

Effect of MnO2 Crystal Type on the Oxidation of Furfural to Furoic Acid


Abstract
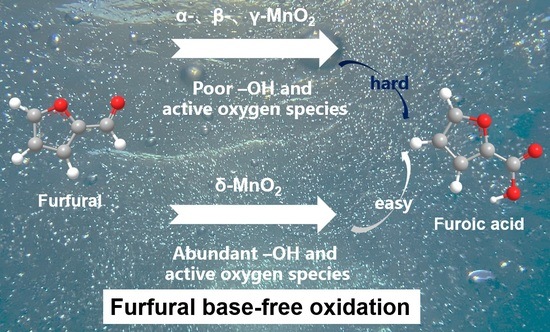
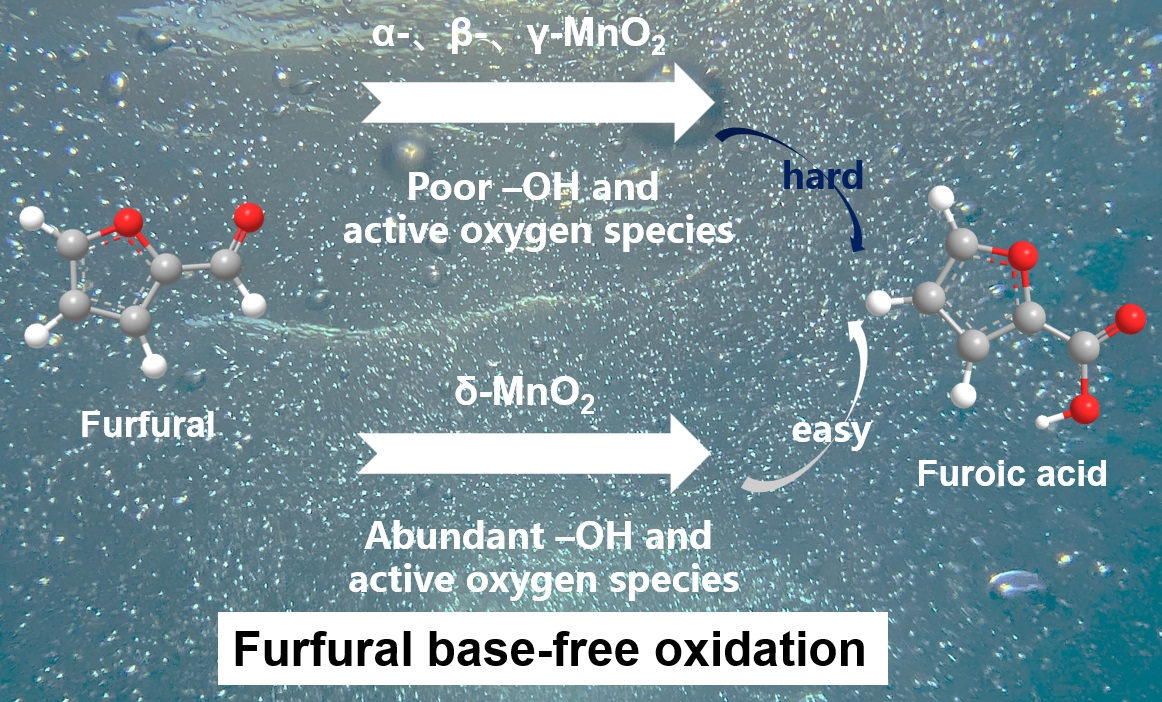
:
1. Introduction
2. Results
2.1. Furfural Oxidation Properties of Different Crystalline MnO2
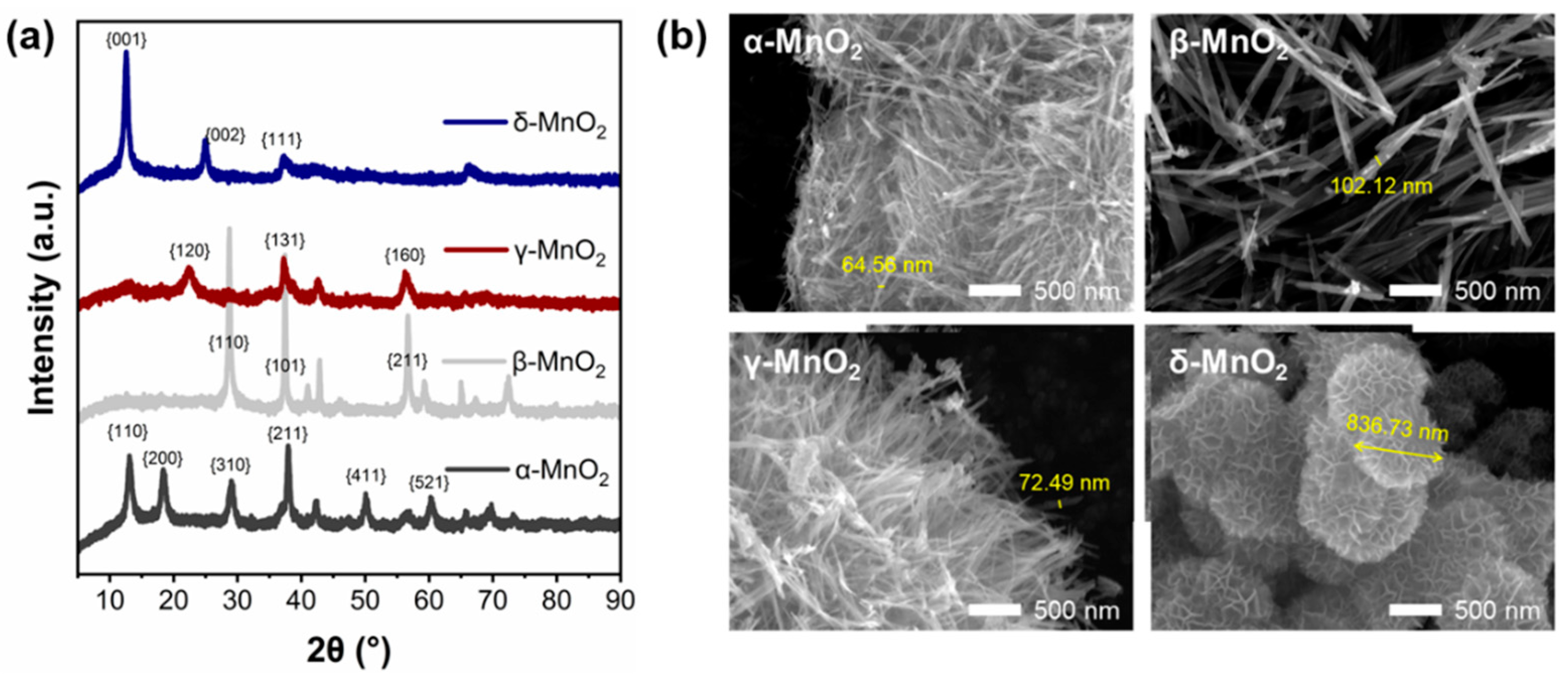
2.2. Structural Properties of Different Crystalline MnO2
2.3. Oxidation Capacity of Different Crystalline MnO2
2.4. Discussion
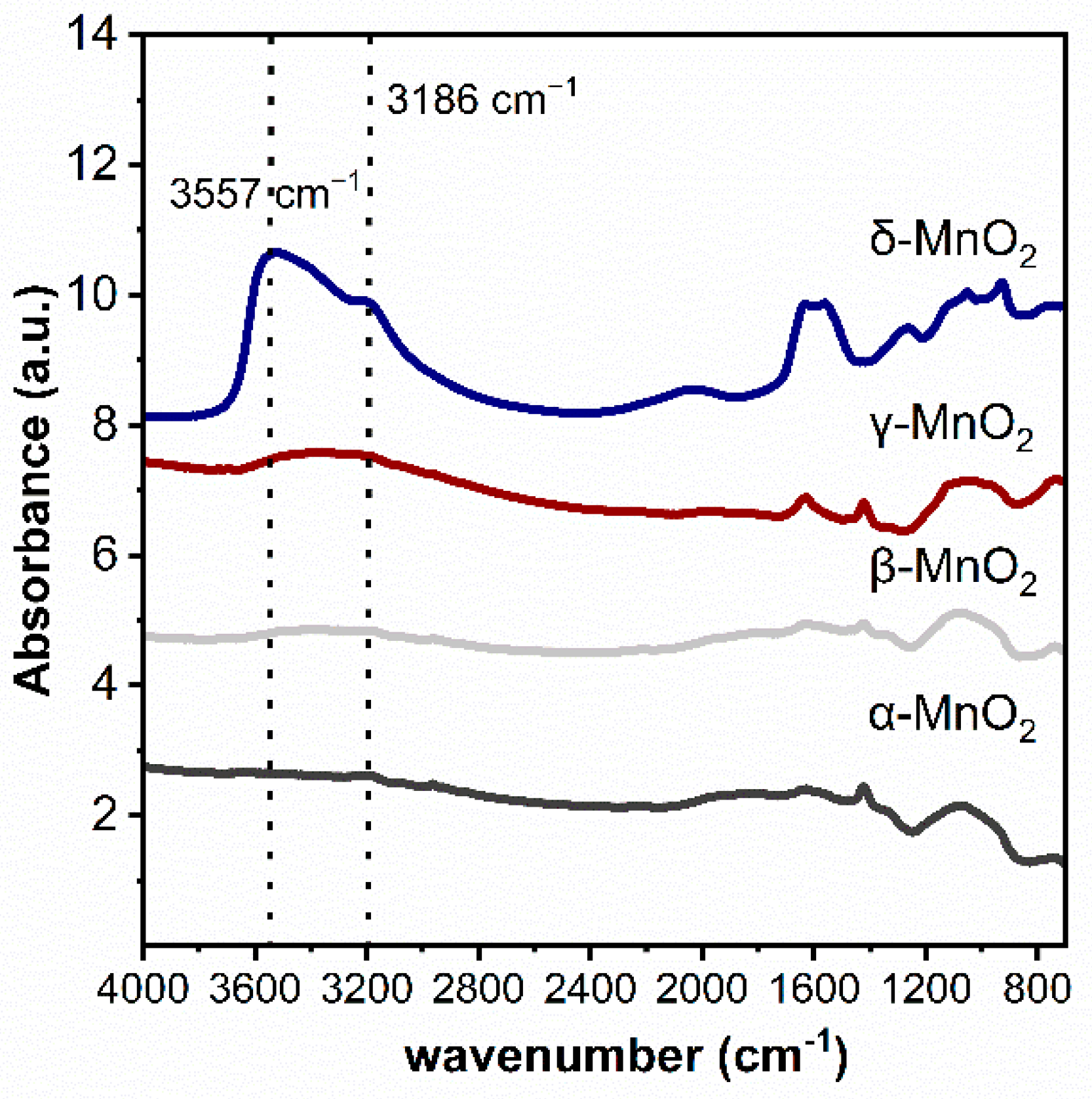
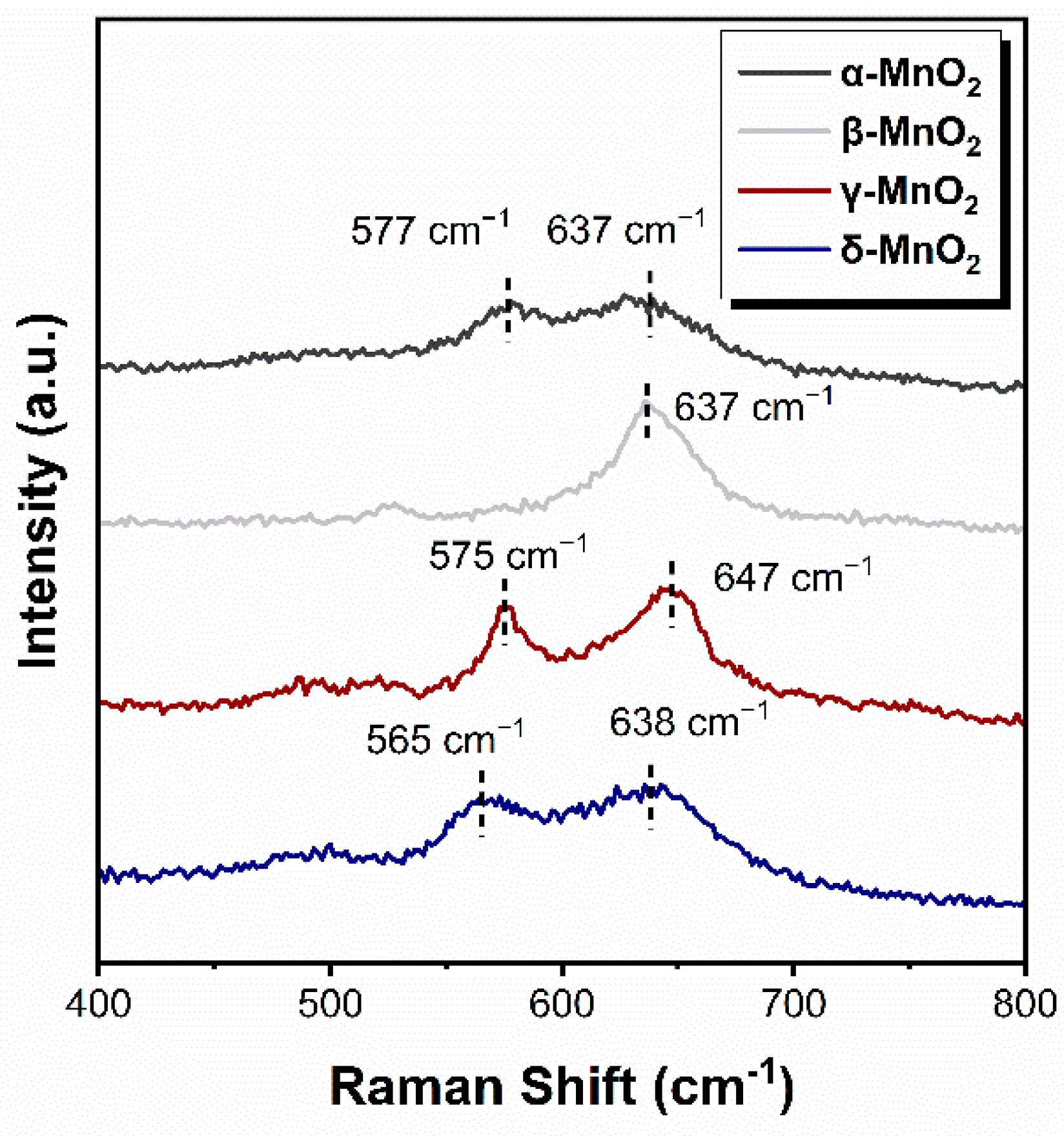
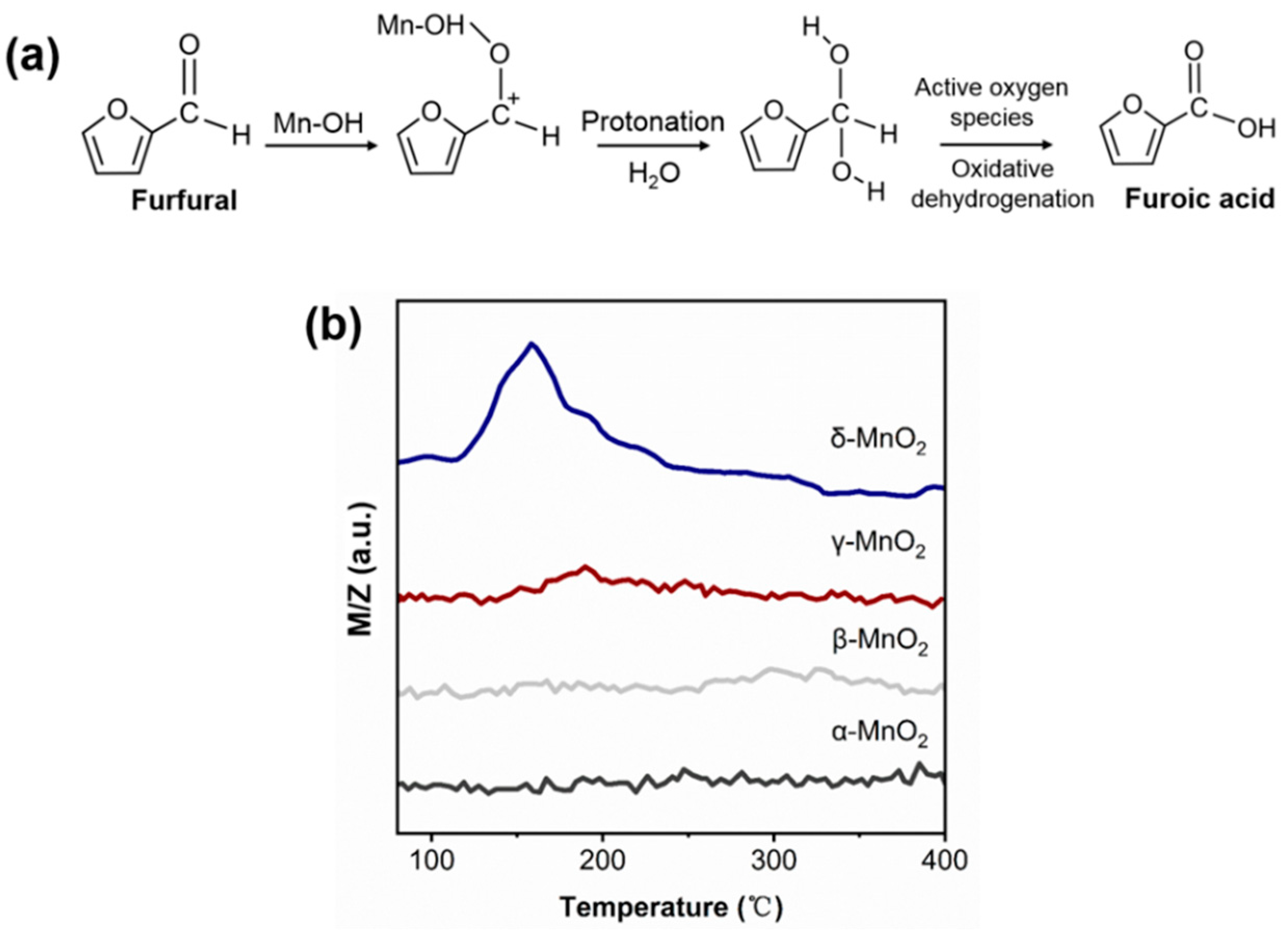
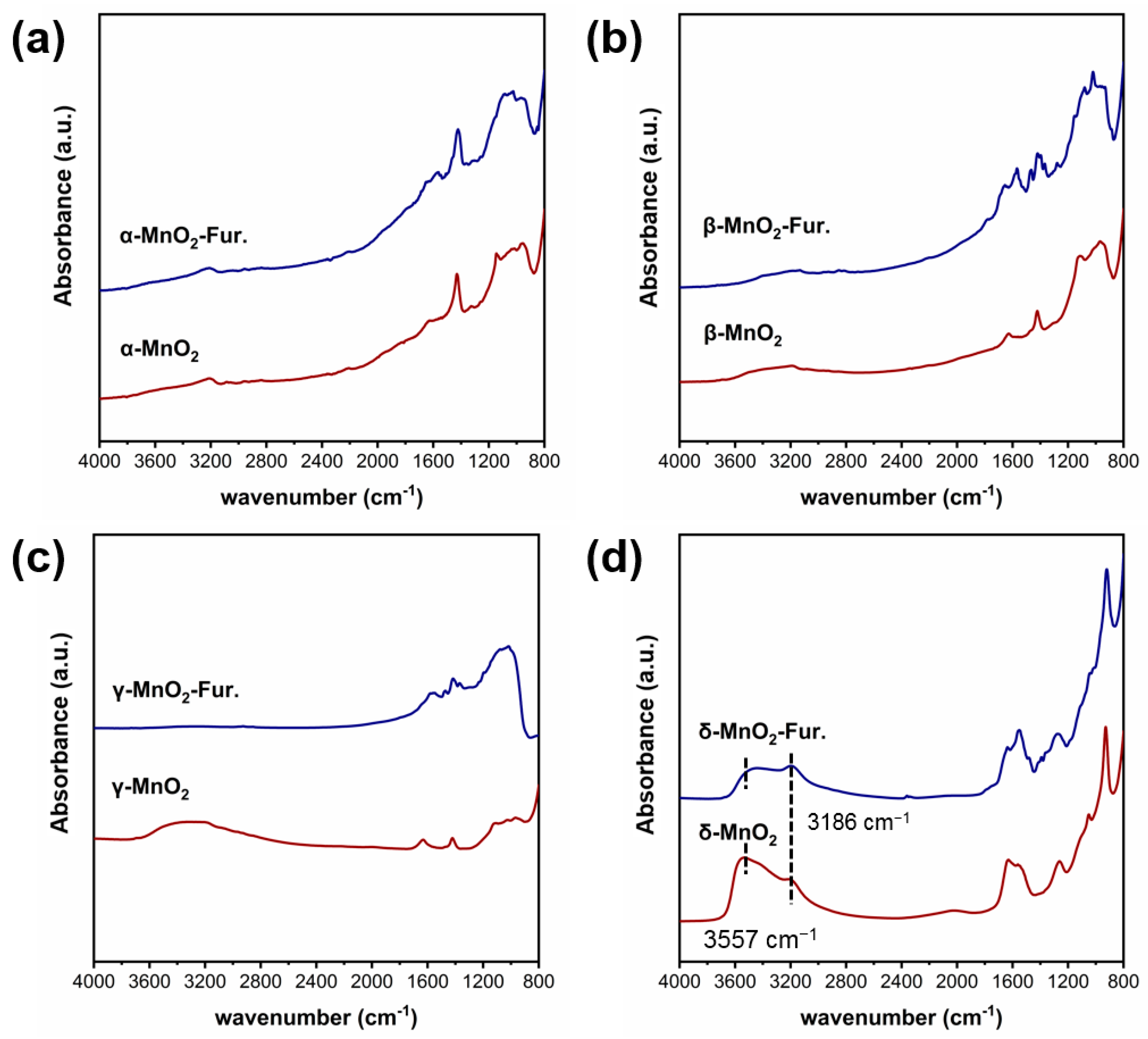
2.4.1. The Role of the Hydroxyl Group of MnO2
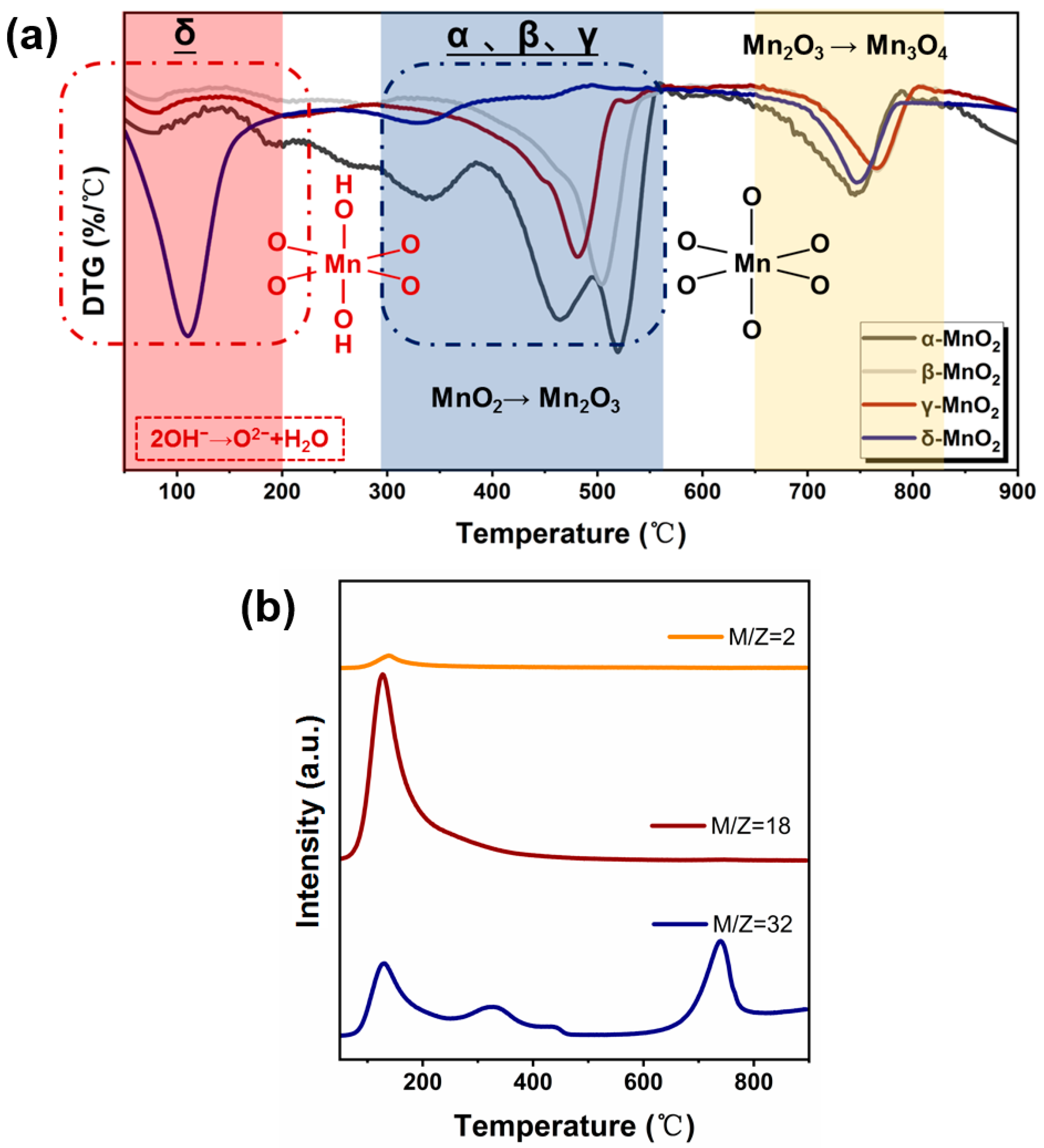
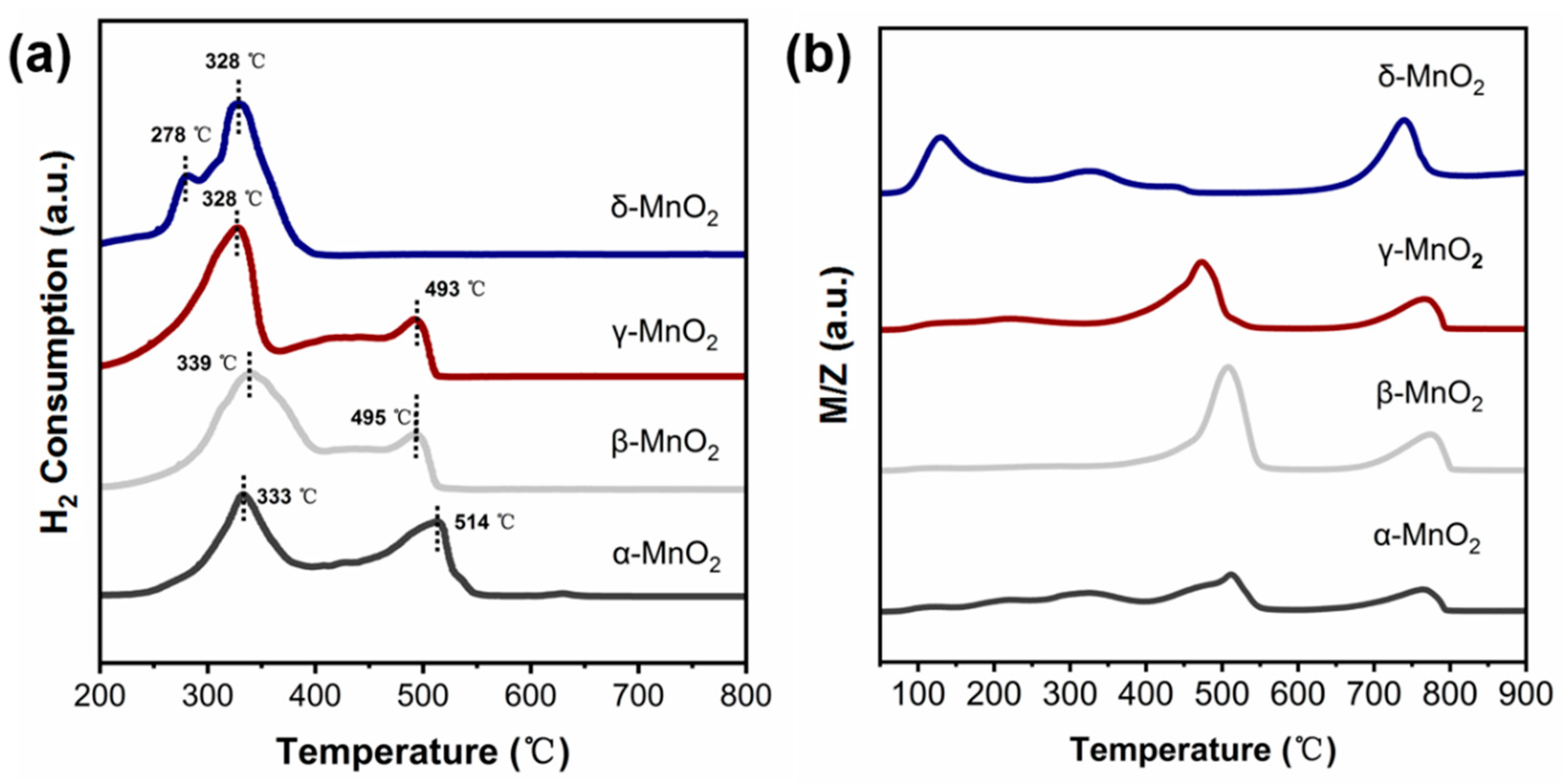
2.4.2. The Role of Active Oxygen Species of MnO2
3. Conclusions
4. Materials and Methods
4.1. Preparation of MnO2 with Different Crystalline Forms
4.2. Evaluation of Catalysts
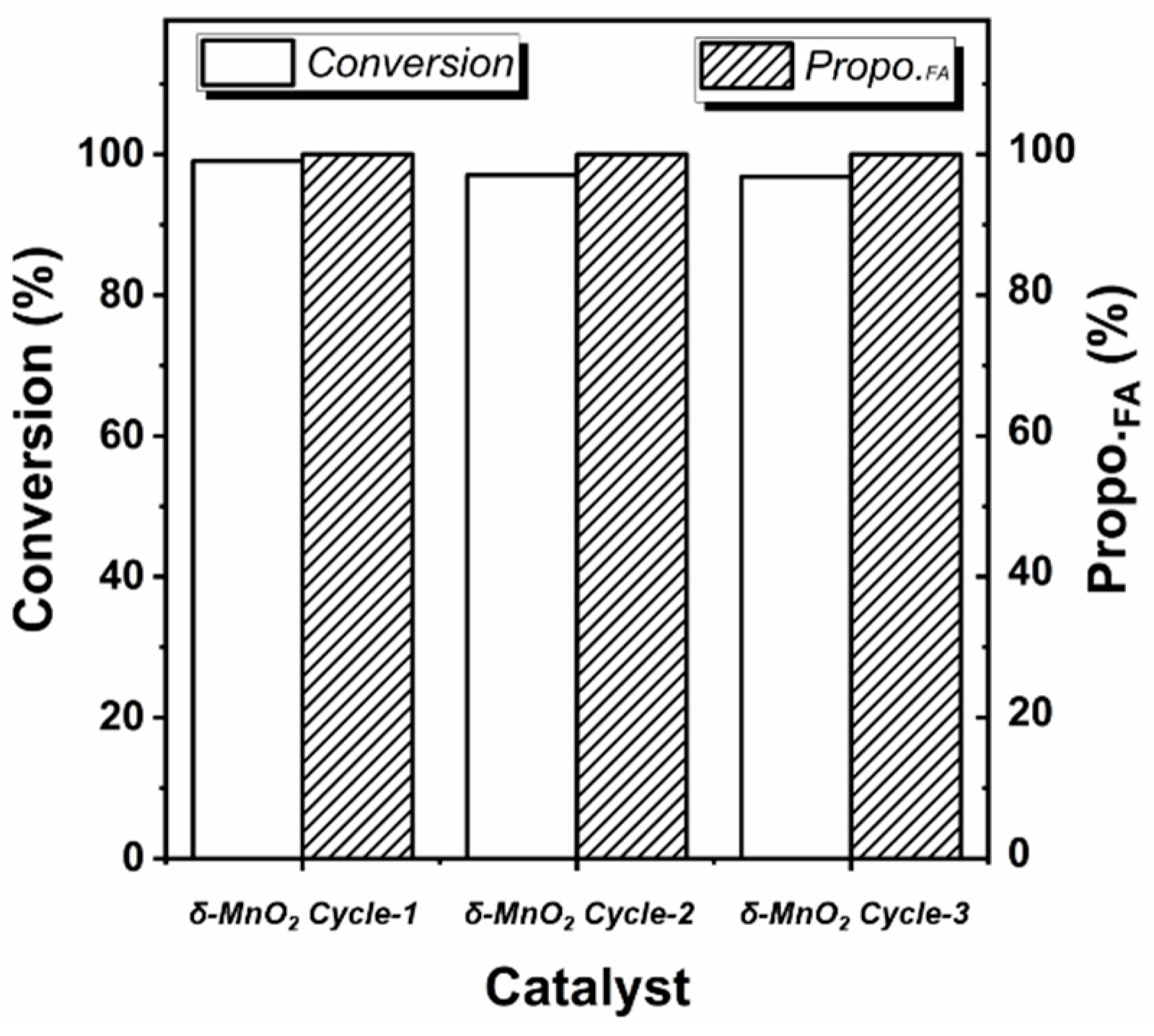
4.3. Catalyst Reuse
4.4. Catalyst Characterization
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mariscal, R.; Maireles-Torres, P.; Ojeda, M.; Sádaba, I.; López Granados, M. Furfural: A renewable and versatile platform molecule for the synthesis of chemicals and fuels. Energy Environ. Sci. 2016, 9, 1144–1189. [Google Scholar] [CrossRef]
- Banerjee, A.; Dick, G.R.; Yoshino, T.; Kanan, M.W. Carbon dioxide utilization via carbonate-promoted C–H carboxylation. Nature 2016, 531, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Gonzalez-Diaz, A.; Ling-Chin, J.; Malik, A.; Roskilly, A.P.; Smallbone, A.J. PEF plastic synthesized from industrial carbon dioxide and biowaste. Nat. Sustain. 2020, 3, 761–767. [Google Scholar] [CrossRef]
- Pinna, F.; Olivo, A.; Trevisan, V.; Menegazzo, F.; Signoretto, M.; Manzoli, M.; Boccuzzi, F. The effects of gold nanosize for the exploitation of furfural by selective oxidation. Catal. Today 2013, 203, 196–201. [Google Scholar] [CrossRef] [Green Version]
