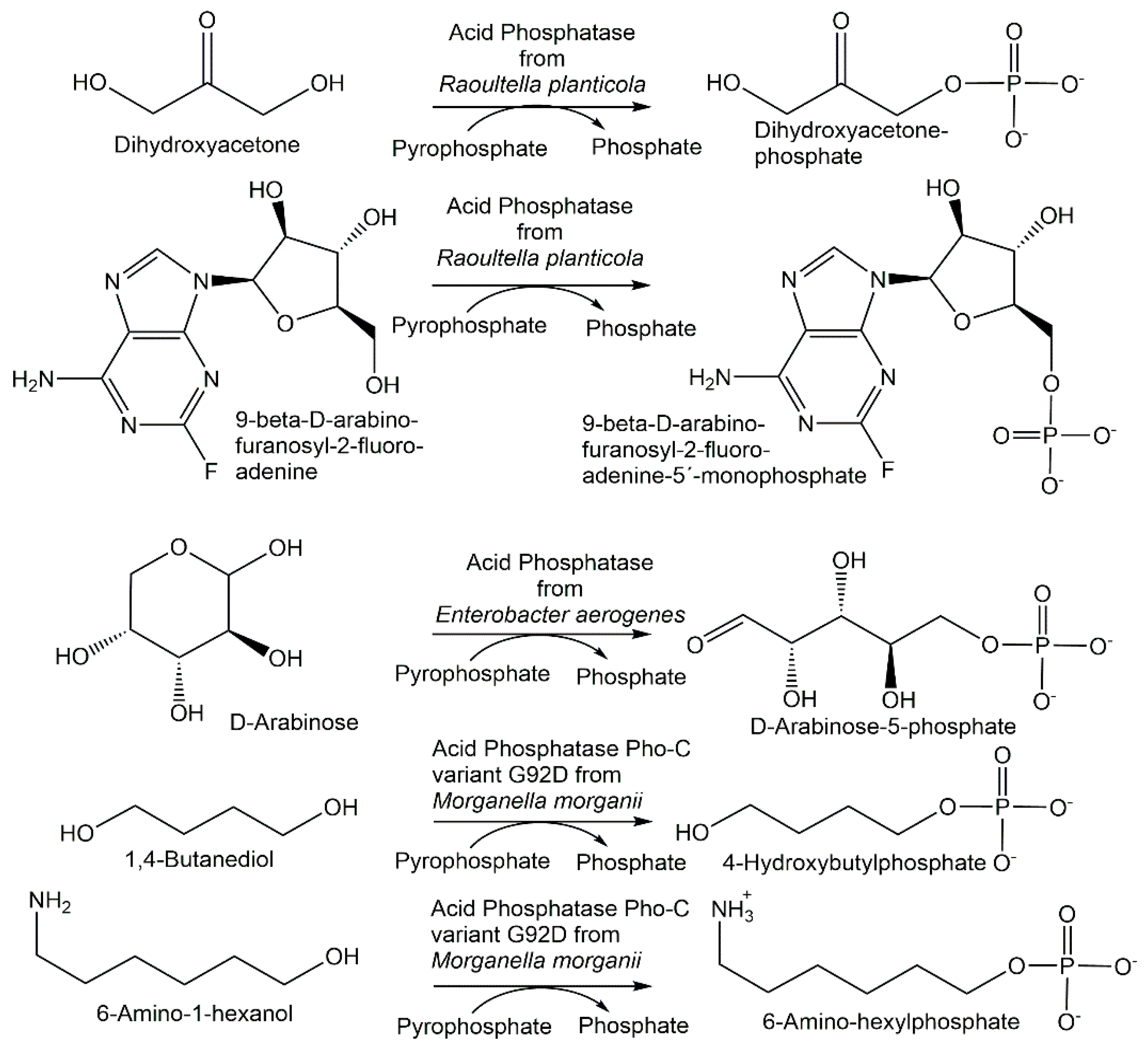{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
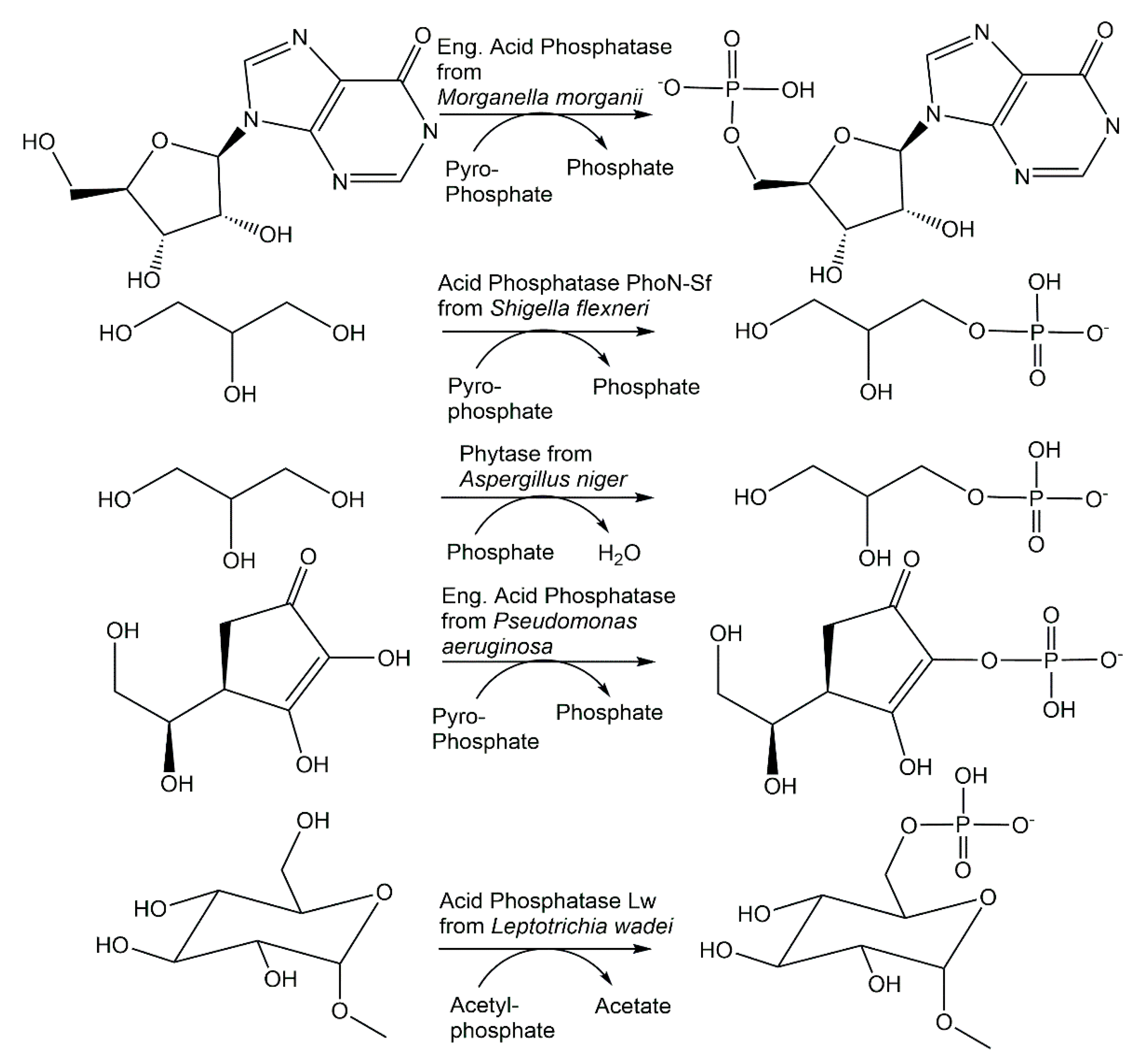{kind=link}
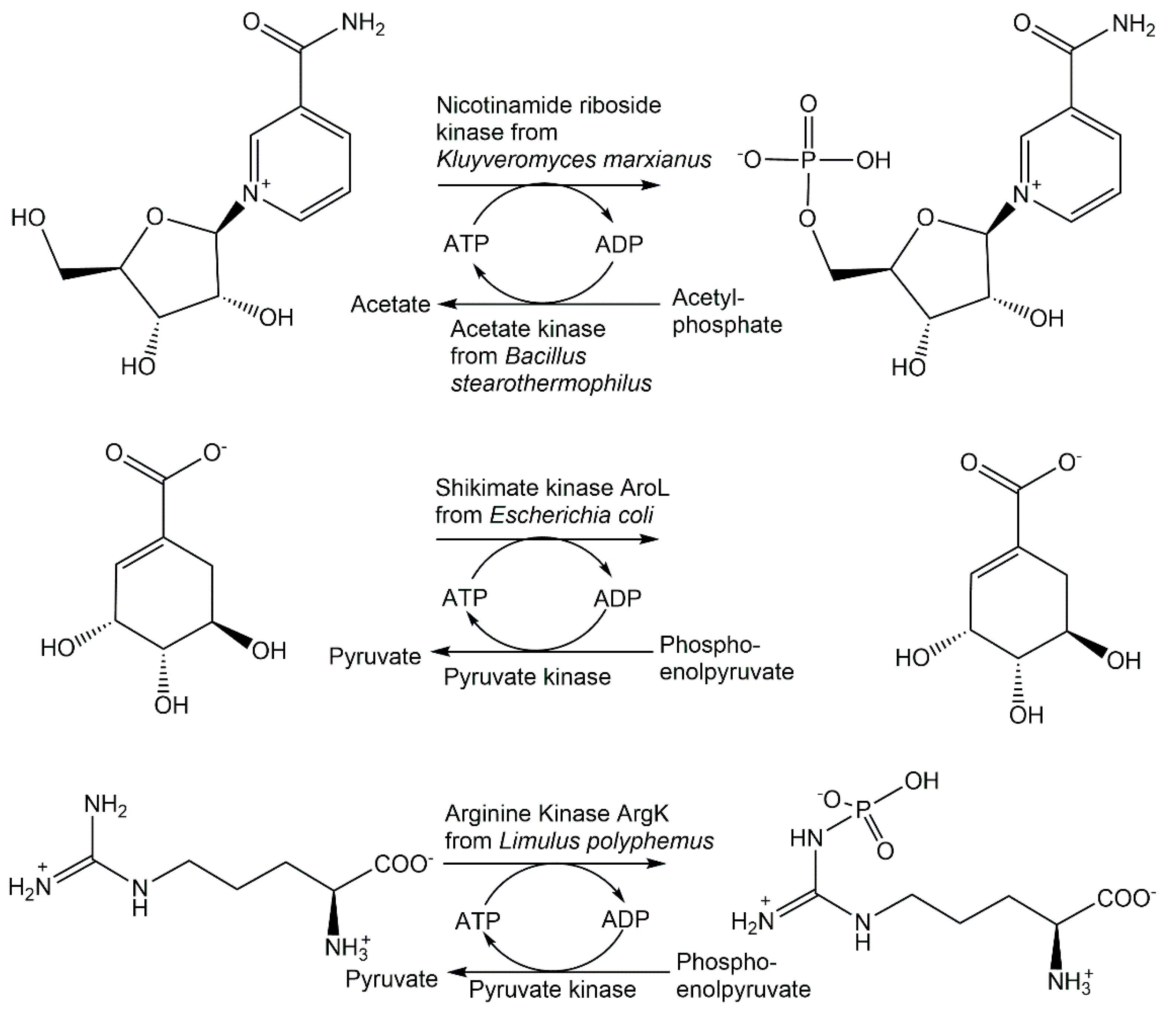{kind=link}
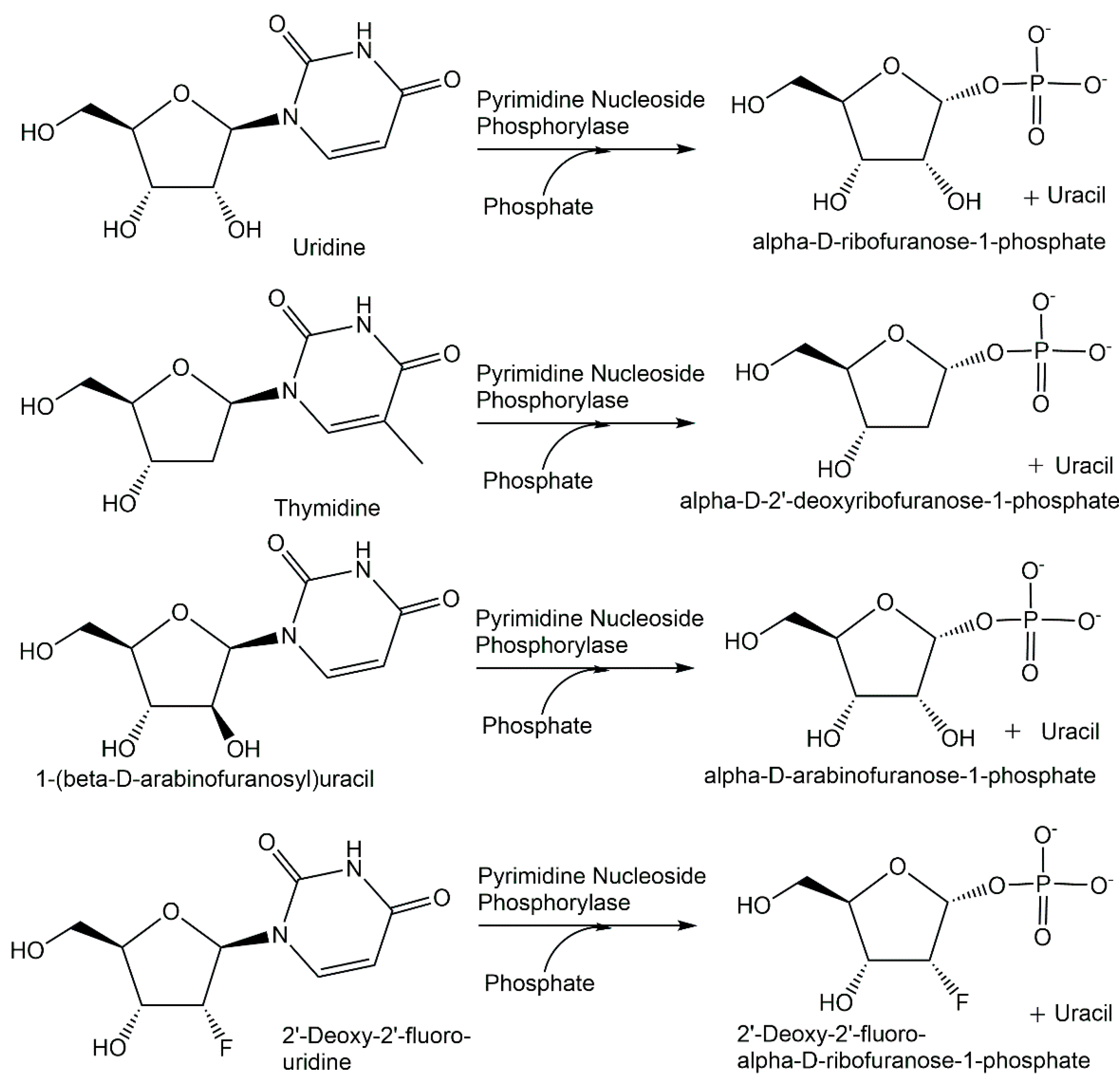{kind=link}
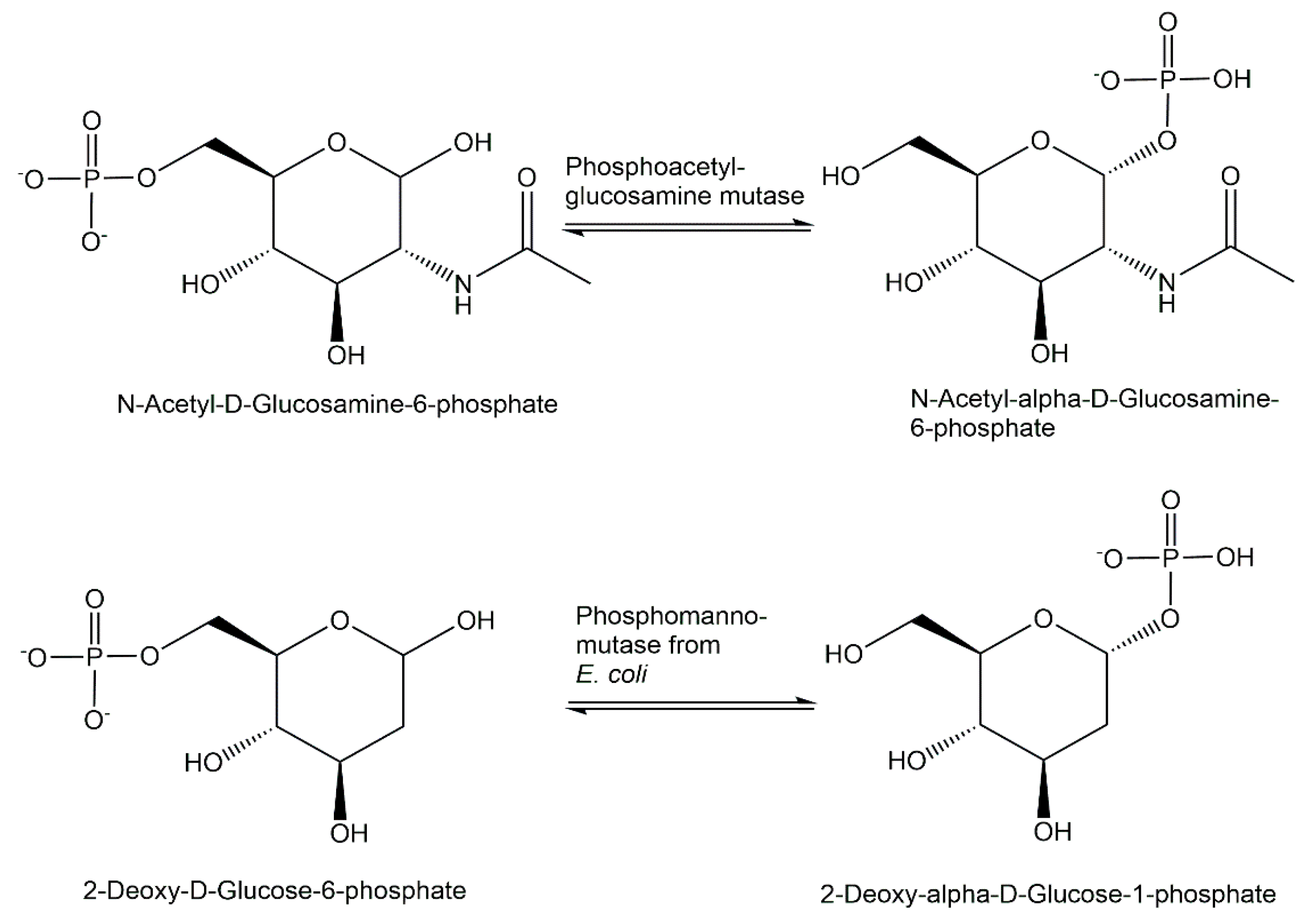{kind=link}
{kind=link}
1. Introduction
2. Structures, Functions and Mechanisms of Phosphorylation Biocatalysts
2.1. Structures of Phosphorylation Biocatalysts
2.1.1. Phosphotransferase Structures
2.1.2. Phosphohydrolase Structures
2.1.3. Phosphorylase Structures
2.1.4. Phosphomutase Structures
2.2. Functions of Phosphorylation Biocatalysts
2.2.1. Phosphotransferase Functions
2.2.2. Phosphohydrolase Functions
2.2.3. Phosphorylase Functions
2.2.4. Phosphomutase Functions
2.3. Mechanisms of Phosphorylation Biocatalysts
2.3.1. Phosphotransferases
2.3.2. Phosphohydrolases
2.3.3. Phosphorylases
2.3.4. Phosphomutases
3. Discovery Methodologies and Tools for Phosphorylation Biocatalysts
4. Novel Phosphorylation Biocatalysts
4.1. Novel Phosphotransferases
4.2. Novel Phosphohydrolases
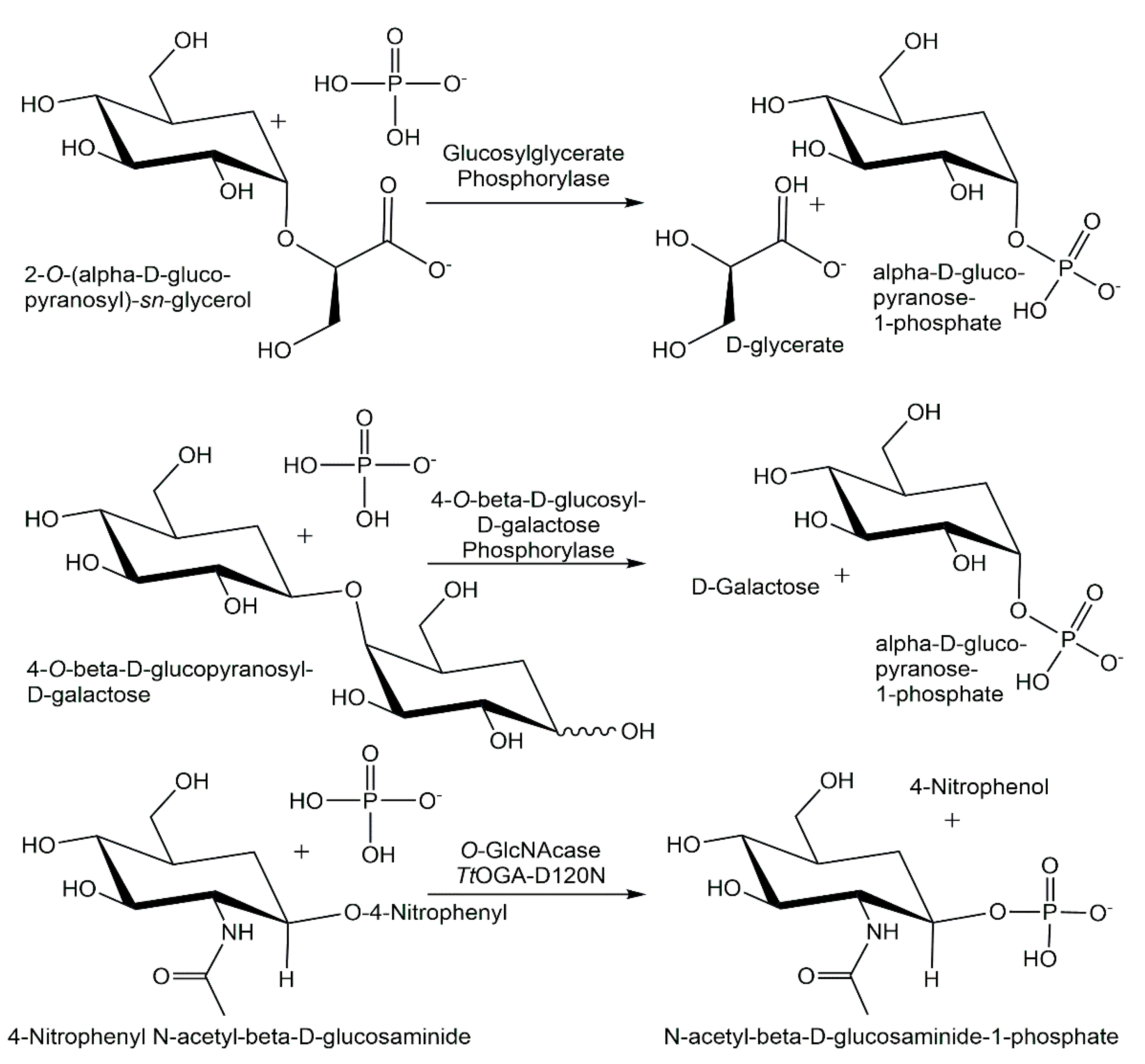
4.3. Novel Phosphorylases
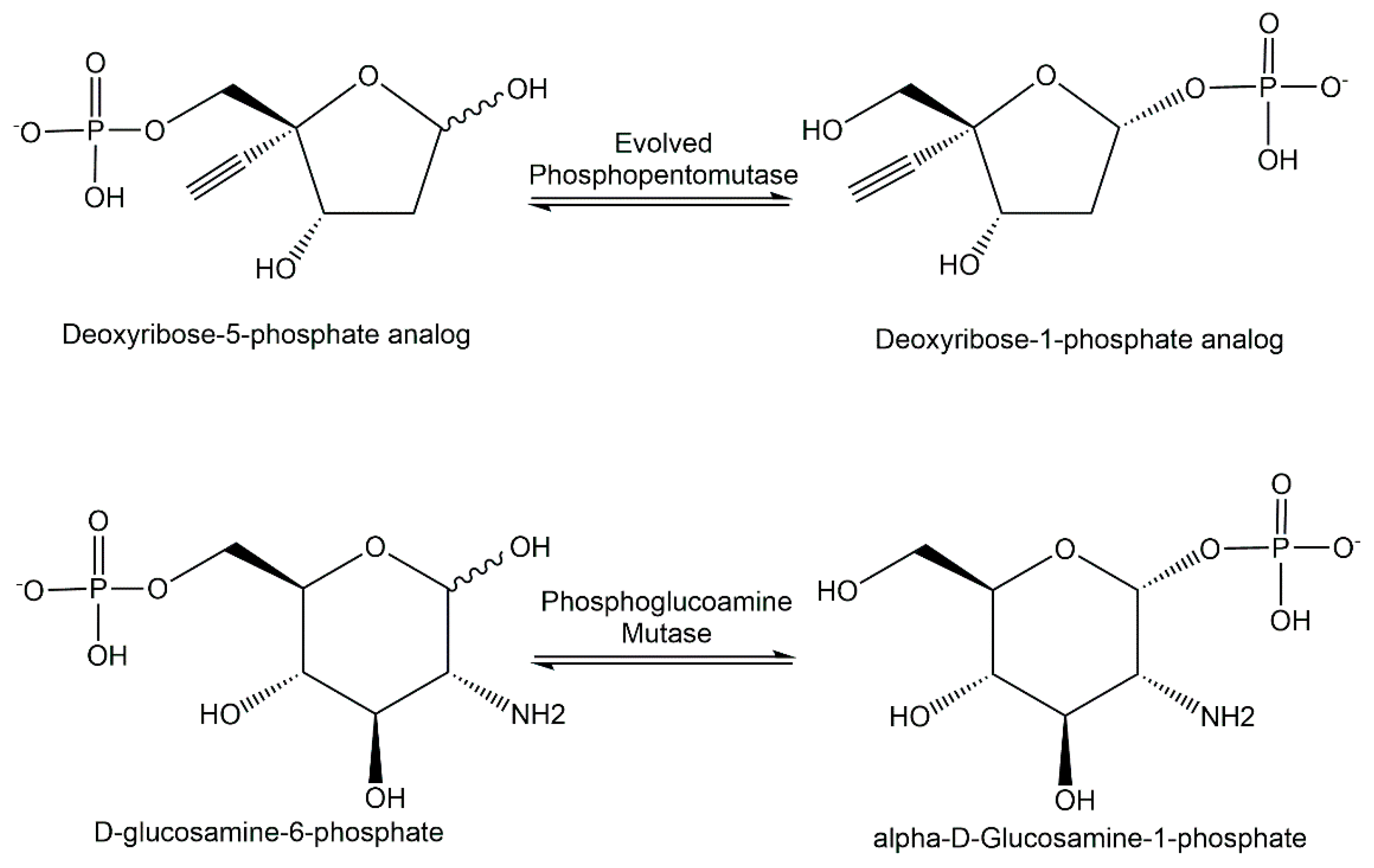
4.4. Novel Phosphomutases
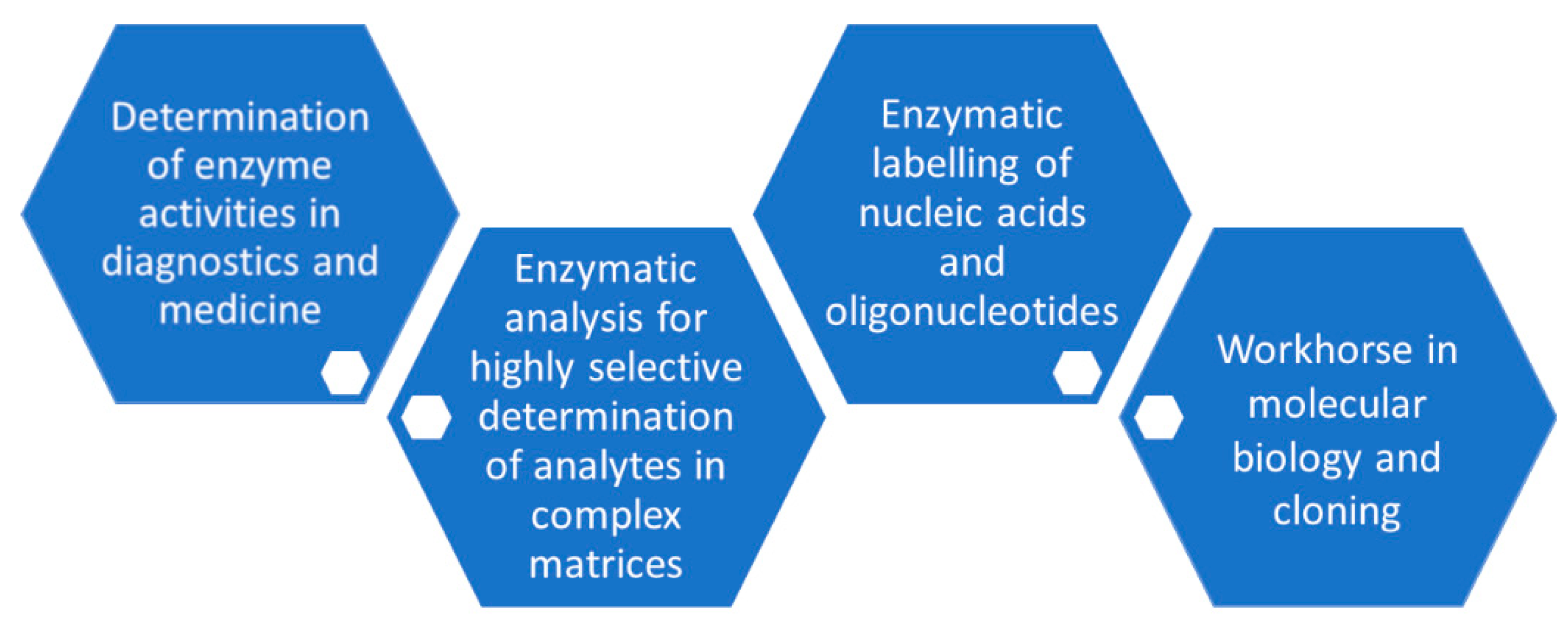
5. Analytical Applications of Phosphorylation Biocatalysts
5.1. Analytical Applications of Phosphotransferases
5.2. Analytical Applications of Phosphohydrolases
5.3. Analytical Applications of Phosphorylases
5.4. Analytical Applications of Phosphomutases
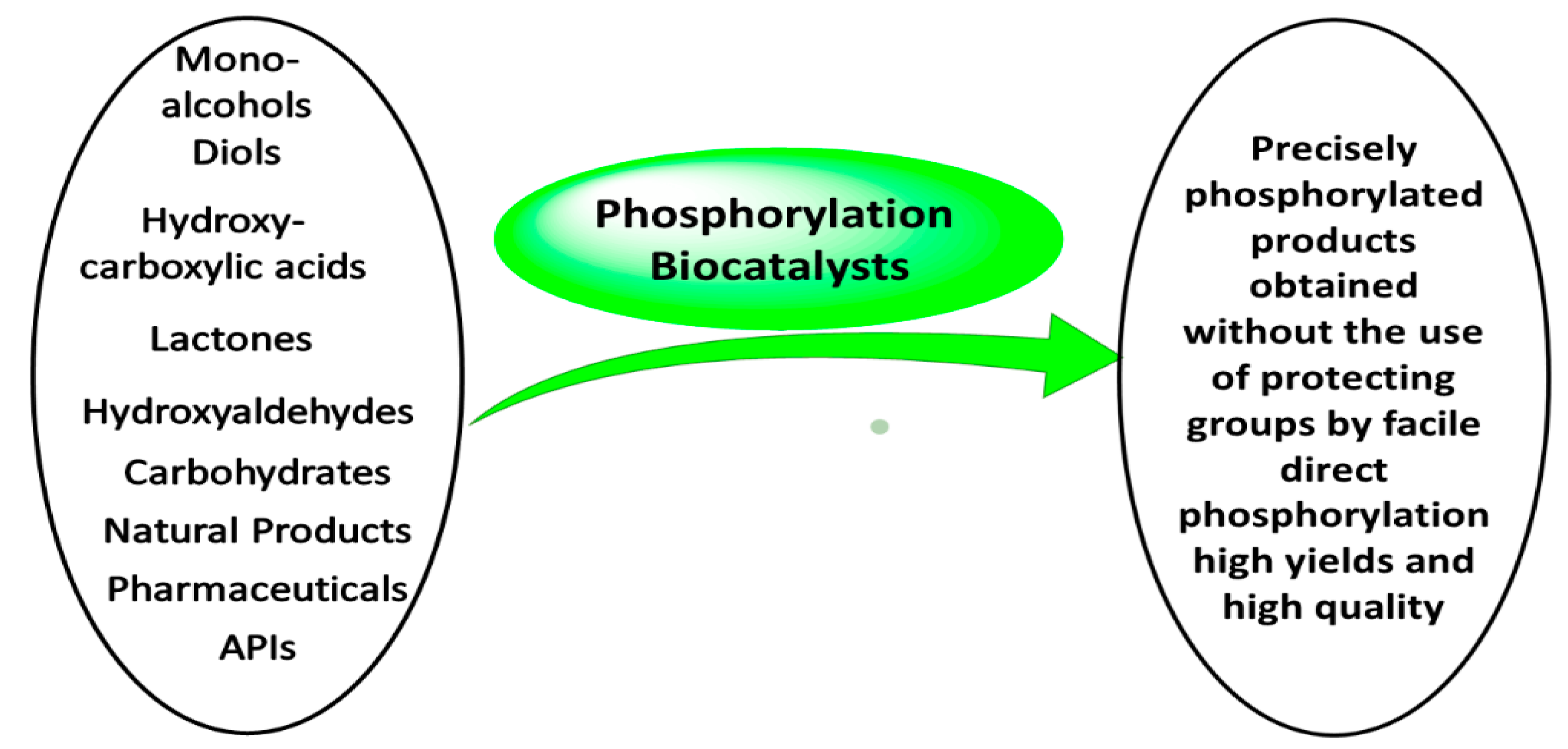
6. Synthetic Applications of Phosphorylation Biocatalysts
6.1. Synthetic Applications of Phosphohydrolases
6.2. Synthetic Applications of Phosphotransferases
6.3. Synthetic Applications of Phosphorylases
6.4. Synthetic Applications of Phosphomutases
6.5. Phosphorylation Biocatalysts in Cascades
6.6. Phosphoryl Donors and Systems for their Regeneration
6.7. Phosphorylation Reaction Engineering
6.8. Product Recovery and Purification
7. Opportunities and Outlook
Funding
Conflicts of Interest
References
- Ruttenberg, K.C. The Global Phosphorus Cycle. Treatise Geochem. 2003, 8, 585–645. [Google Scholar] [CrossRef]
