
CO2 Electroreduction over Metallic Oxide, Carbon-Based, and Molecular Catalysts: A Mini-Review of the Current Advances





Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Electrocatalytic CO2 Reduction
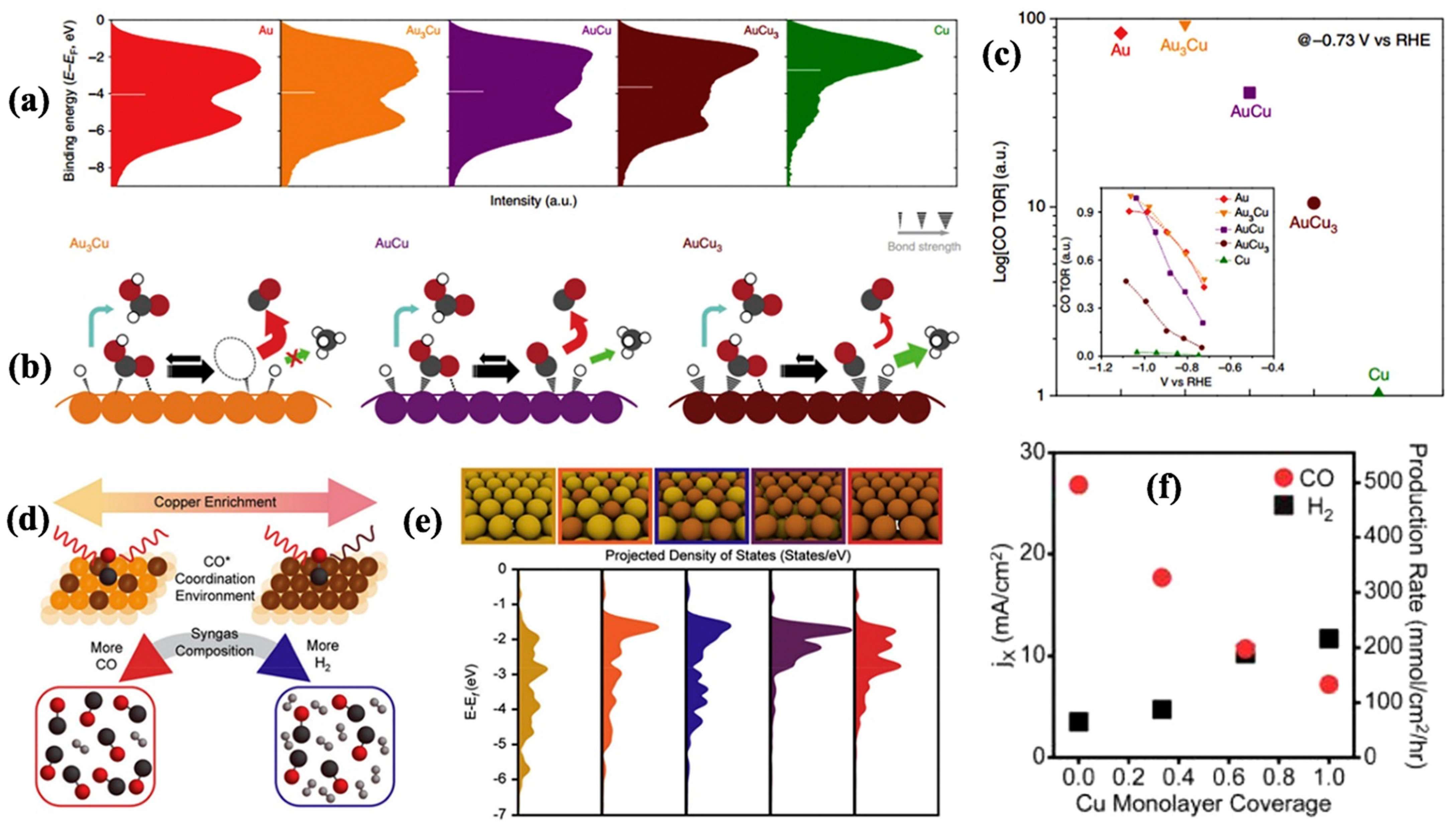
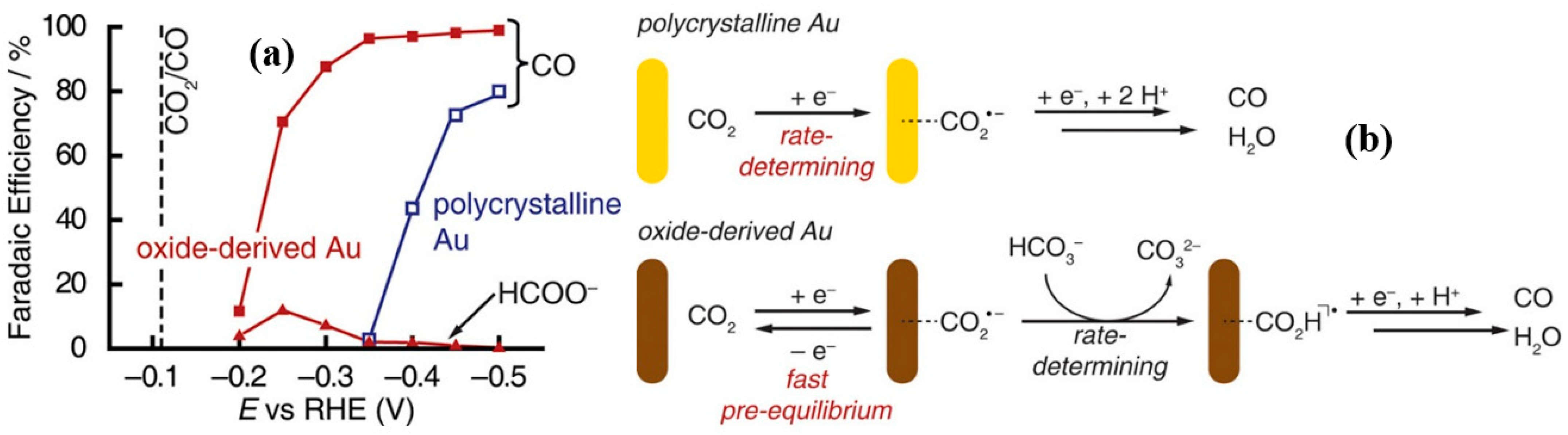
2.1. Oxide, Metallic, and Bimetallic Catalysts
2.2. Single-Atom Catalysts
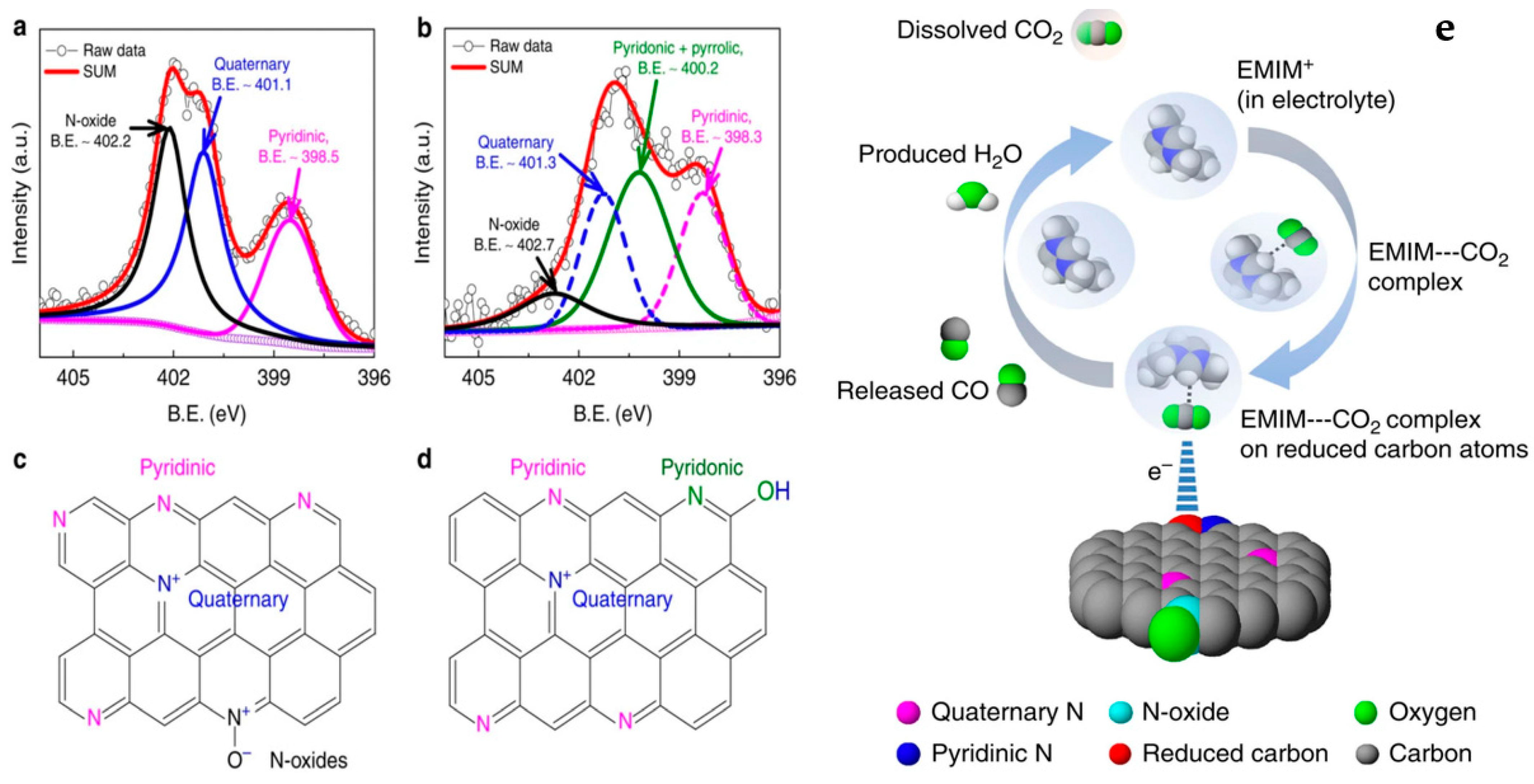
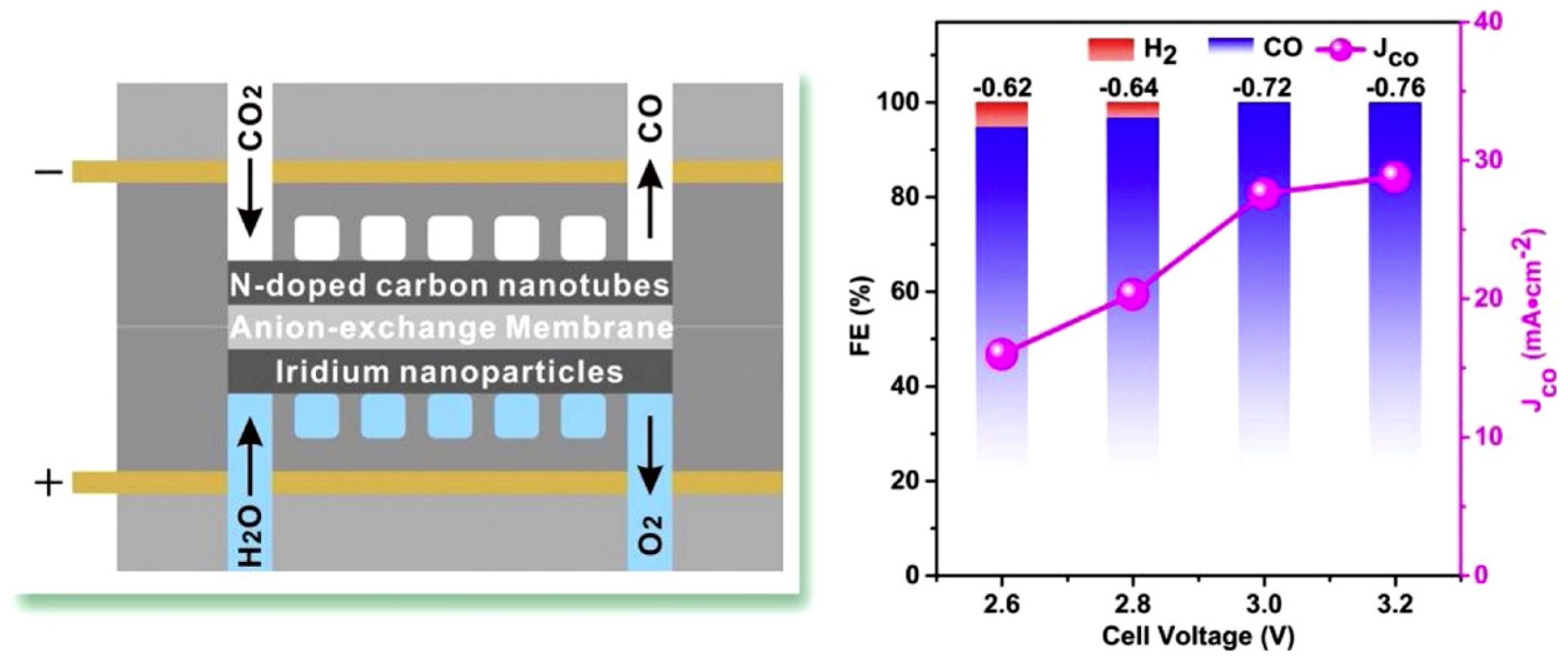
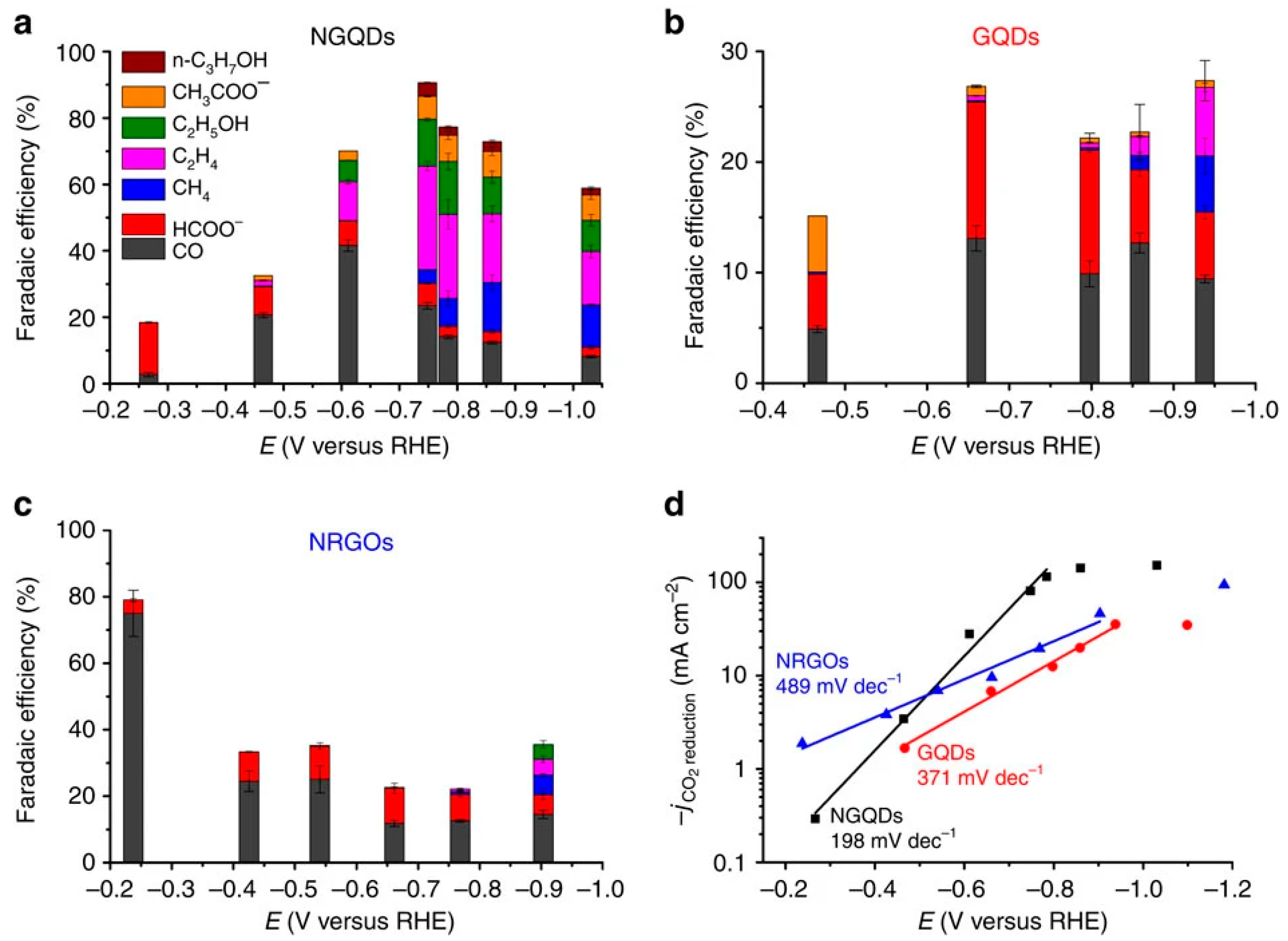
2.3. Carbon-Based Catalysts
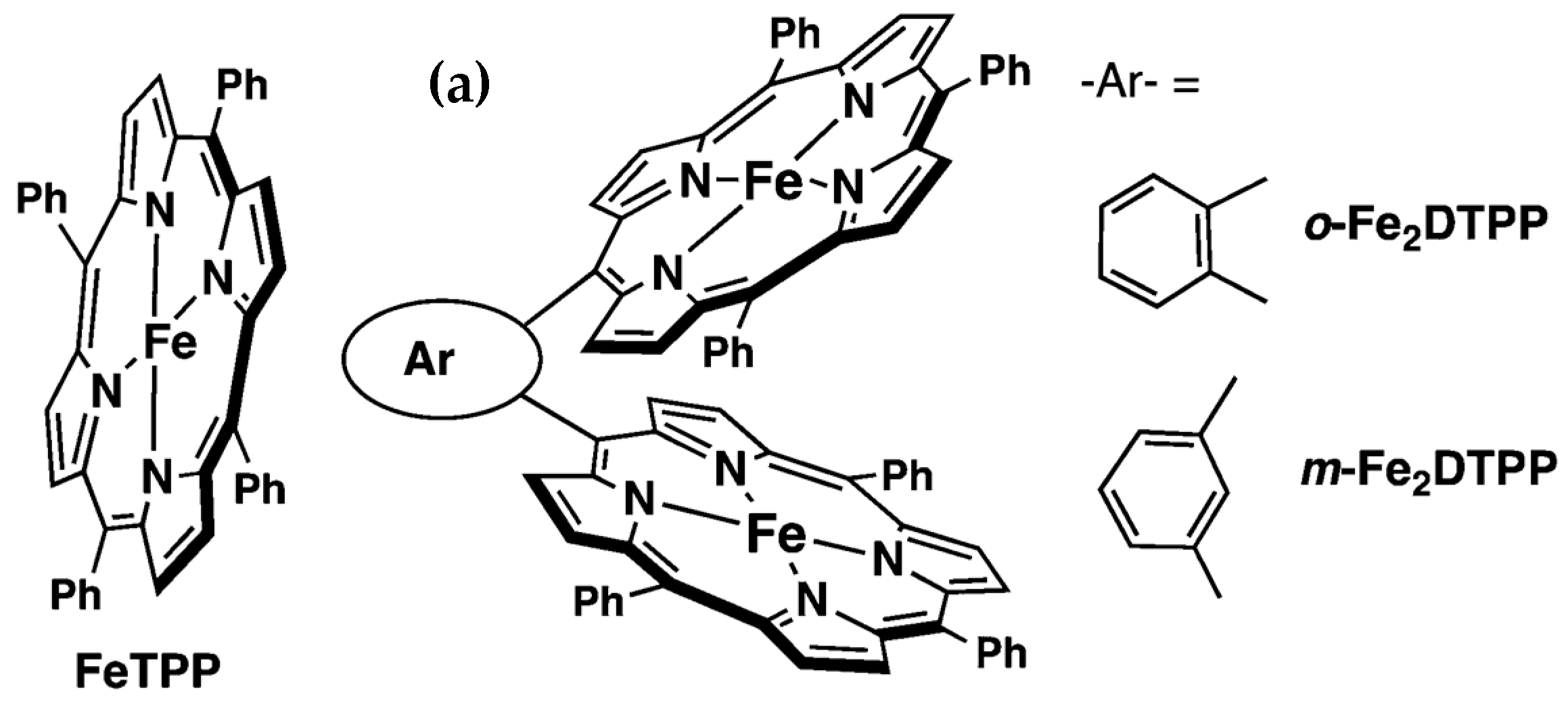
2.4. Porphyrins, Covalent, and Metal-Organic Framework Catalysts
2.5. Phthalocyanines-Based Catalysts
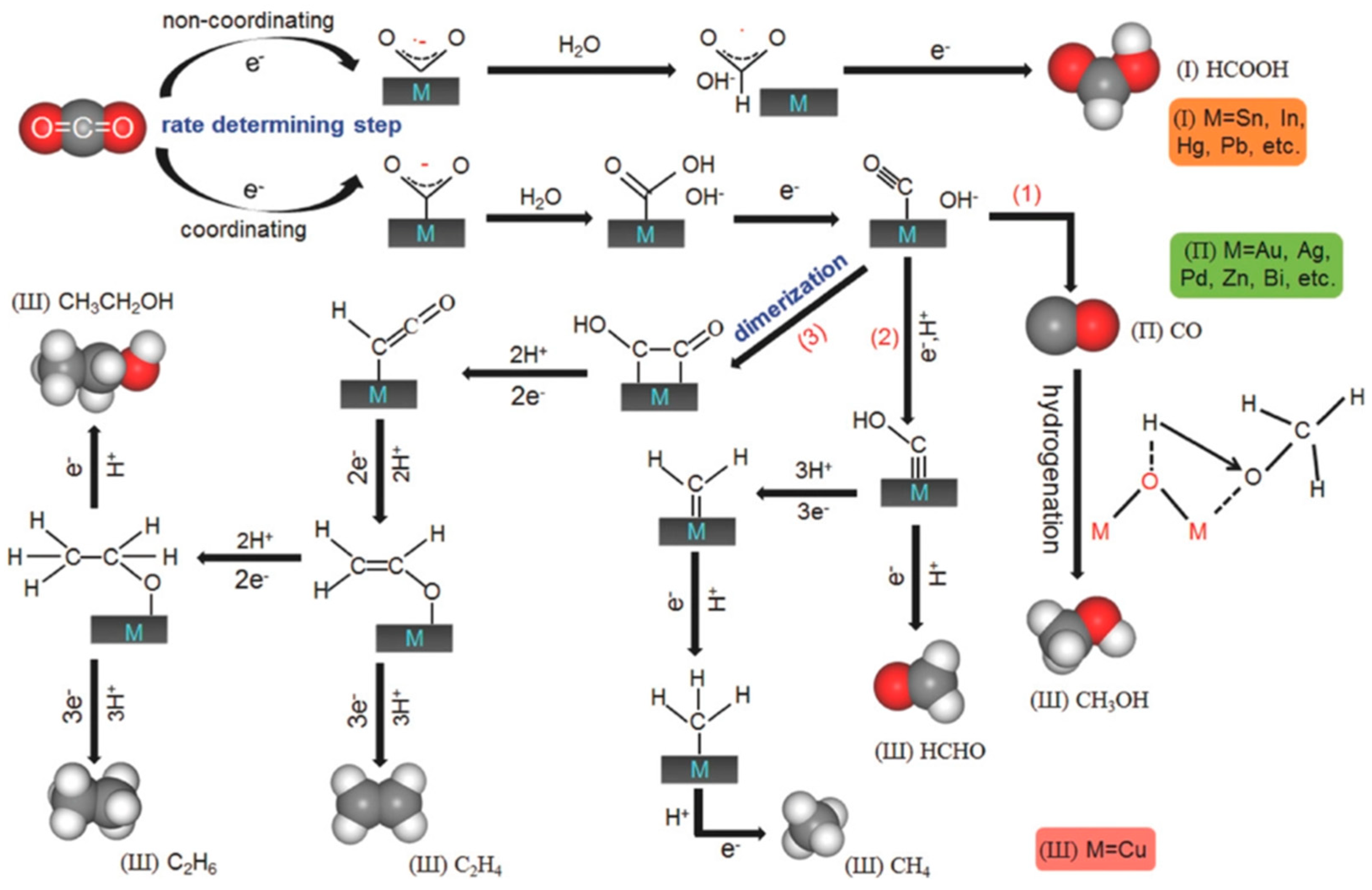
2.6. CO2 Reduction Mechanisms
3. Conclusions and Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef] [PubMed]
- Dincer, I. Renewable energy and sustainable development: A crucial review. Renew. Sustain. Energy Rev. 2000, 4, 157–175. [Google Scholar] [CrossRef]
- Burke, M.J.; Stephens, J.C. Political power and renewable energy futures: A critical review. Energy Res. Soc. Sci. 2018, 35, 78–93. [Google Scholar] [CrossRef]
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, X.; Li, L.; Qi, H.; Yang, C.; Liu, W.; Pan, X.; Liu, X.; Yang, X.; Huang, Y.; et al. Ru/TiO2 catalysts with size-dependent metal/support interaction for tunable reactivity in Fischer-Tropsch synthesis. ACS Catal. 2020, 10, 12967–12975. [Google Scholar] [CrossRef]
- Kang, J.; He, S.; Zhou, W.; Shen, Z.; Li, Y.; Chen, M.; Zhang, Q.; Wang, Y. Single-pass transformation of syngas into ethanol with high selectivity by triple tandem catalysis. Nat. Commun. 2020, 11, 827. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-H.; Himeda, Y.; Muckerman, J.T.; Manbeck, G.F.; Fujita, E. CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction. Chem. Rev. 2015, 115, 12936–12973. [Google Scholar] [CrossRef]
- Li, W.; Wang, H.; Jiang, X.; Zhu, J.; Liu, Z.; Guo, X.; Song, C. A short review of recent advances in CO2 hydrogenation to hydrocarbons over heterogeneous catalysts. RSC Adv. 2018, 8, 7651–7669. [Google Scholar] [CrossRef] [Green Version]
- Albo, J.; Alvarez-Guerra, M.; Castaño, P.; Irabien, A. Towards the electrochemical conversion of carbon dioxide into methanol. Green Chem. 2015, 17, 2304–2324. [Google Scholar] [CrossRef]
- Olah, G.A. Beyond Oil and Gas: The Methanol Economy. Angew. Chemie Int. Ed. 2005, 44, 2636–2639. [Google Scholar] [CrossRef]
- Albo, J.; Sáez, A.; Solla-Gullón, J.; Montiel, V.; Irabien, A. Production of methanol from CO2 electroreduction at Cu2O and Cu2O/ZnO-based electrodes in aqueous solution. Appl. Catal. B Environ. 2015, 176–177, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Guerra, M.; Quintanilla, S.; Irabien, A. Conversion of carbon dioxide into formate using a continuous electrochemical reduction process in a lead cathode. Chem. Eng. J. 2012, 207, 278–284. [Google Scholar] [CrossRef]
- Del Castillo, A.; Alvarez-Guerra, M.; Irabien, A. Continuous electroreduction of CO2 to formate using Sn gas diffusion electrodes. AIChE J. 2014, 60, 3557–3564. [Google Scholar] [CrossRef]
- Wang, Q.; Dong, H.; Yu, H. Fabrication of a novel tin gas diffusion electrode for electrochemical reduction of carbon dioxide to formic acid. RSC Adv. 2014, 4, 59970–59976. [Google Scholar] [CrossRef]
- Wang, Q.; Dong, H.; Yu, H. Development of rolling tin gas diffusion electrode for carbon dioxide electrochemical reduction to produce formate in aqueous electrolyte. J. Power Sources 2014, 271, 278–284. [Google Scholar] [CrossRef]
- Jhong, H.-R. “Molly”; Brushett, F.R.; Kenis, P.J.A. The Effects of Catalyst Layer Deposition Methodology on Electrode Performance. Adv. Energy Mater. 2013, 3, 589–599. [Google Scholar] [CrossRef]
- Vass, A.; Endrődi, B.; Samu, G.F.; Balog, A.; Kormányos, A.; Cherevko, S.; Janáky, C. Local Chemical Environment Governs Anode Processes in CO2 Electrolyzers. ACS Energy Lett. 2021, 6, 3801–3808. [Google Scholar] [CrossRef]
- Vass, A.; Kormányos, A.; Kószó, Z.; Endrődi, B.; Janáky, C. Anode Catalysts in CO2 Electrolysis: Challenges and Untapped Opportunities. ACS Catal. 2022, 12, 1037–1051. [Google Scholar] [CrossRef]
- Zhu, D.D.; Liu, J.L.; Qiao, S.Z. Recent Advances in Inorganic Heterogeneous Electrocatalysts for Reduction of Carbon Dioxide. Adv. Mater. 2016, 28, 3423–3452. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, Z.-J.; Gong, J. Nanostructured Materials for Heterogeneous Electrocatalytic CO2 Reduction and their Related Reaction Mechanisms. Angew. Chem. Int. Ed. 2017, 56, 11326–11353. [Google Scholar] [CrossRef]
- Vasileff, A.; Zheng, Y.; Qiao, S.Z. Carbon Solving Carbon’s Problems: Recent Progress of Nanostructured Carbon-Based Catalysts for the Electrochemical Reduction of CO2. Adv. Energy Mater. 2017, 7, 1700759. [Google Scholar] [CrossRef]
- Lu, Q.; Rosen, J.; Zhou, Y.; Hutchings, G.S.; Kimmel, Y.C.; Chen, J.G.; Jiao, F. A selective and efficient electrocatalyst for carbon dioxide reduction. Nat. Commun. 2014, 5, 3242. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhang, Y.-J.; Zhang, H.; Lv, H.; Li, Q.; Michalsky, R.; Peterson, A.A.; Sun, S. Active and Selective Conversion of CO2 to CO on Ultrathin Au Nanowires. J. Am. Chem. Soc. 2014, 136, 16132–16135. [Google Scholar] [CrossRef]
