
Solvent Effect in Catalytic Lignin Hydrogenolysis



Abstract
:
1. Introduction
2. Solvent Effect in Lignin Depolymerization
2.1. Hydrogenolysis
2.2. Choice of Solvent
2.3. Supercritical Fluids
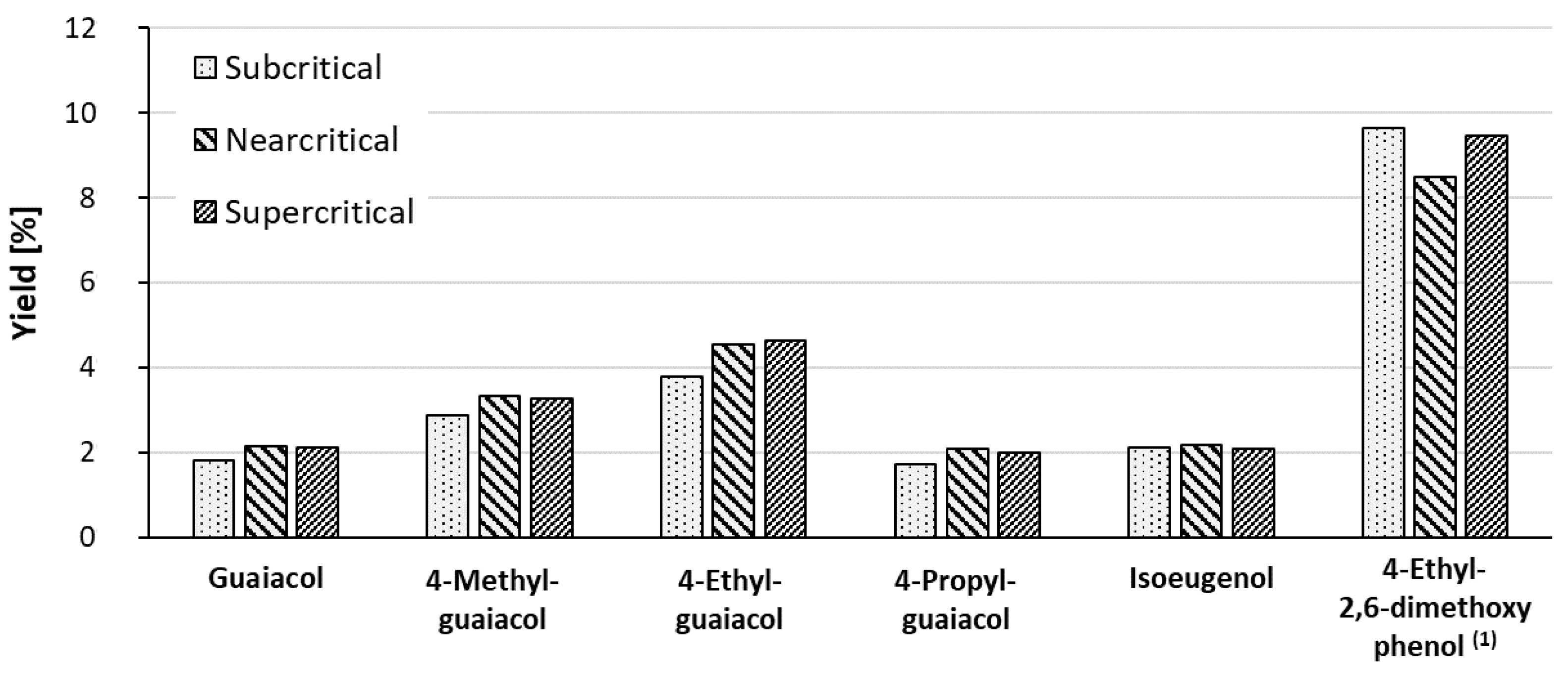
2.4. Examples of Lignin Conversion in Near-Critical or Supercritical Fluids
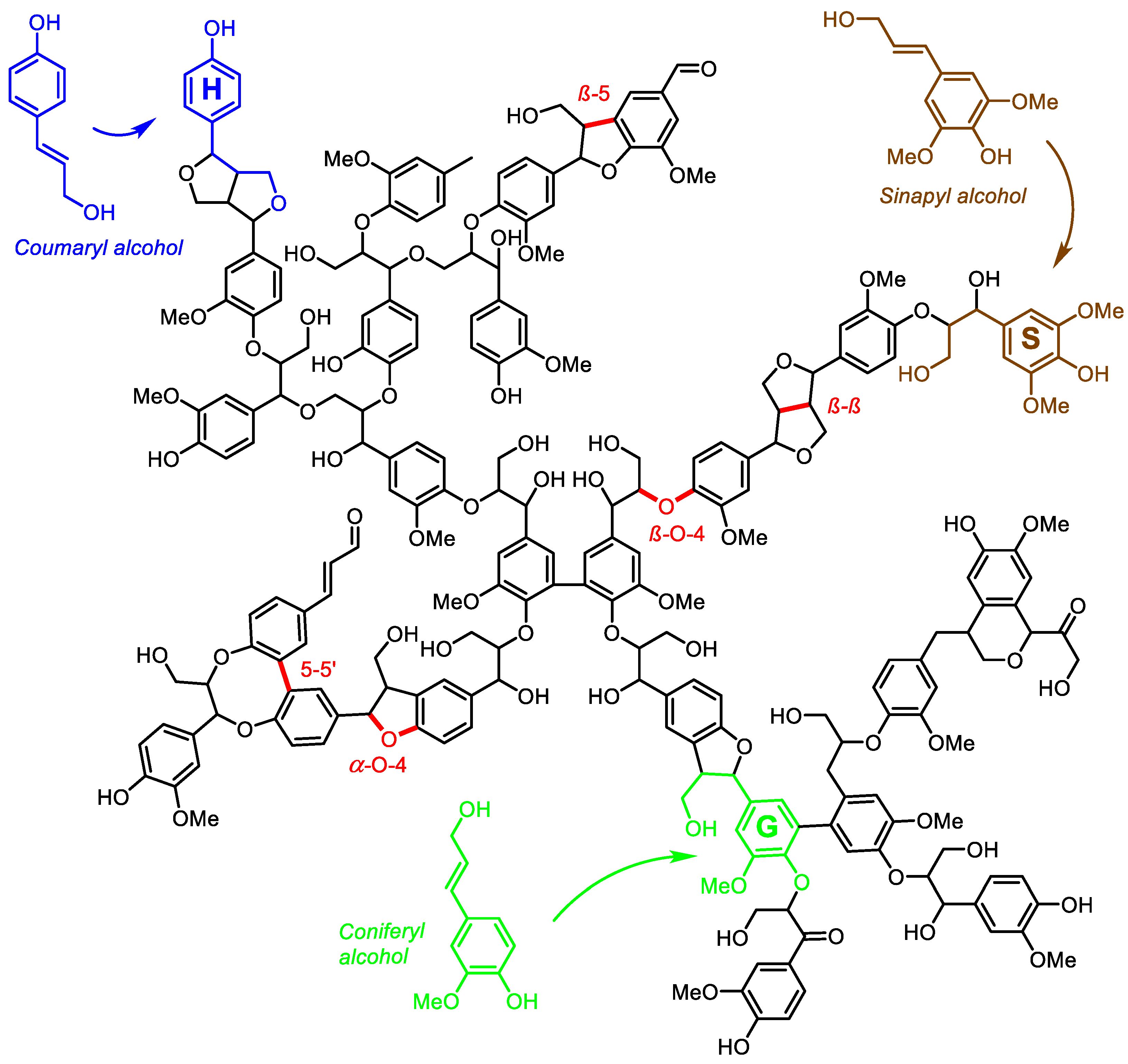
3. Cleavage of Lignin by Hydrogenolysis
3.1. Lignin Depolymerization in Methanol
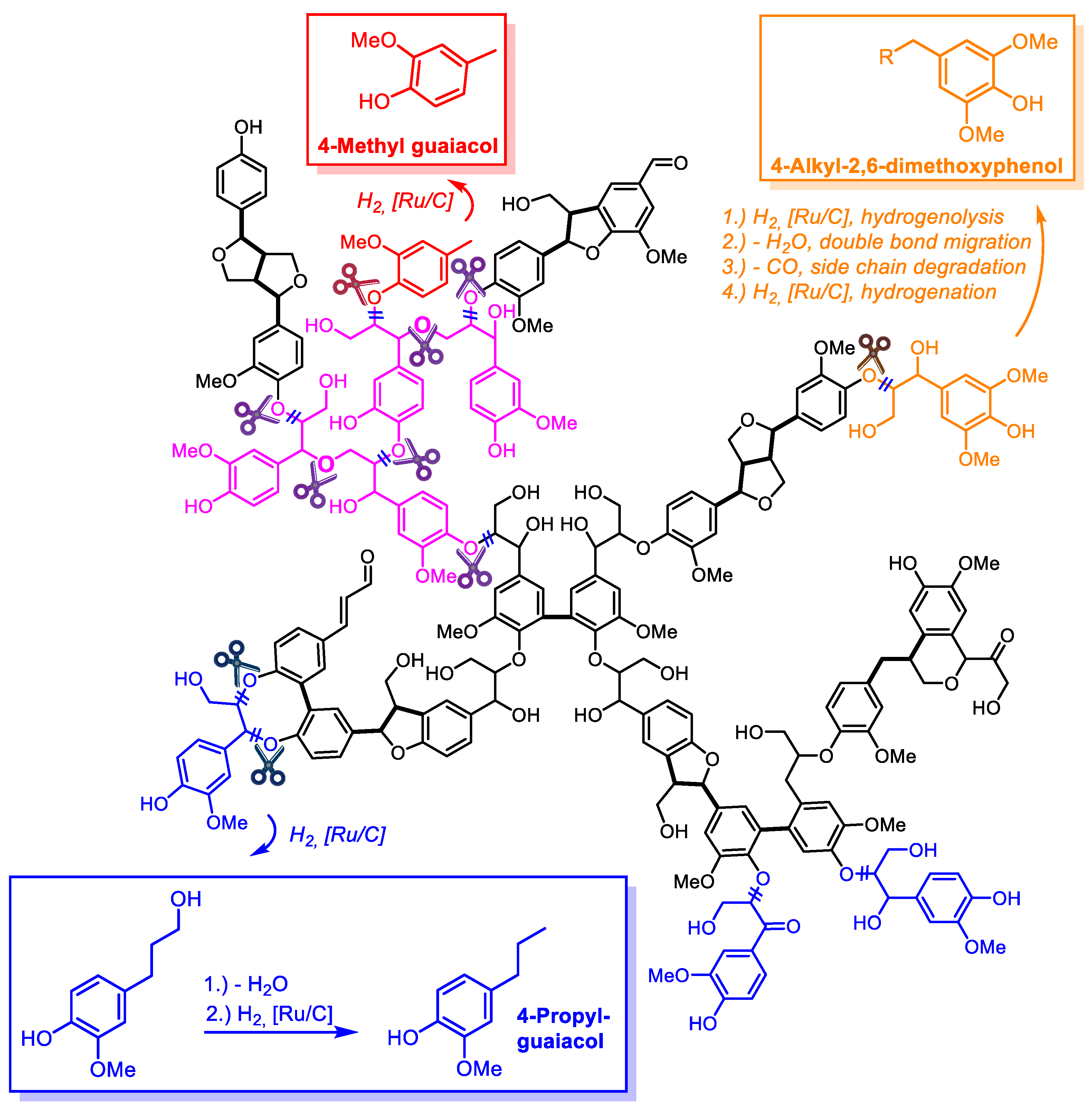
3.2. Product Spectrum of Lignin Depolymerization in Methanol
3.3. Hydrogenolytic Cleavage of Aryl Ether Moieties in Lignin Depolymerization
3.4. Role of Methanol Solvent in the Hydrogenolysis of Lignin
- (i)
- Of all alcohols, methanol has the highest solubility for hydrogen [53]. Nevertheless, the solubility of hydrogen in methanol (5.21×10−5 mol.cm−3 at 1.56 MPa and 278 K, [54]) is very low but increases linearly with pressure. High pressure together with conditions close to the supercritical point (vide supra) ensure high availability of hydrogen necessary for efficient hydrogenolysis reactions and avoid potential, mass transfer limitations from occurring. The rapid saturation of unsaturated entities in the chemical intermediates of lignin degradation, such as aldehyde, keto, and alkene groups, is essential to prevent the repolymerization of lignin fragments and concomitant formation of new hard-to-cleave carbon bonds.
