


Guaiacol to Aromatics: Efficient Transformation over In Situ-Generated Molybdenum and Tungsten Oxides



Abstract
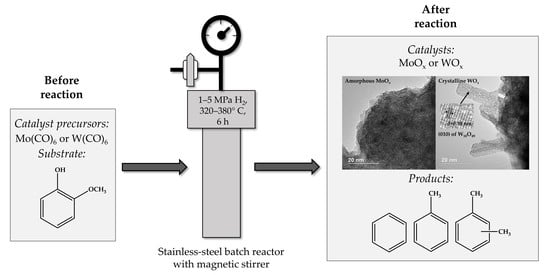
:
1. Introduction
2. Results and Discussion
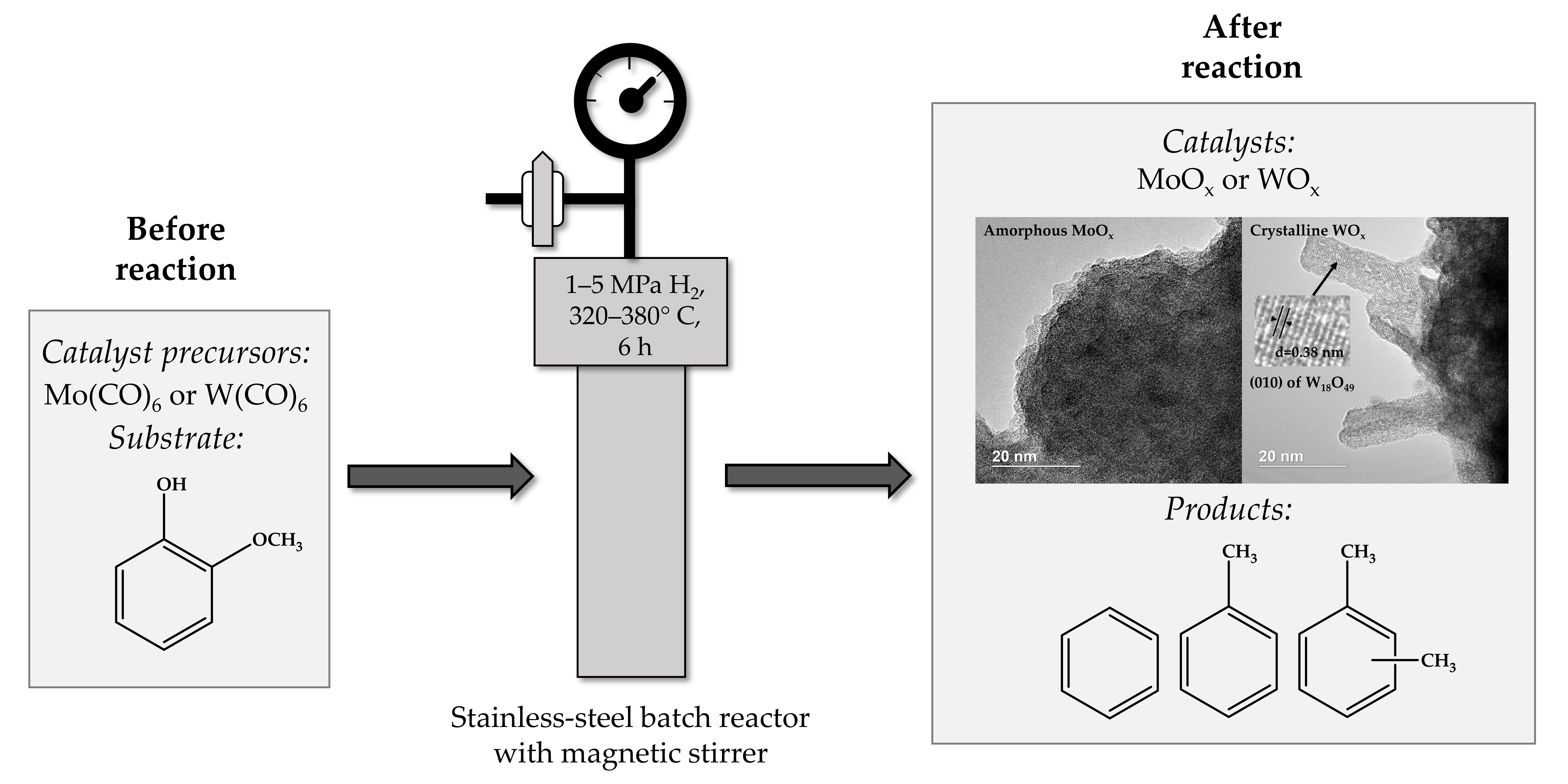
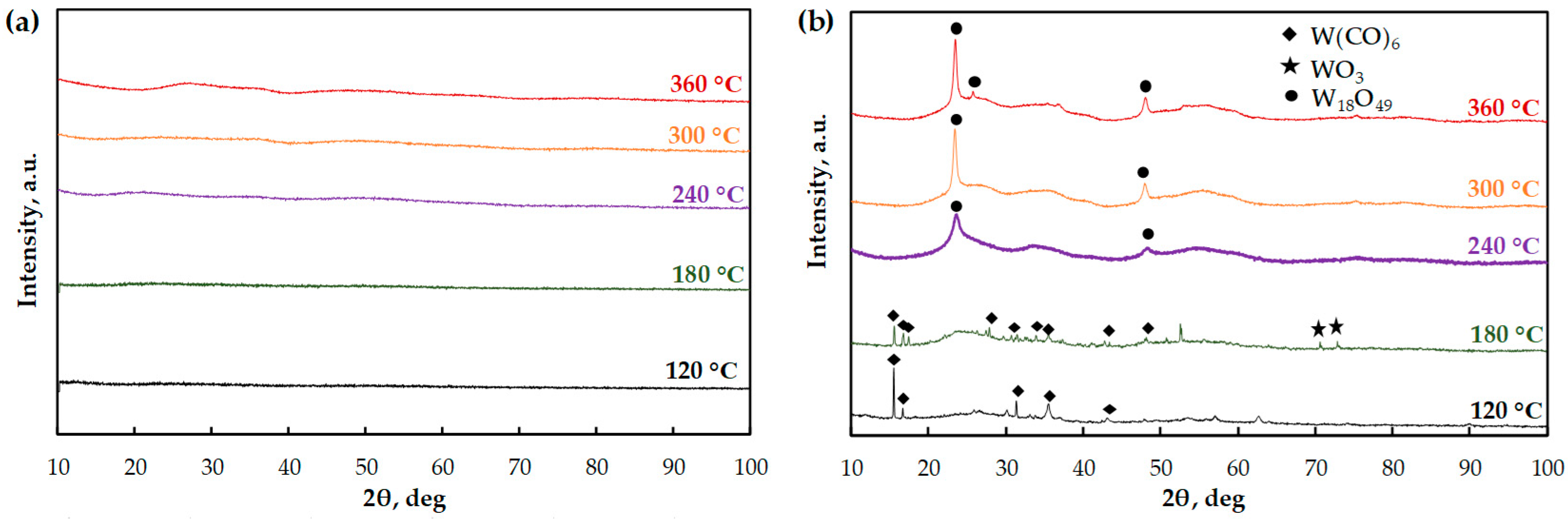
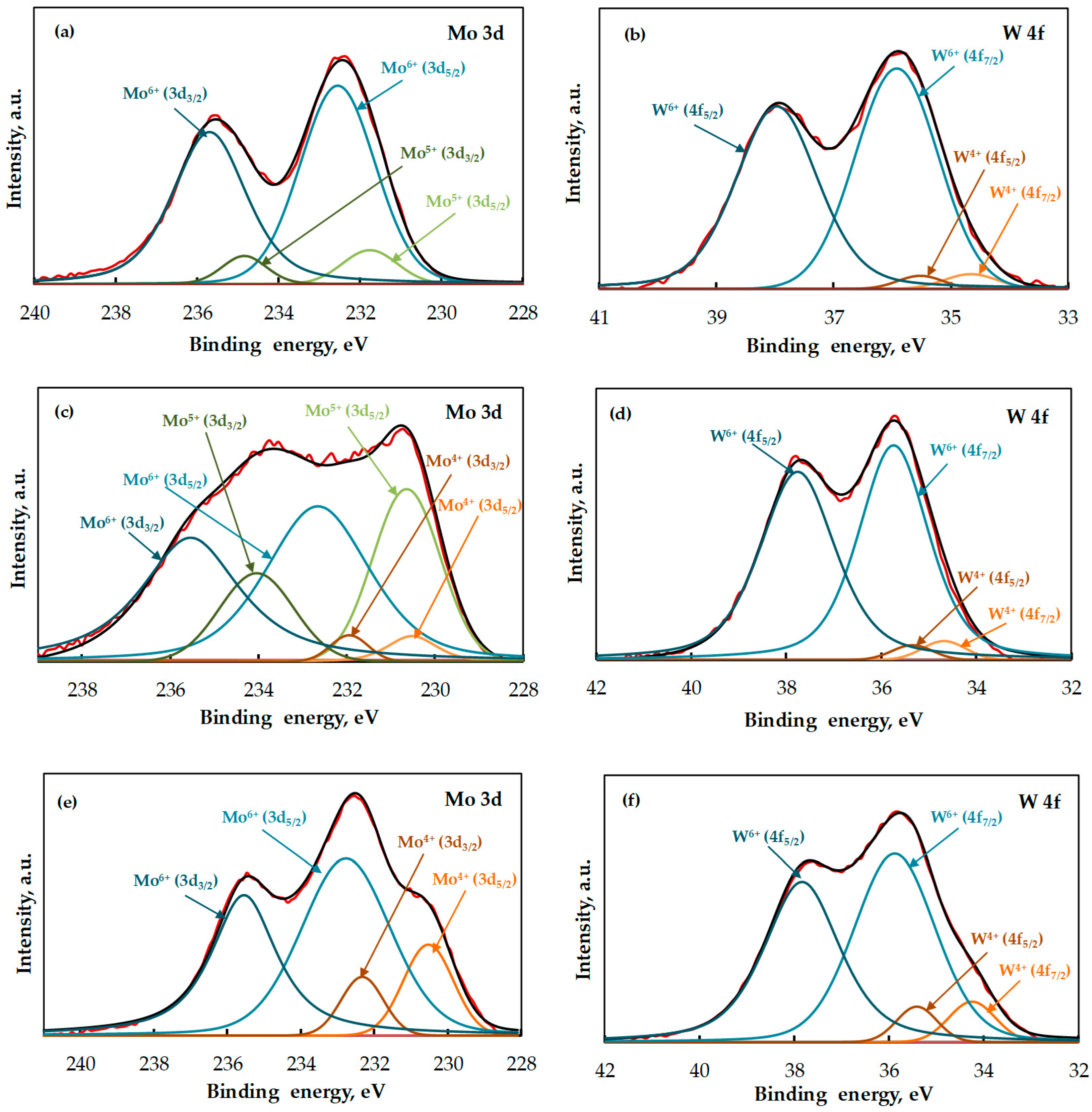
2.1. Catalyst Characterization
2.2. Catalytic Activity
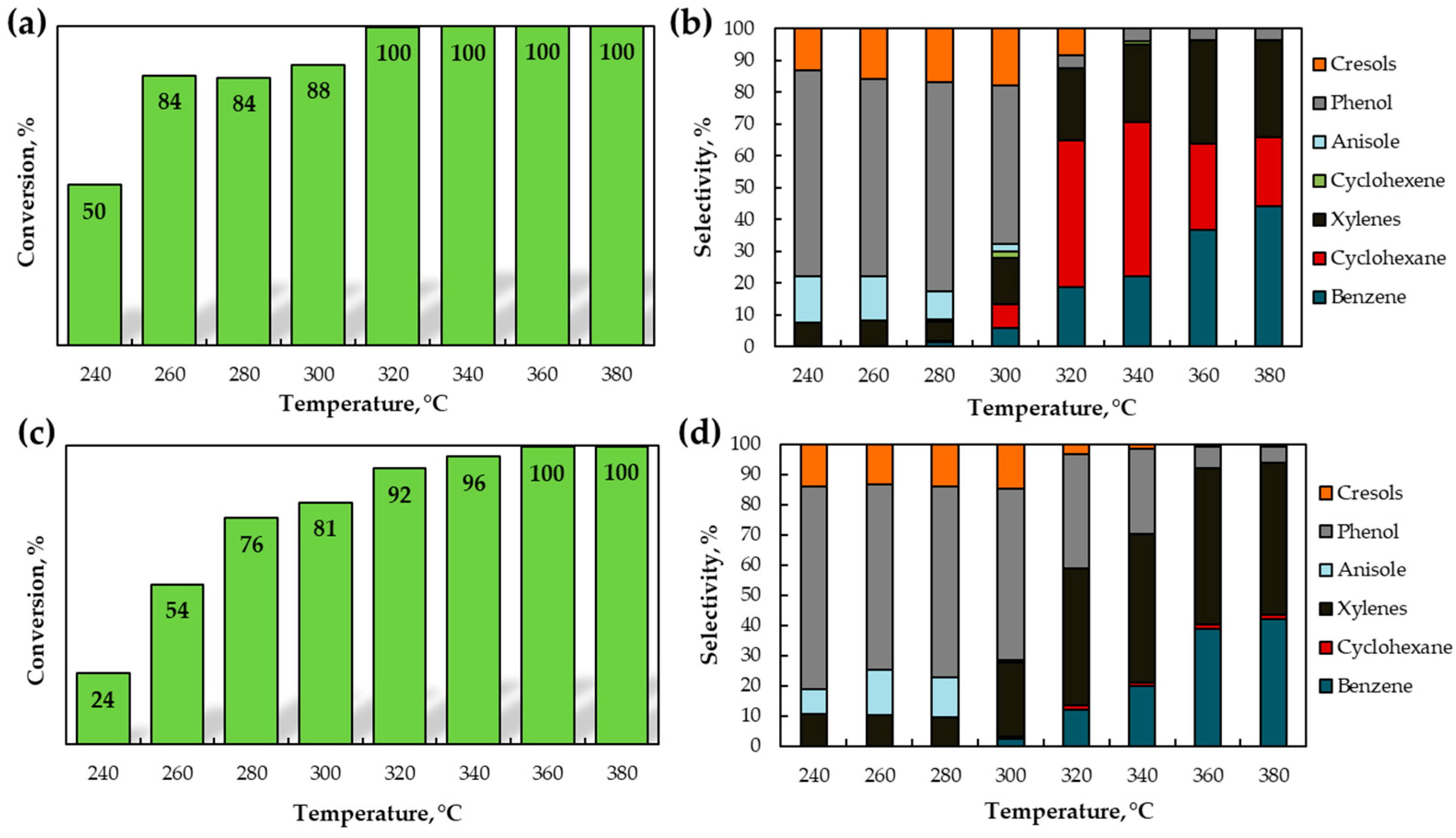
2.2.1. The Effect of Temperature in Toluene
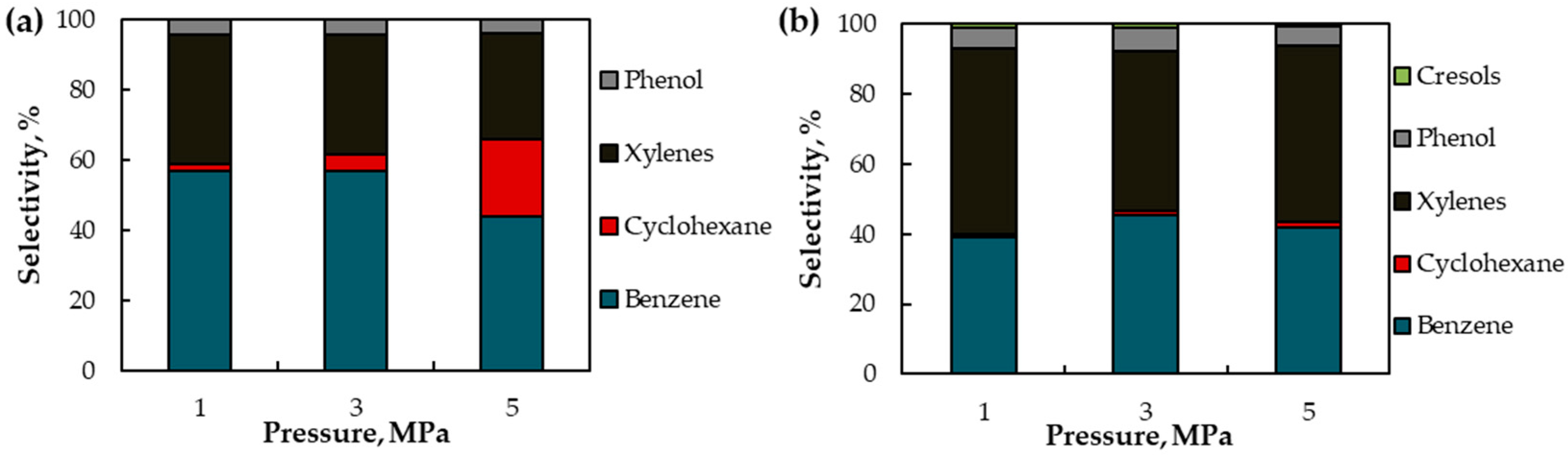
2.2.2. The Effect of Pressure in Toluene
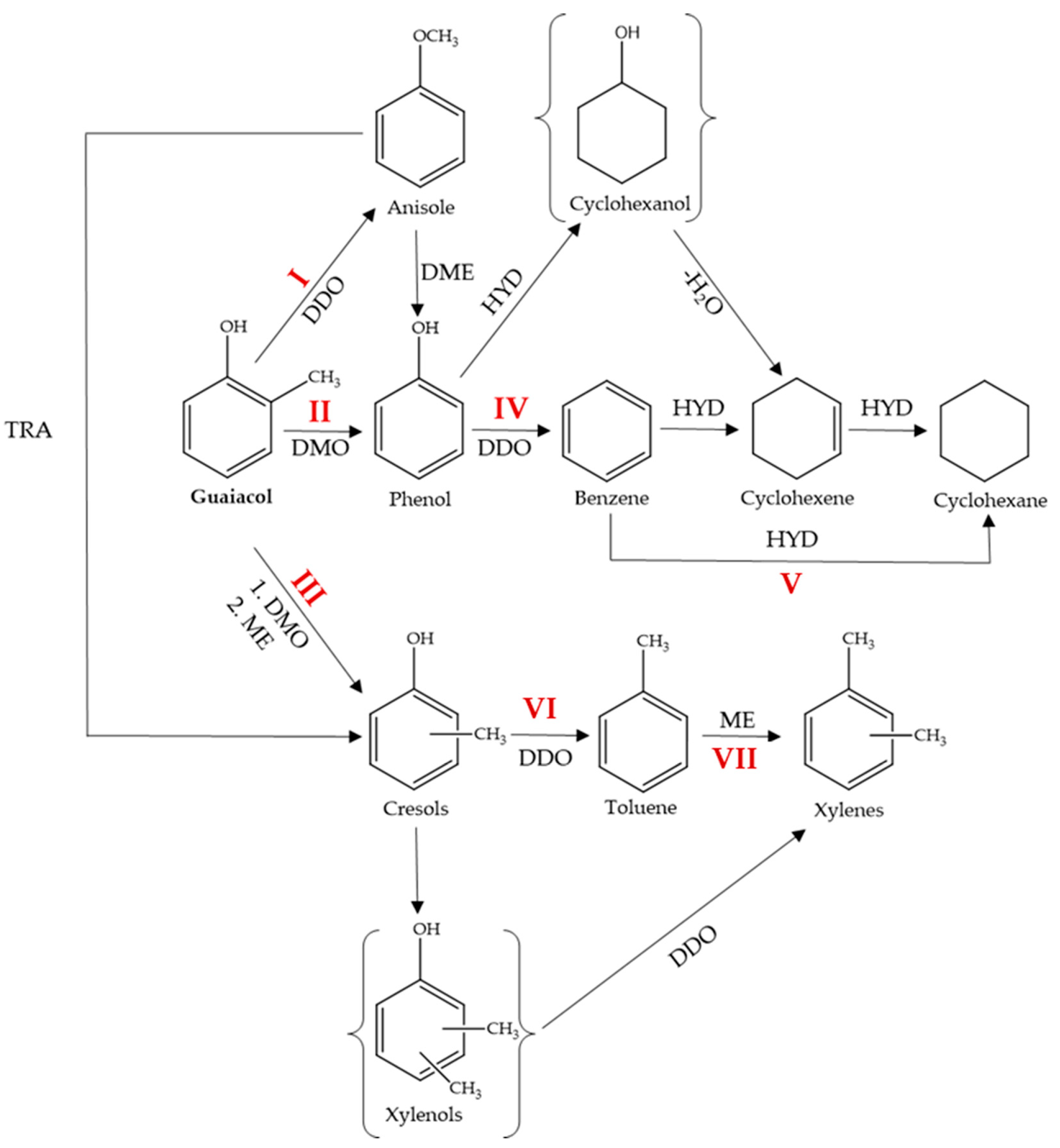
2.2.3. Reaction Pathways
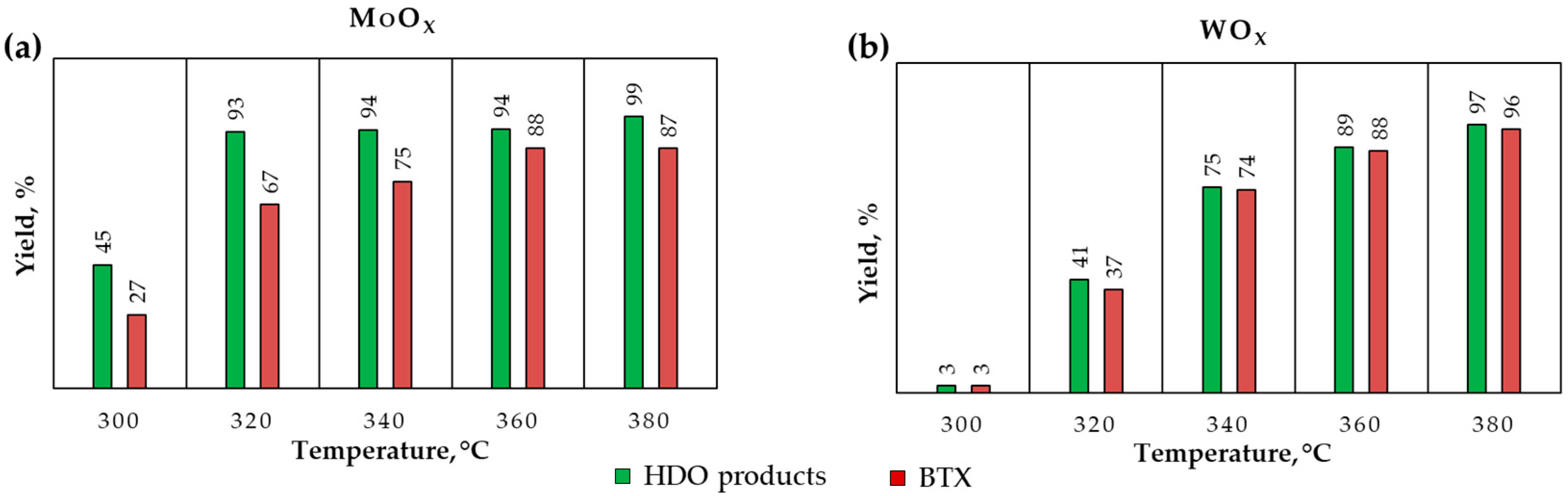
2.2.4. The Effect of Temperature and Pressure in Dodecane
2.2.5. The Recycling Tests
3. Materials and Methods
3.1. Materials
3.2. Catalyst Preparation and Catalytic Tests
3.3. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nanda, S.; Mohammad, J.; Reddy, S.N.; Kozinski, J.A.; Dalai, A.K. Pathways of lignocellulosic biomass conversion to renewable fuels. Biomass Convers. Biorefin. 2014, 4, 157–191. [Google Scholar] [CrossRef]
- Cherubini, F.; Strømman, A.H. Production of Biofuels and Biochemicals from Lignocellulosic Biomass: Estimation of Maximum Theoretical Yields and Efficiencies Using Matrix Algebra. Energy Fuels 2010, 24, 2657–2666. [Google Scholar] [CrossRef]
- Tribot, A.; Amer, G.; Abdou, A.M.; de Baynast, H.; Delattre, C.; Pons, A.; Mathias, J.; Callois, J.; Vial, C.; Michaud, P.; et al. Wood-lignin: Supply, extraction processes and use as bio-based material. Eur. Polym. J. 2019, 112, 228–240. [Google Scholar]
- Zhang, J.; Sun, J.; Wang, Y. Recent Advances in Selectively Catalytic Hydrodeoxygenation of Lignin-derived Oxygenates to Arenes. Green Chem. 2020, 22, 1072–1098. [Google Scholar] [CrossRef]
- Führer, M.; Van Haasterechta, T.; Bitter, J.H. Cinnamaldehyde hydrogenation over carbon supported molybdenum and tungsten carbide catalysts. Chem. Commun. 2022, 58, 13608–13611. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, X.; Xu, Y.; Gao, X.; Dai, Y.; Tang, Y. Palladium-Incorporated α-MoC Mesoporous Composites for Enhanced Direct Hydrodeoxygenation of Anisole. Catalysts 2021, 11, 370. [Google Scholar] [CrossRef]