- Menegazzo, F.; Signoretto, M.; Pinna, F.; Manzoli, M.; Aina, V.; Cerrato, G.; Boccuzzi, F. Oxidative esterification of renewable furfural on gold-based catalysts: Which is the best support? J. Catal. 2014, 309, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Menegazzo, F.; Fantinel, T.; Signoretto, M.; Pinna, F.; Manzoli, M. On the process for furfural and HMF oxidative esterification over Au/ZrO2. J. Catal. 2014, 319, 61–70. [Google Scholar] [CrossRef]
- Ferraz, C.P.; Zieliński, M.; Pietrowski, M.; Heyte, S.; Dumeignil, F.; Rossi, L.M.; Wojcieszak, R. Influence of Support Basic Sites in Green Oxidation of Biobased Substrates Using Au-Promoted Catalysts. ACS Sustain. Chem. Eng. 2018, 6, 16332–16340. [Google Scholar] [CrossRef]
- Douthwaite, M.; Huang, X.; Iqbal, S.; Miedziak, P.J.; Brett, G.L.; Kondrat, S.A.; Edwards, J.K.; Sankar, M.; Knight, D.W.; Bethell, D.; et al. The controlled catalytic oxidation of furfural to furoic acid using AuPd/Mg(OH)2. Catal. Sci. Technol. 2017, 7, 5284–5293. [Google Scholar] [CrossRef] [Green Version]
- Ferraz, C.P.; Navarro-Jaén, S.; Rossi, L.M.; Dumeignil, F.; Ghazzal, M.N.; Wojcieszak, R. Enhancing the activity of gold supported catalysts by oxide coating: Towards efficient oxidations. Green Chem. 2021, 23, 8453–8457. [Google Scholar] [CrossRef]
- Papanikolaou, G.; Lanzafame, P.; Perathoner, S.; Centi, G.; Cozza, D.; Giorgianni, G.; Migliori, M.; Giordano, G. High performance of Au/ZTC based catalysts for the selective oxidation of bio-derivative furfural to 2-furoic acid11This paper in honor of Professor James G. Goodwin, Jr., in the occasion of his 75th birthday, to celebrate his outstanding contribution to catalysis sciences and technology. Catal. Commun. 2021, 149, 106234. [Google Scholar] [CrossRef]