- Steffen, W.; Richardson, K.; Rockström, J.; Cornell, S.E.; Fetzer, I.; Bennett, E.M.; Biggs, R.; Carpenter, S.R.; De Vries, W.; De Wit, C.A.; et al. Planetary boundaries: Guiding human development on a changing planet. Science 2015, 347, 1259855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jupp, A.R.; Beijer, S.; Narain, G.C.; Schipper, W.; Slootweg, J.C. Phosphorus recovery and recycling–closing the loop. Chem. Soc. Rev. 2021, 50, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Geeson, M.B.; Cummins, C.C. Phosphoric acid as a precursor to chemicals traditionally synthesized from white phosphorus. Science 2018, 359, 1383–1385. [Google Scholar] [CrossRef] [Green Version]
- Willey, N.; Timbs, P. Radioactivity in Future Phosphogypsum: New predictions based on estimates of ‘Peak P’ and rock phosphate resources. J. Environ. Radioact. 2022, 244–245, 106828. [Google Scholar] [CrossRef]
- Pasek, M.A. Thermodynamics of Prebiotic Phosphorylation. Chem. Rev. 2020, 120, 4690–4706. [Google Scholar] [CrossRef]
- Westheimer, F.H. Why Nature Chose Phosphates. Science 1987, 235, 1173–1178. [Google Scholar] [CrossRef]
- Bowler, M.W.; Cliff, M.J.; Waltho, J.P.; Blackburn, G.M. Why did Nature select phosphate for its dominant roles in biology? New J. Chem. 2010, 34, 784–794. [Google Scholar] [CrossRef]
- Kamerlin, S.C.; Sharma, P.K.; Prasad, R.B.; Warshel, A. Why Nature Really Chose Phosphate. Q. Rev. Biophys. 2013, 246, 1–132. [Google Scholar] [CrossRef]
- Wohlgemuth, R. Key advances in biocatalytic phosphorylations in the last two decades: Biocatalytic syntheses in vitro and biotransformations in vivo (in humans). Biotechnol. J. 2021, 16, 2000090. [Google Scholar] [CrossRef]
- Nam, I.; Lee, J.K.; Nam, H.G.; Zare, R.N. Abiotic production of sugar phosphates and uridine ribonucleoside in aqueous micro-droplets. PNAS 2017, 114, 12396–12400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, F. Neuere Methoden der präparativen organischen Chemie III 2. Darstellung von Estern, Amiden und Anhydriden der Phosphorsäure. Angew. Chem. 1960, 72, 236–249. [Google Scholar] [CrossRef]
- Cramer, F.; Weimann, G. Imidoester, VII. Trichloracetonitril, ein Reagenz zur selektiven Veresterung von Phosphorsäuren. Chem. Ber. 1961, 94, 996–1007. [Google Scholar] [CrossRef]
- Sakakura, A.; Katsukawa, M.; Ishihara, K. Selective synthesis of phosphate monoesters by dehydrative condensation of phosphoric acid and alcohols promoted by nucleophilic bases. Org. Lett. 2005, 7, 1999–2002. [Google Scholar] [CrossRef] [PubMed]
- Dueymes, C.; Pirat, C.; Pascal, R. Facile synthesis of simple mono-alkyl phosphates from phosphoric acid and alcohols. Tetrahedron Lett. 2008, 49, 5300–5301. [Google Scholar] [CrossRef]