- Kurisingal, J.F.; Lee, S.; Lee, J.G.; An, K. Zeolitic Imidazolate Framework Decorated Molybdenum Carbide Catalysts for Hydrodeoxygenation of Guaiacol to Phenol. Catalysts 2022, 12, 1605. [Google Scholar] [CrossRef]
- Wang, F.; Wen, C.; Lu, M.; Zhang, P.; Zhu, J.; Li, M.; Shan, Y.; Song, C. SBA-15-supported ultrastable Mo2N@CN catalysts for hydrodeoxygenation of guaiacol. Biomass Bioenergy 2023, 168, 106680. [Google Scholar] [CrossRef]
- Golubeva, M.A.; Mukhtarova, M.; Bugaev, A.L.; Naranov, E.R. In Situ Generated Dispersed Catalysts Based on Molybdenum and Tungsten Phosphides in Hydroprocessing of Guaiacol. Pet. Chem. 2023. [Google Scholar] [CrossRef]
- Golubeva, M.A.; Mukhtarova, M.; Sadovnikov, A.A.; Maximov, A.L. Bulk Molybdenum and Tungsten Phosphides for Selective Phenol Production from Guaiacol. ACS Omega 2022, 7, 40586–40595. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, J.; Zhang, Q.; Liu, Q.; Li, Y.; Chen, L.; Wang, C.; Ma, L. Hydrodeoxygenation of lignin-derived phenolic compounds into aromatic hydrocarbons under low hydrogen pressure using Molybdenum oxide as catalyst. Catal. Today 2019, 319, 41–47. [Google Scholar] [CrossRef]
- Jiang, S.; Ji, N.; Diao, X.; Li, H.; Rong, Y.; Lei, Y.; Yu, Z. Vacancy Engineering in Transition Metal Sulfide and Oxide Catalysts for Hydrodeoxygenation of Lignin-Derived Oxygenates. ChemSusChem 2021, 14, 4377–4396. [Google Scholar] [CrossRef] [PubMed]
- Prasomsri, T.; Nimmanwudipong, T.; Román-Leshkov, Y. Effective hydrodeoxygenation of biomass-derived oxygenates into unsaturated hydrocarbons by MoO3 using low H2 pressures. Energy Environ. Sci. 2013, 6, 1732. [Google Scholar] [CrossRef]
- Bagnato, G.; Sanna, A.; Paone, E.; Catizzone, E. Recent catalytic advances in hydrotreatment processes of pyrolysis bio-oil. Catalysts 2021, 11, 157. [Google Scholar] [CrossRef]