- Daniel, S.; Subbiah, V.P.; Ramaganthan, B.; Kanthapazham, R.; Vanaraj, R. Upgrading the Strategy of Multistage Torrefaction Liquid by the Selective Oxidation Reaction Route Using a Reusable MgO-Based Au/Al2O3 Catalyst. Energy Fuel 2021, 35, 15831–15841. [Google Scholar] [CrossRef]
- Ren, Z.; Yang, Y.; Wang, S.; Li, X.; Feng, H.; Wang, L.; Li, Y.; Zhang, X.; Wei, M. Pt atomic clusters catalysts with local charge transfer towards selective oxidation of furfural. Appl. Catal. B Environ. 2021, 295, 120290. [Google Scholar] [CrossRef]
- Gupta, K.; Rai, R.K.; Dwivedi, A.D.; Singh, S.K. Catalytic Aerial Oxidation of Biomass-Derived Furans to Furan Carboxylic Acids in Water over Bimetallic Nickel–Palladium Alloy Nanoparticles. Chemcatchem 2017, 9, 2760–2767. [Google Scholar] [CrossRef]
- Al Rawas, H.K.; Ferraz, C.P.; Thuriot-Roukos, J.; Heyte, S.; Paul, S.; Wojcieszak, R. Influence of Pd and Pt Promotion in Gold Based Bimetallic Catalysts on Selectivity Modulation in Furfural Base-Free Oxidation. Catalysts 2021, 11, 1226. [Google Scholar] [CrossRef]
- Tian, Q.; Shi, D.; Sha, Y. CuO and Ag2O/CuO Catalyzed Oxidation of Aldehydes to the Corresponding Carboxylic Acids by Molecular Oxygen. Molecules 2008, 13, 948. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, H.; Wang, X.; Liu, Y. Convergent production of 2,5-furandicarboxylic acid from biomass and CO2. Green Chem. 2019, 21, 2923–2927. [Google Scholar] [CrossRef]
- Yu, K.; Lou, L.-L.; Liu, S.; Zhou, W. Asymmetric Oxygen Vacancies: The Intrinsic Redox Active Sites in Metal Oxide Catalysts. Adv. Sci. 2020, 7, 1901970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, K.; Liu, Y.; Lei, D.; Jiang, Y.; Wang, Y.; Feng, Y.; Lou, L.-L.; Liu, S.; Zhou, W. M3+O(–Mn4+)2 clusters in doped MnOx catalysts as promoted active sites for the aerobic oxidation of 5-hydroxymethylfurfural. Catal. Sci. Technol. 2018, 8, 2299–2303. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Qin, L.; Xiao, W.; Zeng, C.; Li, N.; Lv, T.; Zhu, H. Oriented growth of layered-MnO2 nanosheets over α-MnO2 nanotubes for enhanced room-temperature HCHO oxidation. Appl. Catal. B Environ. 2017, 207, 233–243. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Jiang, C.; Zhou, P.; Zhang, P.; Yu, J. The effect of manganese vacancy in birnessite-type MnO2 on room-temperature oxidation of formaldehyde in air. Appl. Catal. B Environ. 2017, 204, 147–155. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, P.; Li, J.; Jiang, C.; Yunus, R.; Kim, J. Room-Temperature Oxidation of Formaldehyde by Layered Manganese Oxide: Effect of Water. Environ. Sci. Technol. 2015, 49, 12372–12379. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jia, W.; Yu, X.; Tang, X.; Zeng, X.; Sun, Y.; Lei, T.; Fang, H.; Li, T.; Lin, L. Vitamin C-Assisted Synthesized Mn–Co Oxides with Improved Oxygen Vacancy Concentration: Boosting Lattice Oxygen Activity for the Air-Oxidation of 5-(Hydroxymethyl)furfural. ACS Catal. 2021, 11, 7828–7844. [Google Scholar] [CrossRef]
- Hayashi, E.; Yamaguchi, Y.; Kamata, K.; Tsunoda, N.; Kumagai, Y.; Oba, F.; Hara, M. Effect of MnO2 Crystal Structure on Aerobic Oxidation of 5-Hydroxymethylfurfural to 2,5-Furandicarboxylic Acid. J. Am. Chem. Soc. 2019, 141, 890–900. [Google Scholar] [CrossRef]
- Ferraz, C.P.; Da Silva, A.G.; Rodrigues, T.S.; Camargo, P.H.; Paul, S.; Wojcieszak, R. Furfural Oxidation on Gold Supported on MnO2: Influence of the Support Structure on the Catalytic Performances. Appl. Sci. 2018, 8, 1246. [Google Scholar] [CrossRef] [Green Version]
- Rong, S.; Zhang, P.; Liu, F.; Yang, Y. Engineering Crystal Facet of α-MnO2 Nanowire for Highly Efficient Catalytic Oxidation of Carcinogenic Airborne Formaldehyde. ACS Catal. 2018, 8, 3435–3446. [Google Scholar] [CrossRef]