- (ii)
- Through polar interactions and hydrogen bonding, methanol can bind to the ether moieties and hydroxyl groups of lignin, thereby opening the structure of lignin for coordination with the catalyst. The phenolic oxygen atom has a strong propensity as a hydrogen bond acceptor [55] that matches well with the hydrogen bond donor ability of methanol. Furthermore, attractive O-H…π-interactions between the OH group of methanol and the aromatic rings of lignin [56] may contribute to the solvent interactions. Such weak interactions resemble conventional hydrogen bonds (see also [57]) and due to their high number, can contribute substantially to the solvent interactions.
- (iii)
- Likewise, the reaction intermediates and their generated products are stabilized by the solvent methanol through the formation of polar interactions and hydrogen-bonding interactions. In particular, aldehydes and ketones that are formed as reaction intermediates may be converted to the corresponding ortho esters in a reversible fashion. In this way, the keto groups are protected against nucleophilic reactions until they become hydrogenated or undergo hydrogenolysis on the catalyst surface.
- (iv)
- Similarly, the high dipole moment of methanol may lower the energy of polar transition states, thereby enhancing chemical rates. Also, the formation of solvent cages [58], similar to the solvent cages observed for ionic liquids [59], may occur that encapsulate [60] lignin fragments until these are dismantled in further hydrogenolysis reactions.
- (v)
- The high solubility of reaction intermediates and generated products in methanol enhances easy separation from the catalyst surface. Thus, potential catalyst poisoning resulting from the strong coordination of product molecules to the catalyst surface is reduced. Please note that the product molecules may bind strongly in a chelating fashion through several oxygen atoms to the ruthenium atoms exposed at the catalyst surface.
3.5. Chemical Transformations in the Lignin Depolymerization in Methanol
4. Materials and Methods
4.1. Materials
4.2. General Procedure
4.3. Procedure Base-Catalyzed Reaction
4.4. Instruments and Analytical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tomkins, P.; Müller, T.E. Evaluating the carbon inventory, carbon fluxes and carbon cycles for a long-term sustainable world. Green Chem. 2019, 21, 3994–4013. [Google Scholar] [CrossRef]
- Kätelhön, A.; Meys, R.; Deutz, S.; Suh, S.; Bardow, A. Climate change mitigation potential of carbon capture and utilization in the chemical industry. Proc. Natl. Acad. Sci. USA 2019, 116, 11187–11194. [Google Scholar] [CrossRef] [Green Version]
- Głowacki, E.D.; Voss, G.; Leonat, L.; Irimia-Vladu, M.; Bauer, S.; Sariciftci, N.S. Indigo and Tyrian Purple—From Ancient Natural Dyes to Modern Organic Semiconductors. Isr. J. Chem. 2012, 52, 540–551. [Google Scholar] [CrossRef]
- Sabesan, M.N.; Harper, E.T. Are aromatic residues essential at the “active sites” of peptide hormones? J. Theor. Biol. 1980, 83, 457–467. [Google Scholar] [CrossRef]
- de la Rosa, L.A.; Moreno-Escamilla, J.O.; Rodrigo-García, J.; Alvarez-Parrilla, E. Chapter 12—Phenolic Compounds. In Postharvest Physiology and Biochemistry of Fruits and Vegetables; Yahia, E.M., Ed.; Woodhead Publishing: Sawston, UK, 2019; pp. 253–271. [Google Scholar]
- Calvo-Flores, F.G.; Dobado, J.A.; Isac-García, J.; Martín-MartíNez, F.J. Lignin and Lignans as Renewable Raw Materials: Chemistry, Technology and Applications; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015. [Google Scholar]
- Cheng, K.; Hagiopol, C. Natural Polyphenols from Wood, Tannin and Lignin—An Industrial Perspective; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- L’udmila, H.; Michal, J.; Andrea, Š.; Aleš, H. Lignin, potential products and their market value. Wood Res. 2015, 60, 973–986. [Google Scholar]
- Bajwa, D.S.; Pourhashem, G.; Ullah, A.H.; Bajwa, S.G. A concise review of current lignin production, applications, products and their environmental impact. Ind. Crops Prod. 2019, 139, 111526. [Google Scholar] [CrossRef]
- Gou, M.; Ran, X.; Martin, D.W.; Liu, C.-J. The scaffold proteins of lignin biosynthetic cytochrome P450 enzymes. Nat. Plants 2018, 4, 299–310. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, Q.; Lee, D.-J. Kraft lignin biorefinery: A perspective. Bioresour. Technol. 2018, 247, 1181–1183. [Google Scholar] [CrossRef]
- Lignin, Biosynthesis and Transformation for Industrial Applications; Springer International Publishing: Cham, Switzerland, 2020; 298p.
