
Tunable Late-Transition-Metal-Catalyzed Polymerization for Controlled Polymer Synthesis


Abstract
:1. Introduction
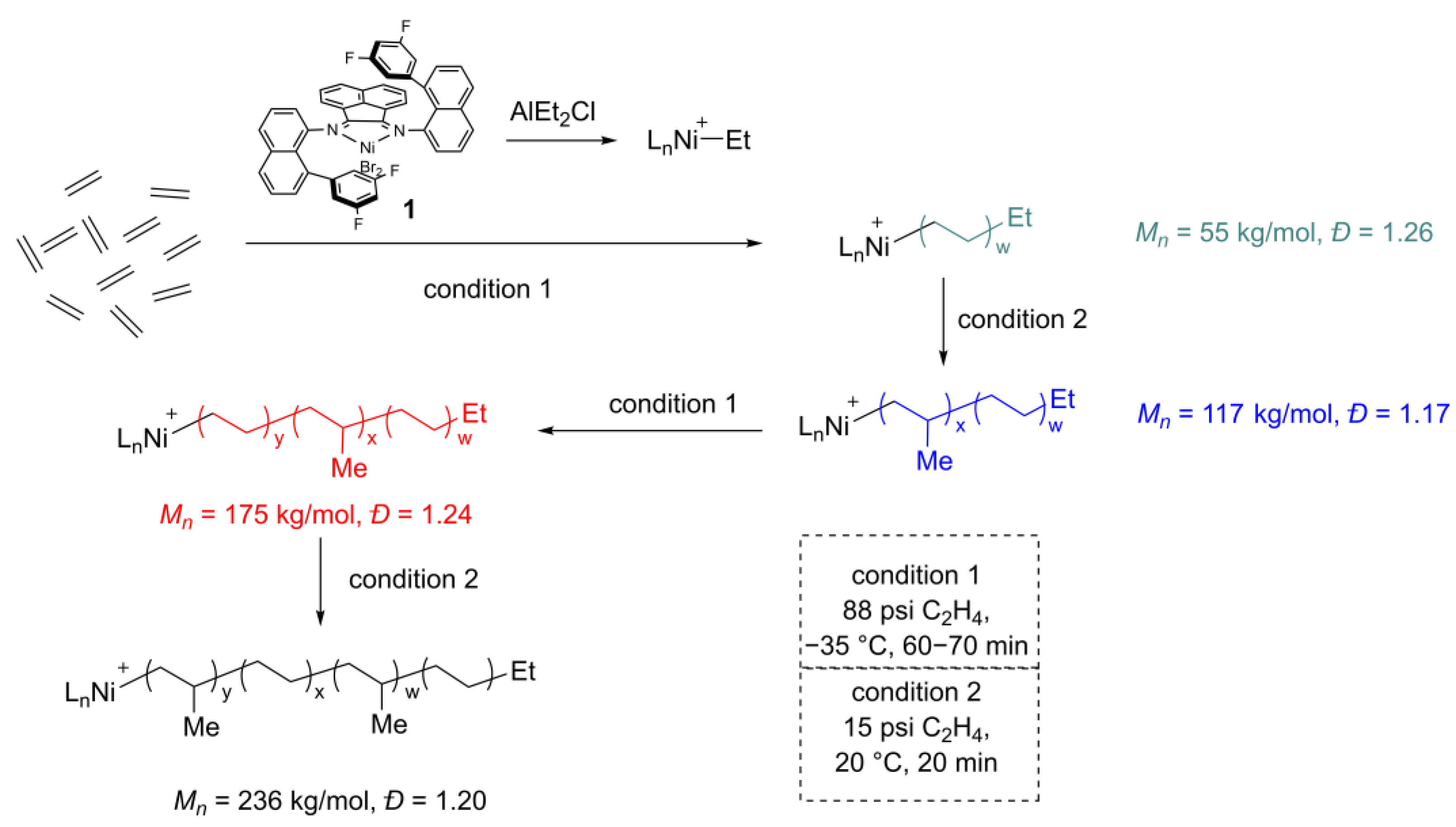
2. Reaction Condition Tuning
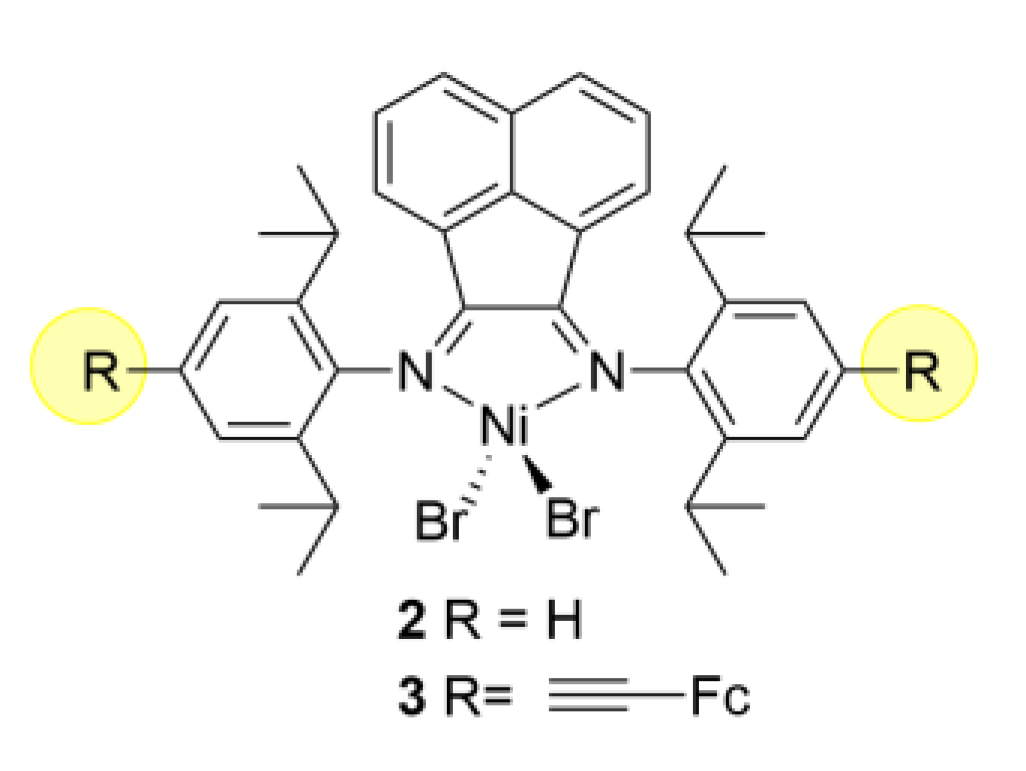
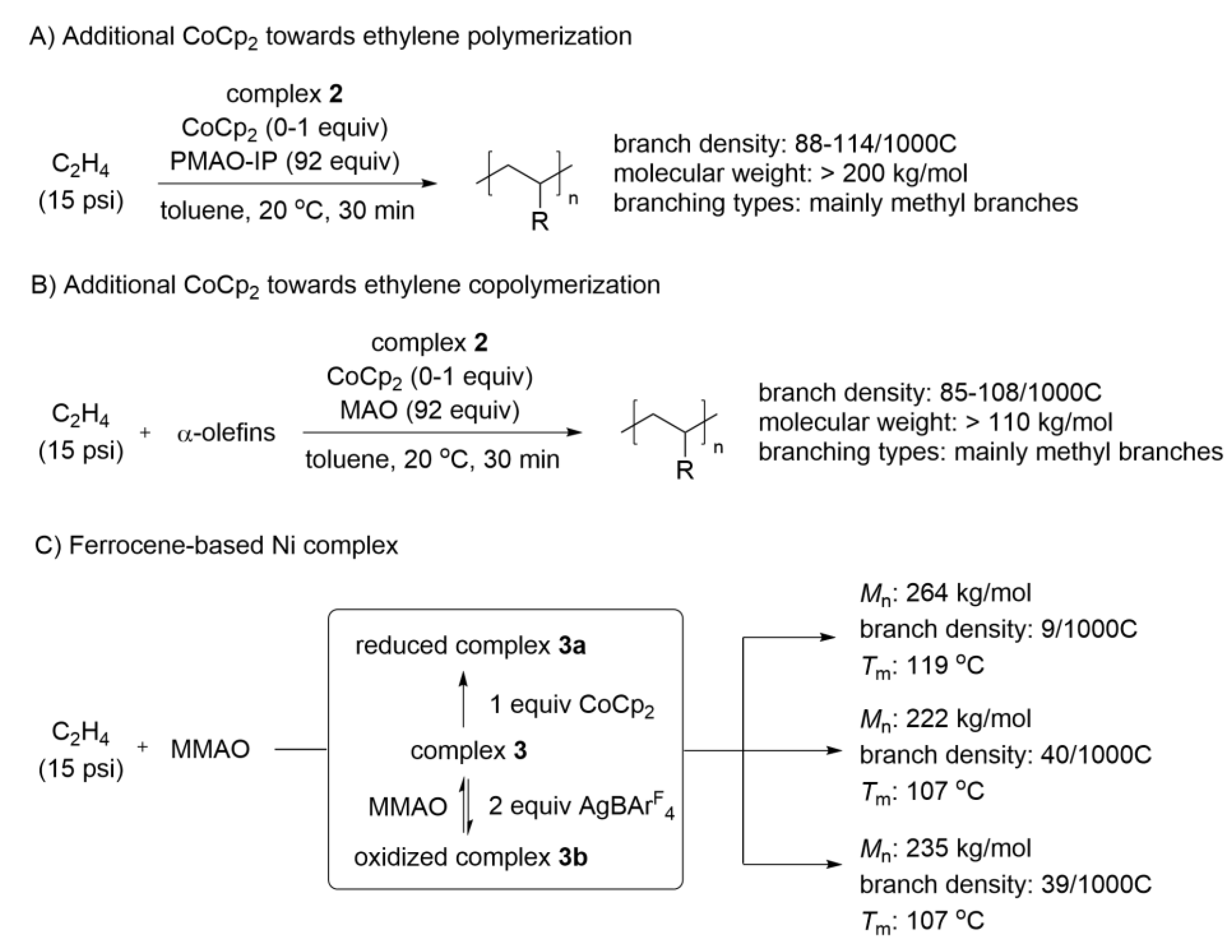
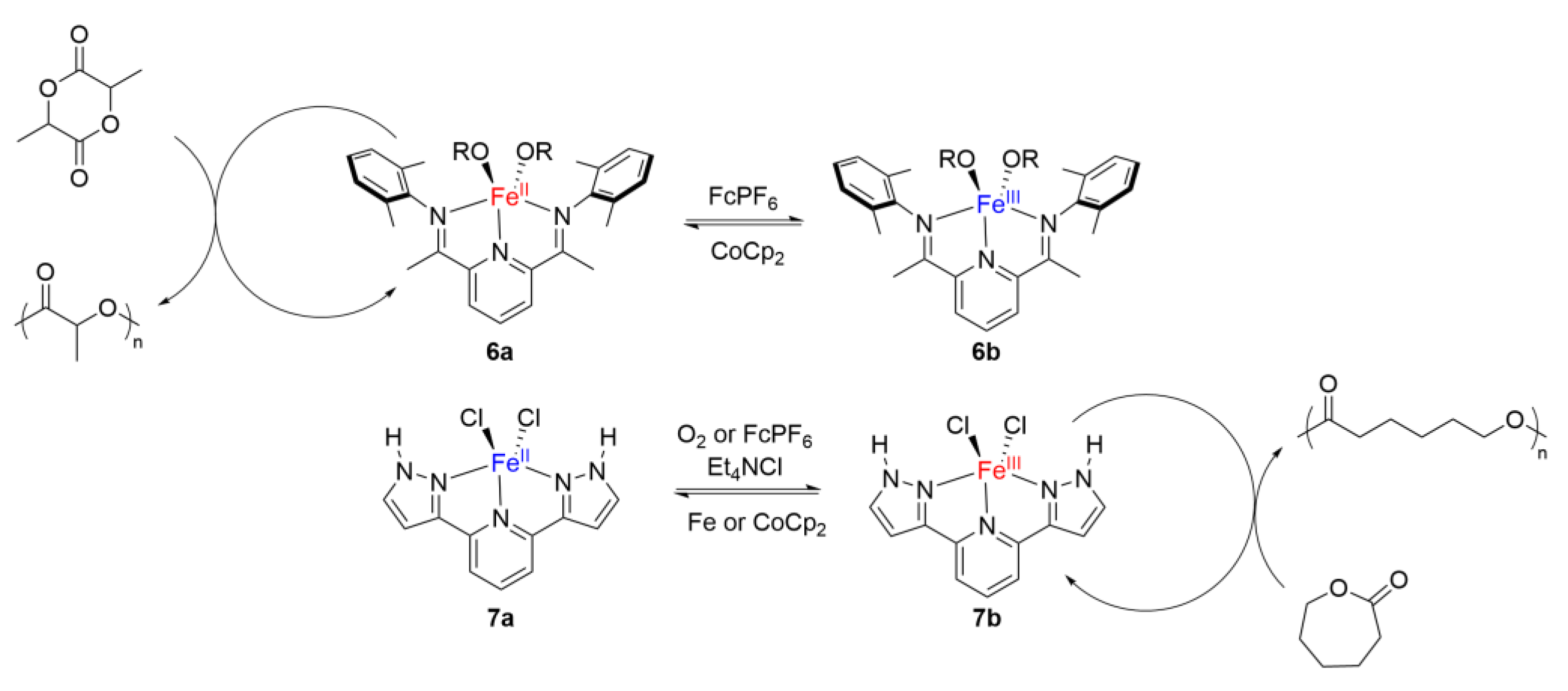
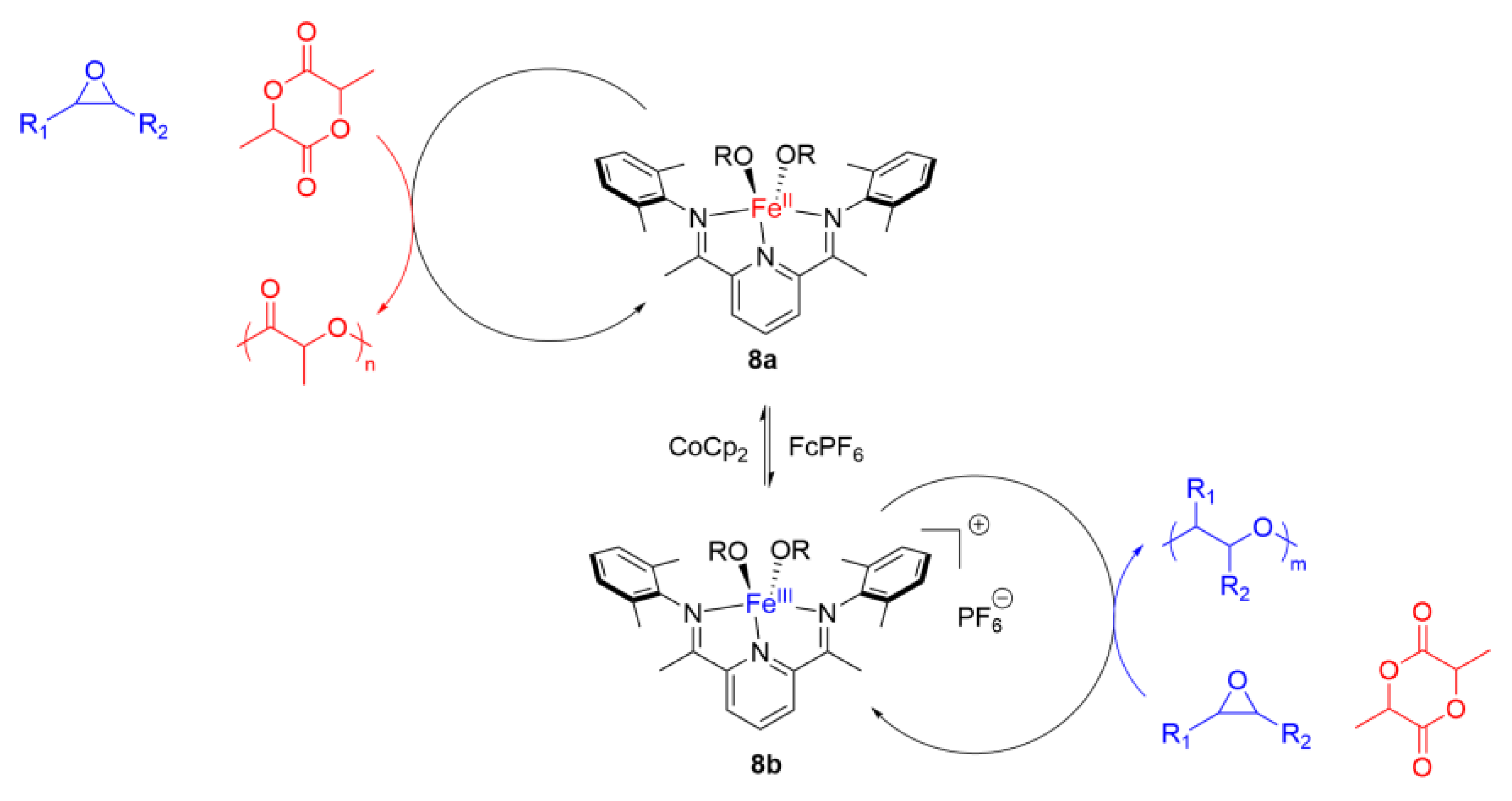
3. Redox Tuning
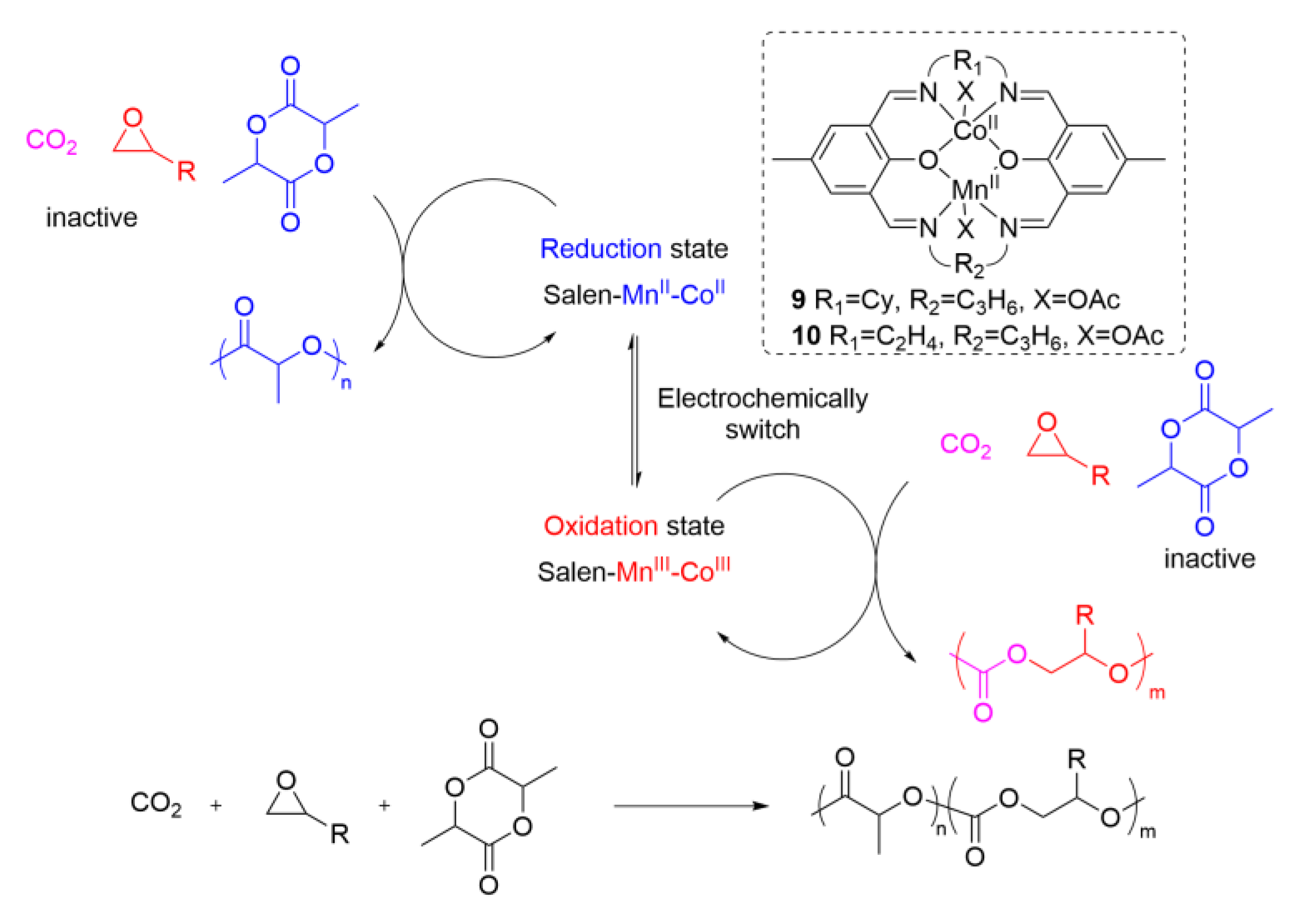
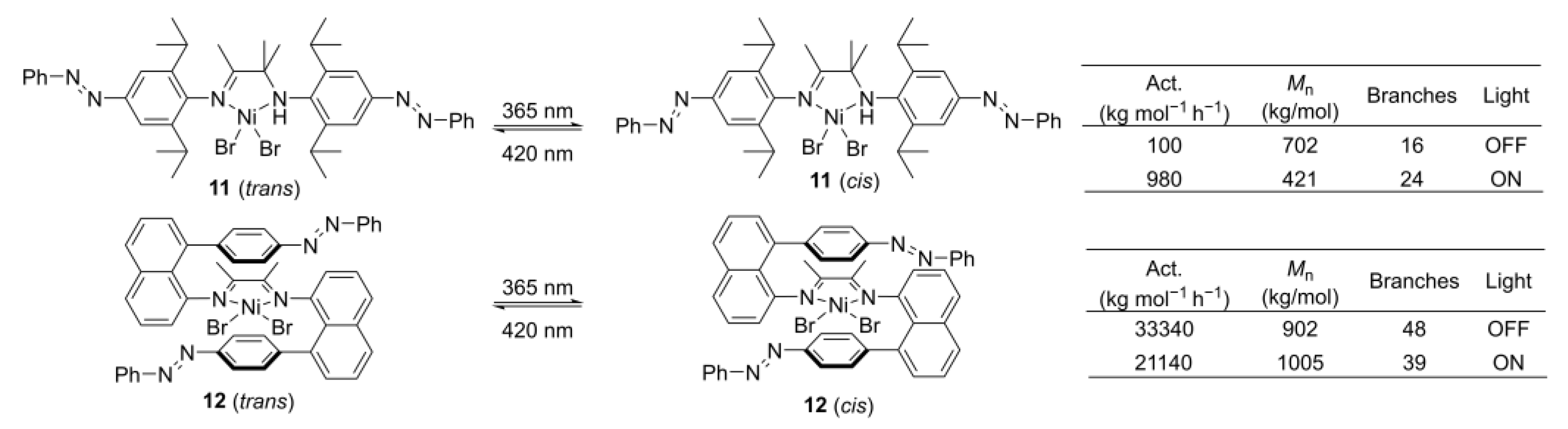
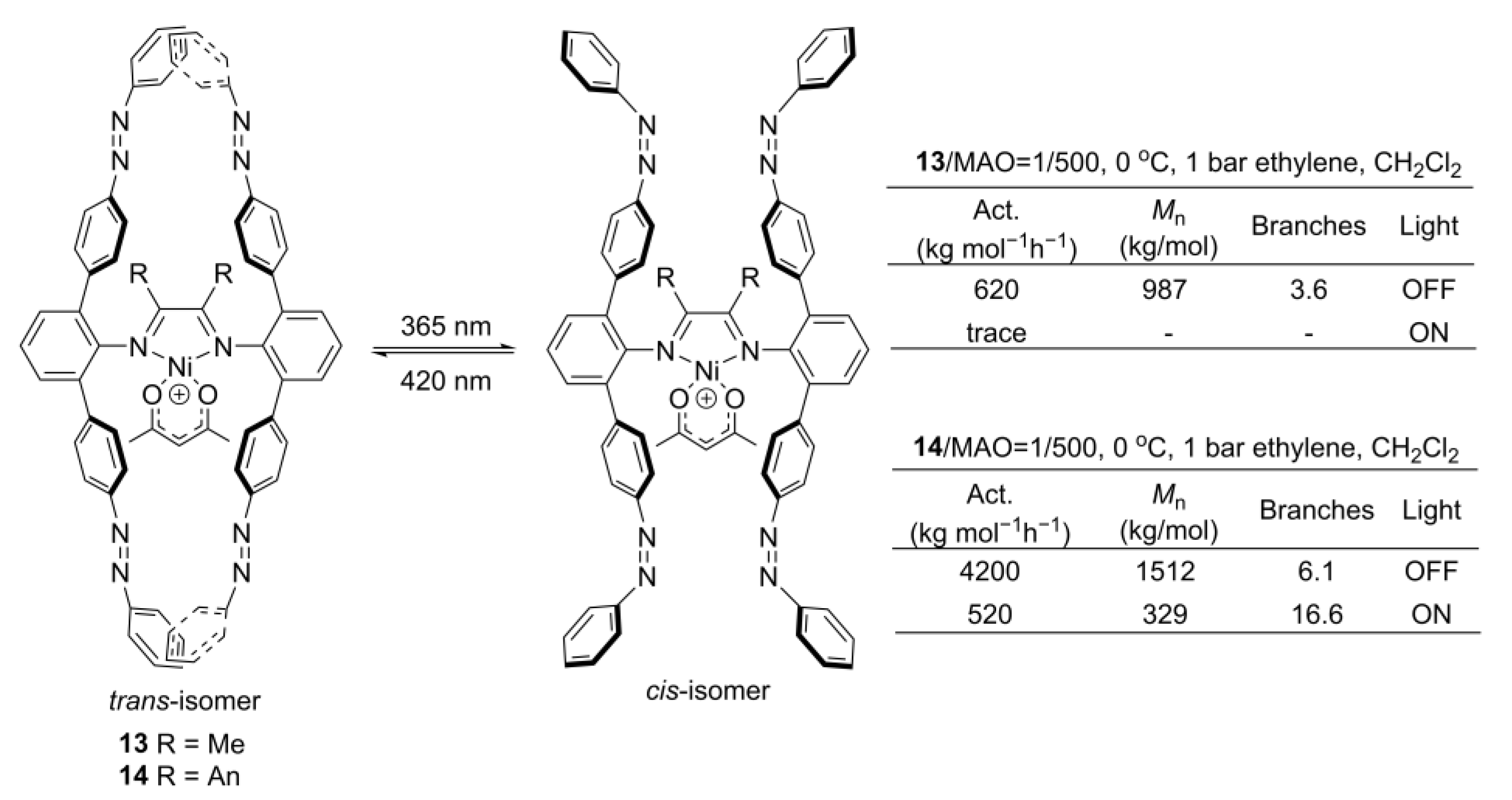
4. Electrochemical or Light Tuning
5. Lewis Acid Tuning
6. Alkali Metal Cation Tuning
7. Summary and Outlook
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ROP | ring-opening polymerization |
| ROCOP | ring-opening copolymerization |
| PE | polyethylene |
| MW | molecular weight |
| Ɖ | molecular weight distribution |
| Tg | glass transition temperature |
| Tm | melting point |
| CoCp2 | cobaltocene |
| AgOTf | silver trifluoromethanesulfonate |
| CO2 | carbon dioxide |
| LA | lactide |
| PLA | polylactide |
| CHO | cyclohexene oxide |
| BIAN | (bis(imino)acenaphthene) |
| Fc | ferrocene |
| MAO | methylaluminoxane |
| Mw | weight-average molecular weight |
| Mn | mass-average molecular weight |
| ROMP | ring-opening metathesis polymerization |
| PEG | polyethylene glycol |
References
- Zubkevich, S.V.; Tuskaev, V.A.; Gagieva, S.C.; Bulychev, B.M. Catalytic oligomerization and polymerization of ethylene with complexes of iron triad metals: Influence of metal nature and new prospects. Russ. Chem. Rev. 2022, 91, RCR5021. [Google Scholar] [CrossRef]
- Wu, R.; Wu, W.K.; Stieglitz, L.; Gaan, S.; Rieger, B.; Heuberger, M. Recent advances on α-diimine Ni and Pd complexes for catalyzed ethylene (co)polymerization: A comprehensive review. Coord. Chem. Rev. 2023, 474, 214844–214876. [Google Scholar] [CrossRef]
- Deng, H.; Zheng, H.; Gao, H.; Pei, L.; Gao, H. Late transition metal catalysts with chelating amines for olefin polymerization. Catalysts 2022, 12, 936. [Google Scholar] [CrossRef]
- Zhang, R.; Gao, R.; Gou, Q.; Lai, J.; Li, X. Recent advances in the copolymerization of ethylene with polar comonomers by nickel catalysts. Polymers 2022, 14, 3809. [Google Scholar] [CrossRef]
- Khoshsefat, M.; Ma, Y.; Sun, W.-H. Multinuclear late transition metal catalysts for olefin polymerization. Coord. Chem. Rev. 2021, 434, 213788–213805. [Google Scholar] [CrossRef]
- Li, P.; Yu, F.; Xu, M.; Li, X.; Li, M.; Xu, G.; Tan, C.; Zhang, S.; Wang, F. Influence of weak Fe–N bond on iron catalyzed ethylene polymerization. Polymer 2021, 220, 4626–4631. [Google Scholar] [CrossRef]
- Qasim, M.; Bashir, M.S.; Iqbal, S.; Mahmood, Q. Recent advancements in α-diimine-nickel and -palladium catalysts for ethylene polymerization. Eur. Polym. J. 2021, 160, 110783. [Google Scholar] [CrossRef]
- Schoeneberger, E.M.; Luinstra, G.A. Investigations on the ethylene polymerization with bisarylimine pyridine iron (BIP) catalysts. Catalysts 2021, 11, 407. [Google Scholar] [CrossRef]
- Wang, M.; Wu, W.; Wang, X.; Huang, X.; Nai, Y.; Wei, X.; Mao, G. Research progress of iron-based catalysts for selective oligomerization of ethylene. RSC Adv. 2020, 10, 43640–43652. [Google Scholar] [CrossRef]
- Zhong, L.; Zheng, H.; Du, C.; Du, W.; Liao, G.; Cheung, C.S.; Gao, H. Thermally robust α-diimine nickel and palladium catalysts with constrained space for ethylene (co)polymerizations. J. Catal. 2020, 384, 208–217. [Google Scholar] [CrossRef]