- Kim, C.; Jeon, H.S.; Eom, T.; Jee, M.S.; Kim, H.; Friend, C.M.; Min, B.K.; Hwang, Y.J. Achieving Selective and Efficient Electrocatalytic Activity for CO2 Reduction Using Immobilized Silver Nanoparticles. J. Am. Chem. Soc. 2015, 137, 13844–13850. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Cheng, T.; Wu, L.; Hu, Y.; Zhou, J.; Maclennan, A.; Jiang, Z.; Gao, Y.; Goddard, W.A.; Wang, Z. Ultrahigh Mass Activity for Carbon Dioxide Reduction Enabled by Gold–Iron Core–Shell Nanoparticles. J. Am. Chem. Soc. 2017, 139, 15608–15611. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kanan, M.W. Tin Oxide Dependence of the CO 2 Reduction Efficiency on Tin Electrodes and Enhanced Activity for Tin/Tin Oxide Thin-Film Catalysts. J. Am. Chem. Soc. 2012, 134, 1986–1989. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Kang, P.; Meyer, T.J. Nanostructured Tin Catalysts for Selective Electrochemical Reduction of Carbon Dioxide to Formate. J. Am. Chem. Soc. 2014, 136, 1734–1737. [Google Scholar] [CrossRef]
- Fan, L.; Xia, Z.; Xu, M.; Lu, Y.; Li, Z. 1D SnO2 with Wire-in-Tube Architectures for Highly Selective Electrochemical Reduction of CO2 to C 1 Products. Adv. Funct. Mater. 2018, 28, 1706289. [Google Scholar] [CrossRef]
- Duan, Y.-X.; Meng, F.-L.; Liu, K.-H.; Yi, S.-S.; Li, S.-J.; Yan, J.-M.; Jiang, Q. Amorphizing of Cu Nanoparticles toward Highly Efficient and Robust Electrocatalyst for CO2 Reduction to Liquid Fuels with High Faradaic Efficiencies. Adv. Mater. 2018, 30, 1706194. [Google Scholar] [CrossRef]
- Li, C.W.; Kanan, M.W. CO2 Reduction at Low Overpotential on Cu Electrodes Resulting from the Reduction of Thick Cu2O Films. J. Am. Chem. Soc. 2012, 134, 7231–7234. [Google Scholar] [CrossRef]
- Gao, S.; Jiao, X.; Sun, Z.; Zhang, W.; Sun, Y.; Wang, C.; Hu, Q.; Zu, X.; Yang, F.; Yang, S.; et al. Ultrathin Co3O4 Layers Realizing Optimized CO2 Electroreduction to Formate. Angew. Chemie Int. Ed. 2016, 55, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Lin, Y.; Jiao, X.; Sun, Y.; Luo, Q.; Zhang, W.; Li, D.; Yang, J.; Xie, Y. Partially oxidized atomic cobalt layers for carbon dioxide electroreduction to liquid fuel. Nature 2016, 529, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Lin, R.; Chen, Y.; Liu, S.; Zhu, W.; Cao, X.; Chen, W.; Wu, K.; Cheong, W.-C.; Wang, Y.; et al. Design of Single-Atom Co–N5 Catalytic Site: A Robust Electrocatalyst for CO2 Reduction with Nearly 100% CO Selectivity and Remarkable Stability. J. Am. Chem. Soc. 2018, 140, 4218–4221. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Chen, W.; Zhao, C.; Li, S.; Wei, W.; Sun, Y. Metal-Free Nitrogen-Doped Mesoporous Carbon for Electroreduction of CO2 to Ethanol. Angew. Chemie 2017, 129, 10980–10984. [Google Scholar] [CrossRef]
- Wu, J.; Ma, S.; Sun, J.; Gold, J.I.; Tiwary, C.; Kim, B.; Zhu, L.; Chopra, N.; Odeh, I.N.; Vajtai, R.; et al. A metal-free electrocatalyst for carbon dioxide reduction to multi-carbon hydrocarbons and oxygenates. Nat. Commun. 2016, 7, 13869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, B.; Asadi, M.; Pisasale, D.; Sinha-Ray, S.; Rosen, B.A.; Haasch, R.; Abiade, J.; Yarin, A.L.; Salehi-Khojin, A. Renewable and metal-free carbon nanofibre catalysts for carbon dioxide reduction. Nat. Commun. 2013, 4, 2819. [Google Scholar] [CrossRef]
- Vasileff, A.; Xu, C.; Jiao, Y.; Zheng, Y.; Qiao, S.-Z. Surface and Interface Engineering in Copper-Based Bimetallic Materials for Selective CO2 Electroreduction. Chem 2018, 4, 1809–1831. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, J.; Wang, Y.; Al-Enizi, A.M.; Zheng, G. Tuning of CO2 Reduction Selectivity on Metal Electrocatalysts. Small 2017, 13, 1701809. [Google Scholar] [CrossRef]
- Feaster, J.T.; Shi, C.; Cave, E.R.; Hatsukade, T.; Abram, D.N.; Kuhl, K.P.; Hahn, C.; Nørskov, J.K.; Jaramillo, T.F. Understanding Selectivity for the Electrochemical Reduction of Carbon Dioxide to Formic Acid and Carbon Monoxide on Metal Electrodes. ACS Catal. 2017, 7, 4822–4827. [Google Scholar] [CrossRef]
- Hatsukade, T.; Kuhl, K.P.; Cave, E.R.; Abram, D.N.; Jaramillo, T.F. Insights into the electrocatalytic reduction of CO2 on metallic silver surfaces. Phys. Chem. Chem. Phys. 2014, 16, 13814–13819. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, B.; Jiang, Y.; Ma, T.; Pan, H.; Sun, W. Single-atom electrocatalysts for multi-electron reduction of CO2. Small 2021, 17, 2101443. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Delmo, E.P.; Li, T.; Qin, X.; Tian, J.; Zhang, L.; Shao, M. Recent advances in catalyst structure and composition engineering strategies for regulating CO2 electrochemical reduction. Adv. Mater. 2021, 33, 2005484. [Google Scholar] [CrossRef] [PubMed]
- Jhong, H.R.; Nwabara, U.O.; Shubert-Zuleta, S.; Grundish, N.S.; Tandon, B.; Reimnitz, L.C.; Staller, C.M.; Ong, G.K.; Cabezas, C.A.S.; Goodenough, J.B.; et al. Efficient Aqueous Electroreduction of CO2 to Formate at Low Overpotential on Indium Tin Oxide Nanocrystals. Chem. Mater. 2021, 33, 7675–7685. [Google Scholar] [CrossRef]
- Wei, F.; Wang, T.; Jiang, X.; Ai, Y.; Cui, A.; Cui, J.; Fu, J.; Cheng, J.; Lei, L.; Hou, Y.; et al. Controllably engineering mesoporous surface and dimensionality of SnO2 toward high-performance CO2 electroreduction. Adv. Funct. Mater. 2020, 30, 2002092. [Google Scholar] [CrossRef]
