

Role of N-Terminal Extensional Long α-Helix in the Arylesterase from Lacticaseibacillus rhamnosus GG on Catalysis and Stability


Abstract
:1. Introduction
2. Results and Discussion
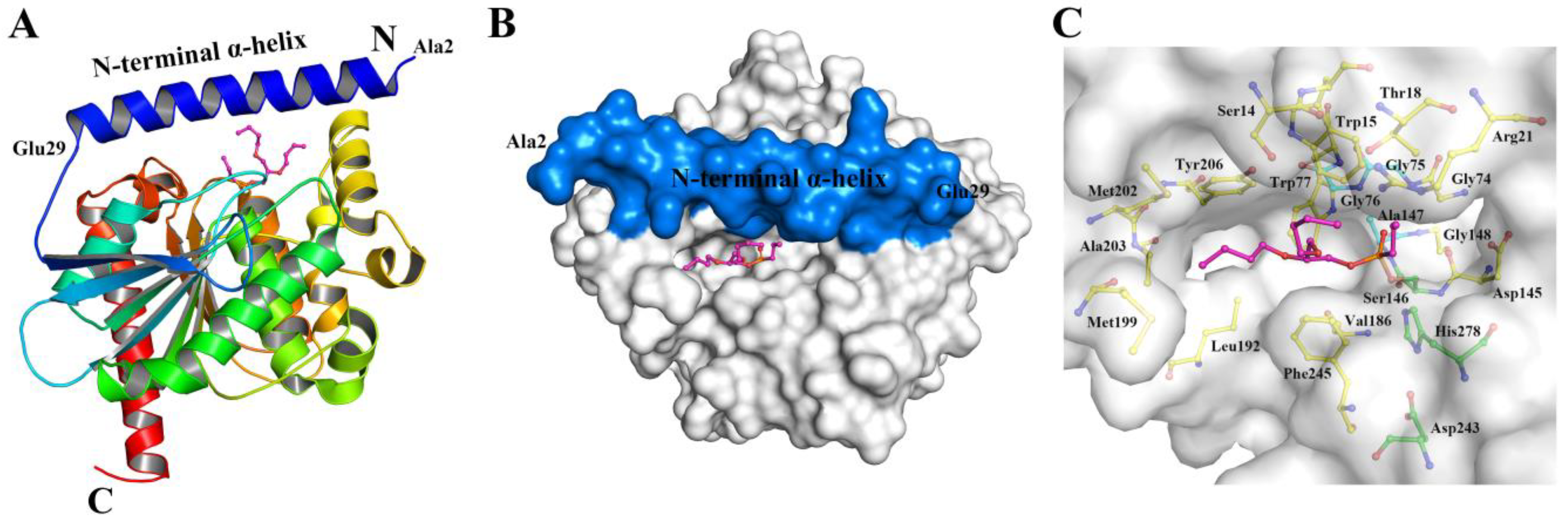
2.1. Location of N-Teminal Extensional Long α-Helix (NEL-Helix)
2.2. Role of the NEL-Helix on Overall Structure
2.3. Role of the NEL-Helix on Optimum pH and Temperature
2.4. Role of the NEL-Helix on Catalysis and Selectivity of Acyl Chain Length
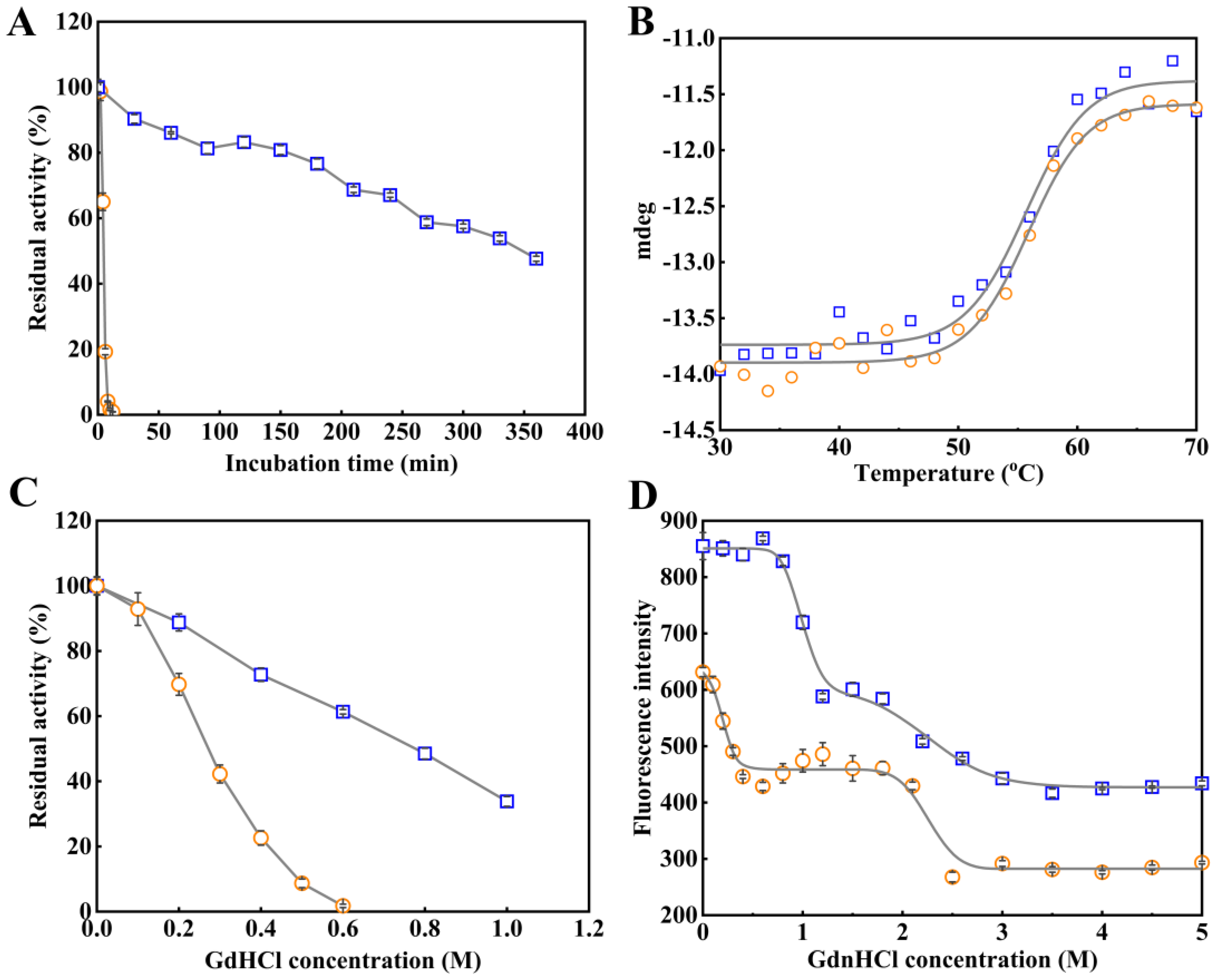
2.5. Role of the NEL-Helix on Thermal and Chemical Stability
2.6. Role of the NEL-Helix on Organic Solvents and DOC Tolerance
3. Materials and Methods
3.1. Bacterial Strains, Enzymes and Reagents
3.2. Gene Cloning, Over-Expression and Purification
3.3. Circular Dichroism (CD) Spectroscopy
3.4. Enzyme Activity Assay
3.5. Effects of pH and Temperature on Activity
3.6. Enzyme Steady-State Kinetics
3.7. Thermal Inactivation and Thermal Unfolding for Arylesterase
3.8. Chemical Inactivation and Chemical Unfolding Induced by GdnHCl
3.9. Effects of Organic Solvents and Sodium Deoxycholate (DOC) on Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nardini, M.; Dijkstra, B.W. α/β Hydrolase fold enzymes: The family keeps growing. Curr. Opin. Struct. Biol. 1999, 9, 732–737. [Google Scholar] [CrossRef]
- Heikinheimo, P.; Goldman, A.; Jeffries, C.; Ollis, D.L. Of barn owls and bankers: A lush variety of α/β hydrolases. Structure 1999, 7, R141–R146. [Google Scholar] [CrossRef] [Green Version]
- Bauer, T.L.; Buchholz, P.C.F.; Pleiss, J. The modular structure of α/β-hydrolases. FEBS J. 2020, 287, 1035–1053. [Google Scholar] [CrossRef] [PubMed]
- Rauwerdink, A.; Kazlauskas, R.J. How the same core catalytic machinery catalyzes 17 different reactions: The serine-histidine-aspartate catalytic triad of α/β-hydrolase fold enzymes. ACS Catal. 2015, 5, 6153–6176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmquist, M. Alpha/beta-hydrolase fold enzymes: Structures, functions and mechanisms. Curr. Protein. Pept. Sci. 2000, 1, 209–235. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.I.; Lan, D.; Durrani, R.; Huan, W.; Zhao, Z.; Wang, Y. The lid domain in lipases: Structural and functional determinant of enzymatic properties. Front. Bioeng. Biotechnol. 2017, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Miled, N.; Bussetta, C.; De caro, A.; Rivière, M.; Berti, L.; Canaan, S. Importance of the lid and cap domains for the catalytic activity of gastric lipases. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2003, 136, 131–138. [Google Scholar] [CrossRef]
