
Catalytic Effect of CO2 and H2O Molecules on •CH3 + 3O2 Reaction



Abstract
:1. Introduction
2. Computational Methodology
2.1. Electronic-Structure Calculations
2.2. Chemical Kinetics Calculations
3. Results and Discussion
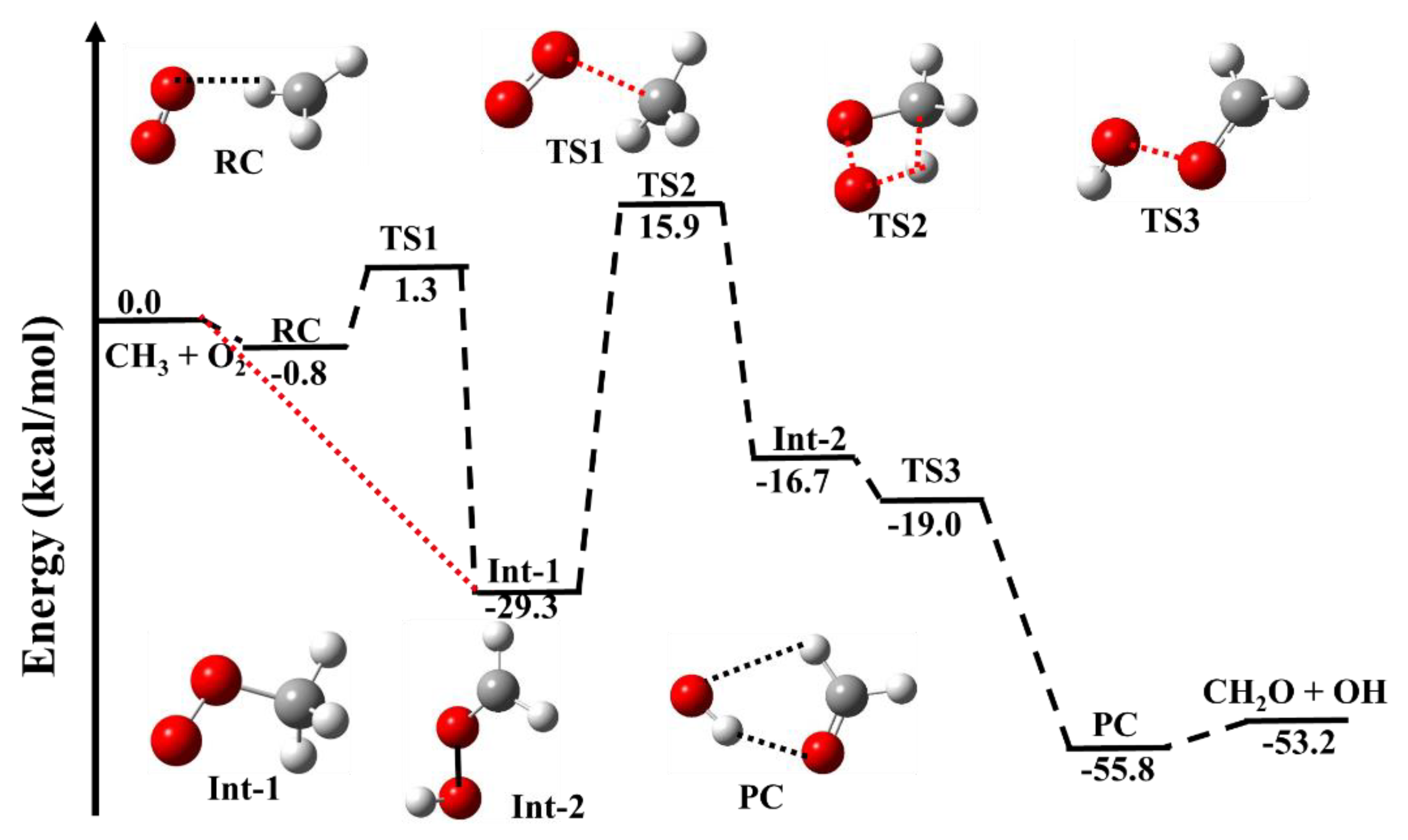
3.1. Reaction Pathways for •CH3 + 3O2
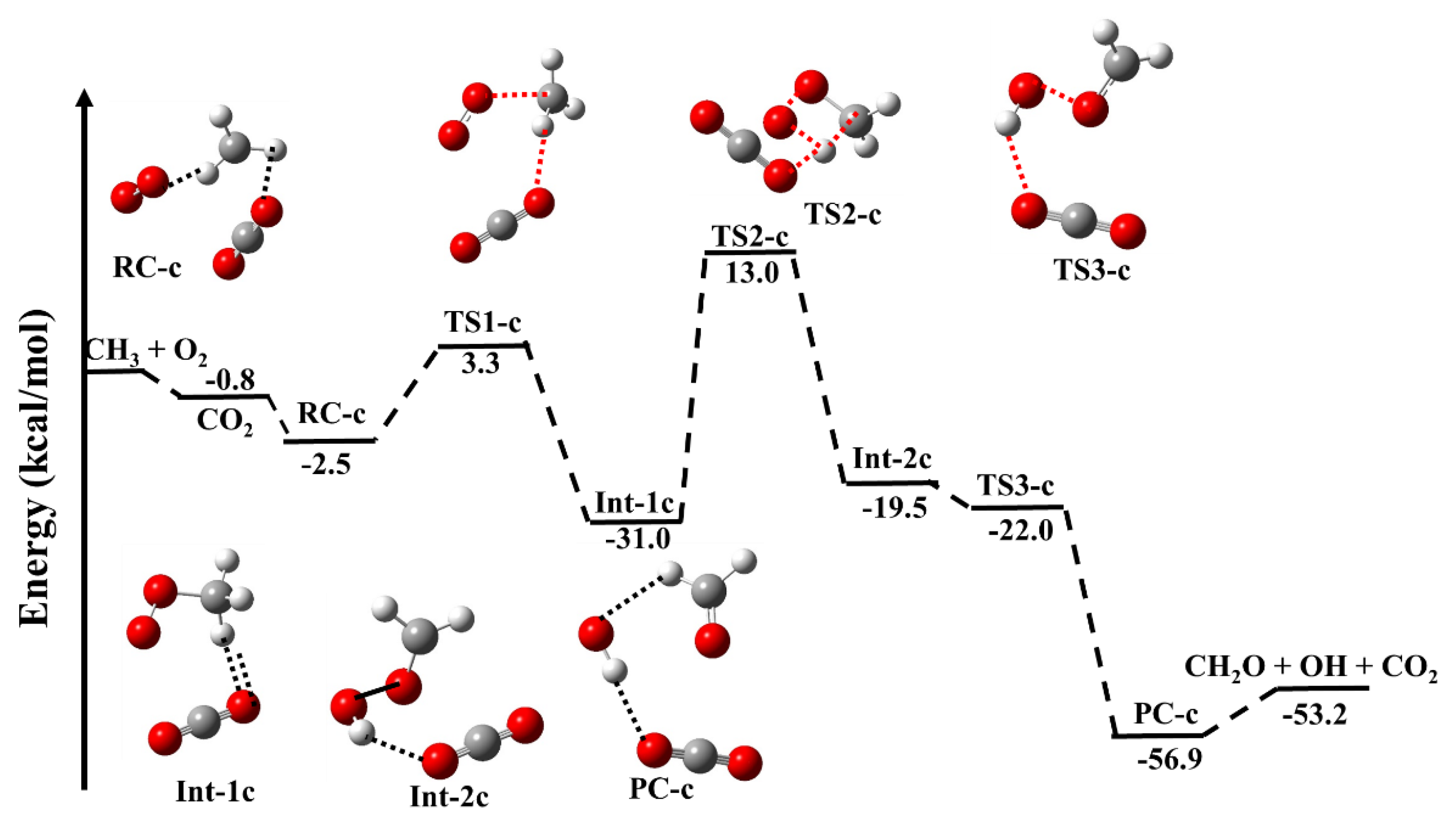
3.1.1. Reaction Pathways for •CH3 + 3O2 (+CO2)
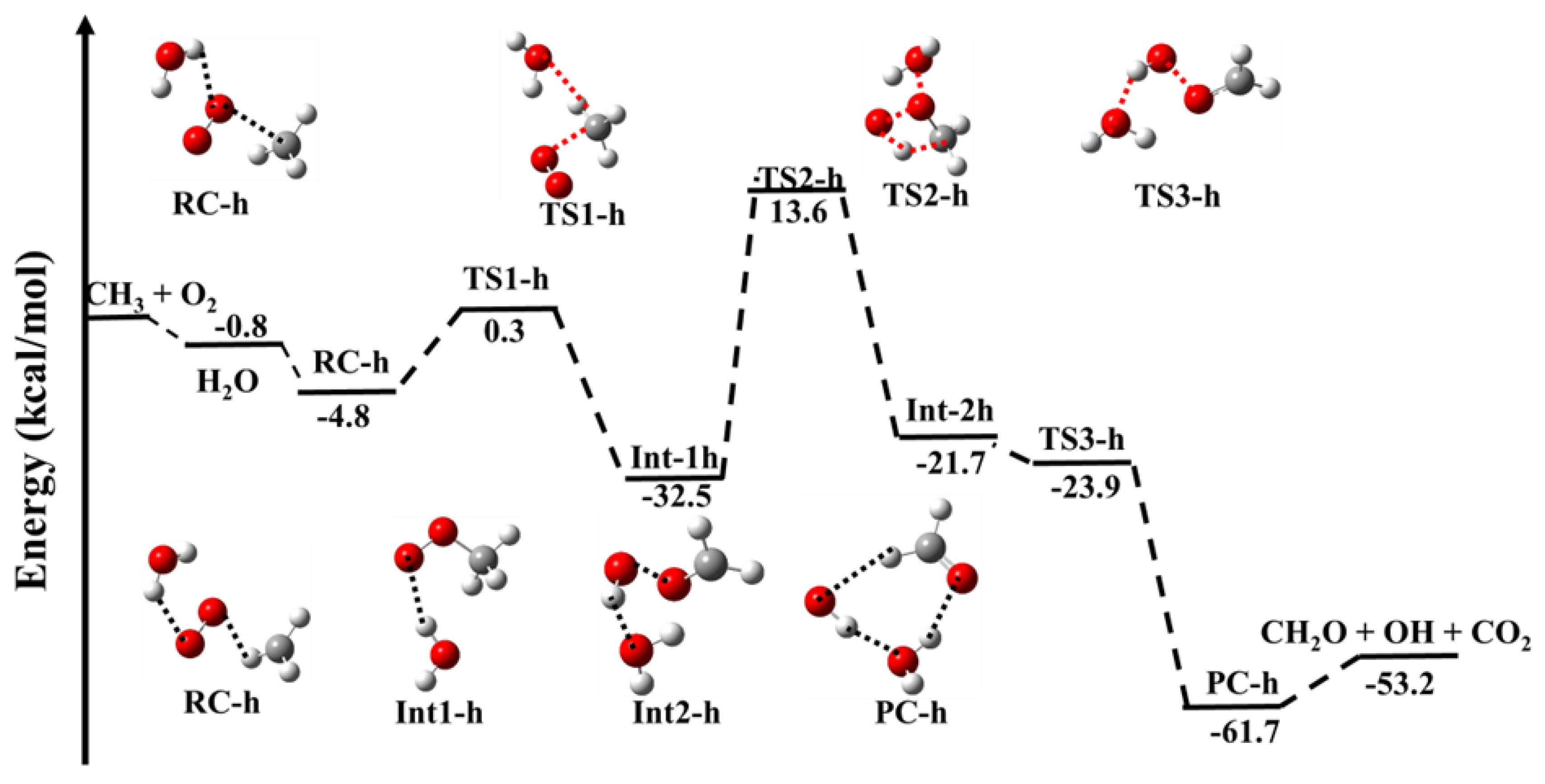
3.1.2. Reaction Pathways for •CH3 + 3O2 (+H2O)
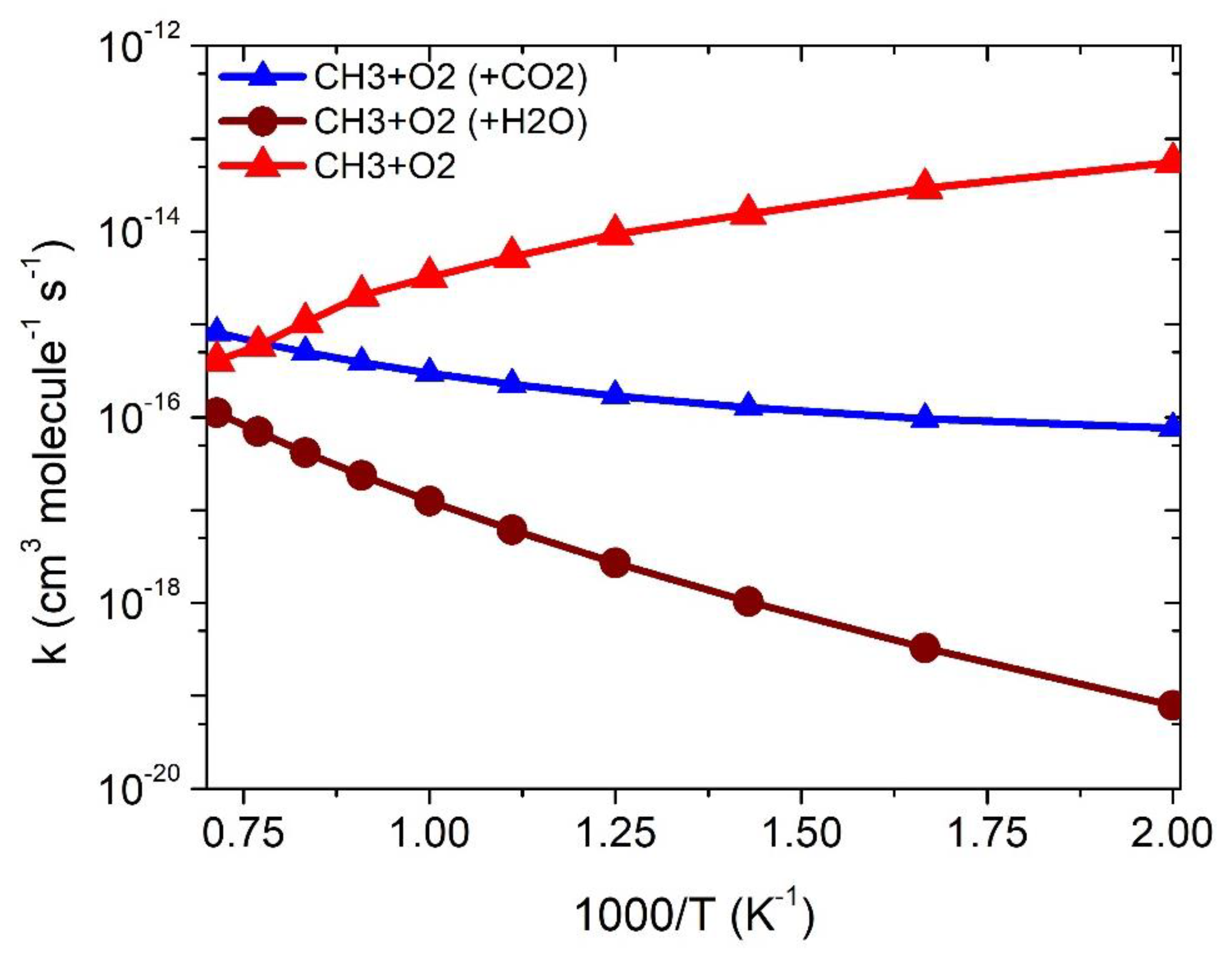
3.2. Rate Constants
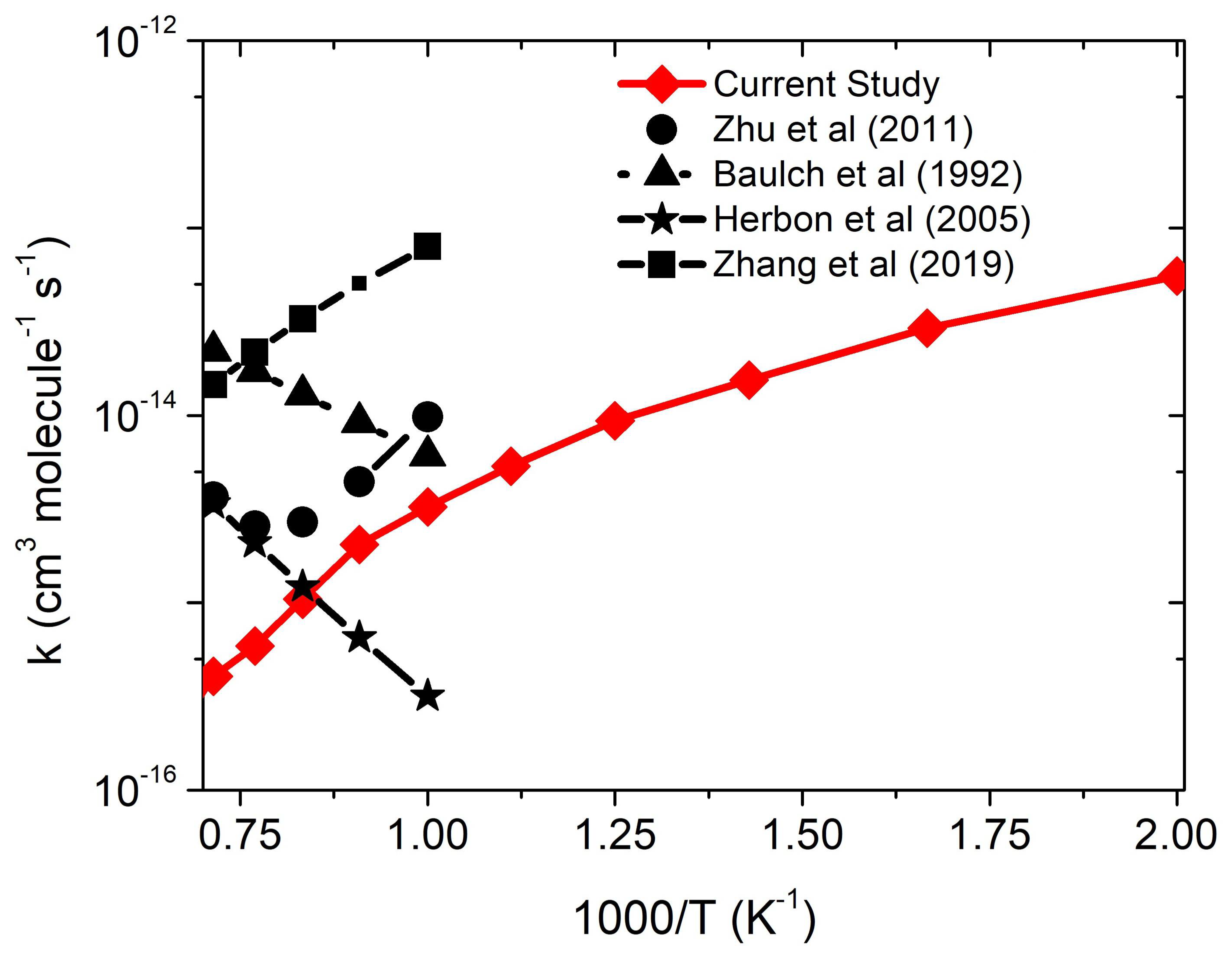
3.2.1. •CH3 + 3O2 Reaction
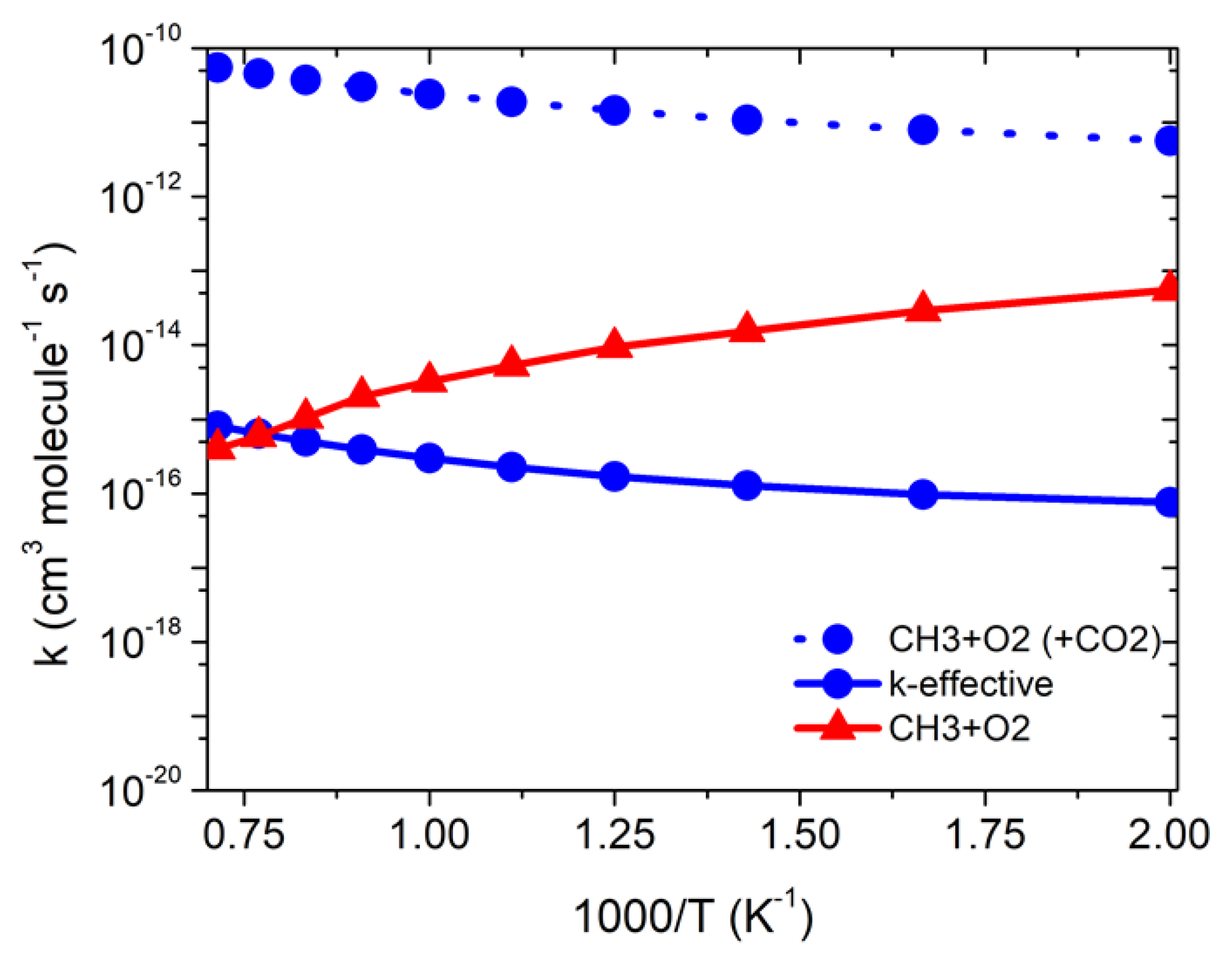
3.2.2. Effect of CO2 on •CH3 + 3O2 Reaction
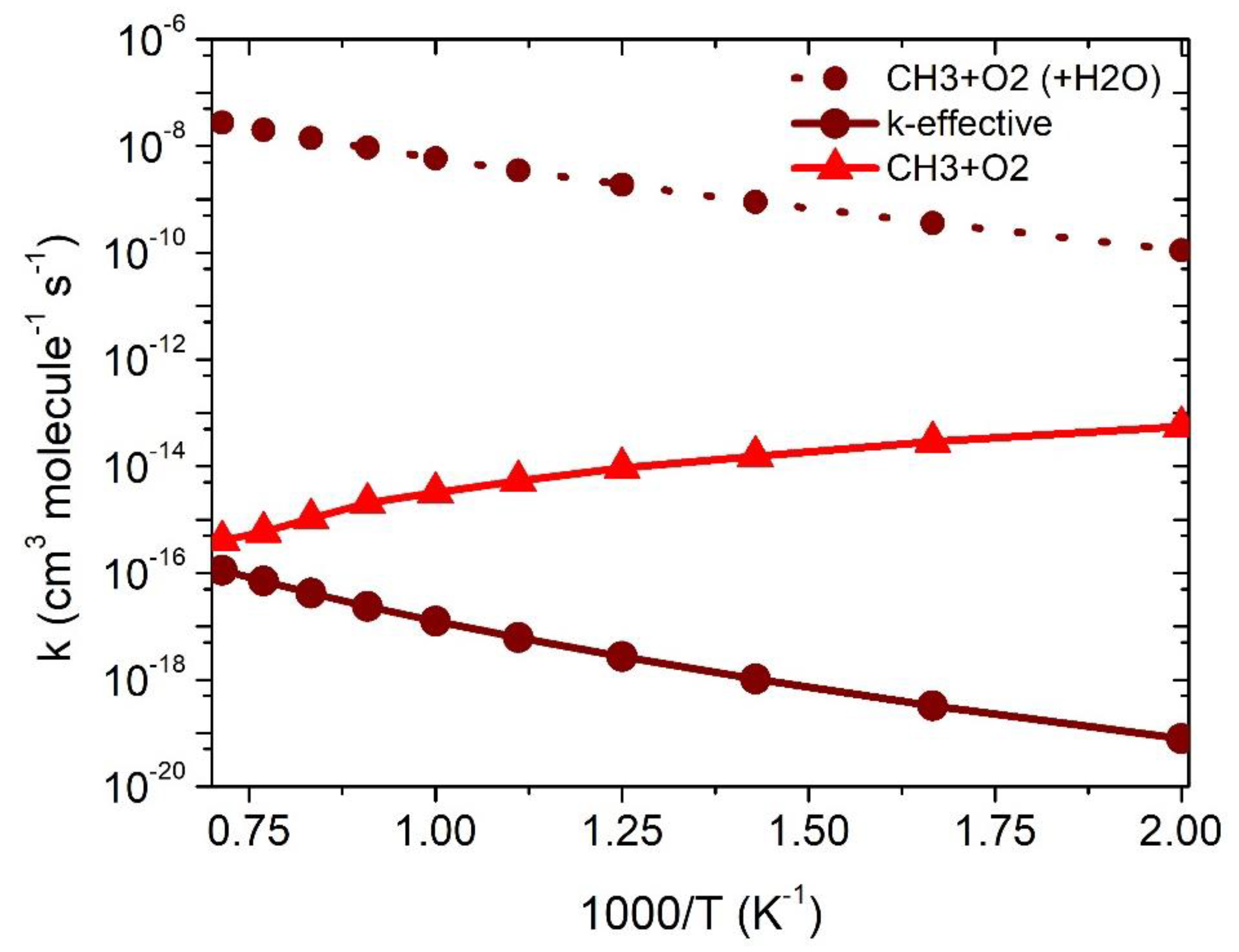
3.2.3. Effect of H2O on •CH3 + 3O2 Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Srinivasan, N.K.; Su, M.-C.; Sutherland, J.W.; Michael, J.V. Reflected Shock Tube Studies of High-Temperature Rate Constants for OH + CH4 → CH3 + H2O and CH3 + NO2 → CH3O + NO. J. Phys. Chem. A 2005, 109, 1857–1863. [Google Scholar] [CrossRef] [PubMed]
- Vaghjiani, G.L.; Ravishankara, A.R. New measurement of the rate coefficient for the reaction of OH with methane. Nature 1991, 350, 406–409. [Google Scholar] [CrossRef]