- Thibodeau, T.J.; Canney, A.S.; DeSisto, W.J.; Wheeler, M.C.; Amar, F.G.; Frederick, B.G. Composition of tungsten oxide bronzes active for Hydrodeoxygenation. Appl. Catal. A 2010, 388, 86–95. [Google Scholar] [CrossRef]
- Whiffen, V.M.L.; Smith, K.J. Hydrodeoxygenation of 4-Methylphenol over Unsupported MoP, MoS2 and MoOx Catalysts. Energy Fuels 2010, 24, 4728–4737. [Google Scholar] [CrossRef]
- Wang, C.; Mironenko, A.V.; Raizada, A.; Chen, T.; Mao, X.; Padmanabhan, A.; Vlachos, D.G.; Gorte, R.J.; Vohs, J.M. Mechanistic Study of the Direct Hydrodeoxygenation of m-Cresol over WOx-Decorated Pt/C Catalysts. ACS Catal. 2018, 8, 7749–7759. [Google Scholar] [CrossRef]
- Wang, C.; Wittreich, G.R.; Lin, C.; Huang, R.; Vlachos, D.G.; Gorte, R.J. Hydrodeoxygenation of m-Cresol Over Pt-WOx/C Using H2 Generated In Situ by n-Hexane Dehydrogenation. Catal. Lett. 2020, 150, 913–921. [Google Scholar] [CrossRef]
- Chen, T.; Kwon, O.; Huang, R.; Lin, C.; Vohs, J.M. WOx promoted nickel catalyst for hydrodeoxygenation of m-cresol. J. Catal. 2021, 400, 294–300. [Google Scholar] [CrossRef]
- Golubeva, M.A.; Maximov, A.L. Hydroprocessing of furfural over in situ generated nickel phosphide based catalysts in different solvents. Appl. Catal. A 2020, 608, 117890. [Google Scholar] [CrossRef]
- Golubeva, M.A.; Maximov, A.L. Investigations on the Formation of Transition Metal Phosphides during the Hydrotreating of Light Cycle Oil Russ. J. Appl. Chem. 2021, 94, 1536–1545. [Google Scholar]
- Vagvala, T.C.; Pandey, S.S.; Ogomi, Y.; Ma, T.; Hayase, S. Investigation of metal xanthates as latent curing catalysts for epoxy resin via formation of in-situ metal sulfides. Inorg. Chim. Acta 2015, 435, 292–298. [Google Scholar] [CrossRef]
- Vutolkina, A.V.; Baygildin, I.G.; Glotov, A.P.; Cherednichenko, K.A.; Maksimov, A.L.; Karakhanov, E.A. Dispersed Ni-Mo sulfide catalysts from water-soluble precursors for HDS of BT and DBT via in situ produced H2 under Water gas shift conditions. Appl. Catal. B 2021, 282, 119616. [Google Scholar] [CrossRef]