- Wei, X.; Wang, Y.L.; Wen, B.T.; Liu, S.J.; Wang, L.; Sun, L.; Gu, T.Y.; Li, Z.; Bao, Y.; Fan, S.L.; et al. The α-helical cap domain of a novel esterase from gut Alistipes shahii shaping the substrate-binding pocket. J. Agric. Food Chem. 2021, 69, 6064–6072. [Google Scholar] [CrossRef]
- Gall, M.G.; Nobili, A.; Pavlidis, I.V.; Bornscheuer, U.T. Improved thermostability of a Bacillus subtilis esterase by domain exchange. Appl. Microbiol. Biotechnol. 2014, 98, 1719–1726. [Google Scholar] [CrossRef]
- Luan, Z.J.; Yu, H.L.; Ma, B.D.; Qi, Y.K.; Chen, Q.; Xu, J.H. Dramatically improved performance of an esterase for cilastatin synthesis by cap domain engineering. Ind. Eng. Chem. Res. 2016, 55, 12167–12172. [Google Scholar] [CrossRef]
- Skjøt, M.; De Maria, L.; Chatterjee, R.; Svendsen, A.; Patkar, S.A.; Ostergaard, P.R.; Brask, J. Understanding the plasticity of the α/β hydrolase fold: Lid swapping on the Candida antarctica lipase B results in chimeras with interesting biocatalytic properties. Chembiochem 2009, 10, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wen, B.; Liu, Y.; Du, G.; Wei, X.; Imam, K.M.S.U.; Zhou, H.; Fan, S.; Wang, F.; Wang, Y.; et al. A reverse catalytic triad Asp containing loop shaping a wide substrate binding pocket of a feruloyl esterase from Lactobacillus plantarum. Int. J. Biol. Macromol. 2021, 184, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Varejão, N.; De-Andrade, R.A.; Almeida, R.V.; Anobom, C.D.; Foguel, D.; Reverter, D. Structural mechanism for the temperature-dependent activation of the hyperthermophilic Pf2001 esterase. Structure 2018, 26, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Noby, N.; Auhim, H.S.; Winter, S.; Worthy, H.L.; Embaby, A.M.; Saeed, H.; Hussein, A.; Pudney, C.R.; Rizkallah, P.J.; Wells, S.A.; et al. Structure and in silico simulations of a cold-active esterase reveals its prime cold-adaptation mechanism. Open Biol. 2021, 11, 210182. [Google Scholar] [CrossRef] [PubMed]
- Romano, D.; Bonomi, F.; de Mattos, M.C.; de Sousa Fonseca, T.; de Oliveira Mda, C.; Molinari, F. Esterases as stereoselective biocatalysts. Biotechnol. Adv. 2015, 33, 547–565. [Google Scholar] [CrossRef]
- Bornscheuer, U.T. Microbial carboxyl esterases: Classification, properties and application in biocatalysis. FEMS Microbiol. Rev. 2002, 26, 73–81. [Google Scholar] [CrossRef]
