1. Introduction
Research into new energy systems has garnered much attention over the past few decades due to the depletion of fossil fuels and the resulting environmental pollution. In this regard, oxygen evolution reactions (OERs) are important for efficient energy conversion and storage, especially in the case of fuel cells [
1,
2,
3,
4,
5]. However, owing to the high overpotential for O–H bond cleavage and O–O bond formation, the kinetics of OERs are sluggish [
6,
7]. Therefore, the use of a catalyst is essential to reduce the massive energy consumption that would otherwise be needed.
The most representative electrocatalysts for OERs are noble metals, such as Ir and Ru, but they are expensive and in limited supply [
8,
9]. Contrarily, transition metal oxides are well-known electrocatalysts with a wide performance range due to the redox couples of their metal centers. In particular, non-noble metal oxides, such as Ni, Co, and Mn oxides, have been studied because of their low cost and high efficiency [
10,
11].
Coordination polymers, including metal–organic frameworks (MOFs), are also considered promising OER electrocatalysts. Their large surface areas, distinctively adjustable porous architectures, and electrical influence of the functional organic components on metal cores make MOFs beneficial [
12,
13].
Recently, we reported on the synthesis and electrocatalytic properties of the 3D porous cobalt cinnamate Co
7(
trans-cinnamate)
14(H
2O)
2, which showed superior electrocatalytic behavior, especially in the OERs of inorganic cobalt oxides [
14,
15]. Previous research revealed that the incorporation of the π-conjugated carboxylate
trans-cinnamate, which can be easily separated from the cobalt cation due to the oxygen atoms in cobalt cinnamate having electron-poor environments compared to simple inorganic cobalt oxides, was responsible for the excellent OER electrocatalytic performance of cobalt cinnamate [
16,
17,
18].
In this work, we presented another cobalt cinnamate complex that incorporates 2-aminopyrimidine. In this case, unique tetrameric Co4 units bridged by H2O and trans-cinnamate anions are linked to each other through a 2-aminopyridmidine ligand, which completes the 1D chain structure. This cobalt complex incorporating π-conjugated trans-cinnamate and 2-aminopyrimidine was expected to exhibit similar electrical behaviors.
In addition, reduced graphene oxide (rGO) was employed as an electrochemical supporter for introducing a catalyst into an electrode surface to enhance the electrocatalytic performance by increasing the electrical contact between the electrode surface and the catalyst. Many previous studies have reported that the use of rGO as a supporter can provide a higher surface area and good electrical conductivity, enabling an efficient electrocatalyst reaction [
19,
20,
21].
In this study, a new cobalt cinnamate complex was developed that incorporates 2-aminopyrimidine (catalyst 1) as an electrocatalyst and rGO as an electrochemical supporter for OERs. The electrocatalytic performances of catalyst 1 with or without the rGO supporter were investigated using linear sweep voltammetry (LSV) and chronoamperometry (CA).
2. Results
Catalyst
1 was prepared by employing a hydrothermal method. By mixing CoCl
2∙6H
2O, 2-aminopyrimidine, and
trans-cinnamic acid in water, we produced catalyst
1 in the form of red, polyhedral-shaped crystals. A powder X-ray diffraction (XRD) analysis confirmed the homogeneity and purity of the bulk products (
FigureS S1 and S2 in the Supporting Information, SI). The structure of
1 was composed of infinite neutral Co
2(
trans-cinnamate)
4(2-aminopyrimidine)
2(H
2O) chains. Heavily disordered lattice water molecules were located between the chains. There were two crystallographically distinct cobalt atoms. The Co(1) atom was coordinated by four
trans-cinnamate ligands to form CoO
4 in a square planar geometry in the range of 2.031(2)–2.133(2) Å. To complete the slightly distorted octahedral coordination of CoNO
4(H
2O)
2, an additional water molecule was coordinated to Co at a distance of 2.207(2) and one nitrogen atom from 2-aminopyrimidine at a distance of 2.244(3) in the
trans position. The coordination environment of the other Co(2) atom was very similar to that of Co(1), except that one of the
trans-cinnamate ligands in the square plane was replaced by 2-aminopyrimidine (
Figure 1a). The bond valence sum (BVS) calculations for Co(1) and Co(2) resulted in values of 1.95 and 1.85, respectively, indicating an oxidation state of +2.00 [
22].