- Saputra, E.; Muhammad, S.; Sun, H.; Ang, H.M.; Tade, M.O.; Wang, S. Different crystallographic one-dimensional MnO2 nanomaterials and their superior performance in catalytic phenol degradation. Environ. Sci. Technol. 2013, 47, 5882–5887. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.; Huang, H.; Dharmarathna, S.; Benbow, E.; Stafford, L.; Suib, S.L. Hydrothermal Synthesis of Manganese Oxide Nanomaterials and Their Catalytic and Electrochemical Properties. Chem. Mater. 2011, 23, 3892–3901. [Google Scholar] [CrossRef]
- Wang, H.; Luo, Q.; Wang, L.; Hui, Y.; Qin, Y.; Song, L.; Xiao, F.-S. Product selectivity controlled by manganese oxide crystals in catalytic ammoxidation. Chin. J. Catal. 2021, 42, 2164–2172. [Google Scholar] [CrossRef]
- Verdeguer, P.; Merat, N.; Gaset, A. Lead/platinum on charcoal as catalyst for oxidation of furfural. Effect of main parameters. Appl. Catal. A Gen. 1994, 112, 1–11. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y. Synthesis and Formation Mechanism of Manganese Dioxide Nanowires/Nanorods. Chem. A Eur. J. 2003, 9, 300–306. [Google Scholar] [CrossRef]
- Luo, J.; Huang, A.; Park, S.H.; Suib, S.L.; O’Young, C.-L. Crystallization of Sodium−Birnessite and Accompanied Phase Transformation. Chem. Mater. 1998, 10, 1561–1568. [Google Scholar] [CrossRef]
- Yang, D.S.; Wang, M.K. Syntheses and Characterization of Well-Crystallized Birnessite. Chem. Mater. 2001, 13, 2589–2594. [Google Scholar] [CrossRef]
- Xu, Z.; Yu, J.; Jaroniec, M. Efficient catalytic removal of formaldehyde at room temperature using AlOOH nanoflakes with deposited Pt. Appl. Catal. B Environ. 2015, 163, 306–312. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Wang, L.; Zhang, C.; He, H. Catalytic oxidation of formaldehyde over manganese oxides with different crystal structures. Catal. Sci. Technol. 2015, 5, 2305–2313. [Google Scholar] [CrossRef]