- Panda, T.; Gowrishankar, B.S. Production and applications of esterases. Appl. Microbiol. Biotechnol. 2005, 67, 160–169. [Google Scholar] [CrossRef]
- Jaeger, K.E.; Eggert, T. Lipases for biotechnology. Curr. Opin. Biotechnol. 2002, 13, 390–397. [Google Scholar] [CrossRef]
- Arpigny, J.L.; Jaeger, K.E. Bacterial lipolytic enzymes: Classification and properties. Biochem. J. 1999, 343, 177–183. [Google Scholar] [CrossRef]
- Wei, Y.; Contreras, J.A.; Sheffield, P.; Osterlund, T.; Derewenda, U.; Kneusel, R.E.; Matern, U.; Holm, C.; Derewenda, Z.S. Crystal structure of brefeldin A esterase, a bacterial homolog of the mammalian hormone-sensitive lipase. Nat. Struct. Biol. 1999, 6, 340–345. [Google Scholar]
- Miguel-Ruano, V.; Rivera, I.; Rajkovic, J.; Knapik, K.; Torrado, A.; Otero, J.M.; Beneventi, E.; Becerra, M.; Sánchez-Costa, M.; Hidalgo, A.; et al. Biochemical and structural characterization of a novel thermophilic esterase EstD11 provide catalytic insights for the HSL family. Comput. Struct. Biotechnol. J. 2021, 19, 1214–1232. [Google Scholar] [CrossRef]
- Mandrich, L.; Merone, L.; Pezzullo, M.; Cipolla, L.; Nicotra, F.; Rossi, M.; Manco, G. Role of the N terminus in enzyme activity, stability and specificity in thermophilic esterases belonging to the HSL family. J. Mol. Biol. 2005, 345, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Foglia, F.; Mandrich, L.; Pezzullo, M.; Graziano, G.; Barone, G.; Rossi, M.; Manco, G.; Del Vecchio, P. Role of the N-terminal region for the conformational stability of esterase 2 from Alicyclobacillus acidocaldarius. Biophys. Chem. 2007, 127, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Santi, C.; Tutino, M.L.; Mandrich, L.; Giuliani, M.; Parrilli, E.; Del Vecchio, P.; de Pascale, D. The hormone-sensitive lipase from Psychrobacter sp. TA144: New insight in the structural/functional characterization. Biochimie 2010, 92, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Li, B.C.; Guo, T.T.; Ding, G.B. Characteration of a novel arylesterase from probiotics Lacticaseibacillus rhamnosus GG with the preference for medium- and long-chain p-nitrophenyl esters. 3 Biotech 2021, 11, 496. [Google Scholar] [CrossRef] [PubMed]
- Velankar, S.; Burley, S.K.; Kurisu, G.; Hoch, J.C.; Markley, J.L. The Protein Data Bank archive. Methods Mol. Biol. 2021, 2305, 3–21. [Google Scholar]