- Baulch, D.L.; Cobos, C.J.; Cox, R.A.; Esser, C.; Frank, P.; Just, T.; Kerr, J.A.; Pilling, M.J.; Troe, J.; Walker, R.W.; et al. Evaluated Kinetic Data for Combustion Modelling. J. Phys. Chem. Ref. Data 1992, 21, 411. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, N.K.; Su, M.-C.; Michael, J.V. CH3 + O2 → H2CO + OH Revisited. J. Phys. Chem. A 2007, 111, 11589–11591. [Google Scholar] [CrossRef] [PubMed]
- Herbon, J.T.; Hanson, R.K.; Bowman, C.T.; Golden, D.M. The reaction of CH3 + O2: Experimental determination of the rate coefficients for the product channels at high temperatures. Proc. Combust. Inst. 2005, 30, 955–963. [Google Scholar] [CrossRef]
- Hsu, D.S.Y.; Shaub, W.M.; Creamer, T.; Gutman, D.; Lin, M.C. Kinetic modeling of co-production from the reaction of CH3 with O2 in shock waves. Ber. Bunsenges. Phys. Chem. 1983, 87, 909–991. [Google Scholar] [CrossRef]
- Cobos, C.J.; Hippler, H.; Luther, K.; Ravishankara, A.R.; Troe, J. High-pressure falloffcurves and specific rate constants for the reaction CH3 + O2 ↔ CH3O2 ↔ CH3 + O. J. Phys. Chem. 1985, 89, 4332–4338. [Google Scholar] [CrossRef]
- Zhang, F.; Huang, C.; Xie, B.; Wu, X. Revisiting the chemical kinetics of CH3 + O2 and its impact on methane ignition. Combust. Flame 2019, 200, 125–134. [Google Scholar] [CrossRef]
- Masunov, A.E.; Wait, E.; Vasu, S.S. Quantum Chemical Study of CH3 + O2 Combustion Reaction System: Catalytic Effects of Additional CO2 Molecule. J. Phys. Chem. A 2017, 121, 5681–5689. [Google Scholar] [CrossRef]
