
A Brønsted Acidic Deep Eutectic Solvent for N-Boc Deprotection

 , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
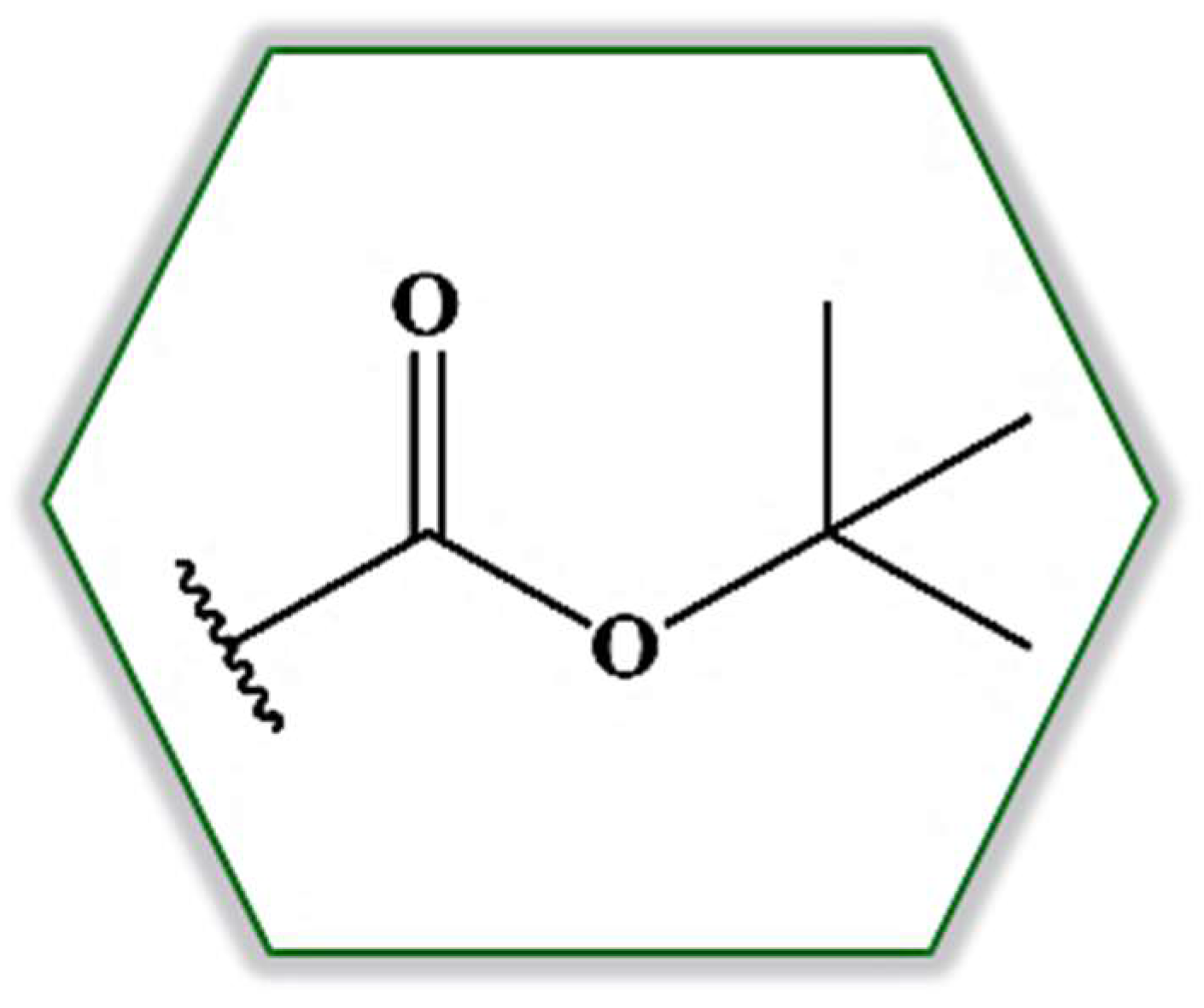
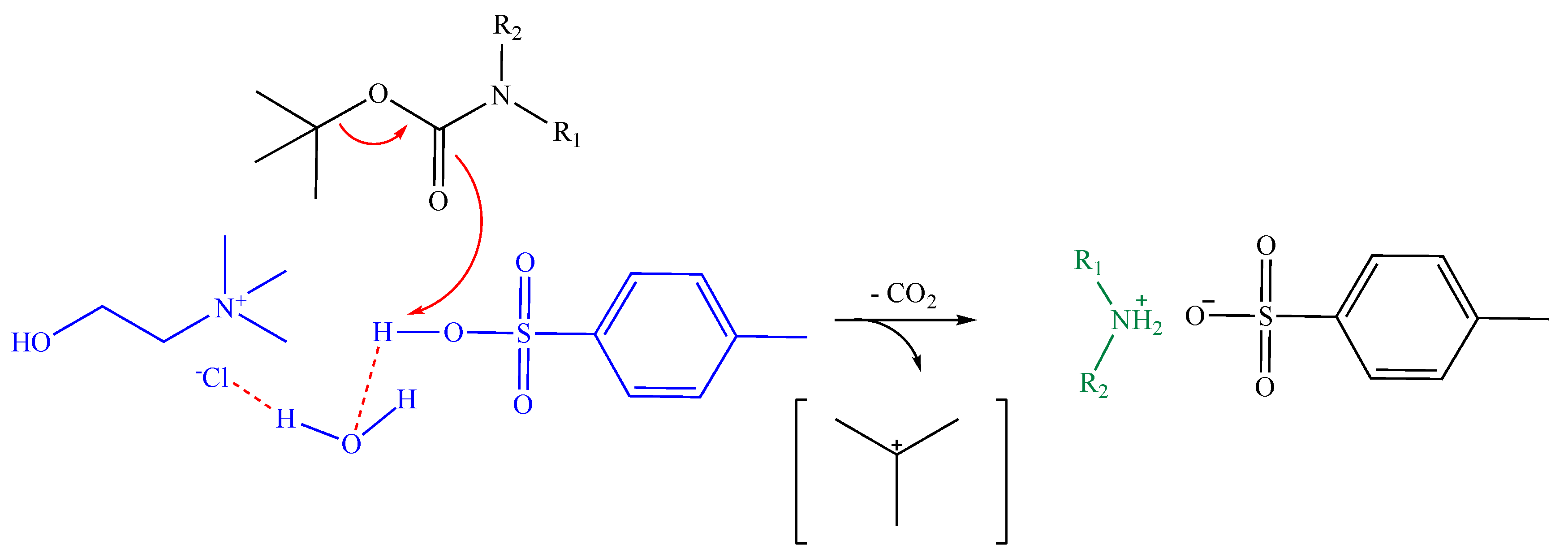
2. Results and Discussion
3. Materials and Methods
3.1. General Informations
3.2. Preparation of DESs
3.3. General Procedure for the N-Boc Deprotection of Amines and Amino Acid Derivatives
3.3.1. Benzylamine (2a)
3.3.2. 2-Phenylethylamine (2b)
3.3.3. Aniline (2c)
3.3.4. p-Toluidine (2d)
3.3.5. p-Anisidine (2e)
3.3.6. Pyridin-4-amine (2f)
3.3.7. 3-Nitroaniline (2g)
3.3.8. Piperidine (2h)
3.3.9. Allylamine (2i)
3.3.10. Cyclopentylamine (2j)
3.3.11. Pentylamine (2k)
3.3.12. (R)-1-Phenylethylamine (2l)
3.3.13. (S)-1-Phenylethylamine (2m)
3.3.14. Ethyl Benzylamine (2n)
3.3.15. N-Isopropylbenzylamine (2o)
3.3.16. l-Alanine Methyl Ester (2p)
3.3.17. d-Alanine Methyl Ester (2q)
3.3.18. l-Valine Methyl Ester (2r)
3.3.19. l-Leucine Methyl Ester (2s)
3.3.20. O-Benzyl-l-Tyrosine Methyl Ester (2t)
3.3.21. Nα-Fmoc-Tryptophane (2u)
3.3.22. l-Phenylalanyl-l-Alanine Methyl Ester (2v)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chen, T.-L.; Kim, H.; Pan, S.Y.; Tsenga, P.C.; Lin, Y.P.; Chiang, P.C. Implementation of green chemistry principles in circular economy system towards sustainable development goals: Challenges and perspectives. Sci. Total Environ. 2020, 716, 136998. [Google Scholar] [CrossRef] [PubMed]
- Procopio, D.; Di Gioia, M.L. An Overview of the Latest Advances in the Catalytic Synthesis of Glycerol Carbonate. Catalysts 2022, 12, 50. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green chemistry: Principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Constable, D.J.C.; Dunn, P.J.; Hayler, J.D.; Humphrey, G.R.; Leazer, J.L.; Linderman, R.J.; Lorenz, K.; Manley, J.; Pearlman, B.A.; Wells, A.; et al. Key green chemistry research areas—A perspective from pharmaceutical manufacturers. Green Chem. 2007, 9, 411–420. [Google Scholar] [CrossRef]
- Đud, M.; Margetić, D. Solvent-free mechanochemical deprotection of N-Boc group. IJOC 2017, 7, 140–144. [Google Scholar]
- Di Gioia, M.L.; Barattucci, A.; Bonaccorsi, P.; Leggio, A.; Minuti, L.; Romio, E.; Temperini, A.; Siciliano, C. Deprotection/reprotection of the amino group in α-amino acids and peptides. A one-pot procedure in [Bmim][BF4] ionic liquid. RSC Adv. 2014, 6, 2678–2686. [Google Scholar] [CrossRef]