- Production of Biofuels and Chemicals from Lignin. In Biofuels and Biorefineries; Springer: Singapore, 2016; Volume XV, 435p.
- del Río, J.C.; Rencoret, J.; Gutiérrez, A.; Elder, T.; Kim, H.; Ralph, J. Lignin Monomers from beyond the Canonical Monolignol Biosynthetic Pathway: Another Brick in the Wall. ACS Sustain. Chem. Eng. 2020, 8, 4997–5012. [Google Scholar] [CrossRef]
- Lignin Chemistry. In Topics in Current Chemistry Collections (TCCC); Springer: Berlin/Heidelberg, Germany, 2020; Volume VI, 271p.
- Balakshin, M.; Capanema, E.A.; Zhu, X.; Sulaeva, I.; Potthast, A.; Rosenau, T.; Rojas, O.J. Spruce milled wood lignin: Linear, branched or cross-linked? Green Chem. 2020, 22, 3985–4001. [Google Scholar] [CrossRef]
- Rinaldi, R.; Jastrzebski, R.; Clough, M.T.; Ralph, J.; Kennema, M.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Paving the Way for Lignin Valorisation: Recent Advances in Bioengineering, Biorefining and Catalysis. Angew. Chem. Int. Ed. 2016, 55, 8164–8215. [Google Scholar] [CrossRef] [Green Version]
- Azadi, P.; Inderwildi, O.R.; Farnood, R.; King, D.A. Liquid fuels, hydrogen and chemicals from lignin: A critical review. Renew. Sustain. Energy Rev. 2013, 21, 506–523. [Google Scholar] [CrossRef]
- Cheremisinoff, N.P.; Rosenfeld, P.E. Chapter 6—Sources of air emissions from pulp and paper mills. In Handbook of Pollution Prevention and Cleaner Production; Cheremisinoff, N.P., Rosenfeld, P.E., Eds.; William Andrew Publishing: Oxford, UK, 2010; pp. 179–259. [Google Scholar]
- Chakar, F.S.; Ragauskas, A.J. Review of current and future softwood kraft lignin process chemistry. Ind. Crops Prod. 2004, 20, 131–141. [Google Scholar] [CrossRef]
- Gruber, E. Grundlagen der Zellstofftechnologie: Vorlesungsskriptum zum Lehrgang “Papiertechnik”; Duale Hochschule Baden-Württemberg Karlsruhe: Karlsruhe, Germany, 2011. [Google Scholar]
- Tomani, P.E.R. The lignoboost process. Cellul. Chem. Technol. 2010, 44, 53–58. [Google Scholar]
- Vishtal, A.; Kraslawski, A. Challenges in industrial applications of technical lignins. Bioresour. Technol. 2011, 6, 3547–3568. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, F. Catalytic Lignin Depolymerization to Aromatic Chemicals. Acc. Chem. Res. 2020, 53, 470–484. [Google Scholar] [CrossRef]
- Wang, H.; Tucker, M.; Ji, Y. Recent Development in Chemical Depolymerization of Lignin: A Review. J. Appl. Chem. 2013, 2013, 838645. [Google Scholar] [CrossRef] [Green Version]
- Roberts, V.M.; Stein, V.; Reiner, T.; Lemonidou , A.; Li, X.; Lercher, J.A. Towards Quantitative Catalytic Lignin Depolymerization. Chem. Eur. J. 2011, 17, 5939–5948. [Google Scholar] [CrossRef]
- Müller, T. Hydrogenation and Hydrogenolysis with Ruthenium Catalysts and Application to Biomass Conversion. In Ruthenium: An Element Loved by Researchers; Ishida, H., Ed.; IntechOpen: London, UK, 2021. [Google Scholar]