The Co octahedra formed dimeric units by sharing the coordinated water molecule and two
trans-cinnamate ligands (
Figure 1). The dimers were connected to each other by bridging two
trans-cinnamate ligands to produce a tetrameric unit. As shown, there were five
trans-cinnamate and three 2-aminopyrimidine ligands in the dimer, two of which were used to form dimers by connecting the Co(1) and Co(2) octahedra, and two other
trans-cinnamate contributed to connecting the dimers to form tetramers. The other
trans-cinnamate and one of the three 2-aminopyrimidine molecules remained terminal, and the other two 2-aminopyrimidines acted as bridging ligands connecting the dimers.
Overall, the Co
4(
trans-cinnamate)
8(2-aminopyrimidine)
4(H
2O)
2 tetrameric units were alternately connected to each other by two 2-aminopyrimidine ligands to produce neutral Co
2(
trans-cinnamate)
4(2-aminopyrimidine)
2(H
2O) chains along the
a-axis (
Scheme 1;
Figure 1b). The interchain packing of
1 is shown in
Figure 1c. The closest interchain contact was found between the
trans-cinnamate ligands (d(C11∙∙∙C14) = 3.483(4) Å), forming a weak π–π stacking. The next closest interchain contact was found between
trans-cinnamate and 2-aminopyrimidine (d(C6∙∙∙C13) = 3.490(4) Å). This implied that the van der Waals interactions were dominant and the π–π stacking between the chains was weak.
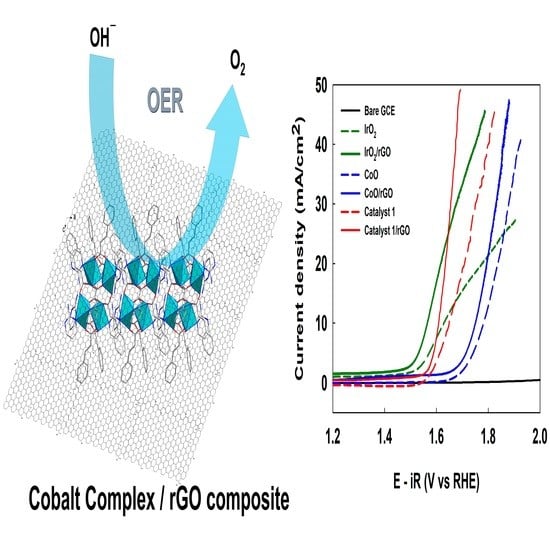
The electrocatalytic properties of catalyst
1 with or without the rGO supporter were investigated with respect to OERs. The linear sweep voltammograms (LSVs) of catalyst
1 and the reference material-modified glassy carbon electrode (GCE) are shown in
Figure 2a. The onset potentials of the electrocatalysts with rGO were ~40 to 60 mV less positive than those of the cases without rGO.
The overpotentials (V
J) at a current density of 10 mA cm
−2 for catalyst
1, CoO, and IrO
2 with the rGO-modified GCEs were 386, 526, and 341 mV, respectively (
Table 1). The overpotential of the catalyst
1/rGO composite was much lower than that of CoO but slightly higher than that of the noble metal catalysts, such as IrO
2.
The kinetic parameters, including the Tafel slope, were also investigated, as shown in
Figure 2b. [
23] The Tafel slopes of the catalyst
1/rGO composite- and catalyst
1-modified GCEs were 64 and 73 mV/dec, respectively (
Table 1). The small Tafel slope of the catalyst
1/rGO composite indicated the best kinetic performance compared to the reference materials. The Tafel slopes of the reference electrocatalysts, which included CoO and IrO
2 with or without rGO, were 119, 157, 116, and 134 mV/dec, respectively.