- Hailu, A.; Tamijani, A.A.; Mason, S.E.; Shaw, S.K. Efficient conversion of CO2 to formate using inexpensive and easily prepared post-transition metal alloy catalysts. Energ. Fuel. 2020, 34, 3467–3476. [Google Scholar] [CrossRef]
- Li, Z.; Feng, Y.; Li, Y.; Chen, X.; Li, N.; He, W.; Liu, J. Fabrication of Bi/Sn bimetallic electrode for high-performance electrochemical reduction of carbon dioxide to formate. Chem. Eng. J. 2022, 428, 130901. [Google Scholar] [CrossRef]
- Wen, J.; Wan, Z.; Hu, X.; Huang, J.; Kang, X. Restructuring of copper catalysts by potential cycling and enhanced two-carbon production for electroreduction of carbon dioxide. J. CO2 Util. 2022, 56, 101846. [Google Scholar] [CrossRef]
- Lee, H.-E.; Yang, K.D.; Yoon, S.M.; Ahn, H.-Y.; Lee, Y.Y.; Chang, H.; Jeong, D.H.; Lee, Y.-S.; Kim, M.Y.; Nam, K.T. Concave Rhombic Dodecahedral Au Nanocatalyst with Multiple High-Index Facets for CO2 Reduction. ACS Nano 2015, 9, 8384–8393. [Google Scholar] [CrossRef]
- Kim, J.-H.; Woo, H.; Choi, J.; Jung, H.-W.; Kim, Y.-T. CO2 Electroreduction on Au/TiC: Enhanced Activity Due to Metal–Support Interaction. ACS Catal. 2017, 7, 2101–2106. [Google Scholar] [CrossRef]
- Won, D.H.; Shin, H.; Koh, J.; Chung, J.; Lee, H.S.; Kim, H.; Woo, S.I. Highly Efficient, Selective, and Stable CO2 Electroreduction on a Hexagonal Zn Catalyst. Angew. Chemie Int. Ed. 2016, 55, 9297–9300. [Google Scholar] [CrossRef]
- Rosen, J.; Hutchings, G.S.; Lu, Q.; Forest, R.V.; Moore, A.; Jiao, F. Electrodeposited Zn Dendrites with Enhanced CO Selectivity for Electrocatalytic CO2 Reduction. ACS Catal. 2015, 5, 4586–4591. [Google Scholar] [CrossRef]
- Cho, M.; Song, J.T.; Back, S.; Jung, Y.; Oh, J. The Role of Adsorbed CN and Cl on an Au Electrode for Electrochemical CO2 Reduction. ACS Catal. 2018, 8, 1178–1185. [Google Scholar] [CrossRef]
- Jiang, K.; Kharel, P.; Peng, Y.; Gangishetty, M.K.; Lin, H.-Y.G.; Stavitski, E.; Attenkofer, K.; Wang, H. Silver Nanoparticles with Surface-Bonded Oxygen for Highly Selective CO2 Reduction. ACS Sustain. Chem. Eng. 2017, 5, 8529–8534. [Google Scholar] [CrossRef]
- Nursanto, E.B.; Jeon, H.S.; Kim, C.; Jee, M.S.; Koh, J.H.; Hwang, Y.J.; Min, B.K. Gold catalyst reactivity for CO2 electro-reduction: From nano particle to layer. Catal. Today 2016, 260, 107–111. [Google Scholar] [CrossRef]
- Verma, S.; Hamasaki, Y.; Kim, C.; Huang, W.; Lu, S.; Jhong, H.-R.M.; Gewirth, A.A.; Fujigaya, T.; Nakashima, N.; Kenis, P.J.A. Insights into the Low Overpotential Electroreduction of CO2 to CO on a Supported Gold Catalyst in an Alkaline Flow Electrolyzer. ACS Energy Lett. 2018, 3, 193–198. [Google Scholar] [CrossRef]
- Mistry, H.; Reske, R.; Zeng, Z.; Zhao, Z.-J.; Greeley, J.; Strasser, P.; Cuenya, B.R. Exceptional Size-Dependent Activity Enhancement in the Electroreduction of CO2 over Au Nanoparticles. J. Am. Chem. Soc. 2014, 136, 16473–16476. [Google Scholar] [CrossRef]
- Yoon, Y.; Hall, A.S.; Surendranath, Y. Tuning of Silver Catalyst Mesostructure Promotes Selective Carbon Dioxide Conversion into Fuels. Angew. Chemie Int. Ed. 2016, 55, 15282–15286. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.S.; Yoon, Y.; Wuttig, A.; Surendranath, Y. Mesostructure-Induced Selectivity in CO2 Reduction Catalysis. J. Am. Chem. Soc. 2015, 137, 14834–14837. [Google Scholar] [CrossRef] [Green Version]
- Mariano, R.G.; Kang, M.; Wahab, O.J.; McPherson, I.J.; Rabinowitz, J.A.; Unwin, P.R.; Kanan, M.W. Microstructural origin of locally enhanced CO2 electroreduction activity on gold. Nat. Mater. 2021, 20, 1000–1006. [Google Scholar] [CrossRef]
- Christophe, J.; Doneux, T.; Buess-Herman, C. Electroreduction of Carbon Dioxide on Copper-Based Electrodes: Activity of Copper Single Crystals and Copper–Gold Alloys. Electrocatalysis 2012, 3, 139–146. [Google Scholar] [CrossRef]
- Jia, F.; Yu, X.; Zhang, L. Enhanced selectivity for the electrochemical reduction of CO2 to alcohols in aqueous solution with nanostructured Cu–Au alloy as catalyst. J. Power Sources 2014, 252, 85–89. [Google Scholar] [CrossRef]
- Kim, D.; Resasco, J.; Yu, Y.; Asiri, A.M.; Yang, P. Synergistic geometric and electronic effects for electrochemical reduction of carbon dioxide using gold–copper bimetallic nanoparticles. Nat. Commun. 2014, 5, 4948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Xie, C.; Becknell, N.; Yu, Y.; Karamad, M.; Chan, K.; Crumlin, E.J.; Nørskov, J.K.; Yang, P. Electrochemical Activation of CO2 through Atomic Ordering Transformations of AuCu Nanoparticles. J. Am. Chem. Soc. 2017, 139, 8329–8336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, M.B.; Dinh, C.T.; Li, Y.; Kim, D.; De Luna, P.; Sargent, E.H.; Yang, P. Tunable Cu Enrichment Enables Designer Syngas Electrosynthesis from CO2. J. Am. Chem. Soc. 2017, 139, 9359–9363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monzó, J.; Malewski, Y.; Kortlever, R.; Vidal-Iglesias, F.J.; Solla-Gullón, J.; Koper, M.T.M.; Rodriguez, P. Enhanced electrocatalytic activity of Au@Cu core@shell nanoparticles towards CO2 reduction. J. Mater. Chem. A 2015, 3, 23690–23698. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Hansen, H.A.; Lausche, A.C.; Nørskov, J.K. Trends in electrochemical CO2 reduction activity for open and close-packed metal surfaces. Phys. Chem. Chem. Phys. 2014, 16, 4720. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, X.; Williams, T.; Bourgeois, L.; MacFarlane, D.R. Electrochemical reduction of CO2 on core-shell Cu/Au nanostructure arrays for syngas production. Electrochim. Acta 2017, 239, 84–89. [Google Scholar] [CrossRef]