- Lira, L.M.; Vasilev, D.; Pilli, R.A.; Wessjohann, L.A. One-pot synthesis of organophosphate monoesters from alcohols. Tetrahedron Lett. 2013, 54, 1690–1692. [Google Scholar] [CrossRef] [Green Version]
- Domon, K.; Puripat, M.; Fujiyoshi, K.; Hatanaka, M.; Kawashima, S.A.; Yamatsugu, K.; Kanai, K. Catalytic Chemoselective O-Phosphorylation of Alcohols. ACS Cent. Sci. 2020, 6, 283–292. [Google Scholar] [CrossRef] [Green Version]
- McDonald, A.G.; Tipton, K.F. Enzyme nomenclature and classification: The state of the art. FEBS J. 2022. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2021, 49, D92–D96. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Chang, A.; Jeske, L.; Ulbrich, S.; Hofmann, J.; Koblitz, J.; Schomburg, I.; Neumann-Schaal, M.; Jahn, D.; Schomburg, D. BRENDA, the ELIXIR core data resource in 2021: New developments and updates. Nucleic Acids Res. 2021, 49, D498–D508. [Google Scholar] [CrossRef] [PubMed]
- Gardossi, L.; Poulsen, P.B.; Ballesteros, A.; Hult, K.; Švedas, V.K.; Vasić-Rački, Đ.; Carrea, G.; Magnusson, A.; Schmid, A.; Wohlgemuth, R.; et al. Guidelines for reporting of biocatalytic reactions. Trends Biotechnol. 2010, 28, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Swainston, N.; Baici, A.; Bakker, B.M.; Cornish-Bowden, A.; Fitzpatrick, P.F.; Halling, P.; Leyh, T.S.; O’Donovan, C.; Raushel, F.M.; Reschel, U.; et al. STRENDA DB: Enabling the validation and sharing of enzyme kinetics data. FEBS J. 2018, 285, 2193–2204. [Google Scholar] [CrossRef]
- Cheek, S.; Zhang, H.; Grishin, N.V. Sequence and structure classification of kinases. J. Mol. Biol. 2002, 320, 855–881. [Google Scholar] [CrossRef] [Green Version]
- Cheek, S.; Ginalski, K.; Zhang, H.; Grishin, N.V. A comprehensive update of the sequence and structure classification of kinases. BMC Struct. Biol. 2005, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Blum, M.; Chang, H.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Kannan, N.; Taylor, S.S.; Zhai, Y.; Venter, J.C.; Manning, G. Structural and Functional Diversity of the Microbial Kinome. PloS Biology 2007, 5, e17. [Google Scholar] [CrossRef]
- Anderson, C.M.; Stenkamp, R.E.; Steitz, T.A. Sequencing a protein by X-ray crystallography: II. Refinement of yeast hexokinase B Co-ordinates and sequence at 2.1 Å resolution. J. Mol. Biol. 1978, 123, 15–33. [Google Scholar] [CrossRef]