Surprisingly, the small onset potential and Tafel slope values of the catalyst 1/rGO composite mean that it exhibits excellent electrocatalytic activity toward OERs from both thermodynamic and kinetic perspectives. The catalyst 1/rGO composite outperformed or was comparable to catalyst 1 alone or other metal oxide reference materials, such as CoO and IrO2, with or without rGO supports.
To test the durability of the electrocatalyst, long-time chronoamperometry (CA) was conducted for the catalyst
1/rGO composite. The CA experiments were performed with an applied potential, where the expected current density was 10 mA cm
−2. As shown in
Figure 3, the current density of the catalyst
1-modified GCE was maintained at ≥90 % for 12 h. However, the current density of the catalyst
1/rGO composite-modified GCE was maintained at ≥64 % for 12 h, which was worse than that of catalyst
1 alone. According to the X-ray photoelectron spectroscopy (XPS) analysis, which will be described in the next section, there are no significant structural changes between the samples with/without rGO and before/after the electrochemical treatments. Therefore, these results suggested that the interaction between catalyst
1 and the electrode was stronger than the one between the catalyst
1/rGO complex and the electrode; thus, this settlement could be maintained even during vigorous gas evolution reactions.
3. Discussion
Recently, we reported a highly active new OER electrocatalyst, cobalt cinnamate, [Co
7(
trans-cinnamate)
14(H
2O)
2], in which the π–π orbital delocalization of the
trans-cinnamate ligand allowed for fast charge transfer through the 3D structure, followed by electron flexibility near the metal center, which improved the OER activity [
14,
19,
20,
21]. Pyridine ligands may have a template effect on the formation of the skeletal structure, depending on the size and shape of the amine used, and may have various levels of structural diversity when used as ligands. Therefore, the addition of a pyridine ligand during the formation of coordination polymers, including MOFs, is expected to bring various structural changes while maintaining electrocatalytic properties.
In this study, a new cobalt complex, abbreviated as
1, was prepared by using
trans-cinnamate and 2-aminopyrimidine ligands. In compound
1, the π–π orbital delocalization was modulated compared to cobalt cinnamate, [Co
7(
trans-cinnamate)
14(H
2O)
2], by introducing 2-aminopyrimidine ligands. The 2-aminopyrimidine ligand is also stabilized by the π–π orbital delocalization. Although the comparison of electrical effects between the two ligands is indirect, considering that pure nitrogen is a better electron-donating element than oxygen, it is predicted that the nitrogen of 2-aminopyrimidine can have a rather strong metal–nitrogen bond compared to the oxygen of
trans-cinnamate. This Co coordination environment of
1 in electron-rich environments was somewhat disadvantageous compared to that of cobalt cinnamate. Therefore, catalyst
1 may show relatively lower activity than the previously reported cobalt cinnamate (with or without rGO with a V
J of 327 and 358 mV, respectively) [
15]. In spite of that, catalyst
1 is still a good electrocatalyst for OERs compared to IrO
2, as shown in
Figure 4 and
Figure 5.
In addition, the rGO supporter, which is a widely used carbon-based supporter, was introduced to reinforce the effectiveness of insufficient π–π stacking interactions and increase stability. As a result of the participation of the rGO supporter, the onset potential was improved, as shown in
Figure 2. The reduced overpotential with the rGO supporter seemed to be due to a strong interaction between the catalyst
1 and rGO, such as additional π–π stacking interactions between the ligand and rGO. On the other hand, a reduction in the overpotential was also seen in the CoO/rGO and IrO
2/rGO composites. This was thought to be because the mixing of rGO increased the electrical conductivity of CoO or IrO
2 on the electrode surface.