- Phillips, A.M.F.; Suo, H.; Guedes da Silva, M.d.F.C.; Pombeiro, A.J.L.; Sun, W.-H. Recent developments in vanadium-catalyzed olefin coordination polymerization. Coord. Chem. Rev. 2020, 416, 213332–213360. [Google Scholar] [CrossRef]
- Purohit, V.B.; Pięta, M.; Pietrasik, J.; Plummer, C.M. Recent advances in the ring-opening polymerization of sulfur-containing monomers. Polym. Chem. 2022, 13, 4858–4878. [Google Scholar] [CrossRef]
- De Hoe, G.X.; Sucu, T.; Shaver, M.P. Sustainability and polyesters: Beyond metals and monomers to function and fate. Acc. Chem. Res. 2022, 55, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Tschan, M.J.; Gauvin, R.M.; Thomas, C.M. Controlling polymer stereochemistry in ring-opening polymerization: A decade of advances shaping the future of biodegradable polyesters. Chem. Soc. Rev. 2021, 50, 13587–13608. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.W.; Li, Y.J.; Wu, J.C. Sequence-controlled alternating copolyesters synthesis via selective ring-opening polymerization. Macromol. Chem. Phys. 2021, 222, 2100323. [Google Scholar] [CrossRef]
- Schafer, P.M.; Herres-Pawlis, S. Robust guanidine metal catalysts for the ring-opening polymerization of lactide under industrially relevant conditions. ChemPlusChem 2020, 85, 1044–1052. [Google Scholar] [CrossRef]
- Santoro, O.; Zhang, X.; Redshaw, C. Synthesis of biodegradable polymers: A review on the use of schiff-base metal complexes as catalysts for the ring opening polymerization (ROP) of cyclic esters. Catalysts 2020, 10, 800. [Google Scholar] [CrossRef]
- Petrus, R.; Sobota, P. Magnesium and zinc alkoxides and aryloxides supported by commercially available ligands as promoters of chemical transformations of lactic acid derivatives to industrially important fine chemicals. Coord. Chem. Rev. 2019, 396, 72–88. [Google Scholar] [CrossRef]
- Nifant’ev, I.; Ivchenko, P. Coordination ring-opening polymerization of cyclic esters: A critical overview of dft modeling and visualization of the reaction mechanisms. Molecules 2019, 24, 4117. [Google Scholar] [CrossRef] [Green Version]
- Plajer, A.J.; Williams, C.K. Heterocycle/heteroallene ring-opening copolymerization: Selective catalysis delivering alternating copolymers. Angew. Chem. Int. Ed. 2022, 61, e202104495. [Google Scholar] [CrossRef]
- Lidston, C.A.L.; Severson, S.M.; Abel, B.A.; Coates, G.W. Multifunctional catalysts for ring-opening copolymerizations. ACS Catal. 2022, 22, 11037–11070. [Google Scholar] [CrossRef]
- Diment, W.T.; Lindeboom, W.; Fiorentini, F.; Deacy, A.C.; Williams, C.K. Synergic heterodinuclear catalysts for the ring-opening copolymerization (ROCOP) of epoxides, carbon dioxide, and anhydrides. Acc. Chem. Res. 2022, 55, 1997–2010. [Google Scholar] [CrossRef] [PubMed]
- Darensbourg, D.J. Switchable catalytic processes involving the copolymerization of epoxides and carbon dioxide for the preparation of block polymers. Inorg. Chem. Front. 2017, 4, 412–419. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Macromolecular engineering: From rational design through precise macromolecular synthesis and processing to targeted macroscopic material properties. Prog. Polym. Sci. 2005, 30, 858–875. [Google Scholar] [CrossRef]
- Walsh, D.J.; Hyatt, M.G.; Miller, S.A.; Guironnet, D. Recent trends in catalytic polymerizations. ACS Catal. 2019, 9, 11153–11188. [Google Scholar] [CrossRef]
- Webster, O.W. Living polymerization methods. Science 1991, 251, 887–893. [Google Scholar] [CrossRef]