- Pilling, M.J.; Smith, M.J.C. A laser flash photolysis study of the reaction CH3 + O2 → CH3O2 at 298 K. J. Phys. Chem. 1985, 89, 4713–4720. [Google Scholar] [CrossRef]
- Zhu, R.; Hsu, C.-C.; Lin, M.C. Ab initio study of the CH3 + O2 reaction: Kinetics, mechanism and product branching probabilities. J. Chem. Phys. 2011, 115, 195. [Google Scholar] [CrossRef]
- Tan, Y.; Douglas, M.A.; Thambimuthu, K.V. CO2 Capture Using Oxygen Enhanced Combustion Strategies for Natural Gas Power Plants. Fuel 2002, 81, 1007–1016. [Google Scholar] [CrossRef]
- Kumar, A.; Mallick, S.; Mishra, B.M.; Kumar, P. Effect of ammonia and formic acid on the CH3O + O2 reaction: A quantum chemical investigation. Phys. Chem. Chem. Phys. 2020, 22, 2405–2413. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Lin, C.Y.; Wang, H.T.; Lin, M.C. A shock tube study of the oxidation of the methyl radical. AIP Conf. Proc. 1990, 208, 450–455. [Google Scholar]
- Zhang, T.; Zhang, Y.; Wen, M.; Tang, Z.; Long, B.; Yu, X.; Zhao, C.; Wang, W. Effects of water, ammonia and formic acid on HO2 + Cl reactions under atmospheric conditions: Competition between a stepwise route and one elementary step. RSC Adv. 2019, 9, 21544–21556. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, N.K.; Su, M.C.; Sutherland, J.W.; Michael, J.V. Reflected shock tube studies of high-temperature rate constants for CH3 + O2, H2CO + O2, and OH + O2. J. Phys. Chem. A 2005, 109, 7902–7914. [Google Scholar] [CrossRef]