- Jia, J.; Zhang, P.; Chen, L. The effect of morphology of α-MnO2 on catalytic decomposition of gaseous ozone. Catal. Sci. Technol. 2016, 6, 5841–5847. [Google Scholar] [CrossRef]
- Hadjiivanov, K. Chapter Two—Identification and Characterization of Surface Hydroxyl Groups by Infrared Spectroscopy. In Advances in Catalysis; Jentoft, F.C., Ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 57, pp. 99–318. [Google Scholar]
- Gu, H.; Liu, X.; Liu, X.; Ling, C.; Wei, K.; Zhan, G.; Guo, Y.; Zhang, L. Adjacent single-atom irons boosting molecular oxygen activation on MnO2. Nat. Commun. 2021, 12, 5422. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Chen, D.; Li, N.; Xu, Q.; Li, H.; He, J.; Lu, J. A highly dispersed Pt/copper modified-MnO2 catalyst for the complete oxidation of volatile organic compounds: The effect of oxygen species on the catalytic mechanism. Green Energy Environ. 2021, 8, 538–547. [Google Scholar] [CrossRef]
- Xu, T.; Zhang, P.; Zhang, H. Ultrathin δ-MnO2 nanoribbons for highly efficient removal of a human-related low threshold odorant—Acetic acid. Appl. Catal. B Environ. 2022, 309, 121273. [Google Scholar] [CrossRef]
- Zhu, G.; Zhu, W.; Lou, Y.; Ma, J.; Yao, W.; Zong, R.; Zhu, Y. Encapsulate α-MnO2 nanofiber within graphene layer to tune surface electronic structure for efficient ozone decomposition. Nat. Commun. 2021, 12, 4152. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, G.; Wang, M.; Li, N.; Xu, Q.; Li, H.; He, J.; Lu, J. Pt/MnO2 Nanoflowers Anchored to Boron Nitride Aerogels for Highly Efficient Enrichment and Catalytic Oxidation of Formaldehyde at Room Temperature. Angew. Chem. Int. Ed. 2021, 60, 6377–6381. [Google Scholar] [CrossRef]