- Kristianto, I.; Limarta, S.O.; Lee, H.; Ha, J.-M.; Suh, D.J.; Jae, J. Effective depolymerization of concentrated acid hydrolysis lignin using a carbon-supported ruthenium catalyst in ethanol/formic acid media. Bioresour. Technol. 2017, 234, 424–431. [Google Scholar] [CrossRef]
- Yan, N.; Zhao, C.; Dyson, P.J.; Wang, C.; Liu, L.-T.; Kou, Y. Selective Degradation of Wood Lignin over Noble-Metal Catalysts in a Two-Step Process. ChemSusChem 2008, 1, 626–629. [Google Scholar] [CrossRef]
- Chio, C.; Sain, M.; Qin, W. Lignin utilization: A review of lignin depolymerization from various aspects. Renew. Sustain. Energy Rev. 2019, 107, 232–249. [Google Scholar] [CrossRef]
- Zhao, C.; Kou, Y.; Lemonidou, A.A.; Li, X.; Lercher, J.A. Highly Selective Catalytic Conversion of Phenolic Bio-Oil to Alkanes. Angew. Chem. Int. Ed. 2009, 48, 3987–3990. [Google Scholar] [CrossRef]
- Dong, L.; Yin, L.-L.; Xia, Q.; Liu, X.; Gong, X.-Q.; Wang, Y. Size-dependent catalytic performance of ruthenium nanoparticles in the hydrogenolysis of a β-O-4 lignin model compound. Catal. Sci. Technol. 2018, 8, 735–745. [Google Scholar] [CrossRef]
- Zakzeski, J.; Jongerius, A.L.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Catalytic Lignin Valorization Process for the Production of Aromatic Chemicals and Hydrogen. ChemSusChem 2012, 5, 1602–1609. [Google Scholar] [CrossRef]
- Shu, R.; Long, J.; Xu, Y.; Ma, L.; Zhang, Q.; Wang, T.; Wang, C.; Yuan, Z.; Wu, Q. Investigation on the structural effect of lignin during the hydrogenolysis process. Bioresour. Technol. 2016, 200, 14–22. [Google Scholar] [CrossRef]
- Xu, W.; Miller, S.J.; Agrawal, P.K.; Jones, C.W. Depolymerization and Hydrodeoxygenation of Switchgrass Lignin with Formic Acid. ChemSusChem 2012, 5, 667–675. [Google Scholar] [CrossRef]
- Ma, R.; Hao, W.; Ma, X.; Tian, Y.; Li, Y. Catalytic Ethanolysis of Kraft Lignin into High-Value Small-Molecular Chemicals over a Nanostructured α-Molybdenum Carbide Catalyst. Angew. Chem. Int. Ed. 2014, 126, 7438–7443. [Google Scholar] [CrossRef]
- Baerns, M.; Behr, A.; Brehm, A.; Gmehling, J.; Hinrichsen, K.-O.; Hofmann, H.; Onken, U.; Palkovits, R.; Renken, A. Technische Chemie, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2014; 762p. [Google Scholar]
- Kouris, P.D.; van Osch, D.J.G.P.; Cremers, G.J.W.; Boot, M.D.; Hensen, E.J.M. Mild thermolytic solvolysis of technical lignins in polar organic solvents to a crude lignin oil. Sustain. Energy Fuels 2020, 4, 6212–6226. [Google Scholar]
- Girard, E.; Tassaing, T.; Marty, J.-D.; Destarac, M. Structure–Property Relationships in CO2-philic (Co)polymers: Phase Behavior, Self-Assembly, and Stabilization of Water/CO2 Emulsions. Chem. Rev. 2016, 116, 4125–4169. [Google Scholar] [CrossRef]
- Aymonier, C.; Philippot, G.; Erriguible, A.; Marre, S. Playing with chemistry in supercritical solvents and the associated technologies for advanced materials by design. J. Supercrit. Fluids 2018, 134, 184–196. [Google Scholar] [CrossRef]