- Stuart, D.I.; Levine, M.; Muirhead, H.; Stammers, D.K. Crystal structure of cat muscle pyruvate kinase at a resolution of 2.6 Å. J. Mol. Biol. 1979, 134, 109–142. [Google Scholar] [CrossRef]
- Watson, H.C.; Walker, N.P.; Shaw, P.J.; Bryant, T.N.; Wendell, P.L.; Fothergill, L.A.; Perkins, R.E.; Conroy, S.C.; Dobson, M.J.; Tuite, M.F. Sequence and structure of yeast phosphoglycerate kinase. EMBO J. 1982, 1, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Deville-Bonne, D.; El Amri, C.; Meyer, P.; Chen, Y.; Agrofoglio, L.A.; Janin, J. Human and viral nucleoside/nucleotide kinases involved in antiviral drug activation: Structural and catalytic properties. Antivir. Res. 2010, 86, 101–120. [Google Scholar] [CrossRef] [PubMed]
- Van Horn, W.D.; Kim, H.-J.; Ellis, C.D.; Hadziselimovic, A.; Sulistijo, E.S.; Karra, M.D.; Tian, C.; Sönnichsen, F.D.; Sanders, C.R. Solution NMR Structure of Membrane-Integral Diacylglycerol Kinase. Science 2009, 324, 1726–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Lyons, J.A.; Pye, V.E.; Vogeley, L.; Araga, D.; Kenyon, C.P.; Shah, S.T.A.; Doherty, C.; Aherne, M.; Caffrey, M. Crystal structure of the integral membrane diacylglycerol kinase. Nature 2013, 497, 521–524. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Shen, Y.; Chen, Y.; Zhang, Z.; Ma, S.; Wan, Q.; Tong, Q.; Glaubitz, C.; Liu, M.; Yang, J. Structure of membrane diacylglycerol kinase in lipid bilayers. Commun. Biol. 2021, 4, 282. [Google Scholar] [CrossRef]
- Knighton, D.R.; Zheng, J.; Ten Eyck, L.F.; Ashford, V.A.; Xuong, N.H.; Taylor, S.S. and Sowadski, J.M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 1991, 253, 407–414. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Arter, C.; Trask, L.; Ward, S.; Yeoh, S. and Bayliss, R. Structural features of the protein kinase domain and targeted binding by small molecule inhibitors. J. Biol. Chem. 2022, 298, 102247. [Google Scholar] [CrossRef]
- Modi, Y.; Dunbrack Jr, R.L. Kincore: A web resource for structural classification of protein kinases and their inhibitors. Nucleic Acids Res. 2022, 50, D654–D664. [Google Scholar] [CrossRef] [PubMed]
- Ohira, T.; Minowa, K.; Sugiyama, K.; Yamashita, S.; Sakaguchi, Y.; Miyauchi, K.; Noguchi, R.; Kaneko, A.; Orita, I.; Fukui, T.; et al. Reversible RNA phosphorylation stabilizes tRNA for cellular thermotolerance. Nature 2022, 605, 372–379. [Google Scholar] [CrossRef] [PubMed]
- SCOP 2 Database. Available online: http://scop.mrc-lmb.cam.ac.uk/ (accessed on 15 August 2022).