- Pander, J.E.; Ren, D.; Huang, Y.; Loo, N.W.X.; Hong, S.H.L.; Yeo, B.S. Understanding the Heterogeneous Electrocatalytic Reduction of Carbon Dioxide on Oxide-Derived Catalysts. ChemElectroChem 2018, 5, 219–237. [Google Scholar] [CrossRef]
- Ren, D.; Deng, Y.; Handoko, A.D.; Chen, C.S.; Malkhandi, S.; Yeo, B.S. Selective Electrochemical Reduction of Carbon Dioxide to Ethylene and Ethanol on Copper(I) Oxide Catalysts. ACS Catal. 2015, 5, 2814–2821. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jung, H.; Kim, N.-K.; Oh, H.-S.; Min, B.K.; Hwang, Y.J. Mixed Copper States in Anodized Cu Electrocatalyst for Stable and Selective Ethylene Production from CO2 Reduction. J. Am. Chem. Soc. 2018, 140, 8681–8689. [Google Scholar] [CrossRef]
- Lee, S.; Kim, D.; Lee, J. Electrocatalytic Production of C3-C4 Compounds by Conversion of CO2 on a Chloride-Induced Bi-Phasic Cu2O-Cu Catalyst. Angew. Chemie Int. Ed. 2015, 54, 14701–14705. [Google Scholar] [CrossRef] [PubMed]
- Won, D.H.; Choi, C.H.; Chung, J.; Chung, M.W.; Kim, E.-H.; Woo, S.I. Rational Design of a Hierarchical Tin Dendrite Electrode for Efficient Electrochemical Reduction of CO2. ChemSusChem 2015, 8, 3092–3098. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, C.W.; Kanan, M.W. Aqueous CO2 Reduction at Very Low Overpotential on Oxide-Derived Au Nanoparticles. J. Am. Chem. Soc. 2012, 134, 19969–19972. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Trześniewski, B.J.; Xie, J.; Smith, W.A. Selective and Efficient Reduction of Carbon Dioxide to Carbon Monoxide on Oxide-Derived Nanostructured Silver Electrocatalysts. Angew. Chem. Int. Ed. 2016, 55, 9748–9752. [Google Scholar] [CrossRef]
- Verdaguer-Casadevall, A.; Li, C.W.; Johansson, T.P.; Scott, S.B.; McKeown, J.T.; Kumar, M.; Stephens, I.E.L.; Kanan, M.W.; Chorkendorff, I. Probing the Active Surface Sites for CO Reduction on Oxide-Derived Copper Electrocatalysts. J. Am. Chem. Soc. 2015, 137, 9808–9811. [Google Scholar] [CrossRef]
- Feng, X.; Jiang, K.; Fan, S.; Kanan, M.W. A Direct Grain-Boundary-Activity Correlation for CO Electroreduction on Cu Nanoparticles. ACS Cent. Sci. 2016, 2, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Kas, R.; Kortlever, R.; Milbrat, A.; Koper, M.T.M.; Mul, G.; Baltrusaitis, J. Electrochemical CO2 reduction on Cu2O-derived copper nanoparticles: Controlling the catalytic selectivity of hydrocarbons. Phys. Chem. Chem. Phys. 2014, 16, 12194–12201. [Google Scholar] [CrossRef]
- Mariano, R.G.; McKelvey, K.; White, H.S.; Kanan, M.W. Selective increase in CO2 electroreduction activity at grain-boundary surface terminations. Science 2017, 358, 1187–1192. [Google Scholar] [CrossRef] [Green Version]
- Eilert, A.; Cavalca, F.; Roberts, F.S.; Osterwalder, J.; Liu, C.; Favaro, M.; Crumlin, E.J.; Ogasawara, H.; Friebel, D.; Pettersson, L.G.M.; et al. Subsurface Oxygen in Oxide-Derived Copper Electrocatalysts for Carbon Dioxide Reduction. J. Phys. Chem. Lett. 2017, 8, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Favaro, M.; Xiao, H.; Cheng, T.; Goddard, W.A.; Yano, J.; Crumlin, E.J. Subsurface oxide plays a critical role in CO2 activation by Cu(111) surfaces to form chemisorbed CO2, the first step in reduction of CO2. Proc. Natl. Acad. Sci. USA 2017, 114, 6706–6711. [Google Scholar] [CrossRef] [Green Version]
- Lum, Y.; Ager, J.W. Stability of Residual Oxides in Oxide-Derived Copper Catalysts for Electrochemical CO2 Reduction Investigated with 18 O Labeling. Angew. Chemie Int. Ed. 2018, 57, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Albo, J.; Irabien, A. Cu2O-loaded gas diffusion electrodes for the continuous electrochemical reduction of CO2 to methanol. J. Catal. 2016, 343, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kim, H.-E.; Lee, H. Single-Atom Catalysts of Precious Metals for Electrochemical Reactions. ChemSusChem 2018, 11, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.B.; Hung, S.-F.; Liu, S.; Yuan, K.; Miao, S.; Zhang, L.; Huang, X.; Wang, H.-Y.; Cai, W.; Chen, R.; et al. Atomically dispersed Ni(i) as the active site for electrochemical CO2 reduction. Nat. Energy 2018, 3, 140–147. [Google Scholar] [CrossRef]
- Jiang, K.; Siahrostami, S.; Zheng, T.; Hu, Y.; Hwang, S.; Stavitski, E.; Peng, Y.; Dynes, J.; Gangisetty, M.; Su, D.; et al. Isolated Ni single atoms in graphene nanosheets for high-performance CO2 reduction. Energy Environ. Sci. 2018, 11, 893–903. [Google Scholar] [CrossRef]
- Jiang, K.; Siahrostami, S.; Akey, A.J.; Li, Y.; Lu, Z.; Lattimer, J.; Hu, Y.; Stokes, C.; Gangishetty, M.; Chen, G.; et al. Transition-Metal Single Atoms in a Graphene Shell as Active Centers for Highly Efficient Artificial Photosynthesis. Chem 2017, 3, 950–960. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, Z.; Han, P.; Du, Y.; Gu, Z.; Xu, X.; Zheng, G. Single-Atomic Cu with Multiple Oxygen Vacancies on Ceria for Electrocatalytic CO2 Reduction to CH4. ACS Catal. 2018, 8, 7113–7119. [Google Scholar] [CrossRef]