- Kuchinskaya, T.; Kniazeva, M.; Samoilov, V.; Maximov, A. In Situ Generated Nanosized Sulfide Ni-W Catalysts Based on Zeolite for the Hydrocracking of the Pyrolysis Fuel Oil into the BTX Fraction. Catalysts 2020, 10, 1152. [Google Scholar] [CrossRef]
- Zhang, K.; McCleese, C.; Lin, P.; Chen, X.; Morales, M.; Cao, W.; Seo, F.J.; Burda, C.; Baumgart, H. Synthesis of ALD Tungsten Trioxide Thin Films from W(CO)6 and H2O Precursors. ECS Trans. 2015, 69, 199–209. [Google Scholar] [CrossRef]
- Davazoglou, D.; Moutsakis, A.; Valamontes, V.; Psychorish, V. Tungsten Oxide Thin Films Chemically Vapor Deposited at Low Pressure by W(CO)6 Pyrolysis. J. Electrochem. Soc. 1997, 144, 595–599. [Google Scholar] [CrossRef]
- Suvanto, M.; Pakkanen, T. Deposition of tungsten hexacarbonyl on alumina: A diffuse reflectance infrared Fourier transform spectroscopy study. J. Mol. Catal. A Chem. 1999, 138, 211–220. [Google Scholar] [CrossRef]
- Ou, N.C.; Su, X.; Bock, D.C.; McElwee-White, L. Precursors for chemical vapor deposition of tungsten oxide and molybdenum oxide. Coord. Chem. Rev. 2020, 421, 213459. [Google Scholar] [CrossRef]
- Dhas, N.A.; Gedanken, A. Characterization of Sonochemically Prepared Unsupported and Silica-Supported Nanostructured Pentavalent Molybdenum Oxide. J. Phys. Chem. B 1997, 101, 9495–9503. [Google Scholar] [CrossRef]
- Dimitrova, Z.; Gogova, D. On the structure, stress and optical properties of CVD tungsten oxide films. Mater. Res. Bull. 2005, 40, 333–340. [Google Scholar] [CrossRef]
- Fu, J.; Liu, S.; Zheng, W.; Huang, R.; Wang, C.; Lawal, A.; Alexopoulos, K.; Liu, S.; Wang, Y.; Yu, K.; et al. Modulating the dynamics of Brønsted acid sites on PtWOx inverse catalyst. Nat. Catal. 2022, 5, 144–153. [Google Scholar] [CrossRef]
- Fang, Z.; Jiao, S.; Kang, Y.; Pang, G.; Feng, S. Photothermal Conversion of W18O49 with a Tunable Oxidation State. ChemistryOpen 2017, 6, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Baltrusaitis, J.; Mendoza-Sanchez, B.; Fernandez, V.; Veenstra, R.; Dukstiene, N.; Roberts, A.; Fairley, N. Generalized molybdenum oxide surface chemical state XPS determination via informed amorphous sample model. Appl. Surf. Sci. 2015, 326, 151–161. [Google Scholar] [CrossRef]
