
Iridium(triNHC)-Catalyzed Transfer Hydrogenation of Glycerol Carbonate without Exogenous Reductants



Abstract
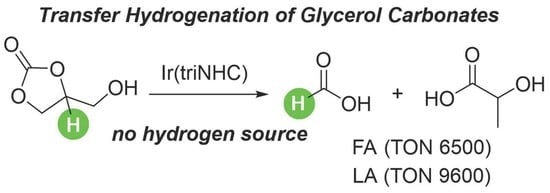
:
1. Introduction
2. Results and Discussion

3. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous catalysis for sustainable hydrogen storage in formic acid and alcohols. Chem. Rev. 2018, 118, 372–433. [Google Scholar] [CrossRef] [PubMed]
- Jessop, P.G.; Ikariya, T.; Noyori, R. Homogeneous hydrogenation of carbon-dioxide. Chem. Rev. 1995, 95, 259–272. [Google Scholar] [CrossRef]
- Jessop, P.G.; Joó, F.; Tai, C.C. Recent advances in the homogeneous hydrogenation of carbon dioxide. Coord. Chem. Rev. 2004, 248, 2425–2442. [Google Scholar] [CrossRef]
- Wang, W.-H.; Himeda, Y.; Muckerman, J.T.; Manbeck, G.F.; Fujita, E. CO2 Hydrogenation to formate and methanol as an alternative to photo- and electrochemical CO2 reduction. Chem. Rev. 2015, 115, 12936–12973. [Google Scholar] [CrossRef]
- Dabral, S.; Schaub, T. The use of carbon dioxide (CO2) as a building block in organic synthesis from an industrial perspective. Adv. Synth. Catal. 2019, 361, 223–246. [Google Scholar] [CrossRef] [Green Version]
- Balaraman, E.; Gunanathan, C.; Zhang, J.; Shimon, L.J.W.; Milstein, D. Efficient hydrogenation of organic carbonates, carbamates and formates indicates alternative routes to methanol based on CO2 and CO. Nat. Chem. 2011, 3, 609–614. [Google Scholar] [CrossRef]
- Krall, E.M.; Klein, T.W.; Andersen, R.J.; Nett, A.J.; Glasgow, R.W.; Reader, D.S.; Dauphinais, B.C.; Mc Ilrath, S.P.; Fischer, A.A.; Carney, M.J.; et al. Controlled hydrogenative depolymerization of polyesters and polycarbonates catalyzed by ruthenium(II) PNN pincer complexes. Chem. Commun. 2014, 50, 4884–4887. [Google Scholar] [CrossRef]
- Han, Z.; Rong, L.; Wu, J.; Zhang, L.; Wang, Z.; Ding, K. Catalytic hydrogenation of cyclic carbonates: A practical approach from CO2 and epoxides to methanol and diols. Angew. Chem. Int. Ed. 2012, 51, 13041–13045. [Google Scholar] [CrossRef]
- Li, Y.; Junge, K.; Beller, M. Improving the efficiency of the hydrogenation of carbonates and carbon dioxide to methanol. ChemCatChem 2013, 5, 1072–1074. [Google Scholar] [CrossRef]
