
Synthetic and DFT Modeling Studies on Suzuki–Miyaura Reactions of 4,5-Dibromo-2-methylpyridazin-3(2H)-one with Ferrocene Boronates, Accompanied by Hydrodebromination and a Novel Bridge-Forming Annulation In Vitro Cytotoxic Activity of the Ferrocenyl–Pyridazinone Products

 ,
,  ,
, 
Abstract
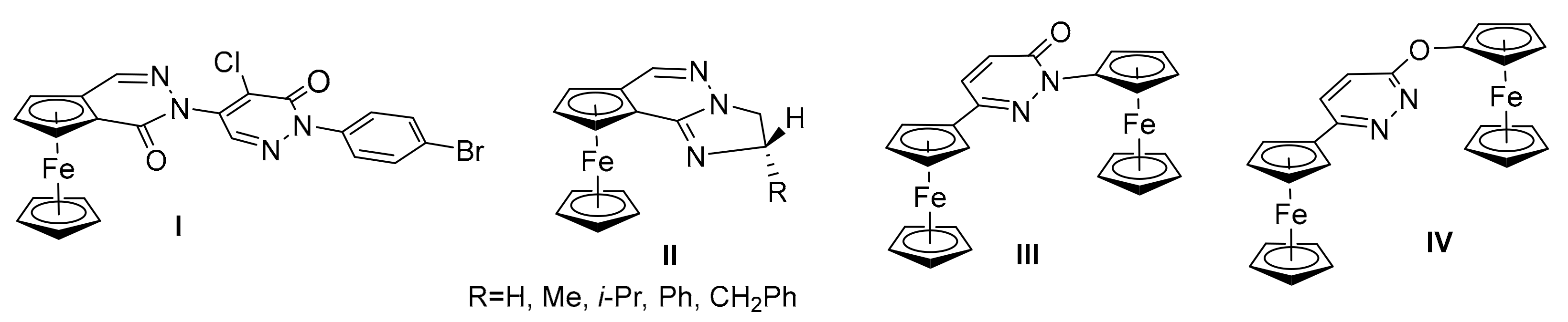
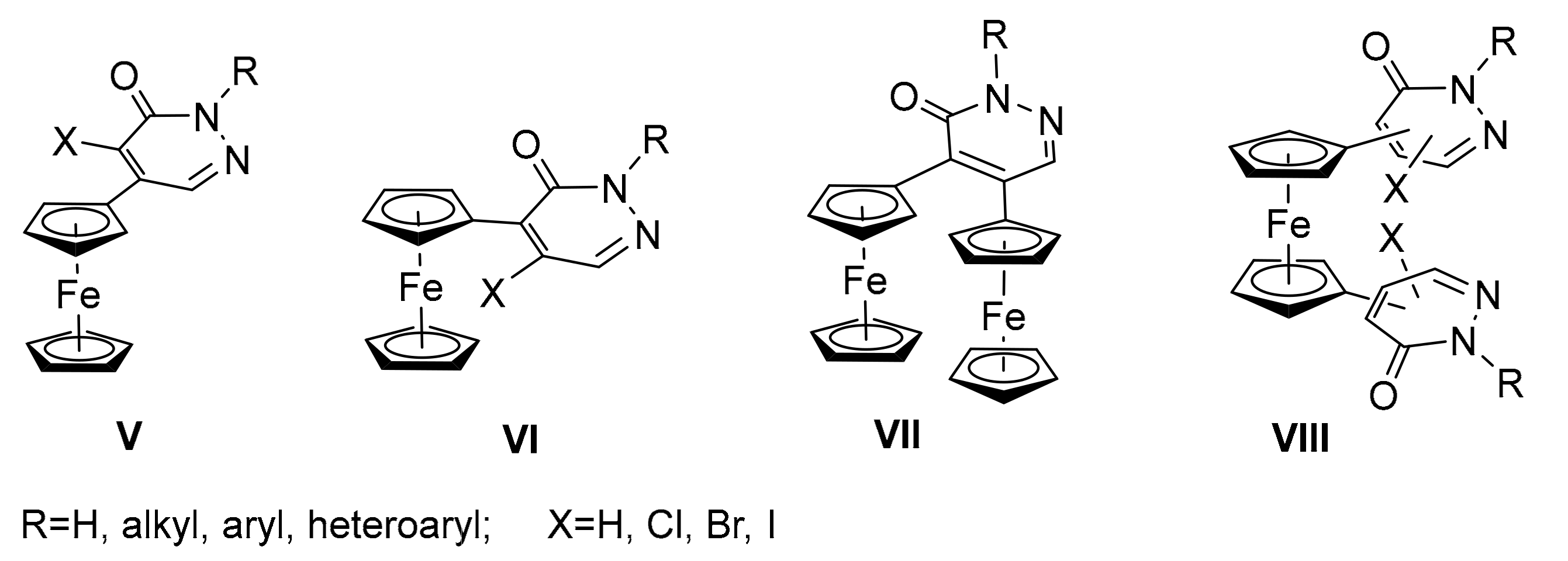
:1. Introduction
2. Results
3. Discussion
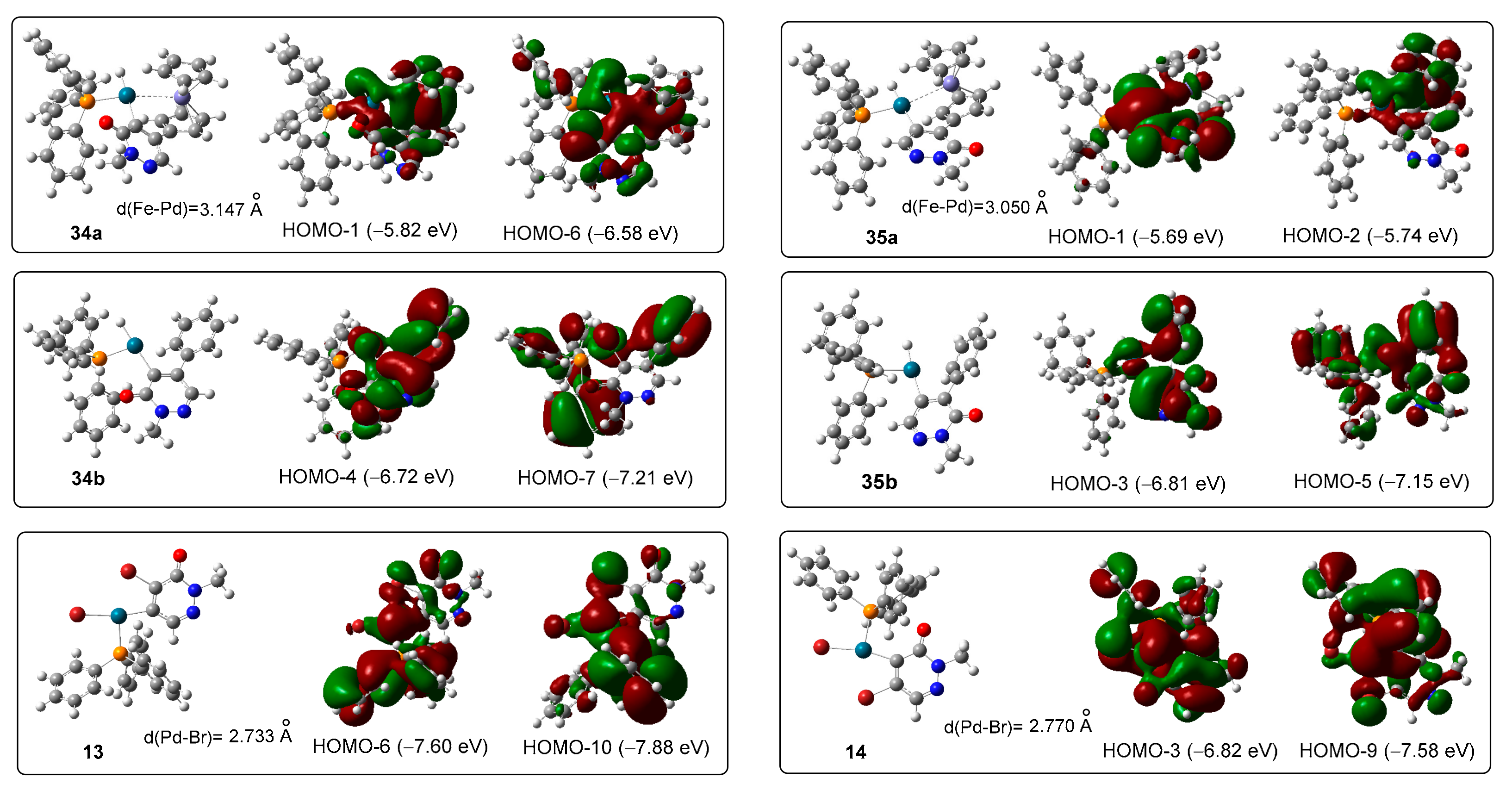
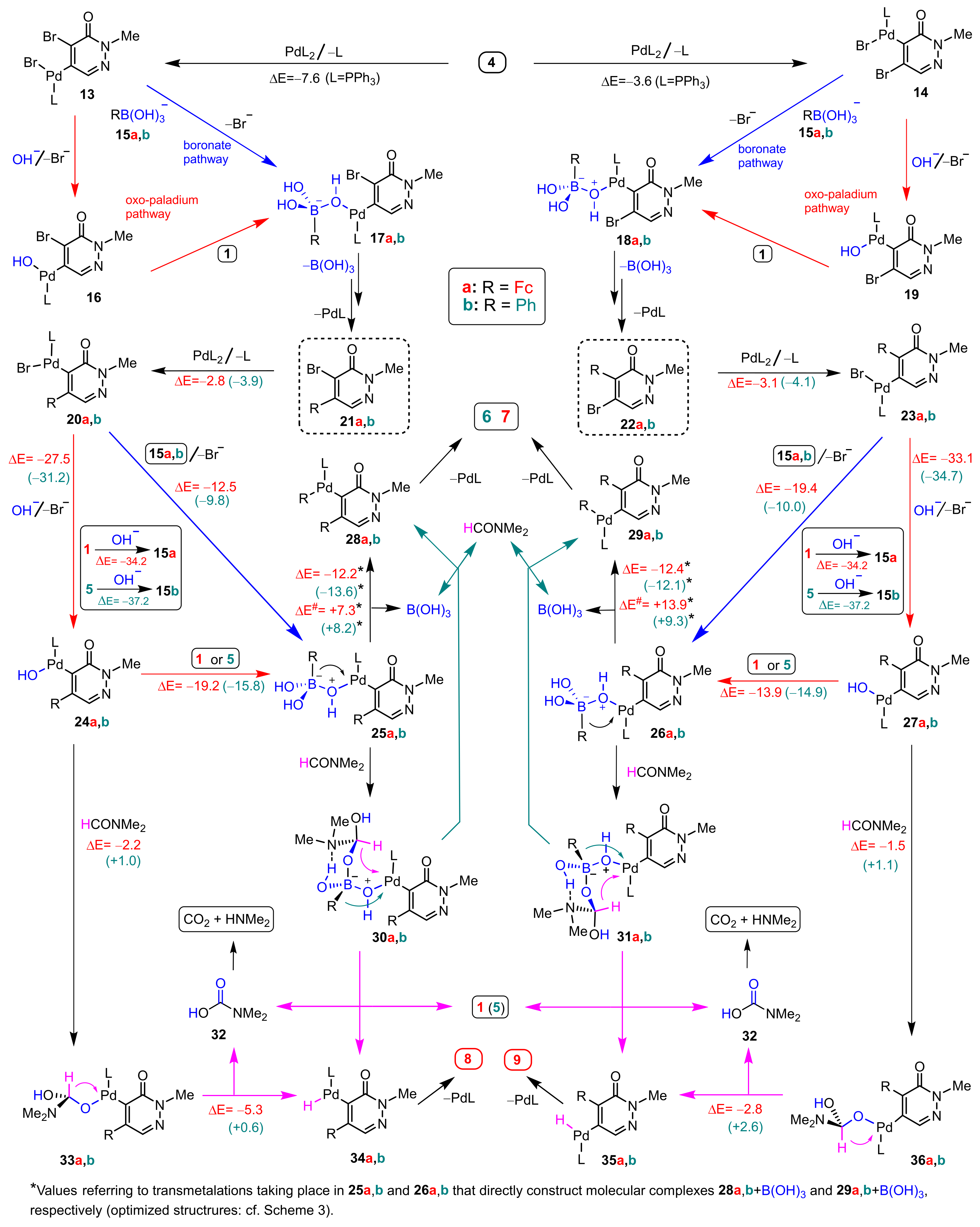
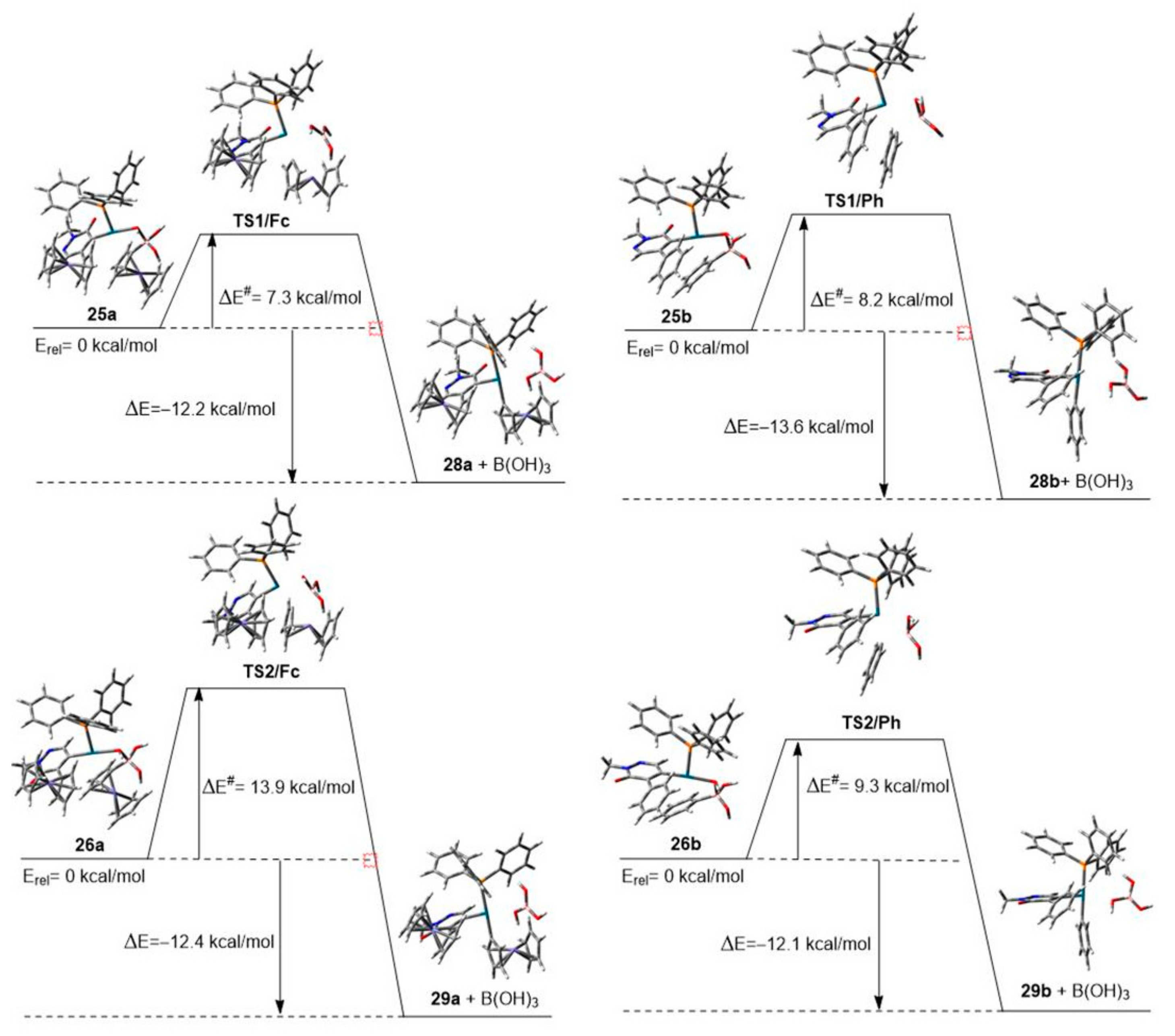
3.1. DFT-Modeled Possible Pathways of Sequential SM Coupling and Hydrodebromination Processes in the Reactions of 1 with Boronic Acids 2 and 5 Taking Place without Single Electron Transfer Steps


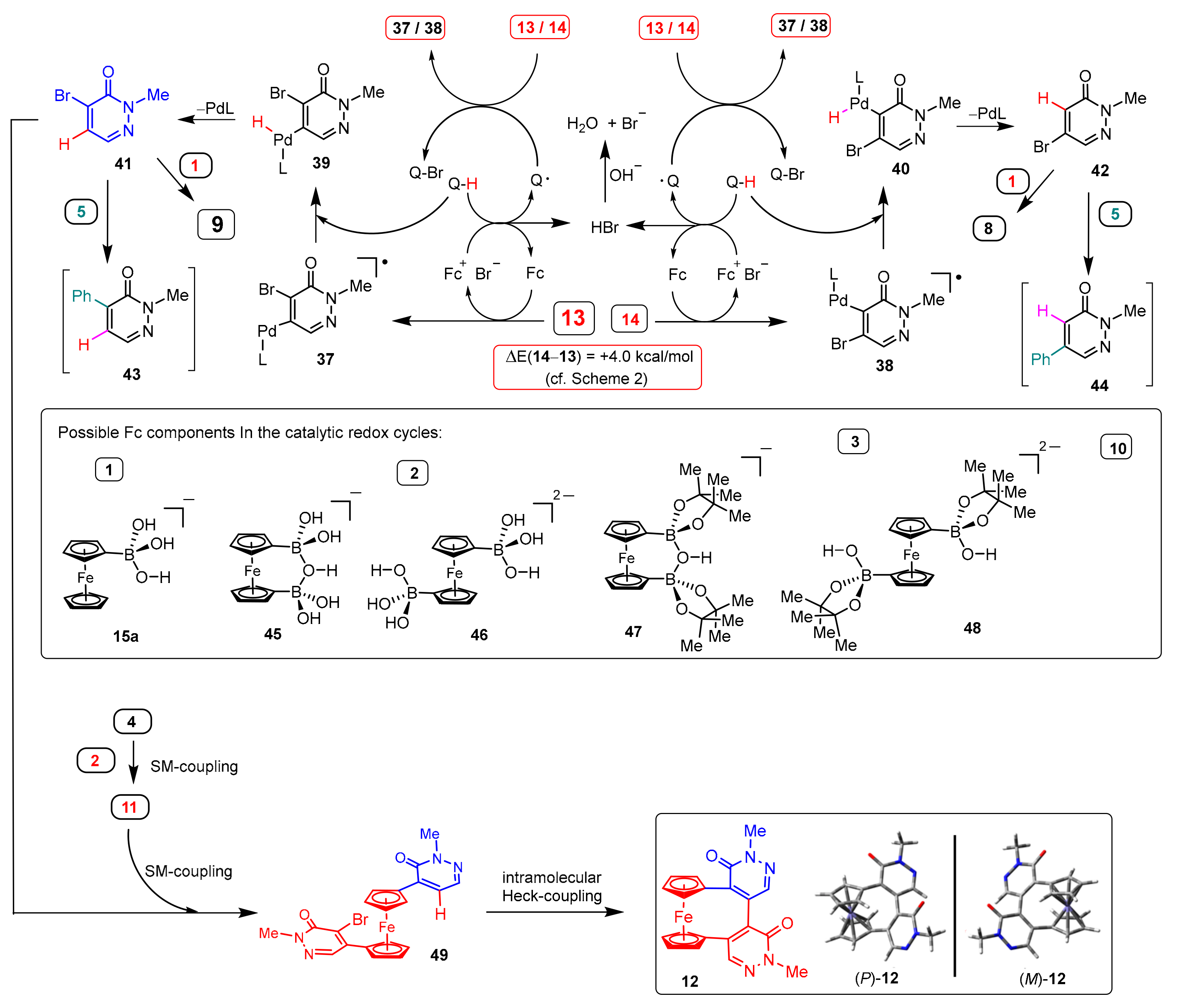
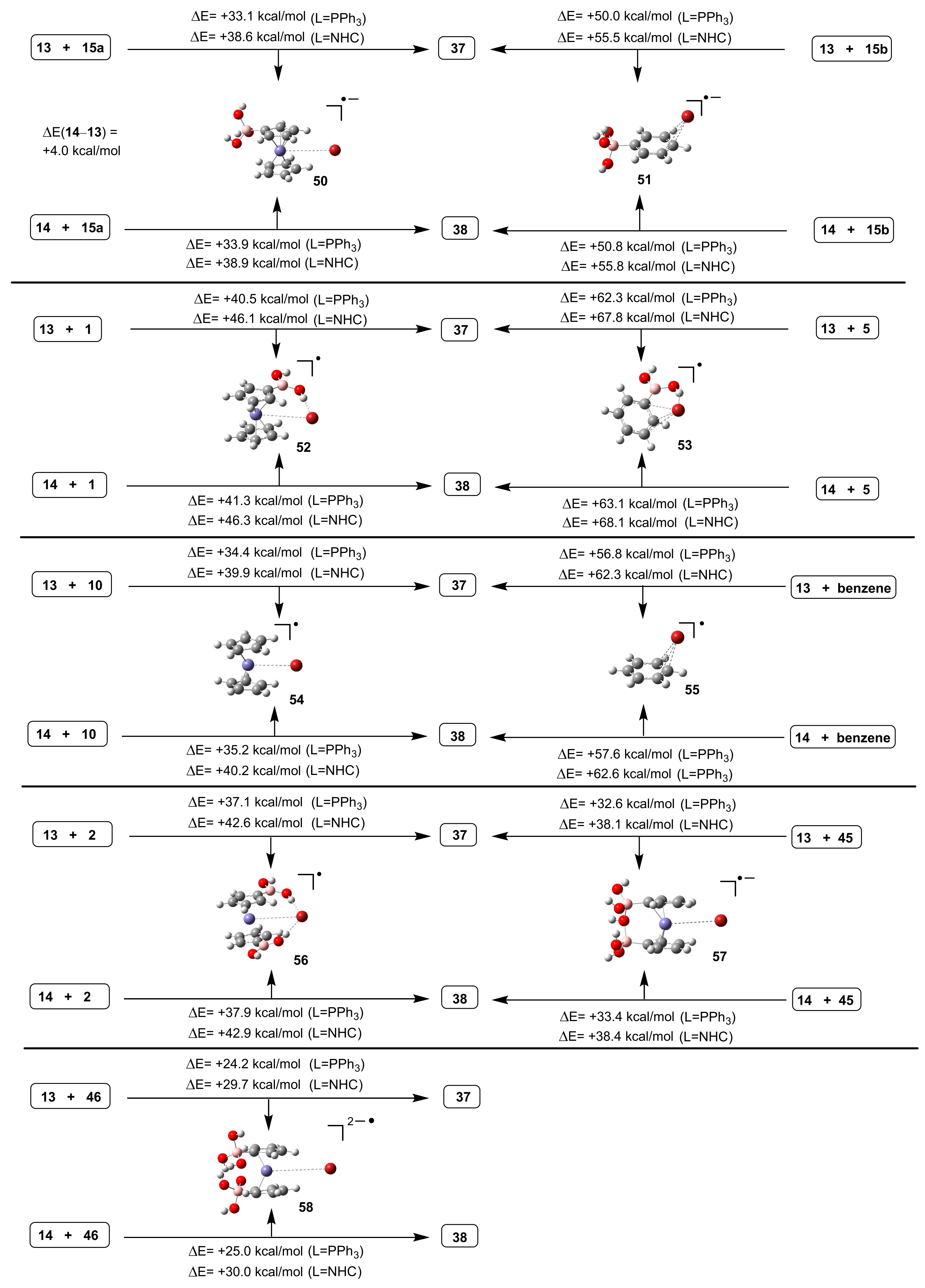
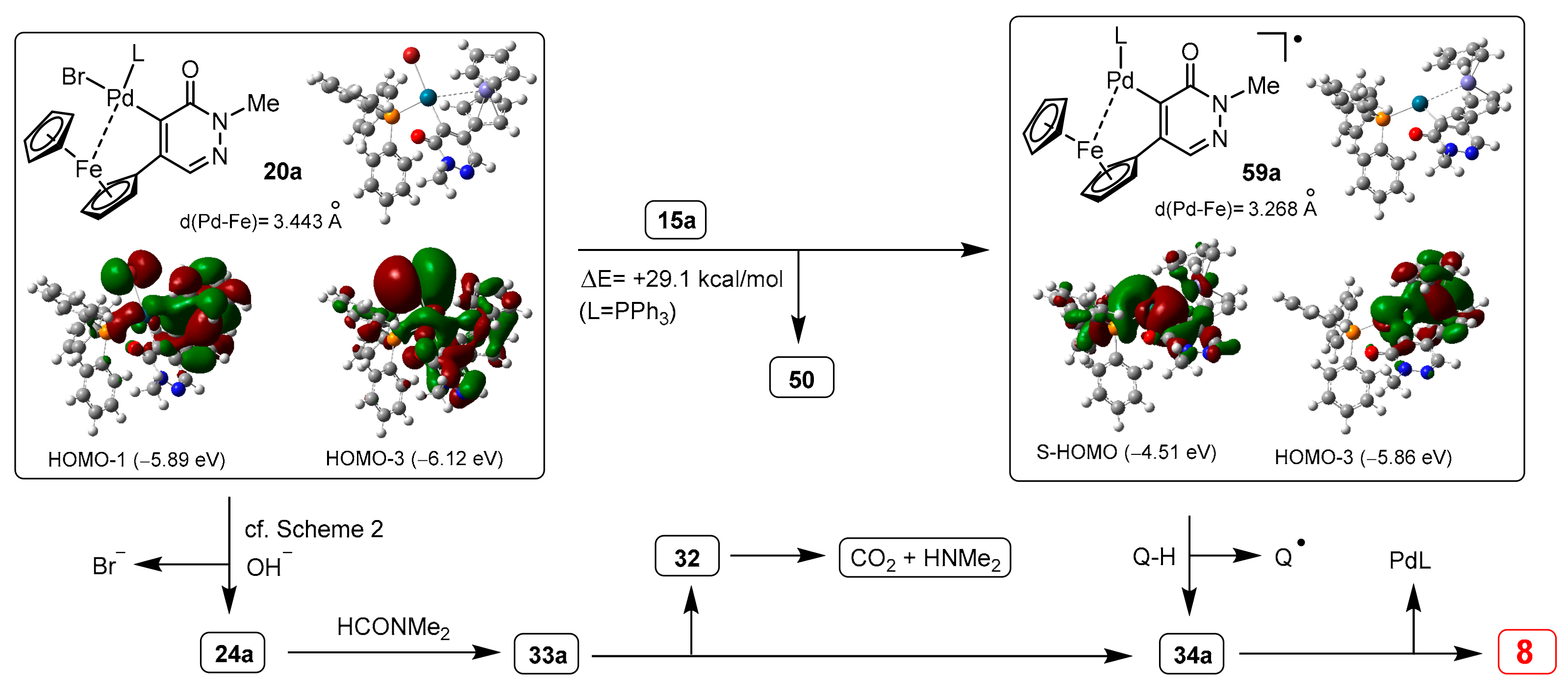
3.2. DFT-Modeled Possible Pathways of Hydrodebromination Processes Taking Place with Single Electron Transfer (SET) Steps Accompanying the Investigated SM Coupling Reactions




3.3. Deuterium Labelling Experiments Disclosing the Implication of DMF as Hydrogen Source in the Hydrodebromination Processes
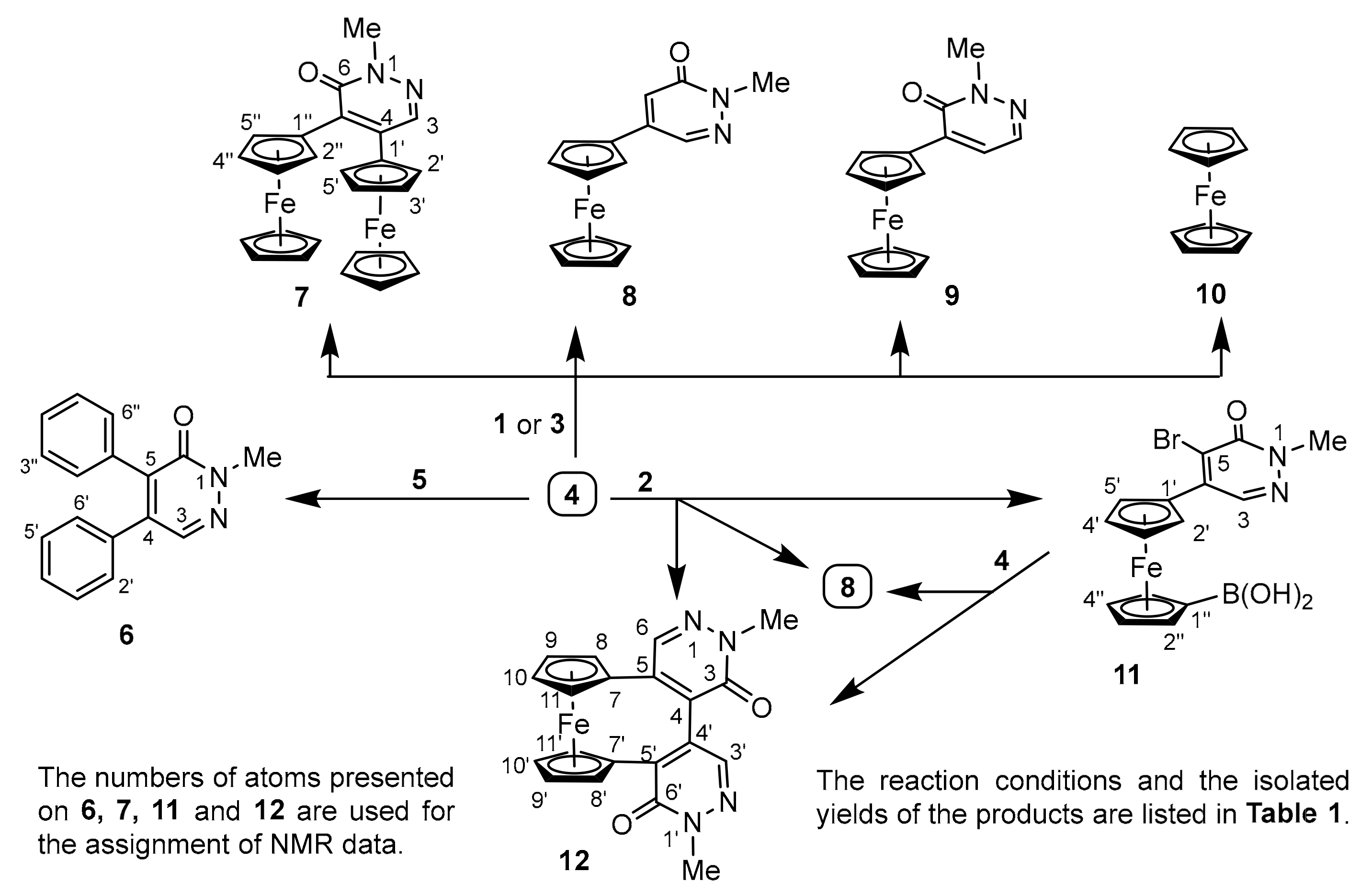
3.4. Structural Elucidation of Ferrocenylpyridazinones 7–9, 11, and Ferrocenophane 12
4. In Vitro Evaluation of the Ferrocene-Containing Pyridazinones for Their Antiproliferative Activity
5. Materials and Methods
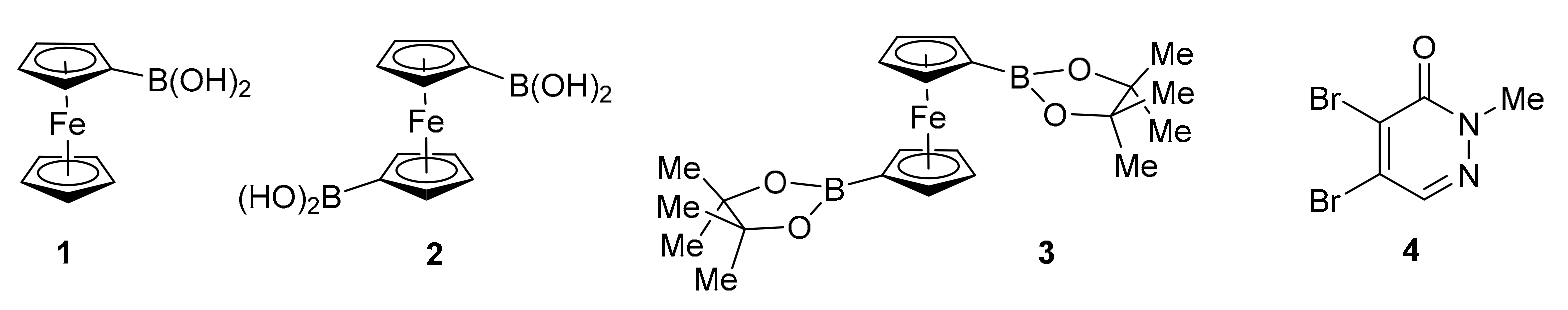
5.1. General Procedure for the Suzuki–Miyaura Reaction of 4,5-Dibromo-2-methylpyridazin-3(2H)-one 4 with Boronic Components 1, 2, 3, 5, and 11
5.1.1. 2-Methyl-4,5-diphenylpyridazin-3(2H)-one (6)
5.1.2. 4,5-Diferrocenyl-2-methylpyridazin-3(2H)-one (7)
5.1.3. 4-Ferrocenyl-2-methylpyridazin-3(2H)-one (8)
5.1.4. 5-Ferrocenyl-2-methylpyridazin-3(2H)-one (9)
5.1.5. [1′-(5-Bromo-1-methyl-6-oxo-1,6-dihydropyridazin-4-yl)ferrocenyl]boronic Acid (11)
5.1.6. 1′,2-Dimethyl-5,5′-(ferrocene-1,1′-diyl)-(4,4′-bipyridazine)-3,6′(1′H,2H)-dione (12)
5.2. Description of In Vitro Cytostasis Experiments
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Togni, A.; Hayashi, T. Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science; VCH Publishers: New York, NY, USA, 1995; ISBN 9783527290482. [Google Scholar] [CrossRef]
- Jaouen, G.; Top, S.; Vessières, A.; Alberto, R. New paradigms for synthetic pathways inspired by bioorganometallic chemistry. J. Organomet. Chem. 2000, 600, 23–36. [Google Scholar] [CrossRef]
- Ornelas, C. Application of ferrocene and its derivatives in cancer research. New J. Chem. 2011, 35, 1973–1985. [Google Scholar] [CrossRef]
- Braga, S.S.; Silva, A.M.S. A New Age for Iron: Antitumoral Ferrocenes. Organometallics 2013, 32, 5626–5639. [Google Scholar] [CrossRef]
- Tamura, H.; Miwa, M. DNA Cleaving Activity and Cytotoxic Activity of Ferricenium Cations. Chem. Lett. 1997, 26, 1177–1178. [Google Scholar] [CrossRef]
- Houlton, A.; Roberts, R.; Silver, J. Studies on the anti-tumour activity of some iron sandwich compounds. J. Organomet. Chem. 1991, 418, 107–112. [Google Scholar] [CrossRef]
- Osella, D.; Ferrali, M.; Zanello, P.; Laschi, F.; Fontani, M.; Nervi, C.; Cavigiolio, G. On the mechanism of the antitumor activity of ferrocenium derivatives. Inorganica Chim. Acta 2000, 306, 42–48. [Google Scholar] [CrossRef]