- Allen, K.N.; Dunaway-Mariano, D. Catalytic scaffolds for phosphoryl group transfer. Curr. Opin. Struct. Biol. 2016, 41, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatzios, S.K.; Iavarone, A.T.; Bertozzi, C.R. Rv2131c from Mycobacterium tuberculosis Is a CysQ 3′-Phosphoadenosine-5′-phosphatase. Biochemistry 2008, 47, 5823–5831. [Google Scholar] [CrossRef] [Green Version]
- Erickson, A.I.; Sarsam, R.D.; Fisher, A.J. Crystal Structures of Mycobacterium tuberculosis CysQ, with Substrate and Products Bound. Biochemistry 2015, 54, 6830–6841. [Google Scholar] [CrossRef]
- Ishikawa, K.; Mihara, Y.; Gondoh, K.; Suzuki, E.; Asano, Y. X-ray structures of a novel acid phosphatase from Escherichia blattae and its complex with the transition-state analog molybdate. EMBO J. 2000, 19, 2412–2423. [Google Scholar] [CrossRef] [Green Version]
- Asano, Y. Overview of screening for new microbial biocatalysts and their uses in organic synthesis – selection and optimization of biocatalysts. J. Biotechnol. 2002, 94, 65–72. [Google Scholar] [CrossRef]
- Ishikawa, K.; Mihara, Y.; Shimba, N.; Ohtsu, N.; Kawasaki, H.; Suzuki, E.-i.; Asano, Y. Enhancement of nucleoside phosphorylation activity in an acid phosphatase. Protein Eng. 2002, 15, 539–543. [Google Scholar] [CrossRef] [Green Version]
- Mihara, Y.; Ishikawa, K.; Suzuki, E.-i.; Asano, Y. Improving the Pyrophosphate-inosine Phosphotransferase Activity of Escherichia blattae Acid Phosphatase by Sequential Site-directed Mutagenesis. Biosci. Biotechnol. Biochem. 2004, 68, 1046–1050. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Yan, S.; Hou, X.; Song, W.; Wang, L.; Wu, T.; Qi, M.; Wu, J.; Rao, Y.; Wang, B.; et al. Local Electric Field Modulated Reactivity of Pseudomonas aeruginosa Acid Phosphatase for Enhancing Phosphorylation of L-Ascorbic Acid. ACS Catal. 2021, 11, 13397–13407. [Google Scholar] [CrossRef]
- Puchart, V. Glycoside phosphorylases: Structure, catalytic properties and biotechnological potential. Biotechnol. Adv. 2015, 33, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Pugmire, M.J.; Ealick, S.E. Structural analyses reveal two distinct families of nucleoside phosphorylases. Biochem. J. 2002, 361, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Drula, E.; Garron, M.-L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The carbohydrate-active enzyme database: Func-tions and literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef]
- Sun, S.; You, C. Disaccharide phosphorylases: Structure, catalytic mechanisms and directed evolution. Synth. Syst. Biotechnol. 2021, 6, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Li, X.; Guo, W.; Wu, B. Crystal structures of a new class of pyrimidine/purine nucleoside phosphorylase revealed a Cupin fold. Proteins 2022, 90, 1233–1241. [Google Scholar] [CrossRef]
- Panosian, T.D.; Nannemann, D.P.; Watkins, G.R.; Phelan, V.V.; McDonald, W.H.; Wadzinski, B.E.; Bachmann, B.O.; Iverson, T.M. Bacillus cereus phosphopentomutase is an alkaline phosphatase family member that exhibits an altered entry point into the catalytic cycle. J. Biol. Chem. 2011, 286, 8043–8054. [Google Scholar] [CrossRef] [Green Version]
- Frasse, P.M.; Miller, J.J.; Polino, A.J.; Soleimani, E.; Zhu, J.S.; Jakeman, D.L.; Jez, J.M.; Goldberg, D.E.; John, A.R.O. 2022. Enzymatic and structural characterization of HAD5, an essential phosphomannomutase of malaria-causing parasites. J. Biol. Chem. 2022, 298, 101550. [Google Scholar] [CrossRef]
- Gauss, D.; Schönenberger, B.; Molla, G.S.; Kinfu, B.M.; Chow, J.; Liese, A.; Streit, W.R.; Wohlgemuth, R. Biocatalytic phosphorylation of metabolites. In Applied Biocatalysis–From Fundamental Science to Industrial Applications; Hilterhaus, L., Liese, A., Kettling, U., Antranikian, G., Eds.; Wiley-VCH: Weinheim, Germany, 2016; pp. 147–177. [Google Scholar]
- Wohlgemuth, R.; Liese, A.; Streit, W. Biocatalytic phosphorylations of metabolites: Past, present, and future. Trends Biotechnol. 2017, 35, 452–465. [Google Scholar] [CrossRef]
- Tsunoda, T.; Samadi, A.; Burade, S.; Mahmud, T. Complete biosynthetic pathway to the antidiabetic drug acarbose. Nat. Commun. 2022, 13, 3455. [Google Scholar] [CrossRef]
- Minagawa, K.; Zhang, Y.; Ito, T.; Bai, L.; Deng, Z.; Mahmud, T. ValC, a New Type of C7-Cyclitol Kinase Involved in the Biosynthesis of the Antifungal Agent Validamycin, A. ChemBioChem 2007, 8, 632–641. [Google Scholar] [CrossRef]
- Smith, C.-I.; Martin, S.R.; and Smith, K.S. Acetate kinase: Not just a bacterial enzyme. Trends Microbiol. 2006, 14, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Bachochin, M.J.; Van Allen, M.; Barber, R.D. Characterization of a Rhodobacter sphaeroides primary fatty acid kinase. Arch. Microbiol. 2021, 203, 861–864. [Google Scholar] [CrossRef] [PubMed]
- Wallimann, T.; Tokarska-Schlattner, M.; Schlattner, U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids 2011, 40, 1271–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.-S.; Wang, Y.; Li, C.; Chen, Z.; Yan, Y.-B.; Zhou, H.-M. Dissecting the key residues crucial for the species-specific thermostability of muscle-type creatine kinase. Int. J. Biol. Macromol. 2010, 47, 366–370. [Google Scholar] [CrossRef]
- Hunter, T. A journey from phosphotyrosine to phosphohistidine and beyond. Mol. Cell 2022, 82, 2190–2220. [Google Scholar] [CrossRef]
- Lu, Z.; Hunter, T. Metabolic kinases moonlighting as protein kinases. Trends Biochem. Sci. 2018, 43, 301–310. [Google Scholar] [CrossRef]
- Berginski, M.E.; Moret, N.; Liu, C.; Goldfarb, D.; Sorger, P.K.; Gomez, S.M. The Dark Kinase Knowledgebase: An online compendium of knowledge and experimental results of understudied kinases. Nucleic Acids Res. 2021, 49, D529–D535. [Google Scholar] [CrossRef]
- Moret, N.; Liu, C.; Gyori, B.M.; Bachman, J.A.; Steppi, A.; Hug, C.; Taujale, R.; Huang, L.C.; Berginski, M.E.; Gomez, S.M.; et al. A resource for exploring the understudied human kinome for research and therapeutic opportunities. BioRxiv 2021. Available online: https://www.biorxiv.org/content/10.1101/2020.04.02.022277v3 (accessed on 15 August 2022).
- Morton, R.K. The Phosphotransferase Activity of Phosphatases. Biochem. J. 1958, 70, 139–155. [Google Scholar] [CrossRef] [Green Version]
- Holden, H.M.; Raushel, F.M. From the Three-Dimensional Structure of Phosphotriesterase. Biochemistry 2021, 60, 3413–3415. [Google Scholar] [CrossRef]
- Asano, Y.; Mihara, Y.; Yamada, H. A novel selective nucleoside phosphorylation enzyme from Morganella morganii. J. Biosci. Bioeng. 1999, 87, 732–738. [Google Scholar] [CrossRef]
- Mihara, Y.; Utagawa, T.; Yamada, H.; Asano, Y. Acid phosphatase/phosphotransferases from enteric bacteria. J. Biosci. Bioeng. 2001, 92, 50–54. [Google Scholar] [CrossRef]
- Tanaka, N.; Hasan, Z.; Hartog, A.F.; van Herk, T.; Wever, R. Phosphorylation and dephosphorylation of polyhydroxy compounds by class A bacterial acid phosphatases. Org. Biomol. Chem. 2003, 1, 2833–2839. [Google Scholar] [CrossRef] [PubMed]
- Tasnádi, G.; Lukesch, M.; Zechner, M.; Jud, W.; Hall, M.; Ditrich, K.; Baldenius, K.; Hartog, A.F.; Wever, R.; Faber, K. Exploiting acid phosphatases in the synthesis of phosphorylated monoalcohols and diols. Eur. J. Org. Chem. 2016, 1, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Ros, S.; Schulze, A. Balancing glycolytic flux: The role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013, 1, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nannemann, D.P.; Kaufmann, K.W.; Meiler, J.; Bachmann, B.O. Design and directed evolution of a dideoxy purine nucleoside phosphorylase. Protein Eng. Des. Sel. 2010, 23, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Birmingham, W.R.; Starbird, C.A.; Panosian, T.D.; Nannemann, D.P.; Iverson, T.M.; Bachmann, B.O. Bioretrosynthetic construction of a didanosine biosynthetic pathway. Nat. Chem. Biol. 2014, 10, 392–399. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Szeker, K.; Janocha, B.; Böhme, T.; Albrecht, D.; Mikhailopulo, I.A.; Neubauer, P. Recombinant purine nucleoside phosphorylases from thermophiles: Preparation, properties and activity towards purine and pyrimidine nucleosides. FEBS J. 2013, 280, 1475–1490. [Google Scholar] [CrossRef]
- Goedl, C.; Schwarz, A.; Minani, A.; Nidetzky, B. Recombinant sucrose phosphorylase: Characterization, kinetic studies of transglucosylation, and application of immobilized enzyme for production of alpha-D-glucose 1-phosphate. J. Biotechnol. 2007, 129, 77–86. [Google Scholar] [CrossRef]
- Van der Borght, J.; Desmet, T.; Soetaert, W. Enzymatic production of β-d-glucose-1-phosphate from trehalose. Biotechnol. J. 2010, 5, 986–993. [Google Scholar] [CrossRef] [Green Version]
- Kulikova, I.V.; Drenichev, M.S.; Solyev, P.N.; Alexeev, C.S.; Mikhailov, C.N. Enzymatic Synthesis of 2-Deoxyribose 1-phosphate and Ribose 1-phosphate and Subsequent Preparation of Nucleosides. Eur. J. Org. Chem. 2019, 6999–7004. [Google Scholar] [CrossRef]
- Fothergill-Gilmore, L.A.; Watson, H.C. The phosphoglycerate mutases. Adv. Enzymol. Relat. Areas Mol. Biol 1989, 62, 227–313. [Google Scholar] [CrossRef]
- Tozzi, M.G.; Camici, M.; Mascia, L.; Sgarrella, F.; Ipata, P.L. Pentose phosphates in nucleoside interconversion and catabolism. FEBS.J. 2006, 273, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- Kedzierski, L.; Malby, R.L.; Smith, B.J.; Perugini, M.A.; Hodder, A.N.; Ilg, T.; Colman, P.M.; Handman, E. Structure of Leishmania mexicana phosphomannomutase highlights similarities with human isoforms. J. Mol. Biol. 2006, 363, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Stiers, K.M.; Muenks, A.G.; Beamer, L.J. Biology, mechanism, and structure of enzymes in the α-D-phosphohexomutase superfamily. Adv. Protein Chem. Struct. Biol. 2017, 109, 265–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Finci, L.; Zhang, C.; Lahiri, S.; Zhang, G.; Peisach, E.; Allen, K.N.; Dunaway-Mariano, D. Analysis of the structural determinants underlying discrimination between substrate and solvent in β-phosphoglucomutase catalysis. Biochemistry 2009, 48, 1984–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Lu, Z.; Han, Y.; Jia, Y.; Howard, A.; Dunaway-Mariano, D.; Herzberg, O. Conformational flexibility of PEP mutase. Biochemistry 2004, 43, 4447–4453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, J.R. Enzyme-Catalyzed Phosphoryl Transfer Reactions. Annu. Rev. Biochem. 1980, 49, 877–919. [Google Scholar] [CrossRef]