- Wu, X.; Ji, L.; Ji, Y.; Elageed, E.H.M.; Gao, G. Hydrogenation of ethylene carbonate catalyzed by lutidine-bridged N-heterocyclic carbene ligands and ruthenium precursors. Catal. Commun. 2016, 85, 57–60. [Google Scholar] [CrossRef]
- Kumar, A.; Janes, T.; Espinosa-Jalapa, N.A.; Milstein, D. Manganese catalyzed hydrogenation of organic carbonates to methanol and alcohols. Angew. Chem. Int. Ed. 2018, 57, 12076–12080. [Google Scholar] [CrossRef] [PubMed]
- Zubar, V.; Levedev, Y.; Azofra, L.M.; Cavallo, L.; El-Sepelgy, O.; Rueping, M. Hydrogenation of CO2-derived carbonates and polycarbonates to methanol and diols by metal-ligand cooperative manganese catalysis. Angew. Chem. Int. Ed. 2018, 57, 13439–13443. [Google Scholar] [CrossRef] [PubMed]
- Kaithal, A.; Hölscher, M.; Leitner, W. Catalytic hydrogenation of cyclic carbonates using manganese complexes. Angew. Chem. Int. Ed. 2018, 57, 13449–13453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahiya, P.; Gangwar, M.K.; Sundararaju, B. Well-defined Cp*Co(III)-catalyzed hydrogenation of carbonates and polycarbonates. ChemCatChem 2021, 13, 934–939. [Google Scholar] [CrossRef]
- Kim, S.H.; Hong, S.H. Transfer hydrogenation of organic formates and cyclic carbonates: An alternative route to methanol from carbon dioxide. ACS Catal. 2014, 4, 3630–3636. [Google Scholar] [CrossRef]
- Liu, X.; de Vries, J.G.; Werner, T. Transfer hydrogenation of cyclic carbonates and polycarbonate to methanol and diols by iron pincer catalysts. Green Chem. 2019, 21, 5248–5255. [Google Scholar] [CrossRef]
- Szewczyk, M.; Magre, M.; Zubar, V.; Rueping, M. Reduction of cyclic and linear organic carbonates using a readily available magnesium catalyst. ACS Catal. 2019, 9, 11634–11639. [Google Scholar] [CrossRef]
- Bobbink, F.D.; Menoud, F.; Dyson, P.J. Synthesis of methanol and diols from CO2 via cyclic carbonates under metal-free, ambient pressure, and solvent-free conditions. ACS Sustain. Chem. Eng. 2018, 6, 12119–12123. [Google Scholar] [CrossRef]
- Feghali, E.; Cantat, T. Room temperature organocatalyzed reductive depolymerization of waste polyethers, polyesters, and polycarbonates. ChemSusChem 2015, 8, 980–984. [Google Scholar] [CrossRef]