- Simon, H.-U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Károlyi, B.I.; Bősze, S.; Orbán, E.; Sohár, P.; Drahos, L.; Gál, E.; Csámpai, A. Acylated mono-, bis- and tris- Cinchona-Based Amines Containing Ferrocene or Organic Residues: Synthesis, Structure and in Vitro Antitumor Activity on Selected Human Cancer Cell Lines. Molecules 2012, 17, 2316–2329. [Google Scholar] [CrossRef] [Green Version]
- Kocsis, L.L.; Szabó, I.; Bosze, S.; Jernei, T.; Hudecz, F.; Csámpai, A. Synthesis, structure and in vitro cytostatic activity of ferrocene—Cinchona hybrids. Bioorg. Med. Chem. Lett. 2016, 26, 946–949. [Google Scholar] [CrossRef] [Green Version]
- Podolski-Renić, A.; Bősze, S.; Dinić, J.; Kocsis, L.; Hudecz, F.; Csámpai, A.; Pešić, M. Ferrocene–cinchona hybrids with triazolyl-chalcone linkers act as pro-oxidants and sensitize human cancer cell lines to paclitaxel. Metallomics 2017, 9, 1132–1141. [Google Scholar] [CrossRef]
- Latif, A.D.; Jernei, T.; Podolski-Renić, A.; Kuo, C.-Y.; Vágvölgyi, M.; Girst, G.; Zupkó, I.; Develi, S.; Ulukaya, E.; Wang, H.-C.; et al. Protoflavone-Chalcone Hybrids Exhibit Enhanced Antitumor Action through Modulating Redox Balance, Depolarizing the Mitochondrial Membrane, and Inhibiting ATR-Dependent Signaling. Antioxidants 2020, 9, 519. [Google Scholar] [CrossRef]
- Bárány, P.; Oláh, R.S.; Kovács, I.; Czuczi, T.; Szabó, C.L.; Takács, A.; Lajkó, E.; Láng, O.; Kőhidai, L.; Schlosser, G.; et al. Ferrocene-Containing Impiridone (ONC201) Hybrids: Synthesis, DFT Modelling, In Vitro Evaluation, and Structure–Activity Relationships. Molecules 2018, 23, 2248. [Google Scholar] [CrossRef] [Green Version]
- Fodor, K.J.; Hutai, D.; Jernei, T.; Takács, A.; Szász, Z.; Sulyok-Eiler, M.; Harmat, V.; Szabó, R.O.; Schlosser, G.; Hudecz, F.; et al. Novel Polycondensed Partly Saturated β-Carbolines Including Ferrocene Derivatives: Synthesis, DFT-Supported Structural Analysis, Mechanism of Some Diastereoselective Transformations and a Preliminary Study of their In Vitro Antiproliferative Effects. Molecules 2020, 25, 1599. [Google Scholar] [CrossRef] [Green Version]
- Akahane, A.; Katayama, H.; Mitsunaga, T.; Kato, T.; Kinoshita, T.; Kita, Y.; Kusunoki, T.; Terai, T.; Yoshida, K.; Shiokawa, Y. Discovery of 6-Oxo-3-(2-phenylpyrazolo[1,5-a]pyridin-3-yl)-1(6H)-pyridazinebutanoic Acid (FK 838): A Novel Non-Xanthine Adenosine A1 Receptor Antagonist with Potent Diuretic Activity. J. Med. Chem. 1999, 42, 779–783. [Google Scholar] [CrossRef]
- Meade, E.A.; Wotring, L.L.; Drach, J.C.; Townsend, L.B. Synthesis and Antiproliferative and Antiviral Activity of Carbohydrate-Modified Pyrrolo[2,3-d]pyridazin-7-one Nucleosides. J. Med. Chem. 1997, 40, 794–801. [Google Scholar] [CrossRef]
- Kimura, T.; Fujihara, Y.; Shibakawa, N.; Fujiwara, H.; Itoh, E.; Matsunobu, K.; Tabata, K.; Yasuda, H. Pyrrolopyridazine Derivatives. U.S. Patent 6063782 A, 17 July 1996. [Google Scholar]
- Siddiqui, A.A.; Mishra, R.; Shaharyar, M. Synthesis, characterization and antihypertensive activity of pyridazinone derivatives. Eur. J. Med. Chem. 2010, 45, 2283–2290. [Google Scholar] [CrossRef]
- Refaat, H.M.; Khalil, O.M.; Kadry, H.H. Synthesis and anti-inflammatory activity of certain piperazinylthienylpyridazine derivatives. Arch. Pharm. Res. 2007, 30, 803–811. [Google Scholar] [CrossRef]
- Malinka, W.; Redzicka, A.; Lozach, O. New derivatives of pyrrolo [3,4-d]pyridazinone and their anticancer effects. Farmaco 2004, 59, 457–462. [Google Scholar] [CrossRef]