- Li, B.; Bemish, R.; Buzon, R.A.; Chiu, C.K.-F.; Colgan, S.T.; Kissel, W.; Le, T.; Leeman, K.R.; Newell, L.; Roth, J. Aqueous phosphoric acid as a mild reagent for deprotection of the t-butoxycarbonyl group. Tetrahedron Lett. 2003, 44, 8113–8115. [Google Scholar] [CrossRef]
- Li, B.; Berliner, M.; Buzon, R.; Chiu, C.K.-F.; Colgan, S.T.; Kaneko, T.; Keene, N.; Kissel, W.; Le, T.; Leeman, K.R.; et al. Aqueous phosphoric acid as a mild reagent for deprotection of tert-butyl carbamates, esters, and ethers. Org. Chem. 2006, 71, 9045–9050. [Google Scholar] [CrossRef]
- Han, G.; Tamaki, M.; Hruby, V.J. Fast, efficient and selective deprotection of the tert-butoxycarbonyl (Boc) group using HCl/dioxane (4 m). J. Pept. Res. 2001, 58, 338–341. [Google Scholar] [CrossRef]
- Coffey, D.S.; Hawk, M.K.N.; Ghera, S.J.; Marler, P.G.; Dodson, P.N.; Lytle, M.L. Large Scale Deprotection of a tert-Butoxycarbonyl (Boc) Group Using Aqueous HCl and Acetone. Org. Proc. Res. Dev. 2004, 8, 945–947. [Google Scholar] [CrossRef]
- Strazzolini, P.; Misuri, N.; Polese, P. Efficient cleavage of carboxylic tert-butyl and 1-adamantyl esters, and N-Boc-amines using H2SO4 in CH2Cl2. Tetrahedron Lett. 2005, 46, 2075–2078. [Google Scholar] [CrossRef]
- Giri, R.S.; Roy, S.; Dolai, G.; Manne, S.R.; Mandal, B. FeCl3-Mediated Boc Deprotection: Mild Facile Boc-Chemistry in Solution and on Resin. ChemistrySelect 2020, 5, 2050–2056. [Google Scholar] [CrossRef]
- López-Soria, J.M.; Pérez, S.J.; Hernández, J.N.; Ramírez, M.A.; Martín, V.S.; Padrón, J.I. A practical, catalytic and selective deprotection of a Boc group in N,N′-diprotected amines using iron (III)-catalysis. RSC Adv. 2015, 5, 6647–6651. [Google Scholar] [CrossRef]
- Bose, D.S.; Kumar, K.K.K.; Reddy, A.V.N. A New Protocol for Selective Deprotection of N-tert-Butoxycarbonyl Protective Group (t-Boc) with Sn(OTf)2. Synth. Commun. 2003, 33, 445–450. [Google Scholar] [CrossRef]
- Tom, N.J.; Simon, W.M.; Frost, H.N.; Ewing, M. Deprotection of a primary Boc group under basic conditions. Tetrahedron Lett. 2004, 45, 905–906. [Google Scholar] [CrossRef]
- Chakrabarty, M.; Kundu, T.; Harigaya, Y. Mild deprotection of tert-butyl carbamates of NH-heteroarenes under basic conditions. Synth. Commun. 2006, 36, 2069–2077. [Google Scholar] [CrossRef]