- Masunov, A.E.; Elizabeth, E.; Wait, S.; Vasu, S. Catalytic Effect of Carbon Dioxide on Reaction OH + CO → H + CO2, in Supercritical Environment: Master Equation Study. J. Phys. Chem. A 2018, 122, 6355–6359. [Google Scholar] [CrossRef]
- Pryor, O.M.; Barak, S.; Koroglu, B.; Ninnemann, E.; Vasu, S.S. Measurements and Interpretation of Shock Tube Ignition Delay Times in Highly CO2 Diluted Mixtures Using Multiple Diagnostics. Combust. Flame 2017, 180, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Koroglu, B.; Pryor, O.; Lopez, J.; Nash, L.; Vasu, S.S. Shock Tube Ignition Delay Times and Methane Time-Histories Measurements During Excess CO2 Diluted Oxy-Methane Combustion. Combust. Flame 2016, 164, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Du, B.; Qin, Z. Catalytic effect of Water, Formic Acid, or Sulfuric Acid on the Reaction of Formaldehyde with OH Radicals. J. Phys. Chem. A 2014, 118, 4797–4807. [Google Scholar] [CrossRef]
- Jara-Toro, R.A.; Hernández, F.J.; Taccone, R.A.; Lane, S.I.; Pino, G.A. Water catalysis of the reaction between methanol and OH at 294 K and the atmospheric implications. Angew. Chem. Int. Ed. 2017, 56, 2166–2170. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Gao, L.G.; Varga, Z.; Xu, X.; Ren, W.; Truhlar, D.G. Water catalysis of the Reaction of Methanol with OH Radical in the Atmosphere is Negligible. Angew. Chem. Int. Ed. 2020, 59, 10826–10830. [Google Scholar] [CrossRef] [PubMed]