- Jiang, J.; Boxer, M.B.; van der Heiden, M.G.; Shen, M.; Skoumbourdis, A.P.; Southall, N.; Veith, H.; Leister, W.; Austin, C.P.; Park, H.W.; et al. Evaluation of thieno [3,2-b]pyrrole [3,2-d]pyridazinones as activators of the tumor cell specific M2 isoform of pyruvate kinase. Bioorg. Med. Chem. Lett. 2010, 20, 3387–3393. [Google Scholar] [CrossRef] [Green Version]
- Abd El-Ghaffar, N.F.; Mohamed, M.K.; Kadah, M.S.; Radwan, A.M.; Said, G.H.; Abd Al, S.N. Synthesis and antitumor activities of some new pyridazinones containing the 2-phenyl-1H-indolyl moiety. J. Chem. Pharm. Res. 2011, 3, 248–259. [Google Scholar]
- Rathish, I.; Javed, K.; Ahmad, S.; Bano, S.; Alam, M.; Akhter, M.; Pillai, K.; Ovais, S.; Samim, M. Synthesis and evaluation of anticancer activity of some novel 6-aryl-2-(p-sulfamylphenyl)-pyridazin-3(2H)-ones. Eur. J. Med. Chem. 2012, 49, 304–309. [Google Scholar] [CrossRef]
- Gutierrez, D.A.; DeJesus, R.E.; Contreras, L.; Rodriguez-Palomares, I.A.; Villanueva, P.J.; Balderrama, K.S.; Monterroza, L.; Larragoity, M.; Varela-Ramirez, A.; Aguilera, R.J. A new pyridazinone exhibits potent cytotoxicity on human cancer cells via apoptosis and poly-ubiquitinated protein accumulation. Cell Biol. Toxicol. 2019, 35, 503–519. [Google Scholar] [CrossRef]
- Csókás, D.; Zupkó, I.; Károlyi, B.I.; Drahos, L.; Holczbauer, T.; Palló, A.; Czugler, M.; Csámpai, A. Synthesis, spectroscopy, X-ray analysis and in vitro antiproliferative effect of ferrocenylmethylene-hydrazinylpyridazin-3(2H)-ones and related ferroceno[d]pyridazin-1(2H)-ones. J. Organomet. Chem. 2013, 743, 130–138. [Google Scholar] [CrossRef]
- Csókás, D.; Károlyi, B.I.; Bősze, S.; Szabó, I.; Báti, G.; Drahos, L.; Csámpai, A. 2,3-Dihydroimidazo[1,2-b]ferroceno[d]pyridazines and a 3,4-dihydro-2H-pyrimido[1,2-b]ferroceno[d]pyridazine: Synthesis, structure and in vitro antiproliferation activity on selected human cancer cell lines. J. Organomet. Chem. 2014, 750, 41–48. [Google Scholar] [CrossRef]
- Jernei, T.; Bősze, S.; Szabó, R.; Hudecz, F.; Majrik, K.; Csámpai, A. N-ferrocenylpyridazinones and new organic analogues: Synthesis, cyclic voltammetry, DFT analysis and in vitro antiproliferative activity associated with ROS-generation. Tetrahedron 2017, 73, 6181–6192. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, T.M.; Crouse, B.A.; Thieu, T.V.; Gebreysus, C.; Finkelstein, B.L.; Sethuraman, M.R.; Dubas-Cordery, C.M.; Piotrowski, D.L. Application of cross-coupling and metalation chemistry of 3(2H)-pyridazinones to fungicide and herbicide discovery. J. Heterocycl. Chem. 2005, 42, 427–435. [Google Scholar] [CrossRef]
- Kadu, B.S. Suzuki–Miyaura cross coupling reaction: Recent advancements in catalysis and organic synthesis. Catal. Sci. Technol. 2021, 11, 1186–1221. [Google Scholar] [CrossRef]
- Shaaban, M.R.; Farghaly, T.A.; Khormi, A.Y.; Farag, A.M. Recent Advances in Synthesis and Uses of Heterocycles-based Palladium(II) Complexes as Robust, Stable, and Low-cost Catalysts for Suzuki-Miyaura Crosscouplings. Curr. Org. Chem. 2019, 23, 1601–1662. [Google Scholar] [CrossRef]
- Pagett, A.B.; Lloyd-Jones, G.C. Suzuki–Miyaura Cross-Coupling. Org. React. 2019, 100, 547–620. [Google Scholar] [CrossRef]