- Back, S.; Jung, Y. TiC- and TiN-Supported Single-Atom Catalysts for Dramatic Improvements in CO2 Electrochemical Reduction to CH 4. ACS Energy Lett. 2017, 2, 969–975. [Google Scholar] [CrossRef]
- Back, S.; Lim, J.; Kim, N.-Y.; Kim, Y.-H.; Jung, Y. Single-atom catalysts for CO 2 electroreduction with significant activity and selectivity improvements. Chem. Sci. 2017, 8, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.-J.; Clark, E.L.; Pham, H.H.; Bell, A.T.; Head-Gordon, M. Quantum Mechanical Screening of Single-Atom Bimetallic Alloys for the Selective Reduction of CO2 to C1 Hydrocarbons. ACS Catal. 2016, 6, 7769–7777. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.Q.; Chen, Z.; Zhao, X.Y.; Yao, T.; Chen, W.X.; You, R.; Zhao, C.M.; Wu, G.; Wang, J.; Huang, W.X.; et al. Regulation of Coordination Number over Single Co Sites: Triggering the Efficient Electroreduction of CO2. Angew. Chem. Int. Ed. 2018, 57, 1944–1948. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Zhang, H.; Liu, K.; Cullen, D.; More, K.; Wang, M.; Feng, Z.; Wang, G.; Wu, G.; Li, Y. Unveiling Active Sites of CO2 Reduction on Nitrogen-Coordinated and Atomically Dispersed Iron and Cobalt Catalysts. ACS Catal. 2018, 8, 3116–3122. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, S.; Wu, J.; Liu, M.; Yazdi, S.; Ren, M.; Sha, J.; Zhong, J.; Nie, K.; Jalilov, A.S.; et al. Electrochemical CO2 reduction with atomic iron-dispersed on nitrogen-doped graphene. Adv. Energy Mater. 2018, 8, 1703487. [Google Scholar] [CrossRef]
- Liu, X.; Dai, L. Carbon-based metal-free catalysts. Nat. Rev. Mater. 2016, 1, 16064. [Google Scholar] [CrossRef]
- Asefa, T. Metal-Free and Noble Metal-Free Heteroatom-Doped Nanostructured Carbons as Prospective Sustainable Electrocatalysts. Acc. Chem. Res. 2016, 49, 1873–1883. [Google Scholar] [CrossRef]
- Duan, X.; Xu, J.; Wei, Z.; Ma, J.; Guo, S.; Wang, S.; Liu, H.; Dou, S. Metal-Free Carbon Materials for CO2 Electrochemical Reduction. Adv. Mater. 2017, 29, 1701784. [Google Scholar] [CrossRef]
- Guo, D.; Shibuya, R.; Akiba, C.; Saji, S.; Kondo, T.; Nakamura, J. Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts. Science 2016, 351, 361–365. [Google Scholar] [CrossRef]
- Xu, J.; Kan, Y.; Huang, R.; Zhang, B.; Wang, B.; Wu, K.-H.; Lin, Y.; Sun, X.; Li, Q.; Centi, G.; et al. Revealing the Origin of Activity in Nitrogen-Doped Nanocarbons towards Electrocatalytic Reduction of Carbon Dioxide. ChemSusChem 2016, 9, 1085–1089. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Yadav, R.M.; Liu, M.; Sharma, P.P.; Tiwary, C.S.; Ma, L.; Zou, X.; Zhou, X.-D.; Yakobson, B.I.; Lou, J.; et al. Achieving Highly Efficient, Selective, and Stable CO2 Reduction on Nitrogen-Doped Carbon Nanotubes. ACS Nano 2015, 9, 5364–5371. [Google Scholar] [CrossRef]
- Sharma, P.P.; Wu, J.; Yadav, R.M.; Liu, M.; Wright, C.J.; Tiwary, C.S.; Yakobson, B.I.; Lou, J.; Ajayan, P.M.; Zhou, X.-D. Nitrogen-Doped Carbon Nanotube Arrays for High-Efficiency Electrochemical Reduction of CO2: On the Understanding of Defects, Defect Density, and Selectivity. Angew. Chem. Int. Ed. 2015, 54, 13701–13705. [Google Scholar] [CrossRef]
- Wu, J.; Liu, M.; Sharma, P.P.; Yadav, R.M.; Ma, L.; Yang, Y.; Zou, X.; Zhou, X.-D.; Vajtai, R.; Yakobson, B.I.; et al. Incorporation of Nitrogen Defects for Efficient Reduction of CO2 via Two-Electron Pathway on Three-Dimensional Graphene Foam. Nano Lett. 2016, 16, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, S.; Quan, X.; Yu, H. Efficient Electrochemical Reduction of Carbon Dioxide to Acetate on Nitrogen-Doped Nanodiamond. J. Am. Chem. Soc. 2015, 137, 11631–11636. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Seredych, M.; Rodríguez-Castellón, E.; Bandosz, T.J. Metal-free Nanoporous Carbon as a Catalyst for Electrochemical Reduction of CO2 to CO and CH4. ChemSusChem 2016, 9, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Sreekanth, N.; Nazrulla, M.A.; Vineesh, T.V.; Sailaja, K.; Phani, K.L. Metal-free boron-doped graphene for selective electroreduction of carbon dioxide to formic acid/formate. Chem. Commun. 2015, 51, 16061–16064. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Wen, J.; Zheng, L.; Yan, H.; Jiao, L.; Wang, X.; Cai, X.; Wu, Y.; Chen, G.; Chen, L.; et al. Single-Atom-Based Heterojunction Coupling with Ion-Exchange Reaction for Sensitive Photoelectrochemical Immunoassay. Nano Lett. 2021, 21, 1879–1887. [Google Scholar] [CrossRef]
- Wu, J.; Sharifi, T.; Gao, Y.; Zhang, T.; Ajayan, P.M. Emerging Carbon-Based Heterogeneous Catalysts for Electrochemical Reduction of Carbon Dioxide into Value-Added Chemicals. Adv. Mater. 2019, 31, 1804257. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, Y.; Hou, X.; Ma, C.; Tan, T. Nitrogen-doped graphenes as efficient electrocatalysts for the selective reduction of carbon dioxide to formate in aqueous solution. Green Chem. 2016, 18, 3250–3256. [Google Scholar] [CrossRef]