- Cheraiet, Z.; Ouarna, S.; Hessainia, S.; Berredjem, M.; Aouf, N. N-tert-Butoxycarbonylation of Structurally Diverse Amines and Sulfamides under Water-Mediated Catalyst-Free Conditions. ISRN Org. Chem. 2012, 8, 404235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, N.; Yurek-George, A.; Ganesan, A. Rapid deprotection of N-Boc amines by TFA combined with freebase generation using basic ion-exchange resins. Mol. Divers. 2005, 9, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Sarko, C.R.; Westbrook, J.; Zhang, Z.; Yu, M.; Hopkins, T.D.; Lowe, M.D. Heterocyclic Carboxylic Acids as Activators of Soluble Guanylate Cyclase. Patent WO2016014463, 28 January 2016. [Google Scholar]
- Cheraiet, Z.; Ouarna, S.; Zoubir, J.; Malika, B.; Nour-Eddine, A. A simple and efficient green method for the deprotection of N-Boc in various structurally diverse amines under water-mediated catalyst-free conditions. Int. J. Chem. 2012, 4, 73. [Google Scholar]
- Verschueren, R.H.; Gilles, P.; Van Mileghem, S.; De Borggraeve, W.M. Solvent-free N-Boc deprotection by ex situ generation of hydrogen chloride gas. Org. Biomol. Chem. 2021, 19, 5782–5787. [Google Scholar] [CrossRef] [PubMed]
- George, N.; Ofori, S.; Parkin, S.; Awua, S.G. Mild deprotection of the N-tert-butyloxycarbonyl (N-Boc) group using oxalyl chloride. RSC Adv. 2020, 10, 24017. [Google Scholar] [CrossRef]
- Wang, G.; Li, C.; Li, J.; Ji, X. Catalyst-free water-mediated N-Boc deprotection. Tetrahedron Lett. 2009, 50, 1438–1440. [Google Scholar] [CrossRef]
- Wang, J.; Liang, Y.L.; Qu, J. Boiling water-catalyzed neutral and selective N-Boc deprotection. Chem. Commun. 2009, 34, 5144–5146. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; De, J.; Chakraborty, A.; Royb, D.; Maitib, D.K. A protic ionic liquid catalyzed strategy for selective hydrolytic cleavage of tert-butyloxycarbonyl amine (N-Boc). RSC Adv. 2015, 5, 3200–3205. [Google Scholar] [CrossRef]
- Procopio, D.; Siciliano, C.; Trombino, S.; Dumitrescu, D.E.; Suciu, F.; Di Gioia, M.L. Green solvents for the formation of amide linkages. Org. Biomol. Chem. 2022, 20, 1137–1149. [Google Scholar] [CrossRef]
- Trombino, S.; Siciliano, C.; Procopio, D.; Curcio, F.; Laganà, A.S.; Di Gioia, M.L.; Cassano, R. Deep Eutectic Solvents for Improving the Solubilization and Delivery of Dapsone. Pharmaceutics 2022, 14, 333. [Google Scholar] [CrossRef]
- Di Gioia, M.L.; Cassano, R.; Costanzo, P.; Cano, N.H.; Maiuolo, L.; Nardi, M.; Nicoletta, F.P.; Oliverio, M.; Procopio, A. Green synthesis of privileged benzimidazole scaffolds using active deep eutectic solvent. Molecules 2019, 24, 2885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Gioia, M.L.; Nardi, M.; Costanzo, P.; De Nino, A.; Maiuolo, L.; Oliverio, M.; Procopio, A. Biorenewable deep eutectic solvent for selective and scalable conversion of furfural into cyclopentenone derivatives. Molecules 2018, 23, 1891. [Google Scholar] [CrossRef] [Green Version]