- Yagci, Y.; Atilla Tasdelen, M. Mechanistic transformations involving living and controlled/living polymerization methods. Prog. Polym. Sci. 2006, 31, 1133–1170. [Google Scholar] [CrossRef]
- Doerr, A.M.; Burroughs, J.M.; Gitter, S.R.; Yang, X.; Boydston, A.J.; Long, B.K. Advances in polymerizations modulated by external stimuli. ACS Catal. 2020, 10, 14457–14515. [Google Scholar] [CrossRef]
- Blanco, V.; Leigh, D.A.; Marcos, V. Artificial switchable catalysts. Chem. Soc. Rev. 2015, 44, 5341–5370. [Google Scholar] [CrossRef] [Green Version]
- Teator, A.J.; Lastovickova, D.N.; Bielawski, C.W. Switchable polymerization catalysts. Chem. Rev. 2016, 116, 1969–1992. [Google Scholar] [CrossRef]
- Leibfarth, F.A.; Mattson, K.M.; Fors, B.P.; Collins, H.A.; Hawker, C.J. External regulation of controlled polymerizations. Angew. Chem. Int. Ed. 2013, 52, 199–210. [Google Scholar] [CrossRef]
- Tran, T.V.; Do, L.H. Tunable modalities in polyolefin synthesis via coordination insertion catalysis. Eur. Polym. J. 2021, 142, 110100–110114. [Google Scholar] [CrossRef]
- Tan, C.; Chen, M.; Chen, C. ‘Catalyst + x’ strategies for transition metal-catalyzed olefin-polar monomer copolymerization. Trends Chem. 2023, 5, 147–159. [Google Scholar] [CrossRef]
- Kaiser, J.M.; Long, B.K. Recent developments in redox-active olefin polymerization catalysts. Coord. Chem. Rev. 2018, 372, 141–152. [Google Scholar] [CrossRef]
- Chen, C. Redox-controlled polymerization and copolymerization. ACS Catal. 2018, 8, 5506–5514. [Google Scholar] [CrossRef]
- Wei, J.; Diaconescu, P.L. Redox-switchable ring-opening polymerization with ferrocene derivatives. Acc. Chem. Res. 2019, 52, 415–424. [Google Scholar] [CrossRef]
- Chen, M.; Chen, C. Controlling the ring-opening polymerization process using external stimuli. Chin. J. Chem. 2020, 38, 282–286. [Google Scholar] [CrossRef]
- Kaler, S.; Jones, M.D. Recent advances in externally controlled ring-opening polymerisations. Dalton Trans. 2022, 51, 1241–1256. [Google Scholar] [CrossRef] [PubMed]
- Shawver, N.M.; Doerr, A.M.; Long, B.K. A perspective on redox-switchable ring-opening polymerization. J. Poly. Sci. 2022, 1, 361–371. [Google Scholar] [CrossRef]
- Li, B.; Hu, C.; Pang, X.; Chen, X. Valence-variable catalysts for redox-controlled switchable ring-opening polymerization. Chem. Asian J. 2023, 18, e202201031. [Google Scholar] [CrossRef] [PubMed]
- Deacy, A.C.; Gregory, G.L.; Sulley, G.S.; Chen, T.T.D.; Williams, C.K. Sequence control from mixtures: Switchable polymerization catalysis and future materials applications. J. Am. Chem. Soc. 2021, 143, 10021–10040. [Google Scholar] [CrossRef]
- Zhang, Y.; Kang, X.; Jian, Z. Selective branch formation in ethylene polymerization to access precise ethylene-propylene copolymers. Nat. Commun. 2022, 13, 725. [Google Scholar] [CrossRef] [PubMed]
- Padilla-Velez, O.; O’Connor, K.S.; LaPointe, A.M.; MacMillan, S.N.; Coates, G.W. Switchable living nickel(Ⅱ) α-diimine catalyst for ethylene polymerisation. Chem. Commun. 2019, 55, 7607–7610. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Dai, S.; Sui, X.; Chen, C. Palladium and nickel catalyzed chain walking olefin polymerization and copolymerization. ACS Catal. 2016, 6, 428–441. [Google Scholar] [CrossRef] [Green Version]
- Guan, Z.; Cotts, P.M.; McCord, E.F.; McLain, S.J. Chain walking: A new strategy to control polymer topology. Science 1999, 283, 2059–2062. [Google Scholar] [CrossRef]