- Zassinovich, G.; Mestroni, G.; Gladiali, S. Asymmetric hydrogen transfer reactions promoted by homogeneous transition metal catalysis. Chem. Rev. 1992, 92, 1051–1069. [Google Scholar] [CrossRef]
- Díaz-Álvarez, A.E.; Cadierno, V. Glycerol: A promising green solvent and reducing agent for metal-catalyzed transfer hydrogenation reactions and nanoparticles formation. Appl. Sci. 2013, 3, 55–69. [Google Scholar] [CrossRef]
- Crabtree, R.H. Transfer hydrogenation with glycerol as H-donor: Catalyst activation, deactivation and homogeneity. ACS Sustain. Chem. Eng. 2019, 7, 15845–15853. [Google Scholar] [CrossRef]
- Cheong, Y.-J.; Sung, K.; Park, S.; Jung, J.; Jang, H.-Y. Valorization of chemical wates: Ir(biscarbene)-catalyzed transfer hydrogenation of inorganic carbonates using glycerol. ACS Sustain. Chem. Eng. 2020, 8, 6972–6978. [Google Scholar] [CrossRef]
- Sung, K.; Lee, M.-h.; Cheong, Y.-J.; Jang, H.-Y. Ir(triscarbene)-catalyzed sustainable transfer hydrogenation of levulinic acid to γ-valerolactone. Appl. Organomet. Chem. 2020, 35, e6105. [Google Scholar] [CrossRef]
- Cheong, Y.-J.; Sung, K.; Kim, J.-A.; Kim, Y.K.; Yoon, W.; Yun, H.; Jang, H.-Y. Iridium(NHC)-catalyzed sustainable transfer hydrogenation of CO2 and inorganic carbonates. Catalysts 2021, 11, 695. [Google Scholar] [CrossRef]
- Christy, S.; Noschese, A.; Lomelí-Rodrigeuz, M.; Greeves, N.; Lopez-Sanchez, J.A. Recent progress in the synthesis and applications of glycerol carbonate. Curr. Opin. Green Sustain. Chem. 2018, 14, 99–107. [Google Scholar] [CrossRef]
- Szöri, M.; Giri, B.R.; Wang, Z.; Dawood, A.E.; Viskolcz, B.; Farooq, A. Glycerol carbonate as a fuel additive for a sustainable future. Sustain. Energy Fuels 2018, 2, 2171–2178. [Google Scholar] [CrossRef] [Green Version]
- Loges, B.; Boddien, A.; Gartner, F.; Junge, H.; Beller, M. Catalytic generation of hydrogen from formic acid and its derivatives: Useful hydrogen storage materials. Top. Catal. 2010, 53, 902–914. [Google Scholar] [CrossRef]
- Grasemann, M.; Laurenczy, G. Formic acid as a hydrogen source-recent developments and future trends. Energy Environ. Sci. 2012, 5, 8171–8181. [Google Scholar] [CrossRef]