- Sun, X.; Kang, X.; Zhu, Q.; Ma, J.; Yang, G.; Liu, Z.; Han, B. Very highly efficient reduction of CO2 to CH4 using metal-free N-doped carbon electrodes. Chem. Sci. 2016, 7, 2883–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Jia, J.; Song, P.; Wang, Q.; Li, D.; Min, S.; Qian, C.; Wang, L.; Li, Y.F.; Ma, C.; et al. Efficient Electrocatalytic Reduction of CO2 by Nitrogen-Doped Nanoporous Carbon/Carbon Nanotube Membranes: A Step Towards the Electrochemical CO2 Refinery. Angew. Chemie Int. Ed. 2017, 56, 7847–7852. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Yang, H.; Huang, X.; Liu, L.; Cai, W.; Gao, J.; Li, X.; Zhang, T.; Huang, Y.; Liu, B. Identifying Active Sites of Nitrogen-Doped Carbon Materials for the CO2 Reduction Reaction. Adv. Funct. Mater. 2018, 28, 1800499. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Y.; Zhu, D.; Zhang, L.; Ma, H.; Yao, N.; Zhang, B. Controllable Growth, Structure, and Low Field Emission of Well-Aligned CN x Nanotubes. J. Phys. Chem. B 2002, 106, 2186–2190. [Google Scholar] [CrossRef]
- Zhang, L.; Xiao, J.; Wang, H.; Shao, M. Carbon-Based Electrocatalysts for Hydrogen and Oxygen Evolution Reactions. ACS Catal. 2017, 7, 7855–7865. [Google Scholar] [CrossRef]
- Zhang, S.; Kang, P.; Ubnoske, S.; Brennaman, M.K.; Song, N.; House, R.L.; Glass, J.T.; Meyer, T.J. Polyethylenimine-Enhanced Electrocatalytic Reduction of CO2 to Formate at Nitrogen-Doped Carbon Nanomaterials. J. Am. Chem. Soc. 2014, 136, 7845–7848. [Google Scholar] [CrossRef] [PubMed]
- Barton Cole, E.; Lakkaraju, P.S.; Rampulla, D.M.; Morris, A.J.; Abelev, E.; Bocarsly, A.B. Using a One-Electron Shuttle for the Multielectron Reduction of CO2 to Methanol: Kinetic, Mechanistic, and Structural Insights. J. Am. Chem. Soc. 2010, 132, 11539–11551. [Google Scholar] [CrossRef] [PubMed]
- Pels, J.R.; Kapteijn, F.; Moulijn, J.A.; Zhu, Q.; Thomas, K.M. Evolution of nitrogen functionalities in carbonaceous materials during pyrolysis. Carbon N. Y. 1995, 33, 1641–1653. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, L.; Xia, Z.; Roy, A.; Chang, D.W.; Baek, J.-B.; Dai, L. BCN Graphene as Efficient Metal-Free Electrocatalyst for the Oxygen Reduction Reaction. Angew. Chem. Int. Ed. 2012, 51, 4209–4212. [Google Scholar] [CrossRef]
- Zhang, L.; Niu, J.; Dai, L.; Xia, Z. Effect of Microstructure of Nitrogen-Doped Graphene on Oxygen Reduction Activity in Fuel Cells. Langmuir 2012, 28, 7542–7550. [Google Scholar] [CrossRef]
- Gong, K.; Du, F.; Xia, Z.; Durstock, M.; Dai, L. Nitrogen-Doped Carbon Nanotube Arrays with High Electrocatalytic Activity for Oxygen Reduction. Science 2009, 323, 760–764. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Xia, Z. Mechanisms of Oxygen Reduction Reaction on Nitrogen-Doped Graphene for Fuel Cells. J. Phys. Chem. C 2011, 115, 11170–11176. [Google Scholar] [CrossRef]
- Winther-Jensen, B.; Winther-Jensen, O.; Forsyth, M.; MacFarlane, D.R. High Rates of Oxygen Reduction over a Vapor Phase–Polymerized PEDOT Electrode. Science 2008, 321, 671–674. [Google Scholar] [CrossRef]
- Ma, C.; Hou, P.; Wang, X.; Wang, Z.; Li, W.; Kang, P. Carbon nanotubes with rich pyridinic nitrogen for gas phase CO2 electroreduction. Appl. Catal. B Environ. 2019, 250, 347–354. [Google Scholar] [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Electrocatalytic reduction of carbon dioxide mediated by Re(bipy)(CO)3 Cl (bipy = 2,2′-bipyridine). J. Chem. Soc. Chem. Commun. 1984, 328–330. [Google Scholar] [CrossRef]
- Becker, J.Y.; Vainas, B.; Eger, R.; Kaufman, L. Electrocatalytic reduction of CO2 to oxalate by Ag II and Pd II porphyrins. J. Chem. Soc. Chem. Commun. 1985, 1471–1472. [Google Scholar] [CrossRef]
- Lu, X.; Ahsaine, H.A.; Dereli, B.; Garcia-Esparza, A.T.; Reinhard, M.; Shinagawa, T.; Li, D.; Adil, K.; Tchalala, M.R.; Kroll, T.; et al. Operando Elucidation on the Working State of Immobilized Fluorinated Iron Porphyrin for Selective Aqueous Electroreduction of CO2 to CO. ACS Catal. 2021, 11, 6499–6509. [Google Scholar] [CrossRef]
- Kornienko, N.; Zhao, Y.; Kley, C.S.; Zhu, C.; Kim, D.; Lin, S.; Chang, C.J.; Yaghi, O.M.; Yang, P. Metal–Organic Frameworks for Electrocatalytic Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2015, 137, 14129–14135. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Diercks, C.S.; Zhang, Y.-B.; Kornienko, N.; Nichols, E.M.; Zhao, Y.; Paris, A.R.; Kim, D.; Yang, P.; Yaghi, O.M.; et al. Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water. Science 2015, 349, 1208–1213. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Kortlever, R.; Kas, R.; Birdja, Y.Y.; Diaz-Morales, O.; Kwon, Y.; Ledezma-Yanez, I.; Schouten, K.J.P.; Mul, G.; Koper, M.T.M. Electrocatalytic reduction of carbon dioxide to carbon monoxide and methane at an immobilized cobalt protoporphyrin. Nat. Commun. 2015, 6, 8177. [Google Scholar] [CrossRef]
- Mohamed, E.A.; Zahran, Z.N.; Naruta, Y. Efficient electrocatalytic CO2 reduction with a molecular cofacial iron porphyrin dimer. Chem. Commun. 2015, 51, 16900–16903. [Google Scholar] [CrossRef]
- Weng, Z.; Jiang, J.; Wu, Y.; Wu, Z.; Guo, X.; Materna, K.L.; Liu, W.; Batista, V.S.; Brudvig, G.W.; Wang, H. Electrochemical CO2 Reduction to Hydrocarbons on a Heterogeneous Molecular Cu Catalyst in Aqueous Solution. J. Am. Chem. Soc. 2016, 138, 8076–8079. [Google Scholar] [CrossRef]