- Knapp, R.; Rehahn, M. Palladium-catalyzed arylation of ferrocene derivatives: A convenient high yield route to 1,1′-bis(halophenyl)ferrocenes. J. Organomet. Chem. 1993, 452, 235–240. [Google Scholar] [CrossRef]
- Imrie, C.; Loubser, C.; Engelbrecht, P.; McCleland, C.W. The use of a modified Suzuki reaction for the synthesis of monoarylferrocenes. J. Chem. Soc. Perkin Trans. 1999, 1, 2513–2523. [Google Scholar] [CrossRef]
- Hua, D.H.; McGill, J.W.; Lou, K.; Ueki, A.; Helfrich, B.; Desper, J.; Zanello, P.; Cinquantini, A.; Corsini, M.; Fontani, M. Parallel and perpendicular stacking of ferrocene rings.: Syntheses, X-ray structures, and electrochemistry of 1,8-bis-[1-(1′-phenylthio)ferrocenyl]naphthalene and 8,8′-bis-[1-(1′-phenylthio)ferrocenyl]-1,1′-binaphthyl. Inorganica Chim. Acta 2003, 350, 259–265. [Google Scholar] [CrossRef]
- Köcher, S.; Lutz, M.; Spek, A.L.; Prasad, R.; van Klink, G.P.; van Koten, G.; Lang, H. Heterobimetallic Fe–Pd and Fe–Pt NCN pincer complexes (NCN=[C6H2(CH2NMe2)2-2,6]−). Inorganica Chim. Acta 2006, 359, 4454–4462. [Google Scholar] [CrossRef] [Green Version]
- Cammidge, A.N.; Scaife, P.J.; Berber, G.; Hughes, D.L. Cofacial Porphyrin−Ferrocene Dyads and a New Class of Conjugated Porphyrin. Org. Lett. 2005, 7, 3413–3416. [Google Scholar] [CrossRef] [PubMed]
- Braga, D.; Polito, M.; Bracaccini, M.; D’Addario, D.; Tagliavini, E.; Sturba, L.; Grepioni, F. Novel Organometallic Building Blocks for Molecular Crystal Engineering. 2. Synthesis and Characterization of Pyridyl and Pyrimidyl Derivatives of Diboronic Acid, [Fe(η5-C5H4-B(OH)2)2], and of Pyridyl Boronic Acid, [Fe(η5-C5H4-4-C5H4N)(η5-C5H4-B(OH)2)]. Organometallics 2003, 22, 2142–2150. [Google Scholar] [CrossRef]
- Braga, D.; D’Addari, D.; Polito, M.; Grepioni, F. Mechanically Induced Expeditious and Selective Preparation of Disubstituted Pyridine/Pyrimidine Ferrocenyl Complexes. Organometallics 2004, 23, 2810–2812. [Google Scholar] [CrossRef]
- Peña-Cabrera, E.; Aguilar-Aguilar, A.; González-Domínguez, M.; Lager, E.; Zamudio-Vázquez, R.; Godoy-Vargas, J.; Villanueva-García, F. Simple, General, and Efficient Synthesis of Meso-Substituted Borondipyrromethenes from a Single Platform. Org. Lett. 2007, 9, 3985–3988. [Google Scholar] [CrossRef]
- Wern, C.; Ehrenreich, C.; Joosten, D.; Stein, T.V.; Buchholz, H.; König, B. Rapid Access to Bi- and Tri-Functionalized Dibenzofurans and their Application in Selective Suzuki-Miyaura Cross Coupling Reactions. Eur. J. Org. Chem. 2018, 2018, 5644–5656. [Google Scholar] [CrossRef]
- Schulz, J.; Horký, F.; Císařová, I.; Štěpnička, P. Synthesis, Structural Characterization and Catalytic Evaluation of Anionic Phosphinoferrocene Amidosulfonate Ligands. Catalysts 2017, 7, 167. [Google Scholar] [CrossRef] [Green Version]
- Waldo, J.P.; Larock, R.C. The Synthesis of Highly Substituted Isoxazoles by Electrophilic Cyclization: An Efficient Synthesis of Valdecoxib. J. Org. Chem. 2007, 72, 9643–9647. [Google Scholar] [CrossRef] [Green Version]
- Karadeniz, E.; Zora, M.; Kılıçaslan, N.Z. Facile synthesis of aryl-substituted pyridines via Suzuki–Miyaura approach. Tetrahedron 2015, 71, 8943–8952. [Google Scholar] [CrossRef]