- Zhang, Y.; Jian, Z. Polar additive triggered branching switch and block polyolefin topology in living ethylene polymerization. Macromolecules 2021, 54, 3191–3196. [Google Scholar] [CrossRef]
- Kernbichl, S.; Reiter, M.; Adams, F.; Vagin, S.; Rieger, B. CO2-controlled one-pot synthesis of AB, ABA block, and statistical terpolymers from β-butyrolactone, epoxides, and CO2. J. Am. Chem. Soc. 2017, 139, 6787–6790. [Google Scholar] [CrossRef]
- Huang, Y.; Hu, C.; Zhou, Y.; Duan, R.; Sun, Z.; Wan, P.; Xiao, C.; Pang, X.; Chen, X. Monomer controlled switchable copolymerization: A feasible route for the functionalization of poly(lactide). Angew. Chem. Int. Ed. 2021, 60, 9274–9278. [Google Scholar] [CrossRef]
- You, H.; Zhuo, C.; Yan, S.; Wang, E.; Cao, H.; Liu, S.; Wang, X. CO2 deprotection-mediated switchable polymerization for precise construction of block copolymers. Macromolecules 2022, 55, 10980–10992. [Google Scholar] [CrossRef]
- Gibson, V.C.; Halliwell, C.M.; Long, N.J.; Oxford, P.J.; Smith, A.M.; White, A.J.P.; Williams, D.J. Synthetic, spectroscopic and olefin oligomerisation studies on nickel and palladium complexes containing ferrocene substituted nitrogen donor ligands. Dalton Trans. 2003, 5, 918–926. [Google Scholar] [CrossRef]
- Bernauer, J.; Polker, J.; Jacobi von Wangelin, A. Redox-active bian-based diimine ligands in metal-catalyzed small molecule syntheses. ChemCatChem 2022, 14, e202101182. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Yu, H.; Wang, L.; Wang, N.; Zhu, L.; Liang, R. The formation of polyethylene using α-diiminonickel precatalyst in the presence of CoCp2 and AgOTf. ChemistrySelect 2021, 6, 7663–7669. [Google Scholar] [CrossRef]
- Anderson, W.C., Jr.; Rhinehart, J.L.; Tennyson, A.G.; Long, B.K. Redox-active ligands: An advanced tool to modulate polyethylene microstructure. J. Am. Chem. Soc. 2016, 138, 774–777. [Google Scholar] [CrossRef]
- Anderson, W.C.; Long, B.K. Modulating polyolefin copolymer composition via redox-active olefin polymerization catalysts. ACS Macro Lett. 2016, 5, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.C.; Park, S.H.; Brown, L.A.; Kaiser, J.M.; Long, B.K. Accessing multiple polyethylene grades via a single redox-active olefin polymerization catalyst. Inorg. Chem. Front. 2017, 4, 1108–1112. [Google Scholar] [CrossRef]
- Popeney, C.; Guan, Z. Ligand electronic effects on late transition metal polymerization catalysts. Organometallics 2005, 24, 1145–1155. [Google Scholar] [CrossRef]
- Chen, M.; Yang, B.; Chen, C. Redox-controlled olefin (co)polymerization catalyzed by ferrocene-bridged phosphine-sulfonate palladium complexes. Angew. Chem. Int. Ed. 2015, 54, 15520–15524. [Google Scholar] [CrossRef]
- Yang, B.; Pang, W.; Chen, M. Redox control in olefin polymerization catalysis by phosphine–sulfonate palladium and nickel complexes. Eur. J. Inorg. Chem. 2017, 2017, 2510–2514. [Google Scholar] [CrossRef]
- Biernesser, A.B.; Li, B.; Byers, J.A. Redox-controlled polymerization of lactide catalyzed by bis(imino)pyridine iron bis(alkoxide) complexes. J. Am. Chem. Soc. 2013, 135, 16553–16560. [Google Scholar] [CrossRef]
- Fang, Y.Y.; Gong, W.J.; Shang, X.J.; Li, H.X.; Gao, J.; Lang, J.P. Synthesis and structure of a ferric complex of 2,6-di(1H-pyrazol-3-yl)pyridine and its excellent performance in the redox-controlled living ring-opening polymerization of ε-caprolactone. Dalton Trans. 2014, 43, 8282–8289. [Google Scholar] [CrossRef]