- Bhugun, I.; Lexa, D.; Savéant, J.-M. Catalysis of the Electrochemical Reduction of Carbon Dioxide by Iron(0) Porphyrins: Synergystic Effect of Weak Brönsted Acids. J. Am. Chem. Soc. 1996, 118, 1769–1776. [Google Scholar] [CrossRef]
- Bhugun, I.; Lexa, D.; Saveant, J.-M. Ultraefficient selective homogeneous catalysis of the electrochemical reduction of carbon dioxide by an iron(0) porphyrin associated with a weak Broensted acid cocatalyst. J. Am. Chem. Soc. 1994, 116, 5015–5016. [Google Scholar] [CrossRef]
- Bhugun, I.; Lexa, D.; Savéant, J.-M. Catalysis of the Electrochemical Reduction of Carbon Dioxide by Iron(0) Porphyrins. Synergistic Effect of Lewis Acid Cations. J. Phys. Chem. 1996, 100, 19981–19985. [Google Scholar] [CrossRef]
- Aoi, S.; Mase, K.; Ohkubo, K.; Fukuzumi, S. Selective electrochemical reduction of CO2 to CO with a cobalt chlorin complex adsorbed on multi-walled carbon nanotubes in water. Chem. Commun. 2015, 51, 10226–10228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashiko, T.; Reed, C.A.; Haller, K.J.; Scheidt, W.R. Nature of iron(I) and iron(0) tetraphenylporphyrin complexes. Synthesis and molecular structure of (dibenzo-18-crown-6)bis(tetrahydrofuran)sodium (meso-tetraphenylporphinato)ferrate and bis[tris(tetrahydrofuran)sodium] (meso-tetraphenylporphinato)ferrate. Inorg. Chem. 1984, 23, 3192–3196. [Google Scholar] [CrossRef]
- Hammouche, M.; Lexa, D.; Savéant, J.M.; Momenteau, M. Catalysis of the electrochemical reduction of carbon dioxide by iron(“0”) porphyrins. J. Electroanal. Chem. Interfacial Electrochem. 1988, 249, 347–351. [Google Scholar] [CrossRef]
- Hammouche, M.; Lexa, D.; Momenteau, M.; Saveant, J.M. Chemical catalysis of electrochemical reactions. Homogeneous catalysis of the electrochemical reduction of carbon dioxide by iron(“0”) porphyrins. Role of the addition of magnesium cations. J. Am. Chem. Soc. 1991, 113, 8455–8466. [Google Scholar] [CrossRef]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 2012, 338, 90–94. [Google Scholar] [CrossRef]
- Jeoung, J.-H.; Dobbek, H. Carbon Dioxide Activation at the Ni,Fe-Cluster of Anaerobic Carbon Monoxide Dehydrogenase. Science 2007, 318, 1461–1464. [Google Scholar] [CrossRef] [Green Version]
- Shin, W.; Lee, S.H.; Shin, J.W.; Lee, S.P.; Kim, Y. Highly Selective Electrocatalytic Conversion of CO2 to CO at −0.57 V (NHE) by Carbon Monoxide Dehydrogenase from Moorella t hermoacetica. J. Am. Chem. Soc. 2003, 125, 14688–14689. [Google Scholar] [CrossRef]
- Naruta, Y.; Sasayama, M.; Sasaki, T. Oxygen Evolution by Oxidation of Water with Manganese Porphyrin Dimers. Angew. Chemie Int. Ed. English 1994, 33, 1839–1841. [Google Scholar] [CrossRef]
- Atoguchi, T.; Aramata, A.; Kazusaka, A.; Enyo, M. Cobalt(II)–tetraphenylporphyrin–pyridine complex fixed on a glassy carbon electrode and its prominent catalytic activity for reduction of carbon dioxide. J. Chem. Soc. Chem. Commun. 1991, 156–157. [Google Scholar] [CrossRef]
- Yoshida, T.; Kamato, K.; Tsukamoto, M.; Iida, T.; Schlettwein, D.; Wöhrle, D.; Kaneko, M. Selective electroacatalysis for CO2 reduction in the aqueous phase using cobalt phthalocyanine/poly-4-vinylpyridine modified electrodes. J. Electroanal. Chem. 1995, 385, 209–225. [Google Scholar] [CrossRef]
- Tanaka, H.; Aramata, A. Aminopyridyl cation radical method for bridging between metal complex and glassy carbon: Cobalt(II) tetraphenylporphyrin bonded on glassy carbon for enhancement of CO2 electroreduction. J. Electroanal. Chem. 1997, 437, 29–35. [Google Scholar] [CrossRef]
- Zhou, X.; Micheroni, D.; Lin, Z.; Poon, C.; Li, Z.; Lin, W. Graphene-Immobilized fac -Re(bipy)(CO)3 Cl for Syngas Generation from Carbon Dioxide. ACS Appl. Mater. Interfaces 2016, 8, 4192–4198. [Google Scholar] [CrossRef]
- Wang, R.; Haspel, H.; Pustovarenko, A.; Dikhtiarenko, A.; Russkikh, A.; Shterk, G.; Osadchii, D.; Ould-Chikh, S.; Ma, M.; Smith, W.A.; et al. Maximizing Ag Utilization in High-Rate CO2 Electrochemical Reduction with a Coordination Polymer-Mediated Gas Diffusion Electrode. ACS Energy Lett. 2019, 4, 2024–2031. [Google Scholar] [CrossRef]
- Sun, X.; Suarez, A.I.O.; Meijerink, M.; van Deelen, T.; Ould-Chikh, S.; Zečević, J.; de Jong, K.P.; Kapteijn, F.; Gascon, J. Manufacture of highly loaded silica-supported cobalt Fischer–Tropsch catalysts from a metal organic framework. Nat. Commun. 2017, 8, 1680. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Olivos-Suarez, A.I.; Osadchii, D.; Romero, M.J.V.; Kapteijn, F.; Gascon, J. Single cobalt sites in mesoporous N-doped carbon matrix for selective catalytic hydrogenation of nitroarenes. J. Catal. 2018, 357, 20–28. [Google Scholar] [CrossRef]
- Sun, X.; Olivos-Suarez, A.I.; Oar-Arteta, L.; Rozhko, E.; Osadchii, D.; Bavykina, A.; Kapteijn, F.; Gascon, J. Metal-Organic Framework Mediated Cobalt/Nitrogen-Doped Carbon Hybrids as Efficient and Chemoselective Catalysts for the Hydrogenation of Nitroarenes. ChemCatChem 2017, 9, 1854–1862. [Google Scholar] [CrossRef]
- Santos, V.P.; Wezendonk, T.A.; Jaén, J.J.D.; Dugulan, A.I.; Nasalevich, M.A.; Islam, H.-U.; Chojecki, A.; Sartipi, S.; Sun, X.; Hakeem, A.A.; et al. Metal organic framework-mediated synthesis of highly active and stable Fischer-Tropsch catalysts. Nat. Commun. 2015, 6, 6451. [Google Scholar] [CrossRef] [Green Version]