- Zhang, L.; Bao, Q.; Zhang, B.; Zhang, Y.; Wan, S.; Wang, S.; Lin, J.; Xiong, H.; Mei, D.; Wang, Y. Distinct Role of Surface Hydroxyls in Single-Atom Pt1/CeO2 Catalyst for Room-Temperature Formaldehyde Oxidation: Acid–Base Versus Redox. JACS Au 2022, 2, 1651–1660. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; He, G.; Li, Y.; Chen, M.; Qin, X.; Zhang, C.; He, H. Identification of a Facile Pathway for Dioxymethylene Conversion to Formate Catalyzed by Surface Hydroxyl on TiO2-Based Catalyst. ACS Catal. 2020, 10, 9706–9715. [Google Scholar] [CrossRef]
- Rong, S.; Zhang, P.; Wang, J.; Liu, F.; Yang, Y.; Yang, G.; Liu, S. Ultrathin manganese dioxide nanosheets for formaldehyde removal and regeneration performance. Chem. Eng. J. 2016, 306, 1172–1179. [Google Scholar] [CrossRef]
- Yang, X.; Makita, Y.; Liu, Z.-h.; Sakane, K.; Ooi, K. Structural Characterization of Self-Assembled MnO2 Nanosheets from Birnessite Manganese Oxide Single Crystals. Chem. Mater. 2004, 16, 5581–5588. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst Name | Temperature /(°C) | O2 Pressure/(MPa) | Reaction Time/(h) | Catalyst Weight/(g) | Conver. 1 /(%) | Propo.FA 2/(%) | Productivity/(mmolfuroic acid/gcat/h) | Carbon Balance 3/(%) |
|---|---|---|---|---|---|---|---|---|
| α-MnO2 | 60 | 1 | 1 | 0.2 | 3.48 | 100 | 0.09 | 96.19 |
| α-MnO2 | 80 | 1 | 1 | 0.2 | 3.37 | 100 | 0.09 | 98.47 |
| α-MnO2 | 100 | 1 | 1 | 0.2 | 4.44 | 100 | 0.12 | 95.57 |
| α-MnO2 | 120 | 1 | 1 | 0.2 | 5.02 | 100 | 0.13 | 91.64 |
| β-MnO2 | 60 | 1 | 1 | 0.2 | 3.43 | 100 | 0.09 | 91.26 |
| β-MnO2 | 80 | 1 | 1 | 0.2 | 3.74 | 100 | 0.10 | 90.20 |
| β-MnO2 | 100 | 1 | 1 | 0.2 | 4.48 | 100 | 0.12 | 94.70 |
| β-MnO2 | 120 | 1 | 1 | 0.2 | 5.89 | 100 | 0.15 | 91.22 |
| γ-MnO2 | 60 | 1 | 1 | 0.2 | 5.79 | 100 | 0.15 | 96.67 |
| γ-MnO2 | 80 | 1 | 1 | 0.2 | 5.80 | 100 | 0.15 | 93.68 |
| γ-MnO2 | 100 | 1 | 1 | 0.2 | 6.76 | 100 | 0.18 | 96.13 |
| γ-MnO2 | 120 | 1 | 1 | 0.2 | 9. 23 | 100 | 0.24 | 99.05 |
| δ-MnO2 | 60 | 1 | 1 | 0.2 | 17.03 | 100 | 0.44 | 98.44 |
| δ-MnO2 | 80 | 1 | 1 | 0.2 | 39.09 | 100 | 1.02 | 85.99 |