- Linstrom, P.J. NIST Standard Reference Database Number 69. In NIST Chemistry WebBook; U.S. Department of Commerce: Washington, DC, USA, 2022. [Google Scholar]
- Shu, R.; Zhang, Q.; Ma, L.; Xu, Y.; Chen, P.; Wang, C.; Wang, T. Insight into the solvent, temperature and time effects on the hydrogenolysis of hydrolyzed lignin. Bioresour. Technol. 2016, 221, 568–575. [Google Scholar] [CrossRef]
- Wang, J.; Li, W.; Wang, H.; Ma, Q.; Li, S.; Chang, H.-m.; Jameel, H. Liquefaction of kraft lignin by hydrocracking with simultaneous use of a novel dual acid-base catalyst and a hydrogenation catalyst. Bioresour. Technol. 2017, 243, 100–106. [Google Scholar] [CrossRef]
- Brand, S.; Susanti, R.F.; Kim, S.K.; Lee, H.-S.; Kim, J.; Sang, B.-I. Supercritical ethanol as an enhanced medium for lignocellulosic biomass liquefaction: Influence of physical process parameters. Energy 2013, 59, 173–182. [Google Scholar] [CrossRef]
- Lee, H.-S.; Jae, J.; Ha, J.-M.; Suh, D.J. Hydro- and solvothermolysis of kraft lignin for maximizing production of monomeric aromatic chemicals. Bioresour. Technol. 2016, 203, 142–149. [Google Scholar] [CrossRef]
- Cheng, S.; D’cruz, I.; Wang, M.; Leitch, M.; Xu, C. Highly Efficient Liquefaction of Woody Biomass in Hot-Compressed Alcohol−Water Co-solvents. Energy Fuels 2010, 24, 4659–4667. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Oh, S.; Hwang, H.; Cho, T.-S.; Choi, I.-G.; Choi, J.W. Effects of various reaction parameters on solvolytical depolymerization of lignin in sub- and supercritical ethanol. Chemosphere 2013, 93, 1755–1764. [Google Scholar] [CrossRef]
- Cheng, S.; Wilks, C.; Yuan, Z.; Leitch, M.; Xu, C. Hydrothermal degradation of alkali lignin to bio-phenolic compounds in sub/supercritical ethanol and water–ethanol co-solvent. Polym. Degrad. Stab. 2012, 97, 839–848. [Google Scholar] [CrossRef]
- Nguyen, T.D.H.; Maschietti, M.; Belkheiri, T.; Åmand, L.-E.; Theliander, H.; Vamling, L.; Olausson, L.; Andersson, S.-I. Catalytic depolymerisation and conversion of Kraft lignin into liquid products using near-critical water. J. Supercrit. Fluids 2014, 86, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Zhang, Y.; Fan, J.; Chang, J. Selective production of 4-ethylphenolics from lignin via mild hydrogenolysis. Bioresour. Technol. 2012, 118, 648–651. [Google Scholar] [CrossRef]
- Kusumoto, S.; Nozaki, K. Direct and selective hydrogenolysis of arenols and aryl methyl ethers. Nat. Commun. 2015, 6, 6296. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Zhang, C.; Han, B.; Wang, F. Catalytic self-transfer hydrogenolysis of lignin with endogenous hydrogen: Road to the carbon-neutral future. Chem. Soc. Rev. 2022, 51, 1608–1628. [Google Scholar] [CrossRef]
- Katayama, T.; Nitta, T. Solubilities of hydrogen and nitrogen in alcohols and n-hexane. J. Chem. Eng. Data 1976, 21, 194–196. [Google Scholar] [CrossRef]