- Oar-Arteta, L.; Wezendonk, T.; Sun, X.; Kapteijn, F.; Gascon, J. Metal organic frameworks as precursors for the manufacture of advanced catalytic materials. Mater. Chem. Front. 2017, 1, 1709–1745. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Wu, Y.; Yuan, X.; Huang, L.; Wu, Z.; Xuan, J.; Wang, Y.; Wang, H. High-Performance Electrochemical CO2 Reduction Cells Based on Non-noble Metal Catalysts. ACS Energy Lett. 2018, 3, 2527–2532. [Google Scholar] [CrossRef] [Green Version]
- Morlanés, N.; Takanabe, K.; Rodionov, V. Simultaneous Reduction of CO2 and Splitting of H2O by a Single Immobilized Cobalt Phthalocyanine Electrocatalyst. ACS Catal. 2016, 6, 3092–3095. [Google Scholar] [CrossRef]
- Kramer, W.W.; McCrory, C.C.L. Polymer coordination promotes selective CO2 reduction by cobalt phthalocyanine. Chem. Sci. 2016, 7, 2506–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, T.; Yoshida, T.; Tokita, S.; Taguchi, F.; Imaya, H.; Kaneko, M. Factors affecting selective electrocatalytic CO2 reduction with cobalt phthalocyanine incorporated in a polyvinylpyridine membrane coated on a graphite electrode. J. Electroanal. Chem. 1996, 412, 125–132. [Google Scholar] [CrossRef]
- Lieber, C.M.; Lewis, N.S. Catalytic reduction of carbon dioxide at carbon electrodes modified with cobalt phthalocyanine. J. Am. Chem. Soc. 1984, 106, 5033–5034. [Google Scholar] [CrossRef]
- Liu, Y.; McCrory, C.C.L. Modulating the mechanism of electrocatalytic CO2 reduction by cobalt phthalocyanine through polymer coordination and encapsulation. Nat. Commun. 2019, 10, 1683. [Google Scholar] [CrossRef]
- Wang, X.; Cai, Z.; Wang, Y.; Feng, Y.; Yan, H.; Wang, D.; Wan, L. In Situ Scanning Tunneling Microscopy of Cobalt-Phthalocyanine-Catalyzed CO2 Reduction Reaction. Angew. Chem. Int. Ed. 2020, 59, 16098–16103. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, Z.; Zhang, X.; Li, L.; Li, Y.; Xu, H.; Li, X.; Yu, X.; Zhang, Z.; Liang, Y.; et al. Highly selective and active CO2 reduction electrocatalysts based on cobalt phthalocyanine/carbon nanotube hybrid structures. Nat. Commun. 2017, 8, 14675. [Google Scholar] [CrossRef] [Green Version]
- Latiff, N.M.; Fu, X.; Mohamed, D.K.; Veksha, A.; Handayani, M.; Lisak, G. Carbon based copper(II) phthalocyanine catalysts for electrochemical CO2 reduction: Effect of carbon support on electrocatalytic activity. Carbon N. Y. 2020, 168, 245–253. [Google Scholar] [CrossRef]
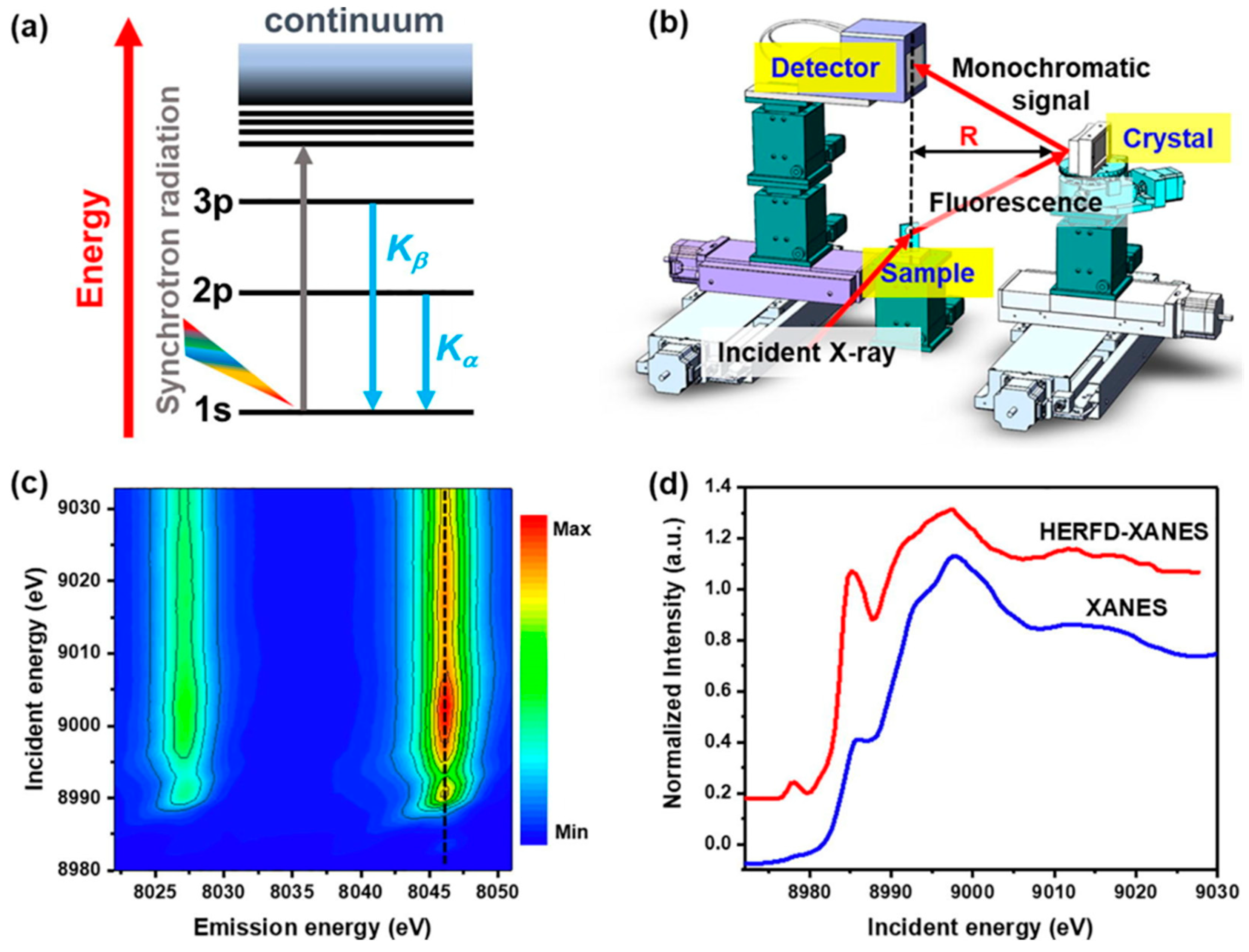
- Mei, B.; Liu, C.; Li, J.; Gu, S.; Du, X.; Lu, S.; Song, F.; Xu, W.; Jiang, Z. Operando HERFD-XANES and surface sensitive Δμ analyses identify the structural evolution of copper(II) phthalocyanine for electroreduction of CO2. J. Energy Chem. 2022, 64, 1–7. [Google Scholar] [CrossRef]
- Kusama, S.; Saito, T.; Hashiba, H.; Sakai, A.; Yotsuhashi, S. Crystalline Copper(II) Phthalocyanine Catalysts for Electrochemical Reduction of Carbon Dioxide in Aqueous Media. ACS Catal. 2017, 7, 8382–8385. [Google Scholar] [CrossRef]
- Acharjya, S.; Chen, J.; Zhu, M.; Peng, C. Elucidating the reactivity and nature of active sites for tin phthalocyanine during CO2 reduction. Greenh. Gases Sci. Technol. 2021, 11, 1191–1197. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Gu, M.; Wang, M.; Zhang, Z.; Pan, W.; Jiang, Z.; Zheng, H.; Lucero, M.; Wang, H.; et al. Molecular engineering of dispersed nickel phthalocyanines on carbon nanotubes for selective CO2 reduction. Nat. Energy 2020, 5, 684–692. [Google Scholar] [CrossRef]
- Li, X.; Chai, G.; Xu, X.; Liu, J.; Zhong, Z.; Cao, A.; Tao, Z.; You, W.; Kang, L. Electrocatalytic reduction of CO2 to CO over iron phthalocyanine-modified graphene nanocomposites. Carbon N. Y. 2020, 167, 658–667. [Google Scholar] [CrossRef]
- Zheng, J.; Li, X.; Qin, Y.; Zhang, S.; Sun, M.; Duan, X.; Sun, H.; Li, P.; Wang, S. Zn phthalocyanine/carbon nitride heterojunction for visible light photoelectrocatalytic conversion of CO2 to methanol. J. Catal. 2019, 371, 214–223. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, Y.; Ma, L.; Zhu, G.; Wang, Y.; Xue, X.; Chen, R.; Yang, S.; Jin, Z. Progress and perspective of electrocatalytic CO2 reduction for renewable carbonaceous fuels and chemicals. Adv. Sci. 2018, 5, 1700275. [Google Scholar] [CrossRef]















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ait Ahsaine, H.; Zbair, M.; BaQais, A.; Arab, M. CO2 Electroreduction over Metallic Oxide, Carbon-Based, and Molecular Catalysts: A Mini-Review of the Current Advances. Catalysts 2022, 12, 450. https://doi.org/10.3390/catal12050450
Ait Ahsaine H, Zbair M, BaQais A, Arab M. CO2 Electroreduction over Metallic Oxide, Carbon-Based, and Molecular Catalysts: A Mini-Review of the Current Advances. Catalysts. 2022; 12(5):450. https://doi.org/10.3390/catal12050450
Chicago/Turabian StyleAit Ahsaine, Hassan, Mohamed Zbair, Amal BaQais, and Madjid Arab. 2022. "CO2 Electroreduction over Metallic Oxide, Carbon-Based, and Molecular Catalysts: A Mini-Review of the Current Advances" Catalysts 12, no. 5: 450. https://doi.org/10.3390/catal12050450
APA StyleAit Ahsaine, H., Zbair, M., BaQais, A., & Arab, M. (2022). CO2 Electroreduction over Metallic Oxide, Carbon-Based, and Molecular Catalysts: A Mini-Review of the Current Advances. Catalysts, 12(5), 450. https://doi.org/10.3390/catal12050450