- Mandrich, L.; Menchise, V.; Alterio, V.; De Simone, G.; Pedone, C.; Rossi, M.; Manco, G. Functional and structural features of the oxyanion hole in a thermophilic esterase from Alicyclobacillus acidocaldarius. Proteins 2008, 71, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Menchise, V.; Manco, G.; Mandrich, L.; Sorrentino, N.; Lang, D.; Rossi, M.; Pedone, C. The crystal structure of a hyper-thermophilic carboxylesterase from the archaeon Archaeoglobus fulgidus. J. Mol. Biol. 2001, 314, 507–518. [Google Scholar] [CrossRef]
- Palm, G.J.; Fernández-Álvaro, E.; Bogdanović, X.; Bartsch, S.; Sczodrok, J.; Singh, R.K.; Böttcher, D.; Atomi, H.; Bornscheuer, U.T.; Hinrichs, W. The crystal structure of an esterase from the hyperthermophilic microorganism Pyrobaculum calidifontis VA1 explains its enantioselectivity. Appl. Microbiol. Biotechnol. 2011, 91, 1061–1072. [Google Scholar] [CrossRef]
- Nam, K.H.; Kim, M.Y.; Kim, S.J.; Priyadarshi, A.; Kwon, S.T.; Koo, B.S.; Yoon, S.H.; Hwang, K.Y. Structural and functional analysis of a novel hormone-sensitive lipase from a metagenome library. Proteins 2009, 74, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Li, P.Y.; Chen, X.L.; Ji, P.; Li, C.Y.; Wang, P.; Zhang, Y.; Xie, B.B.; Qin, Q.L.; Su, H.N.; Zhou, B.C.; et al. Interdomain hydrophobic interactions modulate the thermostability of microbial esterases from the hormone-sensitive lipase family. J. Biol. Chem. 2015, 290, 11188–11198. [Google Scholar] [CrossRef] [Green Version]
- Nam, K.H.; Kim, M.Y.; Kim, S.J.; Priyadarshi, A.; Lee, W.H.; Hwang, K.Y. Structural and functional analysis of a novel EstE5 belonging to the subfamily of hormone-sensitive lipase. Biochem. Biophys. Res. Commun. 2009, 379, 553–556. [Google Scholar] [CrossRef]
- Byun, J.S.; Rhee, J.K.; Kim, N.D.; Yoon, J.; Kim, D.U.; Koh, E.; Oh, J.W.; Cho, H.S. Crystal structure of hyperthermophilic esterase EstE1 and the relationship between its dimerization and thermostability properties. BMC Struct. Biol. 2007, 7, 47. [Google Scholar] [CrossRef] [Green Version]
- Angkawidjaja, C.; Koga, Y.; Takano, K.; Kanaya, S. Structure and stability of a thermostable carboxylesterase from the thermoacidophilic archaeon Sulfolobus tokodaii. FEBS J. 2012, 279, 3071–3084. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.H.; Lee, H.S.; Kim, J.T.; Kim, S.J.; Choi, S.H.; Kang, S.G.; Lee, J.H. Identification of a new subfamily of salt-tolerant esterases from a metagenomic library of tidal flat sediment. Appl. Microbiol. Biotechnol. 2012, 93, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Delano, W.L. The PyMOL Molecular Graphics System. DeLano Scientific 2002. [Google Scholar]