- Biernesser, A.B.; Delle Chiaie, K.R.; Curley, J.B.; Byers, J.A. Block copolymerization of lactide and an epoxide facilitated by a redox switchable iron-based catalyst. Angew. Chem. Int. Ed. 2016, 55, 5251–5254. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lu, H.; Luo, G.; Kang, X.; Luo, Y. Theoretical insight into the opposite redox activity of iron complexes toward the ring opening polymerization of lactide and epoxide. Inorg. Chem. Front. 2021, 8, 1005–1014. [Google Scholar] [CrossRef]
- Delle Chiaie, K.R.; Yablon, L.M.; Biernesser, A.B.; Michalowski, G.R.; Sudyn, A.W.; Byers, J.A. Redox-triggered crosslinking of a degradable polymer. Polym. Chem. 2016, 7, 4675–4681. [Google Scholar] [CrossRef]
- Qi, M.; Dong, Q.; Wang, D.; Byers, J.A. Electrochemically switchable ring-opening polymerization of lactide and cyclohexene oxide. J. Am. Chem. Soc. 2018, 140, 5686–5690. [Google Scholar] [CrossRef]
- Qi, M.; Zhang, H.; Dong, Q.; Li, J.; Musgrave, R.A.; Zhao, Y.; Dulock, N.; Wang, D.; Byers, J.A. Electrochemically switchable polymerization from surface-anchored molecular catalysts. Chem. Sci. 2021, 12, 9042–9052. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hu, C.; Pang, X.; Zhou, Y.; Duan, R.; Sun, Z.; Chen, X. Electrochemically controlled switchable copolymerization of lactide, carbon dioxide, and epoxides. Angew. Chem. Int. Ed. 2022, 61, e202202660. [Google Scholar] [CrossRef]
- Kaiser, J.M.; Anderson, W.C.; Long, B.K. Photochemical regulation of a redox-active olefin polymerization catalyst: Controlling polyethylene microstructure with visible light. Polym. Chem. 2018, 9, 1567–1570. [Google Scholar] [CrossRef]
- Peng, D.; Chen, C. Photoresponsive palladium and nickel catalysts for ethylene polymerization and copolymerization. Angew. Chem. Int. Ed. 2021, 60, 22195–22200. [Google Scholar] [CrossRef]
- Gong, Y.; Li, S.; Gong, Q.; Zhang, S.; Liu, B.; Dai, S. Systematic investigations of ligand steric effects on α-diimine nickel catalyzed olefin polymerization and copolymerization. Organometallics 2019, 38, 2919–2926. [Google Scholar] [CrossRef]
- Muhammad, Q.; Tan, C.; Chen, C. Concerted steric and electronic effects on α-diimine nickel- and palladium-catalyzed ethylene polymerization and copolymerization. Sci. Bull. 2020, 65, 300–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Hu, X.; Jian, Z. Photoresponsive α-diimine nickel modulated ethylene (co)polymerization. Chin. J. Chem. 2022, 40, 2919–2926. [Google Scholar] [CrossRef]
- Lee, B.Y.; Bazan, G.C.; Vela, J.; Komon, Z.J.; Bu, X. Alpha-iminocarboxamidato-nickel(Ⅱ) ethylene polymerization catalysts. J. Am. Chem. Soc. 2001, 123, 5352–5353. [Google Scholar] [CrossRef]
- Chen, Y.; Boardman, B.M.; Wu, G.; Bazan, G.C. A zwitterionic nickel–olefin initiator for the preparation of high molecular weight polyethylene. J. Organomet. Chem. 2007, 692, 4745–4749. [Google Scholar] [CrossRef]
- Shim, C.B.; Kim, Y.H.; Lee, B.Y.; Dong, Y.; Yun, H. [2-(alkylideneamino)benzoato]nickel(Ⅱ) complexes: Active catalysts for ethylene polymerization. Organometallics 2003, 22, 4272–4280. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, G.; Bazan, G.C. Remote activation of nickel catalysts for ethylene oligomerization. Angew. Chem. Int. Ed. 2005, 44, 1108–1112. [Google Scholar] [CrossRef]