- Cleland, W.W.; Hengge, A.C. Enzymatic Mechanisms of Phosphate and Sulfate Transfer. Chem. Rev. 2006, 106, 3252–3278. [Google Scholar] [CrossRef]
- Lassila, J.K.; Zalatan, J.G.; Herschlag, D. Biological Phosphoryl-Transfer Reactions: Understanding Mechanism and Catalysis. Ann. Rev. Biochem. 2011, 80, 669–702. [Google Scholar] [CrossRef] [Green Version]
- Allen, K.N.; Dunaway-Mariano, D. Phosphoryl group transfer: Evolution of a catalytic scaffold. Trends Biochem. Sci. 2004, 29, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Stockbridge, R.B.; Wolfenden, R. The intrinsic reactivity of ATP and the catalytic proficiencies of kinases acting on glucose, N-acetylgalactosamine, and homoserine: A thermodynamic analysis. J. Biol. Chem. 2009, 284, 22747–22757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerns, S.J.; Agafonov, R.V.; Cho, Y.-J.; Pontiggia, F.; Otten, R.; Pachov, D.V.; Kutter, S.; Phung, L.A.; Murphy, P.N.; Thai, V.; et al. The energy landscape of adenylate kinase during catalysis. Nat. Struct. Mol. Biol. 2015, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Parnell, A.E.; Mordhorst, S.; Kemper, F.; Giurrandino, M.; Prince, J.P.; Schwarzer, N.J.; Hofer, A.; Wohlwend, D.; Jessen, H.J.; Gerhardt, S.; et al. Substrate recognition and mechanism revealed by ligand-bound polyphosphate kinase 2 structures. Proc. Natl. Acad. Sci. USA 2018, 115, 3350–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeiffer, M.; Crean, R.M.; Moreira, C.; Parracino, A.; Oberdorfer, G.; Brecker, L.; Hammerschmidt, F.; Lynn Kamerlin, S.C.; Nidetzky, B. Essential Functional Interplay of the Catalytic Groups in Acid Phosphatase. ACS Catal. 2022, 12, 3357–3370. [Google Scholar] [CrossRef]
- Štefanić, Z.; Narczyk, M.; Mikleušević, G.; Kazazić, S.; Bzowska, A.; Luić, M. Crystallographic snapshots of ligand binding to hexameric purine nucleoside phosphorylase and kinetic studies give insight into the mechanism of catalysis. Sci. Rep. 2018, 8, 15427. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Bhattasali, D.; Pellegrini, E.; Forget, S.M.; Baxter, N.J.; Cliff, M.J.; Bowler, M.W.; Jakeman, D.L.; Blackburn, G.M.; Waltho, J.P. α-Fluorophosphonates reveal how a phosphomutase conserves transition state conformation over hexose recognition in its two-step reaction. Proc. Natl. Acad. Sci. USA 2014, 111, 12384–12389. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Figueroa, J.S.; Palmer, D.R.; Horsman, G.P. Phosphoenolpyruvate mutase-catalyzed C-P bond formation: Mechanistic ambiguities and opportunities. ChemBioChem 2022, 23, e202200285. [Google Scholar] [CrossRef]
- Kubota, K.; Anjum, R.; Yu, Y.; Kunz, R.C.; Andersen, J.N.; Kraus, M.; Keilhack, H.; Nagashima, K.; Krauss, S.; Paweletz, C.; et al. Sensitive multiplexed analysis of kinase activities and activity-based kinase identification. Nat. Biotechnol. 2009, 27, 933–940. [Google Scholar] [CrossRef] [Green Version]
- Sévin, D.C.; Fuhrer, T.; Zamboni, N.; Sauer, U. Nontargeted in vitro metabolomics for high-throughput identification of novel enzymes in Escherichia coli. Nat. Methods 2017, 14, 187–194. [Google Scholar] [CrossRef]
- Schastnaya, E.; Raguz Nakic, Z.; Gruber, C.H.; Doubleday, P.F.; Krishnan, A.; Johns, N.I.; Park, J.; Wang, H.H.; Sauer, U. Extensive regulation of enzyme activity by phosphorylation in Escherichia coli. Nat. Commun. 2021, 12, 5650. [Google Scholar] [CrossRef] [PubMed]
- Cravatt, B.F.; Wright, A.T.; Kozarich, J.W. Activity-based protein profiling: From enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 2008, 77, 383–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zallot, R.; Oberg, N.; Gerlt, J.A. Discovery of new enzymatic functions and metabolic pathways using genomic enzymology web tools. Curr. Opin. Biotechnol. 2021, 69, 77–90. [Google Scholar] [CrossRef]
- Zhang, X.; Carter, M.S.; Vetting, M.W.; San Francisco, B.; Zhao, S.; Al-Obaidi, N.F.; Solbiati, J.O.; Thiaville, J.J.; de Crécy-Lagard, V.; Jacobson, M.P.; et al. Assignment of function to a domain of unknown function: DUF1537 is a new kinase family in catabolic pathways for acid sugars. Proc. Natl. Acad. Sci. USA 2016, 113, E4161–E4169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, F.H. Directed evolution: Bringing new chemistry to life. Angew. Chem. Int. Ed. 2018, 57, 4143–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, R.S.; Rix, G.; Mengiste, A.A.; Álvarez, B.; Seo, D.; Chen, H.; Hurtado, J.E.; Zhang, Q.; García-García, J.D.; Heins, Z.J.; et al. In vivo hypermutation and continuous evolution. Nat. Rev. Methods Prim. 2022, 2, 1–22. [Google Scholar] [CrossRef]
- Christians, F.C.; Scapozza, L.; Crameri, A.; Folkers, G.; Stemmer, W.P. Directed evolution of thymidine kinase for AZT phosphorylation using DNA family shuffling. Nat. Biotechnol. 1999, 17, 259–264. [Google Scholar] [CrossRef]
- Miller, D.C.; Athavale, S.V.; Arnold, F.H. Combining chemistry and protein engineering for new-to-nature biocatalysis. Nat. Synth. 2022, 1, 18–23. [Google Scholar] [CrossRef]
- Fryszkowska, A.; Devine, P.N. Biocatalysis in drug discovery and development. Curr. Opin. Chem. Biol. 2020, 55, 151–160. [Google Scholar] [CrossRef]
- Umezawa, H.; Okanishi, M.; Kondo, S.; Hamana, K.; Utahara, R.; Maeda, K.; Mitsuhashi, S. Phosphorylative inactivation of aminoglycosidic antibiotics by Escherichia coli carrying R factor. Science 1967, 157, 1559–1561. [Google Scholar] [CrossRef]
- Crofts, T.S.; Gasparrini, A.J.; Dantas, G. Next-generation approaches to understand and combat the antibiotic resistome. Nat. Rev. Microbiol. 2017, 15, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Surette, M.D.; Spanogiannopoulos, P.; Wright, G.D. The enzymes of the rifamycin antibiotic resistome. Acc. Chem. Res. 2021, 54, 2065–2075. [Google Scholar] [CrossRef] [PubMed]
- Terekhov, S.S.; Mokrushina, Y.A.; Nazarov, A.S.; Zlobin, A.; Zalevsky, A.; Bourenkov, G.; Golovin, A.; Belogurov Jr, A.; Osterman, I.A.; Kulikova, A.A.; et al. A kinase bioscavenger provides antibiotic resistance by extremely tight substrate binding. Sci. Adv. 2020, 6, eaaz9861. [Google Scholar] [CrossRef] [PubMed]
- Stogios, P.J.; Cox, G.; Spanogiannopoulos, P.; Pillon, M.C.; Waglechner, N.; Skarina, T.; Koteva, K.; Guarné, A.; Savchenko, A.; Wright, G.D. Rifampin phosphotransferase is an unusual antibiotic resistance kinase. Nat. Commun. 2016, 7, 11343. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Goswami, A.; Yang, Z.; Liu, X.; Green, K.D.; Barnard-Britson, S.; Baba, S.; Funabashi, M.; Nonaka, K.; Sunkara, M.; et al. The Biosynthesis of Capuramycin-type Antibiotics. J. Biol. Chem. 2015, 290, 13710–13724. [Google Scholar] [CrossRef] [Green Version]
- Kita, A.; Kishimoto, A.; Shimosaka, T.; Tomita, H.; Yokooji, Y.; Imanaka, T.; Atomi, H.; Miki, K. Crystal structure of pantoate kinase from Thermococcus kodakarensis. Proteins 2020, 88, 718–724. [Google Scholar] [CrossRef]
- Hsu, C.; Tsai, H.Y.; Chang, C.F.; Yang, C.C.; Su, N.W. Discovery of a novel phosphotransferase from Bacillus subtilis that phos-phorylates a broad spectrum of flavonoids. Food Chem. 2022, 400, 134001. [Google Scholar] [CrossRef]
- Jacoby, C.; Goerke, M.; Bezold, D.; Jessen, H.; Boll, M. A fully reversible 25-hydroxy steroid kinase involved in oxygen-inde-pendent cholesterol side-chain oxidation. J. Biol. Chem. 2021, 297, 101105. [Google Scholar] [CrossRef]
- Hoffmeister, D.; Yang, J.; Liu, L.; Thorson, J.S. Creation of the first anomeric D/L-sugar kinase by means of directed evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 13184–13189. [Google Scholar] [CrossRef]
- Nagata, R.; Fujihashi, M.; Sato, T.; Atomi, H.; Miki, K. Crystal Structure and Product Analysis of an Archaeal myo-Inositol Kinase Reveal Substrate Recognition Mode and 3-OH Phosphorylation. Biochemistry 2015, 54, 3494–3503. [Google Scholar] [CrossRef]
- Tashiro, R.; Sato, T.; Atomi, H.; Miki, K.; Fujihashi, M. Altering the phosphorylation position of pyrophosphate-dependent myo-inositol-1-kinase based on its crystal structure. ACS Chem. Biol. 2021, 16, 794–799. [Google Scholar] [CrossRef]
- Huang, H.; Parmeggiani, F.; Pallister, E.; Huang, C.-J.; Liu, F.-F.; Li, Q.; Birmingham, W.R.; Both, P.; Thomas, B.; Liu, L.; et al. Characterisation of a Bacterial Galactokinase with High Activity and Broad Substrate Tolerance for Chemoenzymatic Synthesis of 6-Aminogalactose-1-Phosphate and Analogues. ChemBioChem 2018, 19, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.; Parmeggiani, F.; Malassis, J.; Fontenelle, C.Q.; Vendeville, J.B.; Offen, W.; Both, P.; Huang, K.; Marchesi, A.; Heyam, A.; et al. Profiling substrate promiscuity of wild-type sugar kinases for multi-fluorinated monosaccharides. Cell Chem. Biol. 2020, 27, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Huddleston, J.P.; Raushel, F.M. Functional Characterization of YdjH, a Sugar Kinase of Unknown Specificity in Escherichia coli K12. Biochemistry 2019, 58, 3354–3364. [Google Scholar] [CrossRef] [PubMed]
- Taylor, Z.W.; Raushel, F.M. Cytidine Diphosphoramidate Kinase: An Enzyme Required for the Biosynthesis of the O-Methyl Phosphoramidate Modification in the Capsular Polysaccharides of Campylobacter jejuni. Biochemistry 2018, 57, 2238–2244. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, M.F.; Winiger, C.B.; Shaw, R.W.; Kim, M.-J.; Kim, M.-S.; Daugherty, A.B.; Chen, F.P.; Moses, J.D.; Lutz, S.; Benner, S.A. A Single Deoxynucleoside Kinase Variant from Drosophila melanogaster Synthesizes Monophosphates of Nucleosides That Are Components of an Expanded Genetic System. ACS Synth. Biol. 2017, 6, 388–394. [Google Scholar] [CrossRef]
- Makino, Y.; Sato, T.; Kawamura, H.; Hachisuka, S.I.; Takeno, R.; Imanaka, T.; Atomi, H. An archaeal ADP-dependent serine kinase involved in cysteine biosynthesis and serine metabolism. Nat. Commun. 2016, 7, 13446. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Kawamura, H.; Sato, T.; Fujita, T.; Nagata, R.; Fujihashi, M.; Miki, K.; Atomi, H. Identification and Enzymatic Analysis of an Archaeal ATP-Dependent Serine Kinase from the Hyperthermophilic Archaeon Staphylothermus marinus. J. Bact. 2021, 203, e00025-21. [Google Scholar] [CrossRef]
- Thiaville, J.J.; Flood, J.; Yurgel, S.; Prunetti, L.; Elbadawi-Sidhu, M.; Hutinet, G.; Forouhar, F.; Zhang, X.; Ganesan, V.; Reddy, P.; et al. Members of a Novel Kinase Family (DUF1537) Can Recycle Toxic Intermediates into an Essential Metabolite. ACS Chem. Biol. 2016, 11, 2304–2311. [Google Scholar] [CrossRef]