- Perna, F.M.; Vitale, P.; Capriati, V. Deep eutectic solvents and their applications as green solvents. Curr. Opin. Green Sustain. Chem. 2020, 21, 27–33. [Google Scholar] [CrossRef]
- Maiuolo, L.; Russo, B.; Algieri, V.; Nardi, M.; Di Gioia, M.L.; Tallarida, M.A.; De Nino, A. Regioselective synthesis of 1,5-disubstituted 1,2,3-triazoles by 1,3-dipolar cycloaddition: Role of Er(OTf)3, ionic liquid and water. Tetrahedron Lett. 2019, 60, 672–674. [Google Scholar] [CrossRef]
- Allegretti, C.; Gatti, F.G.; Marzorati, S.; Rossato, L.A.M.; Serra, S.; Strini, A.; D’Arrigo, P. Reactive Deep Eutectic Solvents (RDESs): A new tool for phospholipase D-catalyzed preparation of phospholipids. Catalysts 2021, 11, 655. [Google Scholar] [CrossRef]
- Ünlü, A.E.; Arikaya, A.; Takaç, S. Use of deep eutectic solvents as catalyst: A mini-review. Green Proc. Synth. 2019, 8, 355–372. [Google Scholar] [CrossRef]
- Cicco, L.; Dilauro, G.; Perna, F.M.; Vitale, P.; Capriati, V. Advances in deep eutectic solvents and water: Applications in metal-and biocatalyzed processes, in the synthesis of APIs, and other biologically active compounds. Org. Biomol. Chem. 2021, 19, 2558–2577. [Google Scholar] [CrossRef] [PubMed]
- Piemontese, L.; Sergio, R.; Rinaldo, F.; Brunetti, L.; Perna, F.M.; Santos, M.A.; Capriati, V. Deep eutectic solvents as effective reaction media for the synthesis of 2-hydroxyphenylbenzimidazole-based scaffolds en route to donepezil-like compounds. Molecules 2020, 25, 574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryan, M.C.; Dunn, P.J.; Entwistle, D.; Gallou, F.; Koenig, S.G.; Hayler, J.D.; Hickey, M.R.; Hughes, S.; Kopach, M.E.; Moine, G.; et al. Key Green Chemistry research areas from a pharmaceutical manufacturers’ perspective revisited. Green Chem. 2018, 20, 5082. [Google Scholar] [CrossRef] [Green Version]
- Palaniappan, S.; Rajender, B. A Novel Polyaniline-Silver Nitrate-p-Toluenesulfonic Acid Salt as Recyclable Catalyst in the Stereoselective Synthesis of β-Amino Ketones: “One-Pot” Synthesis in Water Medium. Adv. Synth. Catal. 2010, 352, 2507–2514. [Google Scholar] [CrossRef]