- Cai, Z.; Shen, Z.; Zhou, X.; Jordan, R.F. Enhancement of chain growth and chain transfer rates in ethylene polymerization by (phosphine-sulfonate)PdMe catalysts by binding of B(C6F5)3 to the sulfonate group. ACS Catal. 2012, 2, 1187–1195. [Google Scholar] [CrossRef]
- Contrella, N.D.; Jordan, R.F. Lewis acid modification and ethylene oligomerization behavior of palladium catalysts that contain a phosphine-sulfonate-diethyl phosphonate ancillary ligand. Organometallics 2014, 33, 7199–7208. [Google Scholar] [CrossRef]
- Xie, K.; Xu, S.; Hao, W.; Wang, J.; Huang, A.; Zhang, Y. Surface effect of the mgcl2 support in Ziegler–Natta catalyst for ethylene polymerization: A computational study. Appl. Surf. Sci. 2022, 589, 153002. [Google Scholar] [CrossRef]
- Xie, K.; Xu, J.; Liu, P. Effect of ligands in TiCl3(OAr) catalysts for ethylene polymerization: Computational and experimental studies. Appl. Surf. Sci. 2018, 461, 175–181. [Google Scholar] [CrossRef]
- Xie, K.; Hao, W.; Xu, S.; Wang, J.; Wang, X.; Han, Z. Substituent effect of Cp2TiCl2 catalyst for ethylene polymerization: A DFT study. Polyolefins J. 2023, 10, 35–43. [Google Scholar] [CrossRef]
- Kuhn, P.; Semeril, D.; Matt, D.; Chetcuti, M.J.; Lutz, P. Structure-reactivity relationships in SHOP-type complexes: Tunable catalysts for the oligomerisation and polymerisation of ethylene. Dalton Trans. 2007, 5, 515–528. [Google Scholar] [CrossRef]
- Komon, Z.J.A.; Bu, X.; Bazan, G.C. Synthesis of butene−ethylene and hexene−butene−ethylene copolymers from ethylene via tandem action of well-defined homogeneous catalysts. J. Am. Chem. Soc. 2000, 122, 1830–1831. [Google Scholar] [CrossRef]
- Komon, Z.J.A.; Bu, X.; Bazan, G.C. Synthesis, characterization, and ethylene oligomerization action of [(C6H5)2PC6H4C(O-B(C6F5)3)O-κ2P,O]Ni(η3-CH2C6H5). J. Am. Chem. Soc. 2000, 122, 12379–12380. [Google Scholar] [CrossRef]
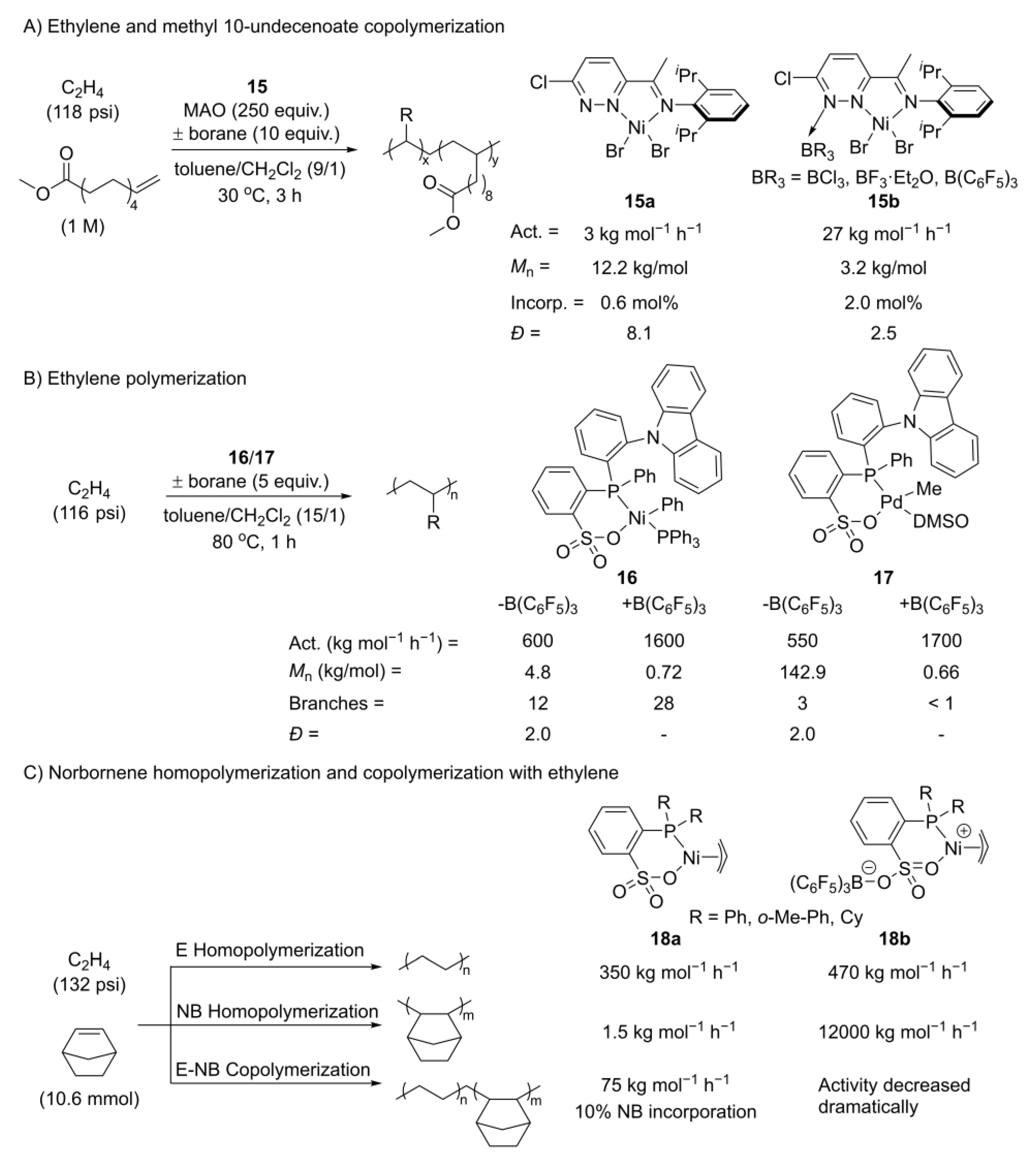
- Wang, G.; Li, M.; Pang, W.; Chen, M.; Tan, C. Lewis acids in situ modulate pyridazine-imine Ni catalysed ethylene (co)polymerisation. New J. Chem. 2019, 43, 13630–13634. [Google Scholar] [CrossRef]
- Tan, C.; Qasim, M.; Pang, W.; Chen, C. Ligand–metal secondary interactions in phosphine–sulfonate palladium and nickel catalyzed ethylene (co)polymerization. Polym. Chem. 2020, 11, 411–416. [Google Scholar] [CrossRef]
- Chen, M.; Zou, W.; Cai, Z.; Chen, C. Norbornene homopolymerization and copolymerization with ethylene by phosphine-sulfonate nickel catalysts. Polym. Chem. 2015, 6, 2669–2676. [Google Scholar] [CrossRef]
- Falivene, L.; Wiedemann, T.; Gottker-Schnetmann, I.; Caporaso, L.; Cavallo, L.; Mecking, S. Control of chain walking by weak neighboring group interactions in unsymmetrical catalysts. J. Am. Chem. Soc. 2018, 140, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Schiebel, E.; Santacroce, S.; Falivene, L.; Göttker-Schnetmann, I.; Caporaso, L.; Mecking, S. Tailored strength neighboring group interactions switch polymerization to dimerization catalysis. ACS Catal. 2019, 9, 3888–3894. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Wang, X.; Luo, Y.; Chen, C. A second-coordination-sphere strategy to modulate nickel- and palladium-catalyzed olefin polymerization and copolymerization. Angew. Chem. Int. Ed. 2017, 56, 11604–11609. [Google Scholar] [CrossRef]
- Wang, Y.; Pang, W.; Zhang, S.; Tan, C. Lewis acids modulation in phosphine-sulfonate palladium catalyzed ethylene polymerization. Appl. Organomet. Chem. 2022, 36, e6834. [Google Scholar] [CrossRef]