- Xie, F.; Choy, W.C.H.; Wang, C.; Li, X.; Zhang, S.; Hou, J. Low-Temperature Solution-Processed Hydrogen Molybdenum and Vanadium Bronzes for an Efficient Hole-Transport Layer in Organic Electronics. Adv. Mater. 2013, 25, 2051–2055. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Cook, J.B.; Lin, H.; Ko, J.S.; Tolbert, S.H.; Ozolins, V.; Dunn, B. Oxygen vacancies enhance pseudocapacitive charge storage properties of MoO3−x. Nat. Mater. 2017, 16, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Apergi, S.; Koch, C.; Brocks, G.; Olthof, S.; Tao, S. Decomposition of Organic Perovskite Precursors on MoO3: Role of Halogen and Surface Defects. Appl. Mater. Interfaces 2022, 14, 34208–34219. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.G.; Thompson, L.T. XPS study of as-prepared and reduced molybdenum oxides. Appl. Surf. Sci. 1996, 93, 143–149. [Google Scholar] [CrossRef]
- Lu, D.Y.; Chen, J.; Zhou, J.; Deng, S.Z.; Xu, N.S.; Xu, J.B. Raman spectroscopic study of oxidation and phase transition in W18O49 nanowires. J. Raman Spectrosc. 2007, 38, 176–180. [Google Scholar] [CrossRef]
- Katoh, M.; Takeda, Y. Chemical State Analysis of Tungsten and Tungsten Oxides Using an Electron Probe Microanalyzer. Jpn. J. Appl. Phys. 2004, 43, 7292–7295. [Google Scholar] [CrossRef]
- Lin, Y.-S.; Lai, J.-Y.; Tsai, T.-H.; Chuang, P.-Y.; Chen, Y.-C. Effects of oxygen addition on electrochromic properties in low temperature plasma-enhanced chemical vapor deposition-synthesized MoOxCy thin films for flexible electrochromic devices. Thin Solid Films 2011, 519, 3875–3882. [Google Scholar] [CrossRef]
- Światowska-Mrowiecka, J.; de Diesbach, S.; Maurice, V.; Zanna, S.; Klein, L.; Briand, E.; Vickridge, I.; Marcus, P. Li-Ion Intercalation in Thermal Oxide Thin Films of MoO3 as Studied by XPS, RBS, and NRA. J. Phys. Chem. C 2008, 112, 11050–11058. [Google Scholar] [CrossRef]