- Palaniappan, S.; Rajender, B.; Umashankar, M. Controllable stereoselective synthesis of cis or trans pyrano and furano tetrahydroquinolines: Polyaniline-p-toluenesulfonate salt catalyzed one-pot aza-Diels–Alder reactions. J. Mol. Catal. A Chem. 2012, 352, 70–74. [Google Scholar] [CrossRef]
- Rajender, B.; Palaniappan, S. Emeraldine Base form of polyaniline nanofibers as new, economical, green, and efficient catalyst for synthesis of Z-Aldoximes. J. Catal. 2014, 2014, 515428. [Google Scholar]
- Wang, L.; Zhou, M.; Chen, Q.; He, M.-Y. Brønsted Acidic Deep Eutectic Solvent Catalysed the One-Pot Synthesis of 2H-indazolo[2,1-b] Phthalazine-Triones. J. Chem. Res. 2013, 37, 598–600. [Google Scholar] [CrossRef]
- Kamarudin, A.F.; Hizaddin, H.F.; El-blidi, L.; Ali, E.; Hashim, M.A.; Hadj-Kali, M.K. Performance of p-Toluenesulfonic Acid–Based Deep Eutectic Solvent in Denitrogenation: Computational Screening and Experimental Validation. Molecules 2020, 25, 5093. [Google Scholar] [CrossRef]
- Gutieŕrez-Hernańdez, A.; Richaud, A.; Chacoń-García, L.; Corteś-García, C.J.; Meńdez, F.; Contreras-Celedoń, C.A.J. Deep Eutectic Solvent Choline Chloride/p-toluenesulfonic Acid and Water Favor the Enthalpy-Driven Binding of Arylamines to Maleimide in Aza-Michael Addition. J. Org. Chem. 2021, 86, 223–234. [Google Scholar] [CrossRef]
- Rodriguez, N.R.; Machiels, L.; Binnemans, K. p-Toluenesulfonic acid-based deep-eutectic solvents for solubilizing metal oxides. ACS Sustain. Chem. Eng. 2019, 7, 3940–3948. [Google Scholar] [CrossRef]
- Zhang, Q.; De Oliveira Vigier, K.; Royer, S.; Jérôme, F. Deep eutectic solvents: Syntheses, properties and applications. Chem. Soc. Rev. 2012, 41, 7108–7146. [Google Scholar] [CrossRef]
- El Achkar, T.; Greige-Gerges, H.; Fourmentin, S. Basics and properties of deep eutectic solvents: A review. Environ. Chem. Lett. 2021, 19, 3397–3408. [Google Scholar] [CrossRef]
- Brinkman, H.R.; Landi, J.J., Jr.; Paterson, J.B., Jr.; Stone, P.J. The Use of P-Toluenesulfonic Acid for Removal of the NT-Butoxy-Carbonyl Protecting Group in Solid Phase Peptide Synthesis. Synth. Commun. 1991, 21, 459–465. [Google Scholar] [CrossRef]
- Kocienski, P.J. Protecting Groups; Georg Thieme: New York, NY, USA, 2004. [Google Scholar]
- Di Gioia, M.L.; Gagliardi, A.; Leggio, A.; Leotta, V.; Romio, E.; Liguori, A. N-Urethane protection of amines and amino acids in an ionic liquid. RSC Adv. 2015, 5, 63407–63420. [Google Scholar] [CrossRef]
- Azizi, N.; Shirdel, F. Sustainable and chemoselective N-Boc protection of amines in biodegradable deep eutectic solvent. Monatsh. Chem. 2017, 148, 1069–1074. [Google Scholar] [CrossRef]