- Zhang, Z.; Zheng, B.; Wang, Y.; Chen, Y.; Manco, G.; Feng, Y. The conserved N-terminal helix of acylpeptide hydrolase from archaeon Aeropyrum pernix K1 is important for its hyperthermophilic activity. Biochim. Biophys. Acta. 2008, 1784, 1176–1183. [Google Scholar] [CrossRef]
- Fuciños, P.; Atanes, E.; López-López, O.; Esperanza Cerdán, M.; Isabel González-Siso, M.; Pastrana, L.; Luisa Rúa, M. Production and characterization of two N-terminal truncated esterases from Thermus thermophilus HB27 in a mesophilic yeast: Effect of N-terminus in thermal activity and stability. Protein Expr. Purif. 2011, 78, 120–130. [Google Scholar] [CrossRef]
- Schneider, B.; Knöchel, T.; Darimont, B.; Hennig, M.; Dietrich, S.; Babinger, K.; Kirschner, K.; Sterner, R. Role of the N-terminal extension of the (βα)8-barrel enzyme indole-3-glycerol phosphate synthase for its fold, stability, and catalytic activity. Biochemistry 2005, 44, 16405–16412. [Google Scholar] [CrossRef]
- Liu, Z.; Cheng, Z.; Ye, S.; Zhou, L.; Zhou, Z. Catalytic ability improvement of phenylalanine hydroxylase from Chromobacterium violaceum by N-terminal truncation and proline introduction. J. Microbiol. Biotechnol. 2019, 29, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.K.; Shivakumaraswamy, S.; Gummadi, S.N.; Manoj, N. Role of an N-terminal extension in stability and catalytic activity of a hyperthermostable α/β hydrolase fold esterase. Protein Eng. Des. Sel. 2017, 30, 559–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, D.; Kang, H. The N-terminal amino acid sequences of the firefly luciferase are important for the stability of the enzyme. Photochem. Photobiol. 1998, 68, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Chen, X.; Zhou, X.; Huang, R.; Li, L.; Miao, Y.; Liu, D.; Zhang, R. Improvement of GH10 family xylanase thermostability by introducing of an extra α-helix at the C-terminal. Biochem. Biophys. Res. Commun. 2019, 515, 417–422. [Google Scholar] [CrossRef]
- You, C.; Zhang, X.Z.; Zhang, Y.H. Simple cloning via direct transformation of PCR product DNA multimer, to Escherichia coli and Bacillus subtilis. Appl. Environ. Microbiol. 2012, 78, 1593–1595. [Google Scholar] [CrossRef] [Green Version]