- Vöhringer-Martinez, E.; Hansmann, B.; Hernandez, H.; Francisco, J.S.; Troe, J.; Abel, B. Water Catalysis of a Radical-Molecule Gas-Phase Reaction. Science 2007, 315, 496–501. [Google Scholar] [CrossRef]
- Thomsen, D.; Kurtén, T.; Jørgensen, S.; Wallington, T.; Baggesen, S.; Aalling, C.; Kjaergaard, H. On the possible catalysis by single water molecules of gas-phase hydrogen abstraction reactions by OH radicals. Phys. Chem. Chem. Phys 2012, 14, 12992–12999. [Google Scholar] [CrossRef] [PubMed]
- Chao, W.; Lin, J.J.; Takahashi, K.; Tomas, A.; Yu, L.; Kajii, Y.; Batut, S.; Schoemaecker, C.; Fittschen, C. Water vapor does not catalyze the reaction between methanol and OH Radicals. Angew. Chem. Int. Ed. 2019, 58, 5013–5017. [Google Scholar] [CrossRef]
- Dash, M.R.; Ali, M.A. Effect of a single water molecule on CH2OH + 3O2 reaction under atmospheric and combustion conditions. Phys. Chem. Chem. Phys. 2022, 24, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Balaganesh, M.; Lin, K.C. Catalytic effect of a single water molecule on the OH + CH2NH reaction. Phys. Chem. Chem. Phys 2018, 20, 4297–4307. [Google Scholar]
- Ali, M.A.; Ali, M.A. Effect of Water and Formic Acid on ·OH + CH4 Reaction: An Ab Initio/DFT Study. Catalysts 2022, 12, 133. [Google Scholar] [CrossRef]