- Ghasempour, R.; Iraji, A. Hybrid multiwalled carbon nanotubes and trioxide tungsten nanoparticles for hydrogen gas sensing. J. Phys. D Appl. Phys. 2009, 42, 165105. [Google Scholar] [CrossRef]
- Li, J.; Zhou, C.; Mu, J.; Yang, E.; Zhao, X. In situ synthesis of molybdenum carbide/N-doped carbon hybrids as an efficient hydrogen-evolution electrocatalyst. RSC Adv. 2018, 8, 17202–17208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajadi, M.; Ranjbar, M.; Rasuli, R. Two-step synthesis of Ag-decorated MoO3 nanotubes, and the effect of hydrogen doping. Appl. Surf. Sci. 2020, 527, 146675. [Google Scholar] [CrossRef]
- Vasilopoulou, M.; Soultati, A.; Georgiadou, D.G.; Stergiopoulos, T.; Palilis, L.C.; Kennou, S.; Stathopoulos, N.A.; Davazoglou, D.; Argitis, P. Hydrogenated under-stoichiometric tungsten oxide anode interlayers for efficient and stable organic photovoltaics. J. Mater. Chem. A 2014, 2, 1738. [Google Scholar] [CrossRef]
- Wang, J.; Gao, Y.; You, Z.; Fan, J.; Zhang, J.; Wang, S.; Xu, J. Laser Induced Nano and Micro Structures of Molybdenum Surface Applied in Multistage Depressed Collector for Secondary Electron Suppression. Appl. Sci. 2019, 9, 4374. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.; Tang, L.; Deng, Y.; Wang, J.; Tang, W.; Liu, Y.; Chen, Z.; Yu, J.; Wang, J.; Liang, Q. Synthesis of branched WO3@W18O49 homojunction with enhanced interfacial charge separation and full-spectrum photocatalytic performance. Chem. Eng. J. 2020, 389, 124474. [Google Scholar] [CrossRef]
- Chen, C.L.; Mori, H. In situ TEM observation of the growth and decomposition of monoclinic W18O49 nanowires. Nanotechnology 2009, 20, 285604. [Google Scholar] [CrossRef]
- Hahn, T.; Bentrup, U.; Armbrüster, M.; Kondratenko, E.V.; Linke, D. The Enhancing Effect of Brønsted Acidity of Supported MoOx Species on their Activity and Selectivity in Ethylene/trans-2-Butene Metathesis. ChemCatChem 2014, 6, 1664–1672. [Google Scholar] [CrossRef]