| δ-MnO2 | 100 | 1 | 1 | 0.2 | 50.07 | 100 | 1.42 | 85.77 |
| δ-MnO2 | 120 | 1 | 1 | 0.2 | 69.68 | 100 | 1.81 | 88.44 |
| δ-MnO2 | 100 | 1 | 3 | 0.2 | 63.52 | 100 | 0.55 | 94.47 |
| δ-MnO2 | 100 | 1 | 6 | 0.2 | 67.06 | 100 | 0.29 | 88.83 |
| δ-MnO2 | 100 | 1 | 12 | 0.2 | 78.37 | 100 | 0.17 | 91.81 |
| δ-MnO2 | 100 | 1 | 24 | 0.2 | 88.54 | 100 | 0.10 | 75.61 |
| δ-MnO2 | 100 | 1 | 12 | 0.05 | 22.23 | 100 | 0.19 | 96.40 |
| δ-MnO2 | 100 | 1 | 12 | 0.2 | 78.37 | 100 | 0.17 | 91.81 |
| δ-MnO2 | 100 | 1 | 12 | 0.3 | 99.04 | 100 | 0.14 | 89.56 |
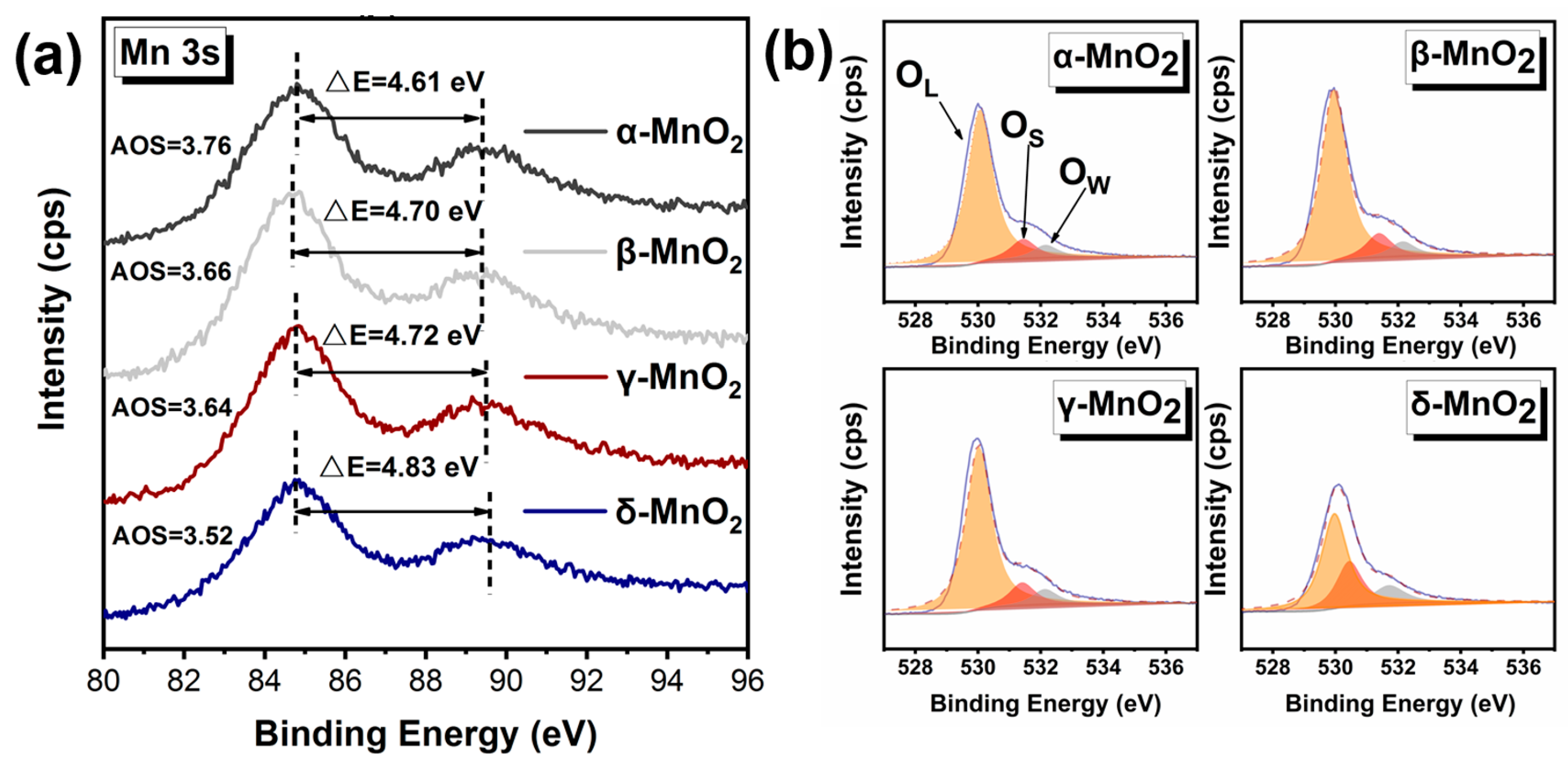
| Catalyst | Oxygen Species Type | Position/Ev | Area Percentage/% |
|---|---|---|---|
| α-MnO2 | OL 1 | 530.06 | 82.6034 |
| OS 2 | 531.44 | 10.5468 | |
| OW 3 | 532.15 | 6.8498 | |
| β-MnO2 | OL | 529.95 | 81.7358 |
| OS | 531.38 | 11.3416 | |
| OW | 532.14 | 6.9227 | |
| γ-MnO2 | OL | 530.02 | 81.0782 |
| OS | 531.42 | 11.3644 | |
| OW | 532.13 | 7.5575 | |
| δ-MnO2 | OL | 529.96 | 58.3633 |
| OS | 530.43 | 27.6973 | |
| OW | 531.70 | 13.9394 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Guo, H.; Jia, L.; Xiao, Y.; Hou, B.; Li, D. Effect of MnO2 Crystal Type on the Oxidation of Furfural to Furoic Acid. Catalysts 2023, 13, 663. https://doi.org/10.3390/catal13040663
Wu X, Guo H, Jia L, Xiao Y, Hou B, Li D. Effect of MnO2 Crystal Type on the Oxidation of Furfural to Furoic Acid. Catalysts. 2023; 13(4):663. https://doi.org/10.3390/catal13040663
Chicago/Turabian StyleWu, Xu, Heqin Guo, Litao Jia, Yong Xiao, Bo Hou, and Debao Li. 2023. "Effect of MnO2 Crystal Type on the Oxidation of Furfural to Furoic Acid" Catalysts 13, no. 4: 663. https://doi.org/10.3390/catal13040663
APA StyleWu, X., Guo, H., Jia, L., Xiao, Y., Hou, B., & Li, D. (2023). Effect of MnO2 Crystal Type on the Oxidation of Furfural to Furoic Acid. Catalysts, 13(4), 663. https://doi.org/10.3390/catal13040663