As shown in
Figure 3, the catalyst
1/rGO composite has relatively low stability compared to the excellent stability of catalyst
1 because the interaction between catalyst
1 and the electrode is stronger than the interaction between the rGO and the electrode.
To identify the enhanced electrocatalytic activity based on the electronic structure modulation of the catalysts, X-ray photoelectron spectroscopy (XPS) analyses before and after the OER measurements were performed.
Figure 4 and
Figure 5 show the XPS spectra for C, N, O, and Co of catalyst
1 with or without an rGO supporter.
As shown in
Figure 4 and
Figure 5, the main C 1s peaks were obtained at ~284.8 eV (blue line) in the catalyst
1/rGO composite and at ~292.0 eV (red line) in catalyst
1 alone. Since the peak at 284.8 eV represents C in highly oriented pyrolytic graphite (HOPG) [
24], this seems to be due to the rGO supporter. The peaks at 292.0 eV represent C connected to highly electronegative atoms (e.g., CF
2, CO
32−) [
24], which seems to be due to the Nafion and cinnamate parts. Therefore, the increment of peak size (counts) in the whole binding energy region and the shift in the main peak from 292.0 to 284.8 eV indicated the successive introduction of the rGO supporter into the catalyst. [
15] However, there was no significant change in the position or size of the peaks before and after the electrochemical measurements. For N, O, Co, and C, there were few differences in the chemical states before and after the electrochemical measurements.
For N, the main N 1s peaks were obtained at ~399.8 eV in both cases and before/after the electrochemical measurements, which seemed to correspond to the N 1s peak at ~398.3 eV, as mentioned in the literature [
25]. The N 1s peaks were not significantly shifted after the composite formation.
The O 1s peaks were observed at ~529.4 (green line) and ~531.1 eV (red line), representing the lattice oxygen and oxygen vacancy (defect), respectively [
26,
27]. For catalyst
1, the strongest peak corresponding to the oxygen vacancy (at ~531.1 eV) was still retained after the rGO composite formation. The existence of a large area of the peak at ~531.1 eV implied the existence of oxygen vacancies and a high electrocatalytic activity of the composite and catalyst
1 [
26]. In the case of catalyst
1 alone, the small peak at ~533.4 eV that appeared more prominent indicated that a different oxidation state of oxygen was available.
The Co 2p peaks of catalyst
1 were observed at ~780.2 and ~794.8 eV with small satellite peaks before and after the electrochemical measurements. For the catalyst
1/rGO composite, the Co 2p peaks appeared at a lower binding energy region, between ~778.3 and ~793.8 eV, than that of catalyst
1, which revealed the electronic interaction between the catalyst and rGO [
28].
Therefore, the XPS analyses suggested that catalyst 1 has enhanced oxygen vacancies and a more exposed Co center, which can improve the electrocatalytic activity toward OERs.
4. Materials and Methods
4.1. Reagents
Cobalt (II) chloride hexahydrate (99.9%) was purchased from Alfa Aesar (Haverhill, MA, USA). Aldrich supplied the 2-aminopyrimidine (97%) reagent (St. Louis, MO, USA), and trans-cinnamic acid (99+%) was obtained from Acros Organics (Geel, Antwerp, Belgium). All chemicals were used as received.