- Zhang, P.; Zhao, M.; Pang, W.; Chen, C. Lewis acid/base modulation in β-diiminate zinc-catalyzed switchable ring-opening polymerization of rac-lactide. Sci. China Chem. 2019, 62, 475–478. [Google Scholar] [CrossRef]
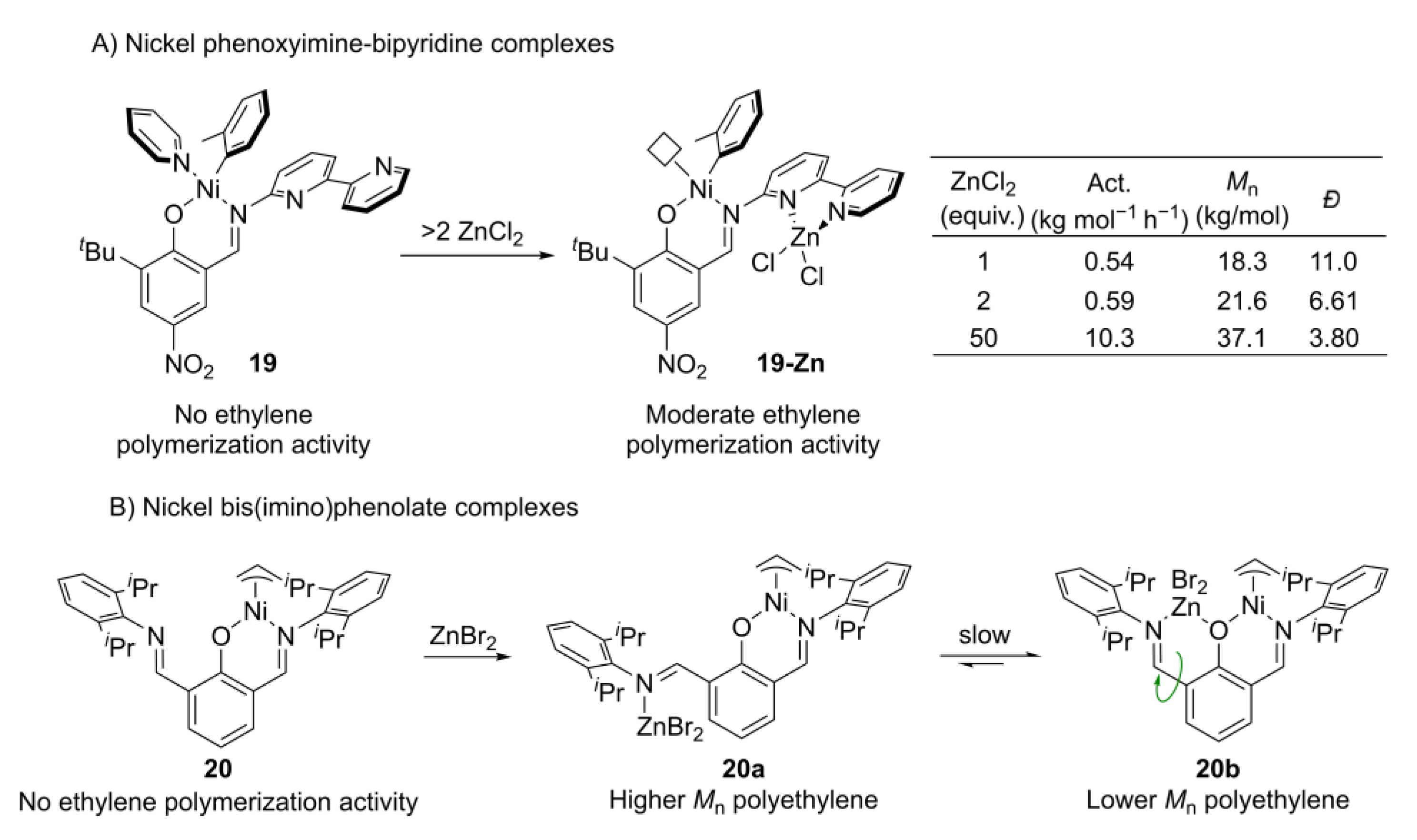
- Smith, A.J.; Kalkman, E.D.; Gilbert, Z.W.; Tonks, I.A. ZnCl2 capture promotes ethylene polymerization by a salicylaldiminato Ni complex bearing a pendent 2,2′-bipyridine group. Organometallics 2016, 35, 2429–2432. [Google Scholar] [CrossRef] [Green Version]
- Chiu, H.C.; Koley, A.; Dunn, P.L.; Hue, R.J.; Tonks, I.A. Ethylene polymerization catalyzed by bridging Ni/Zn heterobimetallics. Dalton Trans. 2017, 46, 5513–5517. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Wang, L.; McLain, S.; Bennett, A.; Dobbs, K.; Hauptman, E.; Ionkin, A.; Ittel, S.; Kunitsky, K.; Marshall, W.; et al. Copolymerization of ethylene and acrylates by nickel catalysts. In Beyond Metallocenes; American Chemical Society: Washington, DC, USA, 2003; pp. 131–142. [Google Scholar]
- Chiu, H.-C.; Pearce, A.J.; Dunn, P.L.; Cramer, C.J.; Tonks, I.A. β-oxo-δ-diimine nickel complexes: A comparison of tautomeric active species in ethylene polymerization catalysis. Organometallics 2016, 35, 2076–2085. [Google Scholar] [CrossRef]
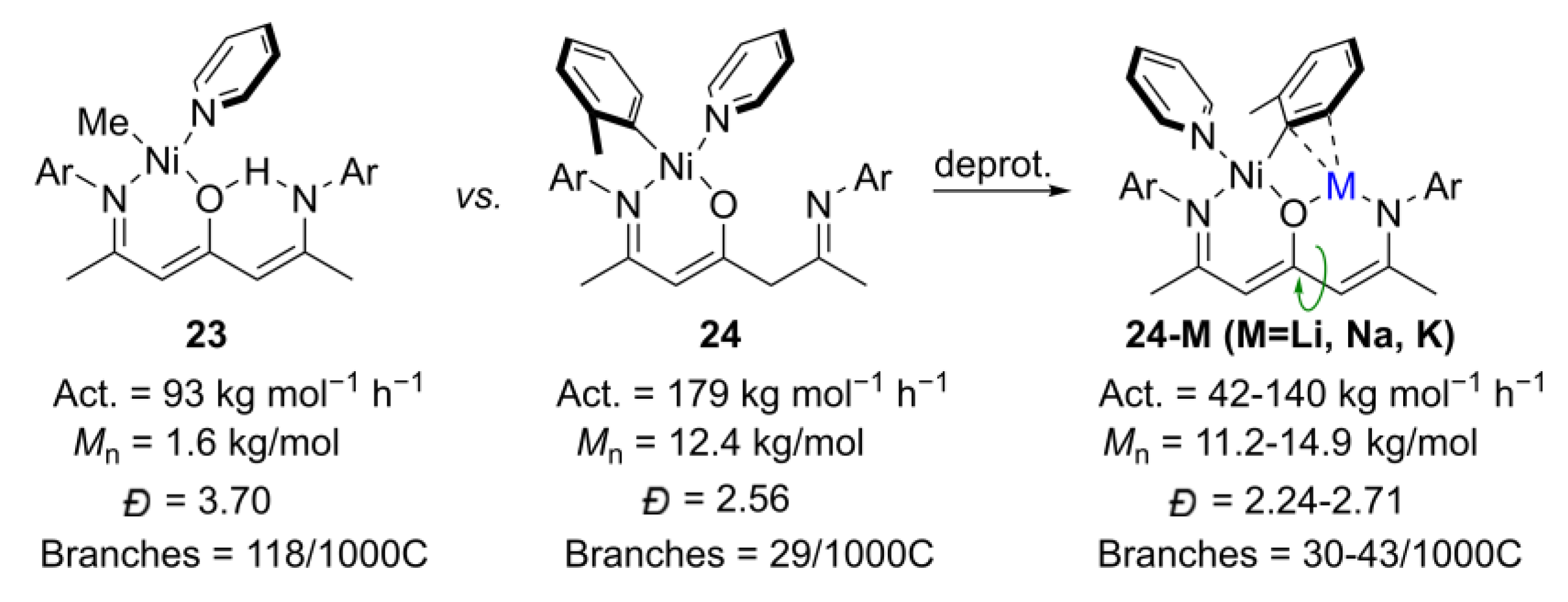
- Cai, Z.; Xiao, D.; Do, L.H. Fine-tuning nickel phenoxyimine olefin polymerization catalysts: Performance boosting by alkali cations. J. Am. Chem. Soc. 2015, 137, 15501–15510. [Google Scholar] [CrossRef]
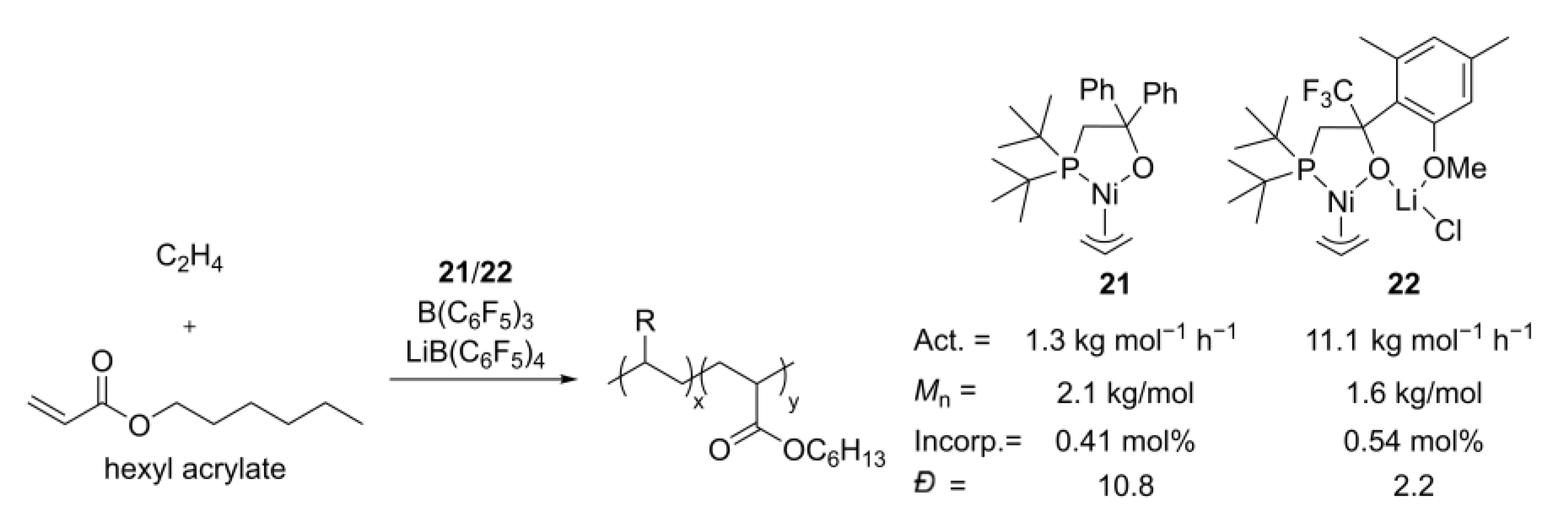
- Zhang, D.; Chen, C. Influence of polyethylene glycol unit on palladium- and nickel-catalyzed ethylene polymerization and copolymerization. Angew. Chem. Int. Ed. 2017, 56, 14672–14676. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, Y.; Jian, Z. Tunable branching and living character in ethylene polymerization using “polyethylene glycol sandwich” α-diimine nickel catalysts. Polym. Chem. 2021, 12, 1236–1243. [Google Scholar] [CrossRef]
- Cai, Z.; Do, L.H. Customizing polyolefin morphology by selective pairing of alkali ions with nickel phenoxyimine-polyethylene glycol catalysts. Organometallics 2017, 36, 4691–4698. [Google Scholar] [CrossRef]
- Tran, T.V.; Nguyen, Y.H.; Do, L.H. Development of highly productive nickel–sodium phenoxyphosphine ethylene polymerization catalysts and their reaction temperature profiles. Polym. Chem. 2019, 10, 3718–3721. [Google Scholar] [CrossRef]
- Tran, T.V.; Karas, L.J.; Wu, J.I.; Do, L.H. Elucidating secondary metal cation effects on nickel olefin polymerization catalysts. ACS Catal. 2020, 10, 10760–10772. [Google Scholar] [CrossRef]
- Tran, T.V.; Lee, E.; Nguyen, Y.H.; Nguyen, H.D.; Do, L.H. Customizing polymers by controlling cation switching dynamics in non-living polymerization. J. Am. Chem. Soc. 2022, 144, 17129–17139. [Google Scholar] [CrossRef]
- Xiao, D.; Cai, Z.; Do, L.H. Accelerating ethylene polymerization using secondary metal ions in tetrahydrofuran. Dalton Trans. 2019, 48, 17887–17897. [Google Scholar] [CrossRef]
- Wilders, A.M.; Contrella, N.D.; Sampson, J.R.; Zheng, M.; Jordan, R.F. Allosteric effects in ethylene polymerization catalysis. Enhancement of performance of phosphine-phosphinate and phosphine-phosphonate palladium alkyl catalysts by remote binding of B(C6F5)3. Organometallics 2017, 36, 4990–5002. [Google Scholar] [CrossRef]
- Cai, Z.; Do, L.H. Thermally robust heterobimetallic palladium–alkali catalysts for ethylene and alkyl acrylate copolymerization. Organometallics 2018, 37, 3874–3882. [Google Scholar] [CrossRef]