- O’Brien, C.J.; Kantchev, E.A.B.; Valente, C.; Hadei, N.; Chass, G.A.; Lough, A.; Hopkinson, A.C.; Organ, M.G. Easily Prepared Air- and Moisture-Stable Pd–NHC (NHC=N-Heterocyclic Carbene) Complexes: A Reliable, User-Friendly, Highly Active Palladium Precatalyst for the Suzuki–Miyaura Reaction. Chem.–Eur. J. 2006, 12, 4743–4748. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Godbout, N.; Salahub, D.R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-type basis sets for local spin density functional calculations. Part I. Boron through neon, optimization technique and validation. Can. J. Chem. 1992, 70, 560–571. [Google Scholar] [CrossRef] [Green Version]
- Paier, J.; Marsman, M.; Kresse, G. Why does the B3LYP hybrid functional fail for metals? J. Chem. Phys. 2007, 127, 024103. [Google Scholar] [CrossRef]
- Ongagna, J.M.; Fouegue, A.D.T.; Amana, B.A.; D’Ambassa, G.M.; Mfomo, J.Z.; Meva’A, L.M.; Mama, D.B. B3LYP, M06 and B3PW91 DFT assignment of nd8 metal-bis-(N-heterocyclic carbene) complexes. J. Mol. Model. 2020, 26, 246. [Google Scholar] [CrossRef]
- Peng, C.; Schlegel, H.B. Combining Synchronous Transit and Quasi-Newton Methods to Find Transition States. Isr. J. Chem. 1993, 33, 449–454. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. THEOCHEM 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Chung, H.-A.; Kang, Y.-J.; Chung, J.-W.; Cho, S.-D.; Yoon, Y.-J. Retro-ene reaction. V. Functionalization of 4,5-dihalopyridazin-6-ones using 1-hydroxymethyl-4,5-dihalopyridazin-6-ones as 1-0, 3-n, 5-0 ene-adducts. J. Heterocycl. Chem. 1999, 36, 277–281. [Google Scholar] [CrossRef]
- Krajsovszky, G.; Károlyhazy, L.; Dunkel, P.; Boros, S.; Grillo, A.; Mátyus, P. Suzuki-aza-Wittig, Suzuki-condensation and aza-Wittig-electrocyclic ring-closure tandem reactions for synthesis of fused nitrogen-containing ring systems. Arkivoc 2011, 2011, 229. [Google Scholar] [CrossRef] [Green Version]
- Ganley, J.M.; Waller, D.L. Synthesis of Furo[2,3-c]pyridazines via Tandem Transition-Metal Catalysis. J. Org. Chem. 2017, 82, 12740–12745. [Google Scholar] [CrossRef]
- Yu, M.; Ledeboer, M.W.; Daniels, M.; Malojcic, G.; Tibbitts, T.T.; Gal, M.C.-L.; Pan-Zhou, X.-R.; Westerling-Bui, A.; Beconi, M.; Reilly, J.F.; et al. Discovery of a Potent and Selective TRPC5 Inhibitor, Efficacious in a Focal Segmental Glomerulosclerosis Model. ACS Med. Chem. Lett. 2019, 10, 1579–1585. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef] [Green Version]
- Amatore, C.; Jutand, A.; Le Duc, G. Kinetic Data for the Transmetalation/Reductive Elimination in Palladium-Catalyzed Suzuki-Miyaura Reactions: Unexpected Triple Role of Hydroxide Ions Used as Base. Chem.–Eur. J. 2011, 17, 2492–2503. [Google Scholar] [CrossRef]
- Molloy, J.J.; Seath, C.P.; West, M.J.; McLaughlin, C.; Fazakerley, N.J.; Kennedy, A.R.; Nelson, D.J.; Watson, A.J.B. Interrogating Pd(II) Anion Metathesis Using a Bifunctional Chemical Probe: A Transmetalation Switch. J. Am. Chem. Soc. 2017, 140, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Miyaura, N.; Yamada, K.; Suginome, H.; Suzuki, A. Novel and convenient method for the stereo- and regiospecific synthesis of conjugated alkadienes and alkenynes via the palladium-catalyzed cross-coupling reaction of 1-alkenylboranes with bromoalkenes and bromoalkynes. J. Am. Chem. Soc. 1985, 107, 972–980. [Google Scholar] [CrossRef]
- Matos, K.; Soderquist, J.A. Alkylboranes in the Suzuki−Miyaura Coupling: Stereochemical and Mechanistic Studies. J. Org. Chem. 1998, 63, 461–470. [Google Scholar] [CrossRef]
- Carrow, B.P.; Hartwig, J.F. Distinguishing Between Pathways for Transmetalation in Suzuki−Miyaura Reactions. J. Am. Chem. Soc. 2011, 133, 2116–2119. [Google Scholar] [CrossRef] [Green Version]