- Inaba, S. Catalytic Role of H2O Molecules in Oxidation of CH3OH in Water. Catalysts 2018, 8, 157. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.A.; Balaganesh, M.; Jang, S. Can a single water molecule catalyze the OH+CH2CH2 and OH+CH2O reactions? Atmos. Environ. 2019, 207, 82–92. [Google Scholar] [CrossRef]
- Ali, M.A.; Balaganesh, M.; Al-Odail, F.A.; Lin, K.C. Effect of ammonia and water molecule on OH + CH3OH reaction under tropospheric condition. Sci. Rep. 2021, 11, 12185. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A. Computational studies on the gas phase reaction of methylenimine (CH2NH) with water molecules. Sci. Rep. 2020, 10, 10995. [Google Scholar] [CrossRef] [PubMed]
- Buszek, R.J.; Torrent-Sucarrat, M.; Anglada, J.M.; Francisco, J.S. Effects of a Single Water Molecule on the OH + H2O2 Reaction. J. Phys. Chem. A 2012, 116, 5821–5829. [Google Scholar] [CrossRef]
- Iuga, C.; Alvarez-Idaboy, J.R. On the Possible Catalytic Role of a Single Water Molecule in the Acetone + OH Gas Phase reaction: A Theoretical Pseudo-Second-order Kinetics Study. Theor. Chem. Acc. 2011, 129, 209–217. [Google Scholar] [CrossRef]
- Iuga, C.; Alvarez-Idaboy, J.R.; Reyes, L.; Vivier-Bunge, A. Can a Single Water Molecule Really Catalyze the Acetaldehyde OH Reaction in Tropospheric Conditions? J. Phys. Chem. Lett. 2010, 1, 3112–3115. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, W.; Li, C.; Du, Y.; Lu, J. Catalytic effect of a single water molecule on the atmospheric reaction of HO2 + OH: Fact or fiction? A mechanistic and kinetic study. RSC Adv. 2013, 3, 7381–7391. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, T., Jr.; Vreven, K.N.; Kudin, J.C.; Burant, J.M.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Barker, J.R.; Nguyen, T.L.; Stanton, J.F.; Aieta, C.; Ceotto, M.; Gabas, F.; Kumar, T.J.D.; Li, C.G.L.; Lohr, L.L.; Maranzana, A.; et al. MultiWell-<Version>-Software Suite; Barker, J.R., Ed.; University of Michigan: Ann Arbor, MI, USA, 2016; Available online: https://multiwell.engin.umich (accessed on 24 May 2022).
- Ruscic, B.; Pinzon, R.E.; Morton, M.L.; von Laszevski, G.; Bittner, S.J.; Nijsure, S.G.; Amin, K.A.; Minkoff, M.; Wagner, A.F. Introduction to Active Thermochemical Tables: Several “Key” Enthalpies of Formation Revisited. J. Phys. Chem. 2004, 108, 9979–9997. [Google Scholar] [CrossRef]
- Ruscic, B.; Pinzon, R.E.; Von Laszewski, G.; Kodeboyina, D.; Burcat, A.; Leahy, D.; Montoy, D.; Wagner, A.F. Active Thermochemical Tables: Thermochemistry for the 21st Century. J. Phys. Conf. Ser. 2005, 16, 561–570. [Google Scholar] [CrossRef]
- Ruscic, B.; Bross, D.H. Active Thermochemical Tables (ATcT) Enthalpies of Formation Values Based on ver. 1.112r of the Thermochemical Network. 2021. Available online: ATcT.anl.gov (accessed on 24 May 2022).