4.2. Preparation of Electrocatalyst
The catalysts were synthesized by mixing cobalt (II) chloride hexahydrate (0.284 g, 1.2 mmol), 2-aminopyrimidine (0.348 g, 3.6 mmol), trans-cinnamic acid (0.088 g, 0.6 mmol), and distilled water (1.0 mL). The obtained solution was sealed in a Pyrex ® tube, heated to 100 °C for 68 h, and finally cooled to room temperature at a rate of 20 °C/h. The pH of the solution before and after the reaction was ~4 and ~7, respectively. The solid products were recovered from the solution by employing vacuum filtration and then washed with ethanol. Red polyhedral-shaped crystals of Co2(trans-cinnamate)4(2-aminopyrimidine)2(H2O)·2.5H2O (referred to as 1) and unidentified pink ribbon-shaped crystals were obtained. Due to their large size, the two types of solid products were manually separated using a microscope (~millimeter scale). Energy-dispersive X-ray spectroscopy (EDS) analyses confirmed the presence of Co in 1. For 1: elemental analysis (%) calculated: C 58.78, H 7.59, N 10.55, and O 12.05; found: C 56.83, H 7.15, N 9.98, and O 11.80. IR(KBr): 3315 s, 3285 s, 3174 m, 3061 w, 3022 w, 2958 m, 2927 m, 2858 m, 1637 m, 1597 m, 1538 s, 1462 m, 1445 m, 1434 m, 1380 s, 1338 m, 1306 w, 1263 w, 1207 w, 1139 s, 1039 w, 1073 w, 1207 s, 1002 w, 984 m, 926 m, 895 s, 862 m, 815 w, 779 s, 745 w, 707 m, 688 m, 674 m, 634 m, 590 m, 555 m, 521 m, and 500 m cm−1.
4.3. Single Crystal Structure Determinations
The single-crystal X-ray analysis was performed using a Siemens SMART diffractometer (Siemens AG, Munich, Germany) outfitted with an Apex II area detector and a graphite-monochromatized Mo Kα radiation. The structures were determined by employing direct methods and then refined by SHELXS and ShelXle with the SHELXL plug-in [
29,
30,
31]. The hydrogen atoms of
trans-cinnamate and 2-aminopyrimidine were generated geometrically and allowed to ride on their respective parent atoms. The oxygen atoms of the lattice water molecules were refined isotopically owing to their heavily disordered behavior and low occupancy. All crystal data are summarized in
Table S1. The Cambridge Crystallographic Data Center (CCDC) reference number for 1 is 2232776. The data can be obtained free of charge from the CCDC. The crystallographic details, atomic coordinates, selected bond distances, and angles are given in
Tables S1–S3.
4.4. Electrochemical Characterization
A glassy carbon electrode (GCE) (with a diameter of 5 mm; Pine Research Instrumentation, Durham, NC, USA) was used as a support for the catalysts. The loading sample solution with a concentration of 2 mg/mL was prepared by dissolving each powder of 1, IrO2, and CoO in an ethanol/water (1:1 v/v) solution. In both cases, 20 µL of Nafion was added and ultrasonicated for 30 min. Finally, 7 µL of the loading sample solution was dropped onto the cleaned GCE and dried for 1 d.
A three-electrode cell consisting of a Pt wire counter electrode, a catalyst-modified GCE, and an Ag/AgCl (1 M KCl; 0.235 V vs. NHE) reference electrode were used for the electrochemical experiments. The electrolyte solution for the OER was O
2-saturated 0.1 M KOH, and its solution resistance obtained from the conductivity measurement was ~42 Ω. The 100 % IR-compensated potentials were reported vs. the reversible hydrogen electrode (RHE) and calculated using the following equation:
4.5. Instruments
The powder X-ray diffraction (XRD) analyses were performed using a Rigaku Ultra X-ray diffractometer (Applied Rigaku Technologies, Austin, TX, USA) equipped with Cu Kα radiation. The infrared spectra (IR) were recorded using the KBr pellet method on a Nicolet 6700 FT-IR spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) in the range of 400–4000 cm−1. The XPS was measured using a Thermo Scientific K-Alpha X-ray photoelectron spectrometer (Waltham, MA, USA). The samples were drop-casted onto an indium tin oxide (ITO) electrode for the electrochemical treatments. The elemental analyses of 1 were performed at Sogang University. All electrochemical measurements with the rotating disk electrode (RDE) were performed using a CHI model 750d potentiostat (CH Instruments, Austin, TX, USA) and an RRDE-3A (ALS Co., Ltd., Tokyo, Japan).


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}