- Zafeiratos, S.; Papakonstantinou, G.; Jacksic, M.M.; Neophytides, S.G. The effect of Mo oxides and TiO2 support on the chemisorption features of linearly adsorbed CO on Pt crystallites: An infrared and photoelectron spectroscopy study. J. Catal. 2005, 232, 127–136. [Google Scholar] [CrossRef]
- Baes, A.U.; Bloom, P.R. Diffuse Reflectance and Transmission Fourier Transform Infrared (DRIFT) Spectroscopy of Humic and Fulvic Acids Soil. Sci. Soc. Am. J. 1989, 53, 695–700. [Google Scholar] [CrossRef]
- Barreto, M.S.C.; Reis, J.V.; Muraoka, T.; Jemo, M.; Vergutz, L.; Alleoni, L.R.F. Diffuse reflectance infrared Fourier transform spectroscopy for a qualitative evaluation of plant leaf pigment extraction. Analyst 2021, 146, 3440. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, C.; Zhang, L.; Gholizadeh, M.; Hu, X. Biochar catalyzing polymerization of the volatiles from pyrolysis of poplar wood. Int. J. Energy. Res. 2021, 45, 13936–13951. [Google Scholar] [CrossRef]
- Ajito, K.; Nagahara, L.A.; Tryk, D.A.; Hashimoto, K.; Fujishima, A. Study of the Photochromic Properties of Amorphous MoO3 Films Using Raman Microscopy. J. Phys. Chem. 1995, 99, 16383–16388. [Google Scholar] [CrossRef]
- Casiraghi, C.; Piazza, F.; Ferrari, A.C.; Grambole, D.; Robertson, J. Bonding in hydrogenated diamond-like carbon by Raman spectroscopy. Diamond Relat. Mater. 2005, 14, 1098–1102. [Google Scholar] [CrossRef]
- Tran, N.T.T.; Uemura, Y.; Chowdhury, S.; Ramli, A. Vapor-phase Hydrodeoxygenation of Guaiacol on Al−MCM−41 Supported Ni and Co Catalysts. Appl. Catal. A 2016, 512, 93–100. [Google Scholar] [CrossRef]
- Bui, V.N.; Laurenti, D.; Afanasiev, P.; Geantet, C. Hydrodeoxygenation of guaiacol with CoMo catalysts. Part I: Promoting effect of cobalt on HDO selectivity and activity. Appl. Catal. B 2011, 101, 239–245. [Google Scholar] [CrossRef]
- Venkatesan, K.; Krishna, J.V.J.; Anjana, S.; Selvam, P.; Vinu, R. Hydrodeoxygenation kinetics of syringol, guaiacol and phenol over H-ZSM-5. Catal. Commun. 2021, 148, 106164. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Feedstock | Reaction Conditions a | Conversion, % | Selectivity, % | Ref. |