- Jahani, F.; Tajbakhsh, M.; Khaksar, S.; Azizi, M.R. An efficient and highly chemoselective N-Boc protection of amines, amino acids, and peptides under heterogeneous conditions. Monatsh. Chem. 2011, 142, 1035–1043. [Google Scholar] [CrossRef]
- Di Gioia, M.L.; Costanzo, P.; De Nino, A.; Maiuolo, L.; Nardi, M.; Olivito, F.; Procopio, A. Simple and efficient Fmoc removal in ionic liquid. RSC Adv. 2017, 758, 36482–36491. [Google Scholar] [CrossRef] [Green Version]
- Nardi, M.; Gioia, M.L.D.; Costanzo, P.; Nino, A.D.; Maiuolo, L.; Oliverio, M.; Olivito, F.; Procopio, A. Selective acetylation of small biomolecules and their derivatives catalyzed by Er(OTf)3. Catalysts 2017, 7, 269. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Li, C.; Bao, M. Deep eutectic solvents (DESs) as powerful and recyclable catalysts and solvents for the synthesis of 3,4-dihydropyrimidin-2(1H)-ones/thiones. Green Proc. Synth. 2019, 8, 568–576. [Google Scholar] [CrossRef]
- Adeeb Hayyan, A.; Hashim, M.A.; Hayyan, M.; Mjalli, F.S.; AlNashef, I.M. A new processing route for cleaner production of biodiesel fuel using a choline chloride based deep eutectic solvent. J. Clean. Prod. 2014, 65, 246–251. [Google Scholar] [CrossRef]
- Sheldon, R.A. Metrics of Green Chemistry and Sustainability: Past, Present, and Future. ACS Sustain. Chem. Eng. 2018, 6, 32–48. [Google Scholar] [CrossRef]
- Monteith, E.R.; Mampuys, P.; Summerton, L.; Clark, J.H.; Maes, B.U.W.; McElroy, C.R. Why we might be misusing process mass intensity (PMI) and a methodology to apply it effectively as a discovery level metric. Green Chem. 2020, 22, 123–135. [Google Scholar] [CrossRef]
- Gawande, M.B.; Branco, P.S. An efficient and expeditious Fmoc protection of amines and amino acids in aqueous media. Green Chem. 2011, 13, 3355–3359. [Google Scholar] [CrossRef]
- Meneses, L.; Santos, F.; Gameiro, A.R.; Paiva, A.; Duarte, A.R.C. Preparation of binary and ternary deep eutectic systems. J. Vis. Exp. 2019, 152, 60326. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Hu, X.; Wang, J.; Cheng, H.; Chen, L.; Qi, Z. Overview of acidic deep eutectic solvents on synthesis, properties and applications. Green Energy Environ. 2020, 5, 8–21. [Google Scholar] [CrossRef]
- Abbott, A.P.; Boothby, D.; Capper, G.; Davies, D.L.; Rasheed, R.K.J. Deep eutectic solvents formed between choline chloride and carboxylic acids: Versatile alternatives to ionic liquids. Am. Chem. Soc. 2004, 9, 29. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