- Taylor, Z.W.; Brown, H.A.; Narindoshvili, T.; Wenzel, C.Q.; Szymanski, C.M.; Holden, H.M.; Raushel, F.M. Discovery of a glutamine kinase required for the biosynthesis of the O-methyl phosphoramidate modifications found in the capsular poly-saccharides of Campylobacter jejuni. J. Am. Chem. Soc. 2017, 139, 9463–9466. [Google Scholar] [CrossRef]
- Taylor, Z.W.; Chamberlain, A.R.; Raushel, F.M. Substrate specificity and chemical mechanism for the reaction catalyzed by glutamine kinase. Biochemistry 2018, 57, 5447–5455. [Google Scholar] [CrossRef] [PubMed]
- Médici, R.; Garaycoechea, J.I.; Valino, A.I.; Pereira, C.A.; Lewkowicz, E.S.; Iribarren, A.M. A comparative study on phosphotransferase activity of acid phosphatases from Raoultella planticola and Enterobacter aerogenes on nucleosides, sugars and related compounds. Appl. Microbiol. Biotechnol. 2014, 98, 3013–3022. [Google Scholar] [CrossRef] [PubMed]
- Mihara, Y.; Utagawa, T.; Yamada, H.; Asano, Y. Phosphorylation of Nucleosides by the Mutated Acid Phosphatase from Morganella morganii. Appl. Environ. Microbiol. 2000, 66, 2811–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasnádi, G.; Zechner, M.; Hall, M.; Baldenius, K.; Ditrich, K.; Faber, K. Investigation of acid phosphatase variants for the synthesis of phosphate monoesters. Biotechnol. Bioeng. 2017, 114, 2187–2195. [Google Scholar] [CrossRef]
- Nagy, F.; Tasnádi, G.; Balogh-Weiser, D.; Bell, E.; Hall, M.; Faber, K.; Poppe, L. Smart nanoparticles for selective immobilization of acid phosphatases. ChemCatChem 2018, 10, 3490–3499. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, S.S.; Armstrong, Z.; Morgan-Lang, C.; Osowiecka, M.; Robinson, K.; Hallam, S.J.; Withers, S.G. Development and Application of a High-Throughput Functional Metagenomic Screen for Glycoside Phosphorylases. Cell Chem. Biol. 2019, 26, 1001–1012. [Google Scholar] [CrossRef]
- Franceus, J.; Pinel, D.; Desmet, T. Glucosylglycerate phosphorylase, an enzyme with novel specificity involved in compatible solute metabolism. Appl. Env. Microbiol. 2017, 83, e01434-17. [Google Scholar] [CrossRef] [Green Version]
- De Doncker, M.; De Graeve, C.; Franceus, J.; Beerens, K.; Křen, V.; Pelantová, H.; Vercauteren, R.; Desmet, T. Exploration of GH94 sequence space for enzyme discovery reveals a novel Glucosylgalactose phosphorylase specificity. ChemBioChem 2021, 22, 3319–3325. [Google Scholar] [CrossRef]
- Teze, D.; Coines, J.; Raich, L.; Kalichuk, V.; Solleux, C.; Tellier, C.; Andre-Miral, C.; Svensson, B.; Rovira, C. A single point mutation converts GH84 O-GlcNac hydrolases into phosphorylases: Experimental and theoretic evidence. J. Am. Chem. Soc. 2020, 142, 2120–2124. [Google Scholar] [CrossRef]
- Novick, S.J.; Dellas, N.; Mitchell, V.; Duan, D.; Nazor, J.; Alvizo, O.; Sowell-Kantz, A.A.; Moore, J.C.; Huffman, M.; Rodriguez-Granillo, A.; et al. Engineered Purine Nucleoside Phosphorylase Variant Enzymes. US 2022/0010316 A1, 2 November 2021. [Google Scholar]
- Kamel, S.; Thiele, I.; Neubauer, P.; Wagner, A. Thermophilic nucleoside phosphorylases: Their properties, characteristics and applications. BBA Proteins Proteom. 2020, 1868, 140304. [Google Scholar] [CrossRef]
- Vroom, J.; Sivaramakrishnan, S.; Hurtak, J.A. Engineered Phosphopentomutase Variant Enzymes. WO 2022/076454 A1, 15 June 2021. [Google Scholar]
- Dadashipour, M.; Iwamoto, M.; Hossain, M.M.; Akutsu, J.-I.; Zhang, Z.; Kawarabayasi, Y. Identification of a Direct Biosyn-thetic Pathway for UDP-N-Acetylgalactosamine from Glucosamine-6-phosphate in Thermophilic Crenarchaeon Sulfolobus tokodaii. J. Bacteriol. 2018, 200, e00048-18. [Google Scholar] [CrossRef] [PubMed]
- Bergmeyer, H.-U. Methods of Enzymatic Analysis; Academic Press: New York, NY, USA, 1965; pp. 407–410. [Google Scholar]
- Wieland, O. An enzymic method for estimating glycerol. Biochem. Ztschr. 1957, 329, 313–319. [Google Scholar]
- Hørder, M.; Elser, R.C.; Gerhardt, W.; Mathieu, M.; Sampson, E.J. International Federation of Clinical Chemistry (IFCC): Scientific Division, Committee on Enzymes. IFCC methods for the measurement of catalytic concentration enzymes. Part 7. IFCC method for creatine kinase (creatine N-phosphotransferase, EC 2.7.3.2). IFCC Recommendation. J. Autom. Chem. 1990, 12, 22–40. [Google Scholar] [CrossRef]
- Galburt, E.A.; Pelletier, J.; Wilson, G.; Stoddard, B.L. Structure of a tRNA repair enzyme and molecular biology workhorse: T4 polynucleotide kinase. Structure 2002, 10, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Green, M.R.; Sambrook, J. Preparation of Labeled DNA, RNA, and Oligonucleotide Probes. Cold Spring Harb. Protoc. 2022, 1, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Anthony, T.M.; Pflum, M.K.H. Kinase-catalyzed biotinylation of DNA. Bioorg. Med. Chem. 2018, 26, 2331–2336. [Google Scholar] [CrossRef]
- Schumann, G.; Klauke, R.; Canalias, F.; Bossert-Reuther, S.; Franck, P.F.H.; Gella, F.-J.; Jørgensen, P.J.; Kang, D.; Lessinger, J.-M.; Panteghini, M.; et al. IFCC primary reference procedures for the measurement of catalytic activity concentrations of enzymes at 37 °C. Part 9: Reference procedure for the measurement of catalytic concentration of alkaline phosphatase. Clin. Chem. Lab. Med. 2011, 49, 1439–1446. [Google Scholar] [CrossRef] [Green Version]
- Shaban, S.M.; Jo, S.B.; Hafez, E.; Cho, J.H.; Kim, D.-H. A comprehensive overview on alkaline phosphatase targeting and reporting assays. Coord. Chem. Rev. 2022, 465, 214567. [Google Scholar] [CrossRef]
- Kitaoka, M.; Aoyagi, C.; Hayashi, K. Colorimetric Quantification of Cellobiose Employing Cellobiose Phosphorylase. Anal. Biochem. 2001, 292, 163–166. [Google Scholar] [CrossRef]
- Zhang, Z.; Jaffrezic-Renault, N.; Bessueille, F.; Leonard, D.; Xia, S.; Wang, X.; Chen, L.; Zhao, J. Development of a conductometric phosphate biosensor based on tri-layer maltose phosphorylase composite films. Ana. Chim. Acta 2008, 615, 73–79. [Google Scholar] [CrossRef]
- Apple, F.S.; Wu, A.H.B.; Mair, J.; Ravkilde, J.; Panteghini, M.; Tate, J.; Pagani, F.; Christenson, R.H.; Mockel, M.; Danne, O.; et al. Future Biomarker for Detection of Ischemia and Risk Stratification in Acute Coronary Syndrome. Clin. Chem 2005, 51, 810–824. [Google Scholar] [CrossRef] [PubMed]
- Hallows, W.C.; Yu, W.; Denu, J.M. Regulation of Glycolytic Enzyme Phosphoglycerate Mutase-1 by Sirt1 Protein-mediated Deacetylation. J. Biol. Chem. 2012, 287, 3850–3856. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Li, S.; Wang, Y.; Peng, H.; Zhang, X.; Zheng, Y.; Li, C.; Li, L.; Chen, R.; Chen, X.; et al. Phosphoglyceric acid mutase-1 contributes to oncogenic mTOR-mediated tumor growth and confers non-small cell lung cancer patients with poor prognosis. Cell Death Diff. 2018, 25, 1160–1173. [Google Scholar] [CrossRef] [Green Version]
- Wohlgemuth, R.; Littlechild, J. Complexity Reduction and Opportunities in the Design, Integration and Intensification of Biocatalytic Processes for Metabolite Synthesis. Front. Bioeng. Biotechnol. Bioprocess. Eng. 2022, 10, 958606. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Mihara, Y.; Yamada, H. A new enzymatic method of selective phosphorylation of nucleosides. J. Mol. Catal. B Enzym. 1999, 6, 271–277. [Google Scholar] [CrossRef]
- Suzuki, E.; Ishikawa, K.; Mihara, Y.; Shimba, N.; Asano, Y. Structural-based engineering for transferases to improve the industrial production of 5′-nucleotides. Bull. Chem. Soc. Jpn. 2007, 80, 276–286. [Google Scholar] [CrossRef] [Green Version]
- Van Herk, T.; Hartog, A.F.; van der Burg, A.M.; Wever, R. Regioselective phosphorylation of carbohydrates and various alcohols by bacterial acid phosphatases; Probing the substrate specificity of the enzyme from Shigella flexneri. Adv. Synth. Catal. 2005, 347, 1155–1162. [Google Scholar] [CrossRef]
- Babich, L.; Hartog, A.F.; van der Horst, M.A.; Wever, R. Continuous-flow reactor-based enzymatic synthesis of phosphorylated compounds on a large scale. Chem. Eur. J. 2012, 18, 6604–6609. [Google Scholar] [CrossRef]
- Van Herk, T.; Hartog, A.F.; Schoemaker, H.E.; Wever, R.; Faber, K. Simple Enzymatic in situ Generation of Dihydroxyacetone phosphate and its Use in a Cascade Reaction for the Production of Carbohydrates: Increased Efficiency by Phosphate Cycling. J. Org. Chem. 2006, 71, 6244–6247. [Google Scholar] [CrossRef]
- Tasnádi, G.; Hall, M.; Baldenius, K.; Ditrich, K.; Faber, K. Biocatalytic functionalization of hydroxyalkyl acrylates and phenoxy-ethanol via phosphorylation. J. Biotechnol. 2016, 233, 219–227. [Google Scholar] [CrossRef]
- Tasnádi, G.; Jud, W.; Hall, M.; Baldenius, K.; Ditrich, K.; Faber, K. Evaluation of Natural and Synthetic Phosphate Donors for the Improved Enzymatic Synthesis of Phosphate Monoesters. Adv. Synth. Catal. 2018, 360, 2394–2401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasnádi, G.; Staśko, M.; Ditrich, K.; Hall, M.; Faber, K. Preparative-Scale Enzymatic Synthesis of rac-Glycerol-1-phosphate from Crude Glycerol Using Acid Phosphatases and Phosphate. ChemSusChem 2020, 13, 1759–1763. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Zheng, K.; Xu, X.; Gao, C.; Guo, L.; Liu, J.; Chen, X.; Liu, L.; Hu, G.; Wu, J. Enzymatic Production of Ascorbic Acid-2-Phosphate by Engineered Pseudomonas aeruginosa Acid Phosphatase. J. Agric. Food Chem. 2021, 69, 14215–14221. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.-L.; Dai, Y.-S.; Li, C.X.; Pan, J.; Xu, J.-H.; Mu, B. Enzymatic synthesis of high-titer nicotinamide mononucleotide with a new nicotinamide riboside kinase and an efficient ATP regeneration system. Bioresour. Bioprocess. 2022, 9, 26. [Google Scholar] [CrossRef]
- Gauss, D.; Schoenenberger, B.; Wohlgemuth, R. Chemical and enzymatic methodologies for the synthesis of enantiomerically pure glyceraldehyde 3-phosphates. Carbohydr. Res. 2014, 389, 18–24. [Google Scholar] [CrossRef]
- Wong, C.H.; Whitesides, G.M. Synthesis of Sugars by Aldolase-Catalyzed Condensation Reactions. J. Org. Chem. 1983, 48, 3199–3205. [Google Scholar] [CrossRef]
- Gauss, D.; Sánchez-Moreno, I.; Oroz-Guinea, I.; García-Junceda, E.; Wohlgemuth, R. Phosphorylation catalyzed by dihydro- xyacetone kinase. Eur. J. Org. Chem. 2018, 2018, 2892–2895. [Google Scholar] [CrossRef]
- Matsumi, R.; Hellriegel, C.; Schoenenberger, B.; Milesi, T.; Van Der Oost, J.; Wohlgemuth, R. Biocatalytic asymmetric phosphorylation of mevalonate. RSC Adv. 2014, 4, 12989–12994. [Google Scholar] [CrossRef]