- Mellmann, D.; Sponholz, P.; Junge, H.; Beller, M. Formic acid as a hydrogen storage material-development of homogeneous catalysts for selective hydrogen release. Chem. Soc. Rev. 2016, 45, 3954–3988. [Google Scholar] [CrossRef]
- Bottari, G.; Barta, K. Lactic acid and hydrogen from glycerol via acceptorless dehydrogenation using homogeneous catalysts. Recycl. Catal. 2015, 2, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Frey, G.D.; Rentzsch, C.F.; von Preysing, D.; Scherg, T.; Mühlhofer, M.; Herdtweck, E.; Herrmann, W.A. Rhodium and iridium complexes of N-heterocyclic carbenes: Structural investigations and their catalytic properties in the borylation reaction. J. Organomet. Chem. 2006, 691, 5725–5738. [Google Scholar] [CrossRef]
- Sharninghausen, L.S.; Campos, J.; Manas, M.G.; Crabtree, R.H. Efficient selective and atom economic catalytic conversion of glycerol to lactic acid. Nat. Commun. 2014, 5, 5084. [Google Scholar] [CrossRef] [PubMed]
- Cheong, Y.-J.; Sung, K.; Kim, J.-A.; Kim, Y.K.; Jang, H.-Y. Highly efficient iridium-catalyzed production of hydrogen and lactate from glycerol: Rapid hydrogen evolution by bimetallic iridium catalysts. Eur. J. Inorg. Chem. 2020, 2020, 4064–4068. [Google Scholar] [CrossRef]
- Sharninghausen, L.S.; Mercado, B.Q.; Crabtree, R.H.; Hazari, N. Selective conversionof glycerol to lactic acid with iron pincer precatalysts. Chem. Commun. 2015, 51, 16201–16204. [Google Scholar] [CrossRef]
- Lu, Z.; Cherepakhin, V.; Demianets, I.; Lauridsen, P.J.; Williams, T.J. Iridium-based hydride transfer catalysts: From hydrogen storage to fine chemicals. Chem. Commun. 2018, 54, 7711–7724. [Google Scholar] [CrossRef]
- Truscott, B.J.; Kruger, H.; Webb, P.B.; Bühl, M.; Nolan, S.P. The mechanism of CO2 insertion into iridium(I) hydroxide and alkoxide bonds: A kinetics and computational study. Chem. Eur. J. 2015, 21, 6930–6935. [Google Scholar] [CrossRef]
- Vummaleti, S.V.C.; Talarico, G.; Nolan, S.P.; Cavallo, L.; Poater, A. Mechanism of CO2 fixation by IrI-X bonds (X = OH, OR, N, C). Eur. J. Inorg. Chem. 2015, 2015, 4653–4657. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