 | ||||
| Entry | DES (1:1 Molar Ratio) | Temp. (°C) | Time b (min.) | Yield (%) |
| 1 | ChCl: pTSA | 25 | 10 | 98 |
| 2 | ChCl: oxalic acid | 25 | 30 | 58 |
| 3 | ChCl: citric acid | 50 a | 30 | 15 |
| 4 | ChCl: malonic acid | 25 | 60 | 26 |
| 5 | ChCl: succinic acid | 25 | 60 | 20 |
| 6 | ChCl: FeCl3 | 25 | 15 | 62 |
 | ||||
| Entry | N-Boc-amine | Product | Time (min) | Yield (%) b |
| 1 |  1b |  2b | 10 | >98 |
| 2 |  1c |  2c | 10 | >98 |
| 3 |  1d |  2d | 15 | >98 |
| 4 |  1e |  2e | 15 | >98 |
| 5 |  1f |  2f | 20 | 90 |
| 6 |  1g |  2g | 20 | 86 |
| 7 |  1h |  2h | 15 | >98 |
| 8 |  1i |  2i | 10 | >98 |
| 9 |  1j |  2j | 10 | >98 |
| 10 |  1k |  2k | 10 | >98 |
| 11 |  1l |  2l | 20 | >98 |
| 12 |  1m |  2m | 20 | >98 |
| 13 |  1n |  2n | 25 | 70 |
| 14 |  1o |  2o | 30 | 56 |
 | ||||
| Entry | N-Boc-amino Acid | Product c | Time (min.) | Yield (%) b |
| 1 |  1p |  2p | 10 | >98 d |
| 2 |  1q |  2q | 10 | >98 d |
| 3 |  1r |  2r | 20 | 63 |
| 4 |  1s |  2s | 15 | 68 |
| 5 |  1t |  2t | 35 | >98 |
| 6 |  1u |  2u | 40 | 90 |
| 7 |  1v |  2v | 15 | >98 |
| Entry | Solvents and Reagents | Yield (%) | PMI | E-Factor | Ref. |
|---|---|---|---|---|---|
| 1 | THF, aqueous H3PO4 | 94 | 91 | 90 | [7] |
| 2 | DCM, aqueous H3PO4 | 91 | 96 | 95 | [8] |
| 3 | DCM, H2SO4 | 93 | 383 | 382 | [11] |
| 4 | DCM, TFA | 98 | 92 | 91 | [18,19] |
| 5 | ChCl:pTSA | 98 | 68 | 67 | Our work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Procopio, D.; Siciliano, C.; De Rose, R.; Trombino, S.; Cassano, R.; Di Gioia, M.L. A Brønsted Acidic Deep Eutectic Solvent for N-Boc Deprotection. Catalysts 2022, 12, 1480. https://doi.org/10.3390/catal12111480
Procopio D, Siciliano C, De Rose R, Trombino S, Cassano R, Di Gioia ML. A Brønsted Acidic Deep Eutectic Solvent for N-Boc Deprotection. Catalysts. 2022; 12(11):1480. https://doi.org/10.3390/catal12111480
Chicago/Turabian StyleProcopio, Debora, Carlo Siciliano, Roberta De Rose, Sonia Trombino, Roberta Cassano, and Maria Luisa Di Gioia. 2022. "A Brønsted Acidic Deep Eutectic Solvent for N-Boc Deprotection" Catalysts 12, no. 11: 1480. https://doi.org/10.3390/catal12111480
APA StyleProcopio, D., Siciliano, C., De Rose, R., Trombino, S., Cassano, R., & Di Gioia, M. L. (2022). A Brønsted Acidic Deep Eutectic Solvent for N-Boc Deprotection. Catalysts, 12(11), 1480. https://doi.org/10.3390/catal12111480