- Hardt, N.; Kinfu, B.M.; Chow, J.; Schoenenberger, B.; Streit, W.R.; Obkircher, M.; Wohlgemuth, R. Biocatalytic Asymmetric Phosphorylation Catalyzed by Recombinant Glycerate-2-Kinase. ChemBioChem 2017, 18, 1518–1522. [Google Scholar] [CrossRef]
- Schoenenberger, B.; Wszolek, A.; Meier, R.; Brundiek, H.; Obkircher, M.; Wohlgemuth, R. Recombinant AroL-Catalyzed Phosphorylation for the Efficient Synthesis of Shikimic Acid 3-Phosphate. Biotechnol. J. 2018, 13, 1700529. [Google Scholar] [CrossRef]
- Hardt, N.; Kind, S.; Schoenenberger, B.; Eggert, T.; Obkircher, M.; Wohlgemuth, R. Facile synthesis of D-xylulose-5-phosphate and L-xylulose-5-phosphate by xylulokinase-catalyzed phosphorylation. Biocat. Biotrans. 2020, 38, 35–45. [Google Scholar] [CrossRef]
- Schoenenberger, B.; Kind, S.; Meier, R.; Eggert, T.; Obkircher, M.; Wohlgemuth, R. Efficient biocatalytic synthesis of D-tagatose 1,6-diphosphate by LacC-catalysed phosphorylation of D-tagatose 6-phosphate. Biocat. Biotrans. 2020, 38, 53–63. [Google Scholar] [CrossRef]
- Wen, L.; Huang, K.; Liu, Y.; Wang, P.G. Facile enzymatic synthesis of phosphorylated ketopentoses. ACS Catal. 2016, 6, 1649–1654. [Google Scholar] [CrossRef]
- Wen, L.; Huang, K.; Wei, M.; Meisner, J.; Liu, Y.; Garner, K.; Zang, L.; Wang, X.; Li, X.; Fang, J.; et al. Facile Enzymatic Synthesis of Ketoses. Angew. Chem., Int. Ed. 2015, 54, 12654–12658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fricke, J.; Kargbo, R.; Regestein, L.; Lenz, C.; Peschel, G.; Rosenbaum, M.A.; Sherwood, A.; Hoffmeister, D. Scalable Hybrid Synthetic/Biocatalytic Route to Psilocybin. Chem. Eur. J. 2020, 26, 8281–8285. [Google Scholar] [CrossRef] [PubMed]
- Schoenenberger, B.; Wszolek, A.; Milesi, T.; Brundiek, H.; Obkircher, M.; Wohlgemuth, R. Synthesis of Nω-Phospho-L-arginine by Biocatalytic Phosphorylation of L-Arginine. ChemCatChem 2017, 9, 121–126. [Google Scholar] [CrossRef]
- De Winter, K.; Cerdobbel, A.; Soetaert, W.; Desmet, T. Operational stability of immobilized sucrose phosphorylase: Continuous production of α-glucose-1-phosphate at elevated temperatures. Proc. Biochem. 2011, 46, 2074–2078. [Google Scholar] [CrossRef]
- Bae, J.; Lee, D.; Kim, D.; Cho, S.-J.; Park, J.E.; Koh, S.; Kim, J.; Park, B.-H.; Choi, Y.; Shin, H.-J.; et al. Facile synthesis of glucose-1-phosphate from starch by Thermus thermophilus GK24 α-glucan phosphorylase. Proc. Biochem. 2005, 40, 3707–3713. [Google Scholar] [CrossRef]
- Kamel, S.; Weiss, M.; Klare, H.F.T.; Mikhailopoulo, I.A.; Neubauer, P.; Wagner, A. Chemo-enzymatic synthesis of α-D-pentofuranose-1-phosphates using thermostable pyrimidine nucleoside phosphorylases. Mol. Catal. 2018, 458, 52–59. [Google Scholar] [CrossRef]
- Heptinstall, J.; Ward, P.J.; Hancock, I.C. The enzymic synthesis of [32P]-N-acetylglucosamine. Anal. Biochem. 1978, 91, 158–165. [Google Scholar] [CrossRef]
- Hassan, M.I.; Lundgren, B.R.; Chaumun, M.; Whitfield, D.M.; Clark, B.; Schoenhofen, I.C.; Boddy, C.N. Total Biosynthesis of Legionaminic Acid, a Bacterial Sialic Acid Analogue. Angew. Chem. Intl. Ed. 2016, 55, 12018–12021. [Google Scholar] [CrossRef] [PubMed]
- Horinouchi, N.; Kawano, T.; Sakai, T.; Matsumoto, S.; Sasaki, M.; Mikami, Y.; Ogawa, J.; Shimizu, S. Screening and characterization of a phosphopentomutase useful for enzymatic production of 2’-deoxynucleoside. New Biotechnol. 2009, 26, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-H.; Kang, Y.-B.; Kim, D.-H.; Lee, T.-H.; Park, S.-H.; Lee, K.; Yoo, D.; Liou, K.-K.; Lee, H.-C.; Sohng, J.-K.; et al. One-pot enzymatic synthesis of deoxy-thymidine-diphosphate (TDP)-2-deoxy-α-D-glucose using phosphomannomutase. J. Mol. Catal. B Enzym. 2010, 62, 282–287. [Google Scholar] [CrossRef] [Green Version]
- Mahour, R.; Klapproth, J.; Rexer, T.F.T.; Schildbach, A.; Klamt, S.; Pietzsch, M.; Rapp, E.; Reichl, U. Establishment of a five-enzyme cell-free cascade for the synthesis of uridine diphosphate N-acetylglucosamine. J. Biotechnol. 2018, 283, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Robescu, M.S.; Serra, I.; Terreni, M.; Ubiali, D.; Bavaro, T. A Multi-Enzymatic Cascade Reaction for the Synthesis of Vidarabine-5′-Monophosphate. Catalysts 2020, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Moreno, I.; Hélaine, V.; Poupard, N.; Charmantray, F.; Légeret, B.; Hecquet, L.; García-Junceda, E.; Wohlgemuth, R.; Guérard-Hélaine, C.; Lemaire, M. One-pot cascade reactions using fructose-6-phosphate aldolase: Efficient synthesis of D-arabinose 5-phosphate, D-fructose 6-phosphate and analogues. Adv. Synth. Catal. 2012, 354, 1725–1730. [Google Scholar] [CrossRef] [Green Version]
- Hélaine, V.; Mahdi, R.; Sudhir Babu, G.V.; de Berardinis, V.; Wohlgemuth, R.; Lemaire, M.; Guérard-Hélaine, C. Straightforward synthesis of terminally phosphorylated L-sugars via multienzymatic cascade reactions. Adv. Synth. Catal. 2015, 357, 1703–1708. [Google Scholar] [CrossRef]
- Huffman, M.A.; Fryszkowska, A.; Alvizo, O.; Borra-Garske, M.; Campos, K.R.; Canada, K.A.; Devine, P.N.; Duan, D.; Forstater, J.H.; Grosser, S.T.; et al. Design of an in vitro biocatalytic cascade for the manufacture of islatravir. Science 2019, 366, 1255–1259. [Google Scholar] [CrossRef]
- Schultheisz, H.L.; Szymczyna, B.R.; Scott, L.G.; Williamson, J.R. Pathway Engineered Enzymatic de Novo Purine Nucleotide Synthesis. ACS Chem. Biol. 2008, 3, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Huffman, M.A.; Whittaker, A.M.; Yang, H.; Nawrat, C.C.; Waterhouse, D.J.; Maloney, K.M.; Strotman, N.A. Synthesis of Isotopically Labeled Anti-HIV Nucleoside Islatravir though a One-Pot Biocatalytic Cascade Reaction. Org. Process Res. Dev. 2021, 25, 516–521. [Google Scholar] [CrossRef]
- McIntosh, J.A.; Benkovics, T.; Silverman, S.M.; Huffman, M.A.; Kong, J.; Maligres, P.E.; Itoh, T.; Yang, H.; Verma, D.; Pan, W.; et al. Engineered ribosyl-1-kinase enables concise synthesis of molnupiravir, an antiviral for COVID-19. ACS Centr. Sci. 2021, 7, 1980–1985. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Liu, Z.; Andresen, B.M.; Marzijarani, N.S.; Moore, J.C.; Marshall, N.M.; Borra-Garske, M.; Obligacion, J.V.; Fier, P.S.; Peng, F.; et al. A kinase-cGAS cascade to synthesize a therapeutic STING activator. Nature 2022, 603, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Uchida, T.; Kato, J.; Chibata, I. Polyphosphate kinase: Distribution, Some Properties and Its Application as an ATP Regeneration System. Agric. Biol. Chem. 1988, 52, 1471–1477. [Google Scholar]
- Murata, K. Polyphosphate-dependent nicotinamide adenine dinucleotide (NAD) kinase: A novel missing link in human mito-chondria. Proc. Jpn. Acad., Ser. B 2021, 97, 479–498. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Uchida, T.; Kato, J.; Chibata, I. A Metaphosphate-dependent Nicotinamide Adenine Dinucleotide Kinase from Brevibacterium ammoniagenes. Agric. Biol. Chem. 1980, 44, 1165–1172. [Google Scholar] [CrossRef]
- Andexer, J.N.; Richter, M. Emerging Enzymes for ATP Regeneration in Biocatalytic Processes. ChemBioChem 2015, 16, 380–386. [Google Scholar] [CrossRef]
- Mordhorst, S.; Andexer, J.N. Round, round we go – strategies for enzymatic cofactor regeneration. Nat. Prod. Rep. 2020, 37, 1316–1333. [Google Scholar] [CrossRef]
- Hirschbein, B.L.; Mazenod, F.P.; Whitesides, G.M. Synthesis of Phosphoenolpyruvate and Its Use in Adenosine Triphosphate Cofactor Regeneration. J. Org. Chem. 1982, 47, 3765–3766. [Google Scholar] [CrossRef]
- Crans, D.C.; Whitesides, G.M. A Convenient Synthesis of Disodium Acetyl Phosphate for Use in in Situ ATP Cofactor Regeneration. J. Org. Chem. 1983, 48, 3130–3132. [Google Scholar] [CrossRef]
- Kazlauskas, R.J.; Whitesides, G.M. Synthesis of Methoxycarbonyl Phosphate, a New Reagent Having High Phosphoryl Donor Potential for Use in ATP Cofactor Regeneration. J. Org. Chem. 1985, 50, 1069–1076. [Google Scholar] [CrossRef]
- Tavanti, M.; Hosford, J.; Lloyd, R.C.; Brown, M.J.B. Recent Developments and Challenges for the Industrial Implementation of Polyphosphate Kinases. ChemCatChem 2021, 13, 3565–3580. [Google Scholar] [CrossRef]
- Kim, D.-M.; Swartz, J.R. Prolonging cell-free protein synthesis with a novel ATP regeneration system. Biotech. Bioeng. 1999, 66, 180–188. [Google Scholar] [CrossRef]
- Ruccolo, S.; Brito, G.; Christensen, M.; Itoh, T.; Mattern, K.; Stone, K.; Strotman, N.A.; Sun, A.C. Electrochemical Recycling of Adenosine Triphosphate in Biocatalytic Reaction Cascades. Chemrxiv 2022. [Google Scholar] [CrossRef]
- Molla, G.S.; Kinfu, B.M.; Chow, J.; Streit, W.; Wohlgemuth, R.; Liese, A. Bioreaction Engineering Leading to Efficient Synthesis of L-Glyceraldehyd-3-phosphate. Biotechnol. J. 2017, 12, 1600625. [Google Scholar] [CrossRef] [PubMed]
- Molla, G.S.; Himmelspach, A.; Wohlgemuth, R.; Haupt, E.T.K.; Liese, A. Mechanistic and kinetic elucidation of Mg2+/ATP molar ratio effect on glycerol kinase. Mol. Catal. 2018, 445, 36–42. [Google Scholar] [CrossRef]
- Wohlgemuth, R. Product Recovery. Compr. Biotechnol. 2011, 2, 591–601. [Google Scholar] [CrossRef]
- Knouse, K.W.; Flood, D.T.; Vantourout, J.C.; Schmidt, M.A.; Mcdonald, I.M.; Eastgate, M.D.; Baran, P.S. Nature chose phosphates and chemists should too: How emerging P (V) methods can augment existing strategies. ACS Cent. Sci. 2021, 7, 1473–1485. [Google Scholar] [CrossRef]
- Alcántara, A.R.; Domínguez de María, P.; Littlechild, J.A.; Schürmann, M.; Sheldon, R.A.; Wohlgemuth, R. Biocatalysis as Key to Sustainable Industrial Chemistry. ChemSusChem 2022, 15, e202102709. [Google Scholar] [CrossRef]

















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wohlgemuth, R. The Power of Biocatalysts for Highly Selective and Efficient Phosphorylation Reactions. Catalysts 2022, 12, 1436. https://doi.org/10.3390/catal12111436
Wohlgemuth R. The Power of Biocatalysts for Highly Selective and Efficient Phosphorylation Reactions. Catalysts. 2022; 12(11):1436. https://doi.org/10.3390/catal12111436
Chicago/Turabian StyleWohlgemuth, Roland. 2022. "The Power of Biocatalysts for Highly Selective and Efficient Phosphorylation Reactions" Catalysts 12, no. 11: 1436. https://doi.org/10.3390/catal12111436
APA StyleWohlgemuth, R. (2022). The Power of Biocatalysts for Highly Selective and Efficient Phosphorylation Reactions. Catalysts, 12(11), 1436. https://doi.org/10.3390/catal12111436