- Lemmon, E.W.; McLinden, M.O.; Friend, D.G. Thermophysical Properties of Fluid Systems. In NIST Chemistry Webbook; NIST Standard Reference Database Number 69. Available online: https://atct.anl.gov/ (accessed on 24 May 2022).
- Metcalfe, W.K.; Burke, M.; Ahmed, S.S.; Curran, H.J. A Hierarchical and Comparative Kinetic Modeling Study of C1−C2 Hydrocarbon and Oxygenated Fuels. Int. J. Chem Kinet. 2013, 45, 638–675. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| •CH3 + O2 → | This Work | Literature |
| OO···CH3 (RC) | −0.8 | −0.4 b |
| OO···CH3 (TS1) | 1.3 | 1.6 b |
| CH3OO (Int-1) | −29.3 | −30.4 a, −33.8 b |
| HOO···CH2 (TS2) | +15.9 | 15.0 a, 13.7 b |
| HO···CH2O (Int-2) | −16.7 | −18.5 a, −20.6 b |
| HO···CH2O (TS3) | −19.0 | −21.3 b |
| HO···CH2O (PC) | −55.8 | −58.2 b |
| CH2O + OH | −53.2 | −53.7, −52.10 c |
| •CH3 + O2 + CO2 → | This Work | Literature |
| CH3···O2 + CO2 | −0.8 | −0.4 b |
| CO2 ···CH3OO (RC-c) | −2.5 | −2.6 b |
| CO2 ···CH3OO (TS1-c) | 3.3 | −0.3 b |
| CO2 ···CH3OO (Int-1c) | −31.0 | −37.0 b |
| CO2 ···CH3OO (TS2-c) | 13.0 | 8.7 b |
| CO2 ···CH3OO (Int-2c) | −19.5 | −25.0 b |
| CO2 ···CH3OO (TS-3c) | −22.0 | −25.4 b |
| HO···CH2O···CO2 (PC-c) | −56.9 | −61.6 b |
| •CH3 + O2 + H2O → | This Work | |
| CH3···O2 + H2O | −0.8 | |
| H2O···CH3OO (RC-h) | −4.8 | |
| H2O···CH3OO (TS1-h) | 0.3 | |
| H2O ···CH3OO (Int-1h) | −32.5 | |
| H2O·····CH3OO (TS2-h) | 13.6 | |
| H2O···CH3OO (Int-2h) | −21.7 | |
| H2O·····CH3OO (TS3-h) | 13.6 | |
| HO···CH2O···H2O (PC-h) | −61.7 |
| Temp | kCH3+O2 | ||
|---|---|---|---|
| 500 | 5.5 × 10−14 | 7.6 × 10−17 | 1.7 × 10−19 |
| 600 | 2.9 × 10−14 | 9.6 × 10−17 | 6.4 × 10−19 |
| 700 | 1.5 × 10−14 | 1.3 × 10−16 | 1.8 × 10−18 |
| 800 | 9.4 × 10−15 | 1.7 × 10−16 | 4.5 × 10−18 |
| 900 | 5.3 × 10−15 | 2.3 × 10−16 | 9.5 × 10−18 |
| 1000 | 3.2 × 10−15 | 3.0 × 10−16 | 1.8 × 10−17 |
| 1100 | 2.1 × 10−15 | 3.9 × 10−16 | 3.4 × 10−17 |
| 1200 | 1.0 × 10−15 | 5.04 × 10−16 | 5.8 × 10−17 |
| 1300 | 5.9 × 10−16 | 6.41 × 10−16 | 9.5 × 10−17 |
| 1400 | 4.0 × 10−16 | 8.2 × 10−16 | 1.5 × 10−16 |
| 1500 | 2.1 × 10−16 | 1.02 × 10−15 | 2.3 × 10−16 |
| k = ATn exp(−B/T) | A = 2.8 × 1014 n = −9.1 B = 3516 | A = 2.5 × 10–33 n = 4.4 B = −1711 | A = 3.3 × 10–33 n = 5.4 B = 1338 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.A.; Dash, M.R.; Al Maieli, L.M. Catalytic Effect of CO2 and H2O Molecules on •CH3 + 3O2 Reaction. Catalysts 2022, 12, 699. https://doi.org/10.3390/catal12070699
Ali MA, Dash MR, Al Maieli LM. Catalytic Effect of CO2 and H2O Molecules on •CH3 + 3O2 Reaction. Catalysts. 2022; 12(7):699. https://doi.org/10.3390/catal12070699
Chicago/Turabian StyleAli, Mohamad Akbar, Manas Ranjan Dash, and Latifah Mohammed Al Maieli. 2022. "Catalytic Effect of CO2 and H2O Molecules on •CH3 + 3O2 Reaction" Catalysts 12, no. 7: 699. https://doi.org/10.3390/catal12070699
APA StyleAli, M. A., Dash, M. R., & Al Maieli, L. M. (2022). Catalytic Effect of CO2 and H2O Molecules on •CH3 + 3O2 Reaction. Catalysts, 12(7), 699. https://doi.org/10.3390/catal12070699