- Gemo, N.; Biasi, P.; Salmi, T.O.; Canu, P. H2 solubility in methanol in the presence of CO2 and O2. J. Chem. Thermodyn. 2012, 54, 1–9. [Google Scholar] [CrossRef]
- Beller, M.; Riermeier, T.H.; Mägerlein, W.; Müller, T.E.; Scherer, W. Chemistry of chelate-stabilized aryloxopalladium(II) complexes: Syntheses, X-ray crystal structures and formation of C≡H…O hydrogen-bonds. Polyhedron 1998, 17, 1165–1176. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond: In Structural Chemistry and Biology International Union of Crystallography Monographs on Crystallography; Oxford University Press: Oxford, UK, 2001; 528p. [Google Scholar]
- Müller, T.E.; Mingos, D.M.P.; Williams, D.J. T-shaped intermolecular CH⋯π(C≡C) interactions in chloroform solvates of gold(I) ethyne complexes. J. Chem. Soc. Chem. Commun. 1994, 1787–1788. [Google Scholar] [CrossRef]
- Li, Y.; Ruan, W. From Solvent Cage to Molecular Cage. Univ. Chem. 2012, 27, 76–81. [Google Scholar] [CrossRef]
- Sievers, C.; Jimenez, O.; Müller, T.E.; Steuernagel, S.; Lercher, J.A. Formation of Solvent Cages around Organometallic Complexes in Thin Films of Supported Ionic Liquid. J. Am. Chem. Soc. 2006, 128, 13990–13991. [Google Scholar] [CrossRef]
- Tsuji, Y.; Richard, J.P. Reactions of ion-pair intermediates of solvolysis. Chem. Rec. 2005, 5, 94–106. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Building Block | Hardwood [%] | Softwood [%] |
|---|---|---|
| H | 0–8 | 5 |
| G | 25–50 | 95 |
| S | 54–75 | 0 |
| Bond Type | Hardwood [%] | Softwood [%] | BDE [kJ/mol] |
|---|---|---|---|
| α-O-4 | 4–8 | 6–12 | 222 |
| ß-1 (C-C) | 5–7 | 3–10 | 285 |
| β-O-4 | 45–65 | 42–50 | 313 |
| 4-O-5 | 6–7 | 3.5–8 | 335 |
| 5-5 | 4–10 | 9.5–25 | 481 |
| ß-5 | 3–11 | 9–12 | 523 |
| ß-1 (C=C) | minor | minor | 690 |
| Descriptor | Value | Unit |
|---|---|---|
| Molar mass | 1500–5000 1 | [g/mol] |
| Polydispersity | 2.5–3.5 | [-] |
| Acid-soluble lignin fraction | 1–4.9 | [wt.-%] |
| Carbohydrate fraction | 1–2.3 | [wt.-%] |
| Water content (often referred to as humidity) | 3–6 | [wt.-%] |
| Nitrogen content | 0.05 | [wt.-%] |
| Sulfur content | 1.8 | [wt.-%] |
| Incombustible residue (ash) | 0.5–3 | [wt.-%] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panke, D.; Bechthold, G.; Müller, T.E. Solvent Effect in Catalytic Lignin Hydrogenolysis. Catalysts 2022, 12, 664. https://doi.org/10.3390/catal12060664
Panke D, Bechthold G, Müller TE. Solvent Effect in Catalytic Lignin Hydrogenolysis. Catalysts. 2022; 12(6):664. https://doi.org/10.3390/catal12060664
Chicago/Turabian StylePanke, Dennis, German Bechthold, and Thomas E. Müller. 2022. "Solvent Effect in Catalytic Lignin Hydrogenolysis" Catalysts 12, no. 6: 664. https://doi.org/10.3390/catal12060664
APA StylePanke, D., Bechthold, G., & Müller, T. E. (2022). Solvent Effect in Catalytic Lignin Hydrogenolysis. Catalysts, 12(6), 664. https://doi.org/10.3390/catal12060664