|---|---|---|---|---|---|
| MoxC/CNF | 0.68 g cinnamaldehyde in 50 mL toluene | 200 °C, 2 MPa H2, ~2.75 h a | 90 | 40.9 for hydrocinnamaldehyde 25.8 for β-methylstyrene | [5] |
| WxC/CNF | 200 °C, 2 MPa H2, ~5.75 h a | 90 | 43 for hydrocinnamaldehyde 34 for β-methylstyrene | ||
| Pd/α-MoC | anisole | 200 °C b | 21.5 | 94 for benzene | [6] |
| Mo2C@ BMZIF-700 °C (4 h) | 0.12 g guaiacol in 20 mL n-decane | 330 °C, 4 MPa H2, 4 h a | 97 | 70 for phenol | [7] |
| Mo2N@ NC500/SBA-15 | 3 wt. % guaiacol in n-decane | 380 °C, 2 MPa H2 c | 100 | 92 to benzene + toluene | [8] |
| in situ formed MoP | 10 wt. % guaiacol in n-dodecane | 360 °C, 5 MPa H2, 6 h a | 90 | 80 for phenol | [9] |
| in situ formed WP | 340 °C, 5 MPa H2, 1 h a | 53 | 78 for phenol | ||
| MoP | 340 °C, 5 MPa H2, 6 h a | 90 | 66 for phenol | [10] | |
| WP | 380 °C, 5 MPa H2, 6 h a | 89 | 84 for phenol | ||
| in situ formed MoOx | 380 °C, 1 MPa H2, 6 h a | 100 | 89 for BTX | This work | |
| in situ formed WOx | 380 °C, 5 MPa H2, 6 h a | 100 | 96 for BTX |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukhtarova, M.; Golubeva, M.; Sadovnikov, A.; Maximov, A. Guaiacol to Aromatics: Efficient Transformation over In Situ-Generated Molybdenum and Tungsten Oxides. Catalysts 2023, 13, 263. https://doi.org/10.3390/catal13020263
Mukhtarova M, Golubeva M, Sadovnikov A, Maximov A. Guaiacol to Aromatics: Efficient Transformation over In Situ-Generated Molybdenum and Tungsten Oxides. Catalysts. 2023; 13(2):263. https://doi.org/10.3390/catal13020263
Chicago/Turabian StyleMukhtarova, Mariyam, Maria Golubeva, Alexey Sadovnikov, and Anton Maximov. 2023. "Guaiacol to Aromatics: Efficient Transformation over In Situ-Generated Molybdenum and Tungsten Oxides" Catalysts 13, no. 2: 263. https://doi.org/10.3390/catal13020263
APA StyleMukhtarova, M., Golubeva, M., Sadovnikov, A., & Maximov, A. (2023). Guaiacol to Aromatics: Efficient Transformation over In Situ-Generated Molybdenum and Tungsten Oxides. Catalysts, 13(2), 263. https://doi.org/10.3390/catal13020263