- Tahmouresilerd, B.; Xiao, D.; Do, L.H. Rigidifying cation-tunable nickel catalysts increases activity and polar monomer incorporation in ethylene and methyl acrylate copolymerization. Inorg. Chem. 2021, 60, 19035–19043. [Google Scholar] [CrossRef]
- Deacy, A.C.; Moreby, E.; Phanopoulos, A.; Williams, C.K. Co(Ⅲ)/alkali-metal(Ⅰ) heterodinuclear catalysts for the ring-opening copolymerization of CO2 and propylene oxide. J. Am. Chem. Soc. 2020, 142, 19150–19160. [Google Scholar] [CrossRef] [PubMed]
- Trott, G.; Garden, J.A.; Williams, C.K. Heterodinuclear zinc and magnesium catalysts for epoxide/CO2 ring opening copolymerizations. Chem. Sci. 2019, 10, 4618–4627. [Google Scholar] [CrossRef] [Green Version]
- Deacy, A.C.; Kilpatrick, A.F.R.; Regoutz, A.; Williams, C.K. Understanding metal synergy in heterodinuclear catalysts for the copolymerization of CO2 and epoxides. Nat. Chem. 2020, 12, 372–380. [Google Scholar] [CrossRef]
- Diment, W.T.; Gregory, G.L.; Kerr, R.W.F.; Phanopoulos, A.; Buchard, A.; Williams, C.K. Catalytic synergy using Al(Ⅲ) and group Ⅰ metals to accelerate epoxide and anhydride ring-opening copolymerizations. ACS Catal. 2021, 11, 12532–12542. [Google Scholar] [CrossRef]
- Plajer, A.J.; Williams, C.K. Heterotrinuclear ring-opening copolymerization catalysis: Structure–activity relationships. ACS Catal. 2021, 11, 14819–14828. [Google Scholar] [CrossRef]
- Saini, P.K.; Romain, C.; Williams, C.K. Dinuclear metal catalysts: Improved performance of heterodinuclear mixed catalysts for CO2-epoxide copolymerization. Chem. Commun. 2014, 50, 4164–4167. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Xiao, D.; Do, L.H. Cooperative heterobimetallic catalysts in coordination insertion polymerization. Comments Inorg. Chem. 2019, 39, 27–50. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
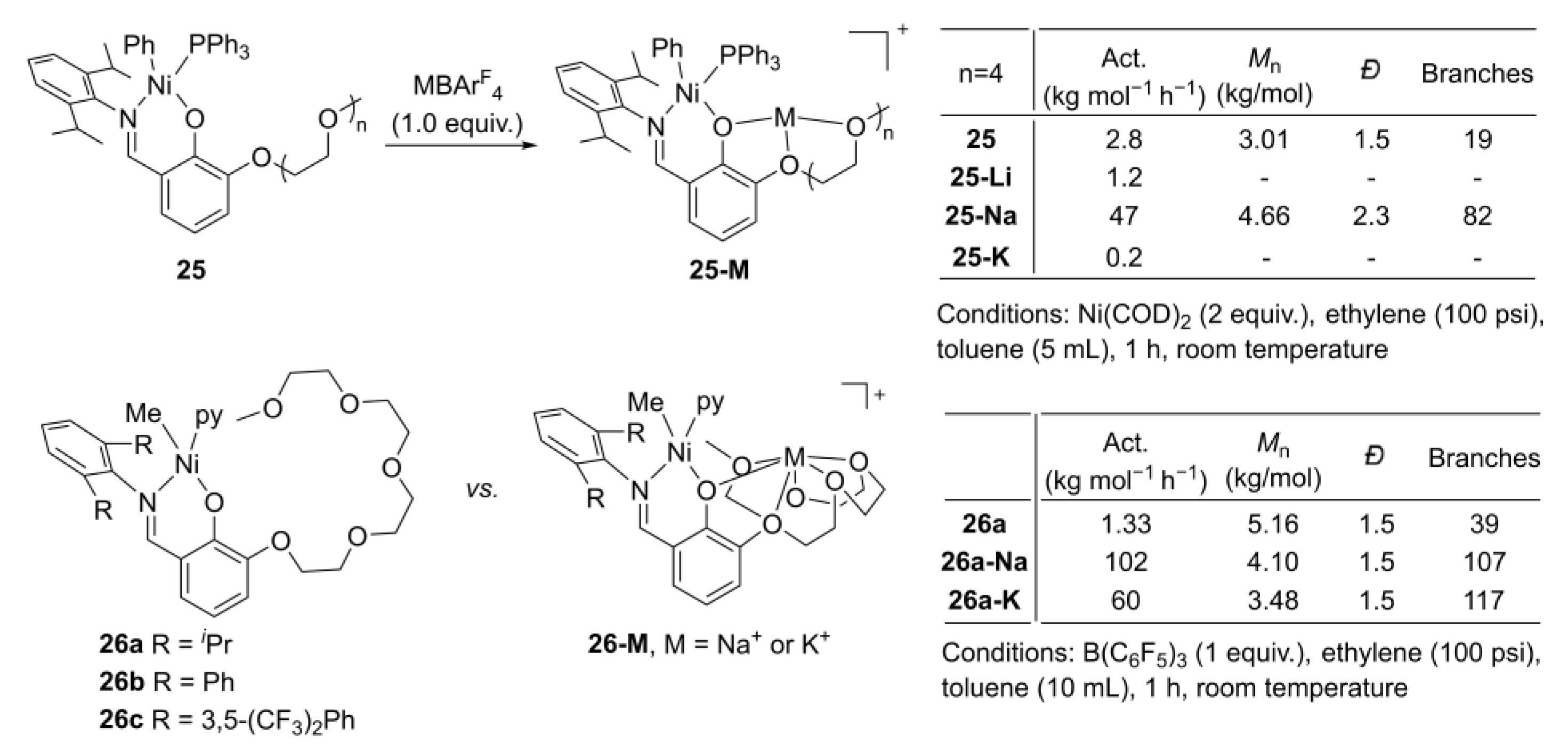{kind=link}
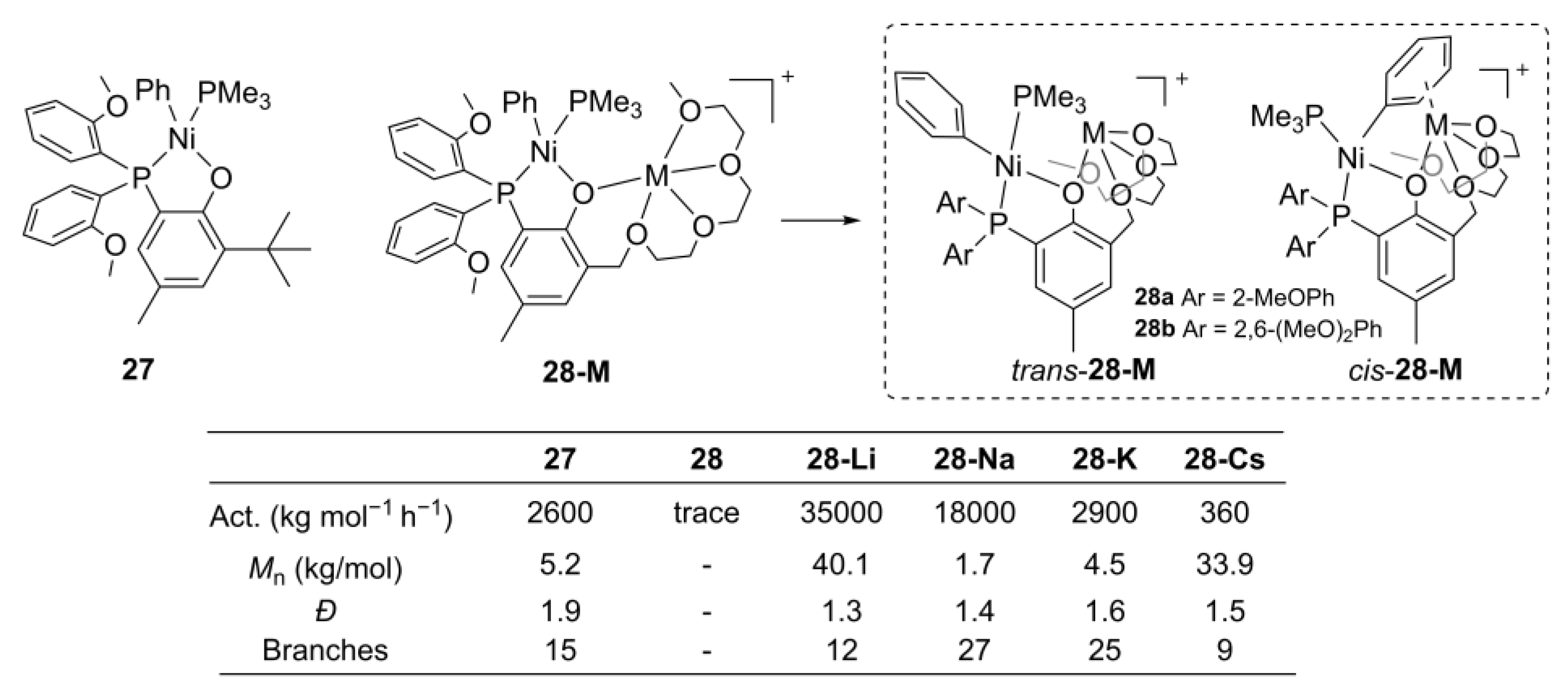{kind=link}
{kind=link}
{kind=link}
| Entry | CoCp2/Ni | AgOTf/Ni | Activity b | Tm/°C | Branches c |
|---|---|---|---|---|---|
| 1 | 0 | 0 | 1060 | −3.00 | 93 |
| 2 | 0.5 | 0 | 750 | 6.68 | 85 |
| 3 | 1.0 | 0 | 610 | 39.69 | 81 |
| 4 | 0 | 0.5 | 1020 | −3.47 | 93 |
| 5 | 0 | 1.0 | 1040 | −6.94 | 95 |
| Reaction State | Catalyst 9 | Catalyst 10 | ||
|---|---|---|---|---|
| Mn (kg/mol) | Đ | Mn (kg/mol) | Đ | |
| Redox state (4 h) | 8.4 | 1.15 | 9.6 | 1.17 |
| Oxidation state (8 h) | 12.1 | 1.16 | 10.4 | 1.20 |
| Redox state (4 h) | 15.1 | 1.15 | 13.1 | 1.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suo, H.; Zhang, Z.; Qu, R.; Gu, Y.; Qin, Y. Tunable Late-Transition-Metal-Catalyzed Polymerization for Controlled Polymer Synthesis. Catalysts 2023, 13, 670. https://doi.org/10.3390/catal13040670
Suo H, Zhang Z, Qu R, Gu Y, Qin Y. Tunable Late-Transition-Metal-Catalyzed Polymerization for Controlled Polymer Synthesis. Catalysts. 2023; 13(4):670. https://doi.org/10.3390/catal13040670
Chicago/Turabian StyleSuo, Hongyi, Zisheng Zhang, Rui Qu, Yanan Gu, and Yusheng Qin. 2023. "Tunable Late-Transition-Metal-Catalyzed Polymerization for Controlled Polymer Synthesis" Catalysts 13, no. 4: 670. https://doi.org/10.3390/catal13040670
APA StyleSuo, H., Zhang, Z., Qu, R., Gu, Y., & Qin, Y. (2023). Tunable Late-Transition-Metal-Catalyzed Polymerization for Controlled Polymer Synthesis. Catalysts, 13(4), 670. https://doi.org/10.3390/catal13040670