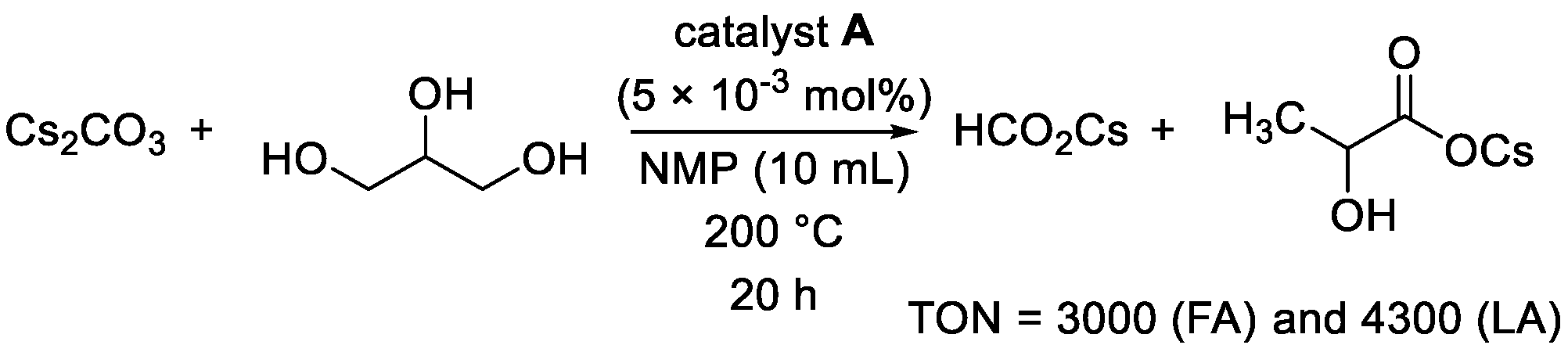{kind=link}
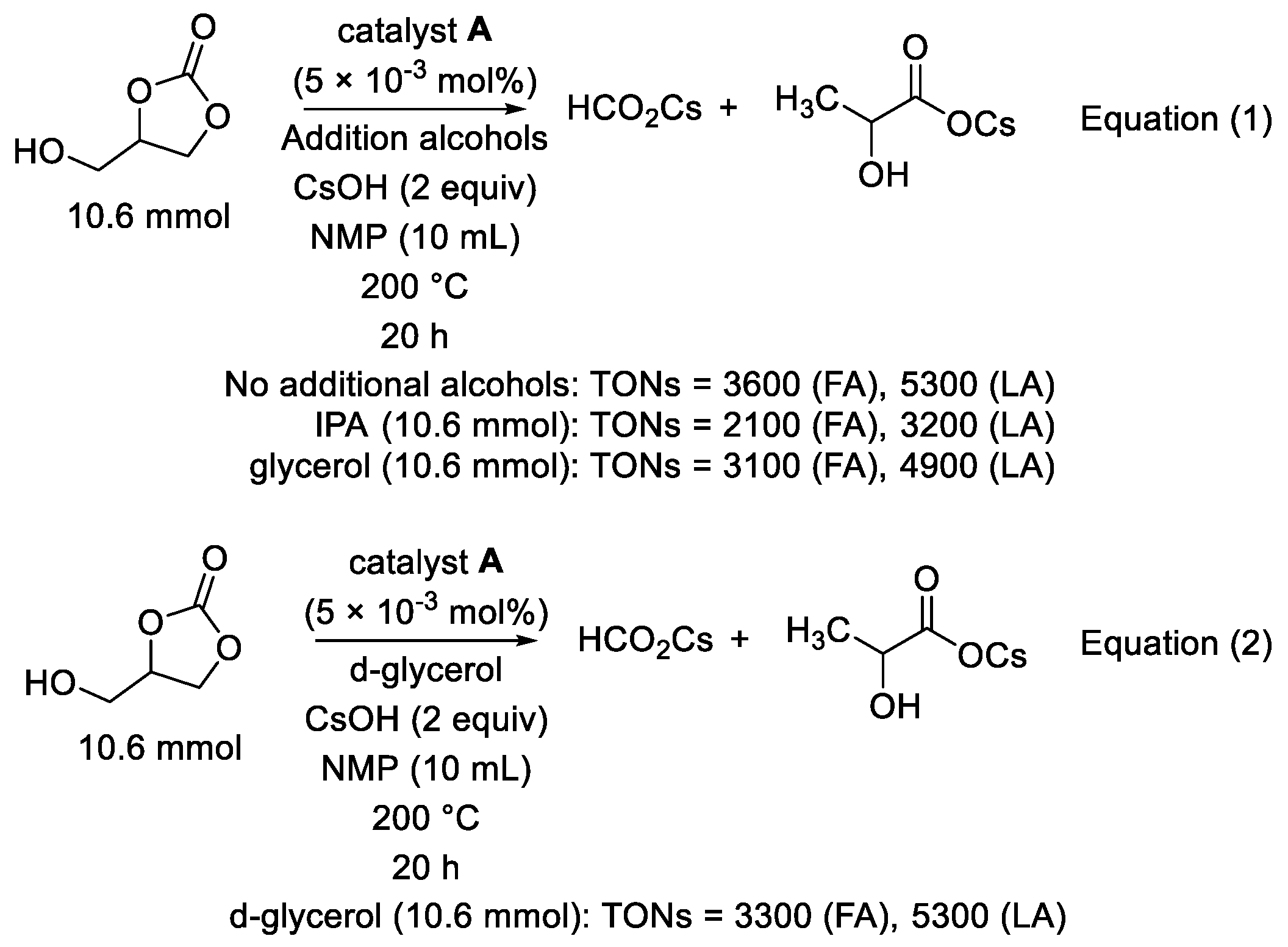{kind=link}
{kind=link}
 | |||||
| Entry | Catalyst (mol%) | Base (Equiv.) | Solvent | Formate (TON) | Lactate (TON) |
|---|---|---|---|---|---|
| 1 | A (2.5 × 10−3) | CsOH⋅H2O (2) | NMP | 6500 | 9600 |
| 2 | A (5.0 × 10−3) | CsOH⋅H2O (2) | NMP | 3600 | 5300 |
| 3 | A (1.0 × 10−2) | CsOH⋅H2O (2) | NMP | 980 | 1900 |
| 4 | A (5.0 × 10−3) | CsOH⋅H2O (2) | H2O | 21 | 89 |
| 5 | A (5.0 × 10−3) | KOH (2) | NMP | 12 | 65 |
| 6 | A (5.0 × 10−3) | NaOH (2) | NMP | 160 | 130 |
| 7 | A (5.0 × 10−3) | CsOH⋅H2O (3) | NMP | 930 | 4800 |
| 8 | A (5.0 × 10−3) | -- | NMP | -- | -- |
| 9 | -- | CsOH⋅H2O (2) | NMP | -- | -- |
| 10 | B (1.0 × 10−2) | CsOH⋅H2O (2) | NMP | 2400 | 3600 |
| 11 | B (5.0 × 10−3) | CsOH⋅H2O (2) | NMP | 5200 | 7400 |
| 12 | C (1.0 × 10−2) | CsOH⋅H2O (2) | NMP | 2600 | 4200 |
| 13 | D (1.0 × 10−2) | CsOH⋅H2O (2) | NMP | 2100 | 3400 |
| 14 | E (5.0 × 10−3) | CsOH⋅H2O (2) | NMP | 580 | 2400 |
| 15 | F (1.0 × 10−2) | CsOH⋅H2O (2) | NMP | 310 | 1700 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheong, Y.-J.; Lee, M.-h.; Byeon, H.; Park, J.; Yu, S.; Jang, H.-Y. Iridium(triNHC)-Catalyzed Transfer Hydrogenation of Glycerol Carbonate without Exogenous Reductants. Catalysts 2022, 12, 656. https://doi.org/10.3390/catal12060656
Cheong Y-J, Lee M-h, Byeon H, Park J, Yu S, Jang H-Y. Iridium(triNHC)-Catalyzed Transfer Hydrogenation of Glycerol Carbonate without Exogenous Reductants. Catalysts. 2022; 12(6):656. https://doi.org/10.3390/catal12060656
Chicago/Turabian StyleCheong, Yeon-Joo, Mi-hyun Lee, Heemin Byeon, Jiyong Park, Sungju Yu, and Hye-Young Jang. 2022. "Iridium(triNHC)-Catalyzed Transfer Hydrogenation of Glycerol Carbonate without Exogenous Reductants" Catalysts 12, no. 6: 656. https://doi.org/10.3390/catal12060656
APA StyleCheong, Y. -J., Lee, M. -h., Byeon, H., Park, J., Yu, S., & Jang, H. -Y. (2022). Iridium(triNHC)-Catalyzed Transfer Hydrogenation of Glycerol Carbonate without Exogenous Reductants. Catalysts, 12(6), 656. https://doi.org/10.3390/catal12060656