- Li, B.C.; Peng, B.; Zhang, T.; Li, Y.Q.; Ding, G.B. A spectrophotometric method for high-throughput screening of α-L-rhamnosidase activity on rutin coupled with a β-D-glucosidase assay. 3 Biotech 2019, 9, 227. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yang, G.; Wu, L.; Feng, Y. Role of the NC-loop in catalytic activity and stability in lipase from Fervidobacterium changbaicum. PLoS ONE 2012, 7, e46881. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LggEst Wild Type | LggEst Deleted Mutant | |||||||
|---|---|---|---|---|---|---|---|---|
| Substrate | Vmax (µmol min−1 mg−1) | kcat (s−1) | KM (µM) | kcat/KM (M−1 s−1) | Vmax (µmol min−1 mg−1) | kcat (s−1) | KM (µM) | kcat/KM (M−1 s−1) |
| pNPC2 | 61.6 ± 2.3 | 37.6 ± 1.4 | 7.8 × 103 ± 0.6 × 103 | 4.8 × 103 | 16.1 ± 0.9 | 9.0 ± 0.5 | 11.2 × 103 ± 1.1 × 103 | 8.0 × 102 |
| pNPC4 | 52.5 ± 2.6 | 32.0 ± 1.6 | 1.2 × 103 ± 0.1 × 103 | 2.7 × 104 | 14.7 ± 0.8 | 8.2 ± 0.4 | 1.5 × 103 ± 0.2 × 103 | 5.5 × 103 |
| pNPC6 | 87.2 ± 7.5 | 53.2 ± 4.5 | 247.1 ± 32.2 | 2.2 × 105 | 25.5 ± 2.6 | 14.2 ± 1.5 | 433.4 ± 58.3 | 3.3 × 104 |
| pNPC8 | 102.8 ± 5.1 | 62.7 ± 3.1 | 19.6 ± 2.6 | 3.2 × 106 | 7.8 ± 0.2 | 4.4 ± 0.1 | 12.7 ± 1.3 | 3.5 × 105 |
| pNPC10 | 28.6 ± 0.8 | 17.4 ± 0.5 | 4.5 ± 0.5 | 3.9 × 106 | 2.7 ± 0.1 | 1.5 ± 0.1 | 3.9 ± 0.4 | 3.8 × 105 |
| pNPC12 | 3.5 ± 0.1 | 2.0 ± 0.1 | 4.0 ± 0.2 | 5.0 × 105 | 4.5 × 10−1 ± 0.1 × 10−1 | 2.5 × 10−1 ± 0.1 × 10−1 | 3.1 ± 0.2 | 8.1 × 104 |
| Enzymes | C50 (M) | Tm (°C) | Cm1 (M) | Cm2 (M) |
|---|---|---|---|---|
| LggEst wild type | 0.76 | 55.7 | 1.2 | 2.3 |
| LggEst deleted mutant | 0.24 | 55.9 | 0.2 | 1.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.-C.; Guo, T.; Li, X.; Hou, X.; Ding, G.-B. Role of N-Terminal Extensional Long α-Helix in the Arylesterase from Lacticaseibacillus rhamnosus GG on Catalysis and Stability. Catalysts 2023, 13, 441. https://doi.org/10.3390/catal13020441
Li B-C, Guo T, Li X, Hou X, Ding G-B. Role of N-Terminal Extensional Long α-Helix in the Arylesterase from Lacticaseibacillus rhamnosus GG on Catalysis and Stability. Catalysts. 2023; 13(2):441. https://doi.org/10.3390/catal13020441
Chicago/Turabian StyleLi, Bin-Chun, Tongtong Guo, Xue Li, Xueting Hou, and Guo-Bin Ding. 2023. "Role of N-Terminal Extensional Long α-Helix in the Arylesterase from Lacticaseibacillus rhamnosus GG on Catalysis and Stability" Catalysts 13, no. 2: 441. https://doi.org/10.3390/catal13020441
APA StyleLi, B. -C., Guo, T., Li, X., Hou, X., & Ding, G. -B. (2023). Role of N-Terminal Extensional Long α-Helix in the Arylesterase from Lacticaseibacillus rhamnosus GG on Catalysis and Stability. Catalysts, 13(2), 441. https://doi.org/10.3390/catal13020441