- Jover, J.; Fey, N.; Purdie, M.; Lloyd-Jones, G.; Harvey, J.N. A computational study of phosphine ligand effects in Suzuki–Miyaura coupling. J. Mol. Catal. A Chem. 2010, 324, 39–47. [Google Scholar] [CrossRef]
- Braga, A.; Morgon, N.H.; Ujaque, G.; Lledos, A.; Maseras, F. Computational study of the transmetalation process in the Suzuki–Miyaura cross-coupling of aryls. J. Organomet. Chem. 2006, 691, 4459–4466. [Google Scholar] [CrossRef]
- Thomas, A.A.; Wang, H.; Zahrt, A.F.; Denmark, S.E. Structural, Kinetic, and Computational Characterization of the Elusive Arylpalladium(II)boronate Complexes in the Suzuki−Miyaura Reaction. J. Am. Chem. Soc. 2017, 139, 3805–3821. [Google Scholar] [CrossRef] [PubMed]
- Cornelio, B.; Saunders, A.R.; Solomonsz, W.A.; Laronze-Cochard, M.; Fontana, A.; Sapi, J.; Khlobystov, A.N.; Rance, G.A. Palladium nanoparticles in catalytic carbon nanoreactors: The effect of confinement on Suzuki–Miyaura reactions. J. Mater. Chem. A 2015, 3, 3918–3927. [Google Scholar] [CrossRef] [Green Version]
- Navarro, O.; Kaur, H.; Mahjoor, P.; Nolan, S.P. Cross-coupling and dehalogenation reactions catalyzed by (N-heterocyclic carbene)Pd(allyl)Cl complexes. J. Org. Chem. 2004, 69, 3173–3180. [Google Scholar] [CrossRef] [PubMed]
- Viciu, M.S.; Grasa, G.A.; Nolan, S.P. Catalytic Dehalogenation of Aryl Halides Mediated by a Palladium/Imidazolium Salt System. Organometallics 2001, 20, 3607–3612. [Google Scholar] [CrossRef]
- Ahmadi, Z.; McIndoe, J.S. A mechanistic investigation of hydrodehalogenation using ESI-MS. Chem. Commun. 2013, 49, 11488–11490. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.; Lee, S. Palladium catalyzed-dehalogenation of aryl chlorides and bromides using phosphite ligands. J. Organomet. Chem. 2009, 694, 473–477. [Google Scholar] [CrossRef]
- Zeng, M.; Du, Y.; Shao, L.; Qi, C.; Zhang, X.-M. Palladium-Catalyzed Reductive Homocoupling of Aromatic Halides and Oxidation of Alcohols. J. Org. Chem. 2010, 75, 2556–2563. [Google Scholar] [CrossRef]
- Vikse, K.L.; Ahmadi, Z.; Manning, C.C.; Harrington, D.A.; McIndoe, J.S. Powerful Insight into Catalytic Mechanisms through Simultaneous Monitoring of Reactants, Products, and Intermediates. Angew. Chem. Int. Ed. 2011, 50, 8304–8306. [Google Scholar] [CrossRef]
- Sato, M.; Shigeta, H.; Sekino, M.; Akabori, S. Synthesis, some reactions, and molecular structure of the Pd(BF4)2 complex of 1,1′-bis(diphenylphosphino)ferrocene. J. Organomet. Chem. 1993, 458, 199–204. [Google Scholar] [CrossRef]
- Klapp, L.R.R.; Bruhn, C.; Leibold, M.; Siemeling, U. Ferrocene-Based Bis(guanidines): Superbases for Tridentate N,Fe,N-Coordination. Organometallics 2013, 32, 5862–5872. [Google Scholar] [CrossRef]
- Bárta, O.; Gyepes, R.; Císařová, I.; Alemayehu, A.; Štěpnička, P. Synthesis and study of Fe → Pd interactions in unsymmetric Pd(ii) complexes with phosphinoferrocene guanidine ligands. Dalton Trans. 2020, 49, 4225–4229. [Google Scholar] [CrossRef]
- Metallinos, C.; Tremblay, D.; Barrett, F.B.; Taylor, N.J. 1,1′-Bis(phosphoranylidenamino)ferrocene palladium(II) complexes: An unusual case of dative Fe → Pd bonding. J. Organomet. Chem. 2006, 691, 2044–2047. [Google Scholar] [CrossRef]
- Abubekerov, M.; Khan, S.I.; Diaconescu, P.L. Ferrocene-bis(phosphinimine) Nickel(II) and Palladium(II) Alkyl Complexes: Influence of the Fe–M (M = Ni and Pd) Interaction on Redox Activity and Olefin Coordination. Organometallics 2017, 36, 4394–4402. [Google Scholar] [CrossRef]
- Jess, K.; Baabe, D.; Bannenberg, T.; Brandhorst, K.; Freytag, M.; Jones, P.G.; Tamm, M. Ni–Fe and Pd–Fe Interactions in Nickel(II) and Palladium(II) Complexes of a Ferrocene-Bridged Bis(imidazolin-2-imine) Ligand. Inorg. Chem. 2015, 54, 12032–12045. [Google Scholar] [CrossRef]
- Fabricant, R.N.; De Larco, J.E.; Todaro, G.J. Nerve growth factor receptors on human melanoma cells in culture. Proc. Natl. Acad. Sci. USA 1977, 74, 565–569. [Google Scholar] [CrossRef] [Green Version]
- Giard, D.J.; Aaronson, S.A.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Dosik, H.; Parks, W.P. In Vitro Cultivation of Human Tumors: Establishment of Cell Lines Derived From a Series of Solid Tumors2. JNCI J. Natl. Cancer Inst. 1973, 51, 1417–1423. [Google Scholar] [CrossRef]
- Pontén, J.; Macintyre, E.H. LONG TERM CULTURE OF NORMAL AND NEOPLASTIC HUMAN GLIA. Acta Pathol. Microbiol. Scand. 1968, 74, 465–486. [Google Scholar] [CrossRef]
- Knowles, B.B.; Aden, D.P. Human Hepatoma Derived Cell Line, Process for Preparation Thereof, and Uses Therefor. U.S. Patent 4393133A, 12 July 1983. [Google Scholar]
- Frisch, G.W.; Trucks, H.B.; Schlegel, G.E.; Scuseria, M.A.; Robb, J.R.; Cheeseman, G.; Scalmani, V.; Barone, G.A.; Petersson, H.; Nakatsuji, X. Gaussian; Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Slater, T.F.; Sawyer, B.; Sträuli, U. Studies on succinate-tetrazolium reductase systems: III. Points of coupling of four different tetrazolium salts III. Points of coupling of four different tetrazolium salts. Biochim. Biophys. Acta (BBA)-Bioenerg. 1963, 77, 383–393. [Google Scholar] [CrossRef]
- Liu, Y.; Peterson, D.A.; Kimura, H.; Schubert, D. Mechanism of Cellular 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) Reduction. J. Neurochem. 2002, 69, 581–593. [Google Scholar] [CrossRef]
- Altman, F.P. Tetrazolium salts and formazans. Prog. Histochem. Cytochem. 1976, 9, 1–56. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolated Products [%] | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Reactants | Catalyst | Base | Solvent | Temp | Time | Method | 6 | 7 | 8 | 9 | 10/X | 11 | 12 |
| 1 | 4 + 5a | PdCl2(PPh3)2 (4 mol%) | Na2CO3 (2.8 eq.) | DME–H2O (4:1) | 100 °C | 10 h | A | 82 | - | - | - | - | - | - |
| 2 | 4 + 1a | PdCl2(PPh3)2 (4 mol%) | Na2CO3 (2.8 eq.) | DME–H2O (4:1) | 100 °C | 10 h | A | - | 26 | 6 | 18 | 7/15 c | - | - |
| 3 | 4 + 1a | Pd(PPh3)4 (4 mol%) | Na2CO3 (3.0 eq.) | Toluene–H2O (3:1) | 100 °C | 8 h | B | 21 | 5 | 23s | 5/20 c | - | - | |
| 4 | 4 + 1a | Pd(PPh3)4 (4 mol%) | Na2CO3 (3.0 eq.) | Toluene–H2O (3:1) | 100 °C | 12 h | C | 16 | 10 | 22 | 7/20 c | - | ||
| 5 | 4 + 1a | PdCl2(PPh3)2 (4 mol%) | K2CO3 (3.0 eq.) | DMF–H2O (4:1) | 80 °C | 12 h | D | 38 | 6 | 24 | 9/10 c | - | - | |
| 6 | 4 + 1a | Pd–PEPPSIiP (4 mol%) | K2CO3 (3.0 eq.) | DMF–H2O (4:1) | 80 °C | 12 h | E | 45 | 7 | 18 | 12/12 c | - | - | |
| 7 | 4 + 1a | PdCl2dppf (4 mol%) | K2CO3 (3.0 eq.) | DMF–H2O (4:1) | 80 °C | 12 h | F | 5 | 18 | 50 | 7/- | - | ||
| 8 | 4 + 5a | PdCl2dppf (4 mol%) | K2CO3 (3.0 eq.) | DMF–H2O (4:1) | 80 °C | 12 h | F | 79 | - | - | - | -/- | - | - |
| 9 | 4 + 2b | PdCl2(PPh3)2 (4 mol%) | Na2CO3 (2.8 eq.) | DME–H2O (4:1) | 100 °C | 10 h | A | - | - | - | 19 | -/- | 36 | 15 |
| 10 | 4 + 2b | PdCl2(PPh3)2 (4 mol%) | K2CO3 (3.0 eq.) | DMF–H2O (4:1) | 80 °C | 12 h | D | - | - | - | 14 | -/- | 56 | 20 |
| 11 | 4 + 2b | Pd–PEPPSIiP (4 mol%) | K2CO3 (3.0 eq.) | DMF–H2O (4:1) | 80 °C | 12 h | E | - | - | 15 | -/- | 44 | 28 | |
| 12 | 4 + 2b | PdCl2dppf (4 mol%) | K2CO3 (3.0 eq.) | DMF–H2O (4:1) | 80 °C | 12 h | F | - | - | 47 | -/- | 7 | 9 | |
| 13 | 4 + 11a | PdCl2(PPh3)2 (4 mol%) | K2CO3 (3.0 eq.) | DMF–H2O (4:1) | 80 °C | 12 h | D | - | - | 34 | - | -/- | 27 d | 24 |
| 14 | 4 + 11a | Pd–PEPPSIiP (4 mol%) | K2CO3 (3.0 eq.) | DMF–H2O (4:1) | 80 °C | 12 h | E | - | 14 | - | -/- | 10 d | 21 | |
| 15 | 4 + 11a | PdCl2dppf (4 mol%) | K2CO3 (3.0 eq.) | DMF–H2O (4:1) | 80 °C | 12 h | F | - | 55 | - | -/- | 12 d | 10 | |
| 16 | 4 + 3b | PdCl2(PPh3)2 (4 mol%) | K2CO3 (3.0 eq.) | DMF–H2O (4:1) | 80 °C | 12 h | D | - | 22 | 7 | 24 | 8/12 c | - | - |
| 17 | 4 + 3b | Pd–PEPPSIiP (4 mol%) | K2CO3 (3.0 eq.) | DMF–H2O (4:1) | 80 °C | 12 h | E | 52 | 6 | 15 | 6/17 c | - | - | |
| 18 | 4 + 3b | PdCl2dppf (4 mol%) | K2CO3 (3.0 eq.) | DMF–H2O (4:1) | 80 °C | 12 h | F | 5 | 9 | 42 | 14/8 c | - | - | |
| Compounds | IC50 [µM] ± SD | |||
|---|---|---|---|---|
| A2058 | A431 | U87 | HepG2 | |
| 7 | 12.20 ± 0.28 | 1.82 ± 0.01 | >50 | 2.57 ± 0.33 |
| 8 | >50 | 45.68 ± 4.84 | >50 | >50 |
| 9 | >50 | 5.16 ± 0.34 | >50 | 3.61 ± 0.62 |
| 11 | >50 | 10.38 ± 0.52 | >50 | >50 |
| 12 | >50 | 2.93 ± 0.11 | >50 | 2.49 ± 0.19 |
| 6 (reference) | >50 | >50 | >50 | >50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alaoui, N.-E.E.; Boulhaoua, M.; Hutai, D.; Oláh-Szabó, R.; Bősze, S.; Hudecz, F.; Csámpai, A. Synthetic and DFT Modeling Studies on Suzuki–Miyaura Reactions of 4,5-Dibromo-2-methylpyridazin-3(2H)-one with Ferrocene Boronates, Accompanied by Hydrodebromination and a Novel Bridge-Forming Annulation In Vitro Cytotoxic Activity of the Ferrocenyl–Pyridazinone Products. Catalysts 2022, 12, 578. https://doi.org/10.3390/catal12060578
Alaoui N-EE, Boulhaoua M, Hutai D, Oláh-Szabó R, Bősze S, Hudecz F, Csámpai A. Synthetic and DFT Modeling Studies on Suzuki–Miyaura Reactions of 4,5-Dibromo-2-methylpyridazin-3(2H)-one with Ferrocene Boronates, Accompanied by Hydrodebromination and a Novel Bridge-Forming Annulation In Vitro Cytotoxic Activity of the Ferrocenyl–Pyridazinone Products. Catalysts. 2022; 12(6):578. https://doi.org/10.3390/catal12060578
Chicago/Turabian StyleAlaoui, Nour-Eddine El, Mohammed Boulhaoua, Dániel Hutai, Rita Oláh-Szabó, Szilvia Bősze, Ferenc Hudecz, and Antal Csámpai. 2022. "Synthetic and DFT Modeling Studies on Suzuki–Miyaura Reactions of 4,5-Dibromo-2-methylpyridazin-3(2H)-one with Ferrocene Boronates, Accompanied by Hydrodebromination and a Novel Bridge-Forming Annulation In Vitro Cytotoxic Activity of the Ferrocenyl–Pyridazinone Products" Catalysts 12, no. 6: 578. https://doi.org/10.3390/catal12060578
APA StyleAlaoui, N. -E. E., Boulhaoua, M., Hutai, D., Oláh-Szabó, R., Bősze, S., Hudecz, F., & Csámpai, A. (2022). Synthetic and DFT Modeling Studies on Suzuki–Miyaura Reactions of 4,5-Dibromo-2-methylpyridazin-3(2H)-one with Ferrocene Boronates, Accompanied by Hydrodebromination and a Novel Bridge-Forming Annulation In Vitro Cytotoxic Activity of the Ferrocenyl–Pyridazinone Products. Catalysts, 12(6), 578. https://doi.org/10.3390/catal12060578