
A Mild and Sustainable Procedure for the Functionalization of Morpholin-2-Ones by Oxidative Imidation Reactions



Abstract
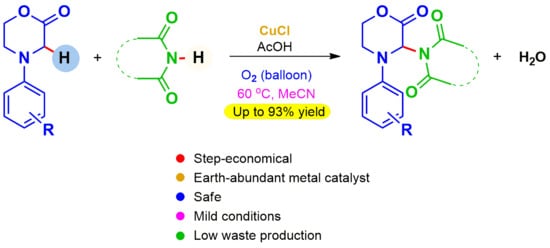
:
1. Introduction
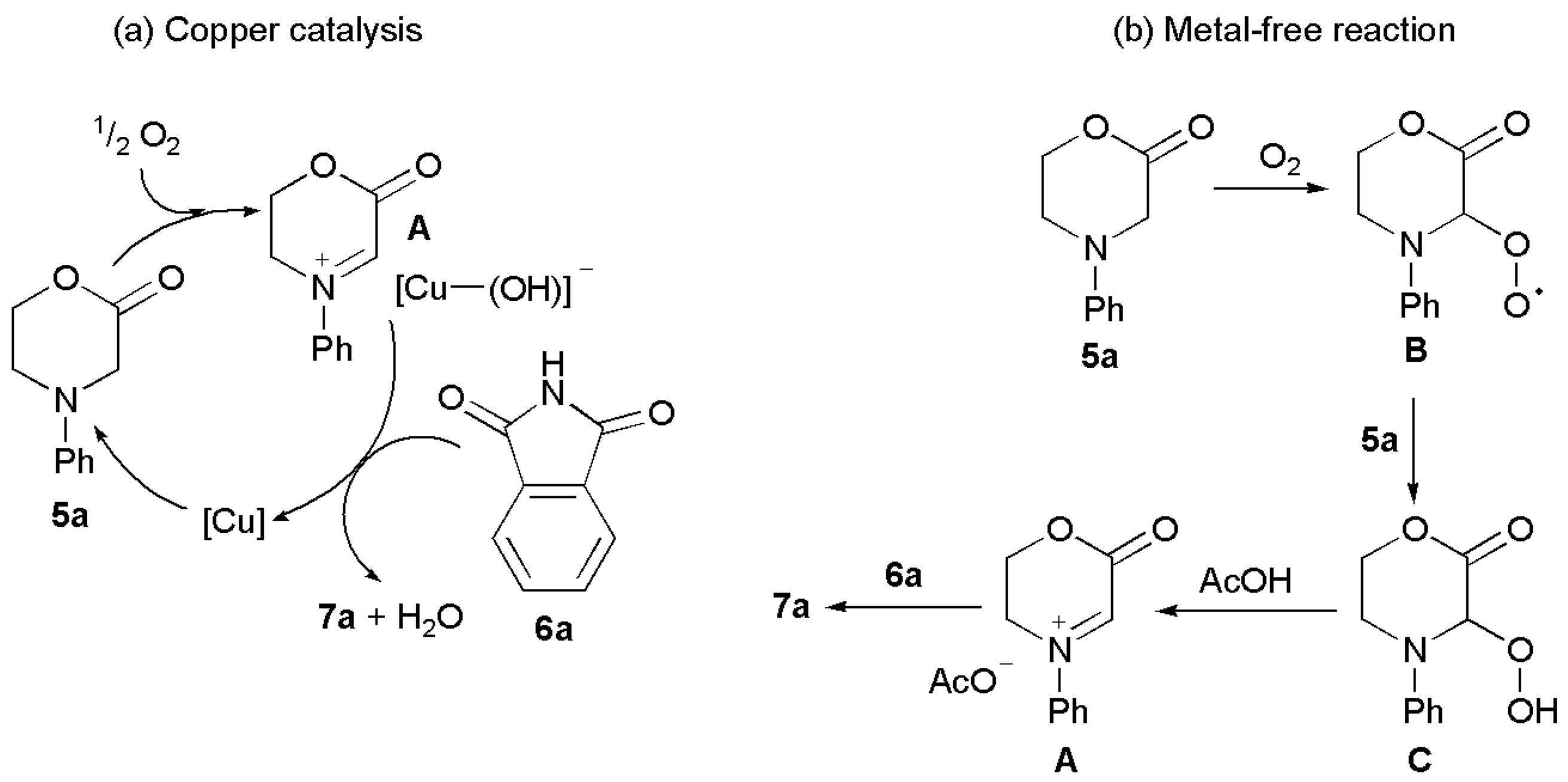
2. Results
3. Materials and Methods
3.1. General Methods
3.2. General Procedure for the CDC Reaction between Morpholinones and Imides
3.3. Characterization Data of Products and Starting Materials
- 3-(1,3-Dioxoindolin-2-yl)-4-phenylmorpholin-2-one (7a): Prepared from N-phenyl morpholine-2-one and phthalimide according to the general procedure, in a reaction performed at 60 °C. Purified by plate chromatography (silica gel) with dichloromethane/ethyl acetate (8:1) as eluent. Obtained as buff-colored crystals, 57.8 mg, 68% yield, m.p. 166 °C. 1H NMR (CDCl3): δ 3.758 (dt, 1 H, J 2.5, 13.4 Hz, C(5)HH), 4.249 (overlapp ddd, 1 H, J 1.3, 10.8, 13.6 Hz, C(5)HH), 4.62–4.79 (m, 2 H, 2 × H-6), 6.516 (s, 1 H, H-3), 6.911 (t, 1 H, J 7.3 Hz, p-Ph-H), 7.020 (d, 2 H, J 8.5 Hz, 2 × o-Ph-H), 7.279 (overlapp. t, 1 H, J 6.9 Hz, m-Ph-H), 7.282 (overlapp. t, 1 H, J 7.4 Hz, m-Ph-H), 7.734 (dd, 2 H, J 3.1 Hz, 5.5 Hz, H-6′ and H-7′), 7.835 (dd, 2 H, J 3.0, 5.5 Hz, H-5′, H-8′) ppm. 13C NMR (CDCl3): δ 42.27 (CH2, C-5), 61.72 (CH, C-3), 69.32 (CH2, C-6), 115.78 (CH, 2 × o-Ph-H), 121.13 (CH, p-C, Ph), 123.78 (CH, C-5′ and C-8′), 129.41 (CH, 2 × m-Ph-H), 131.55 (Cq, i-C, Ph), 134.42 (CH, C-6′ and C-7′), 144.83 (Cq, C-4′ and C-9′), 164.41 (Cq, CO, C-2), 167.43 (Cq, 2 × Phth-CO) ppm. IR (neat): ṽ 2957, 1742, 1714, 1600, 1500, 1380, 1221, 1186, 1080, 1000, 979, 886, 765, 714, 693, 656, 521 cm−1. Elemental analysis: Calcd for C18H14N2O4·¼H2O: C, 66.15; H, 4.47; N, 8.57. Found C, 66.19; H, 4.12; N, 8.49. HRMS (ESI): Calcd for C18H14N2NaO4: 345.0847. Found: 345.0846. Calcd for C18H14KN2O4: 361.0589. Found: 361.0585.
- 2-(2-Oxo-4-phenylmorpholin-3-yl)-3a,4,7,7a-tetrahydroisoindole-1,3-dione (7b): Prepared from N-phenyl morpholine-2-one and cis-1,2,3,6-tetrahydrophthalimide according to the general procedure, in a reaction performed at 60 °C. Purified by plate chromatography (silica gel) with dichloromethane/ethyl acetate (8:1) as eluent. Obtained as buff-colored crystals, 38.2 mg, 93% yield, m.p. 131–133 °C. 1H NMR (CDCl3): δ 2.129 (dd, 2 H, J 6.6, 15.0 Hz, C(5′)HH and C(8′)HH), 2.363 (d, 2 H, J 13.9 Hz, C(5′)HH and C(8′)HH), 2.94–3.11 (m, 2 H, H-4′ and H-9′), 3.584 (apparent. d, 1 H, 13.0 Hz, C(5)HH), 4.136 (appar. t, 1 H, J 12.7 Hz, C(5)HH), 4.53–4.71 (m, 2 H, 2 × H-6), 5.536 (apparent. s, 2 H, 6′ and 7′), 6.327 (s, 1 H, H-3), 6.880 (d, 2 H, J 7.8 Hz, 2 × o-Ph-H), 6.934 (t, 1 H, J 7.0 Hz, p-Ph-H), 7.258 (t, 2 H, J 7.4 Hz, 2 × m-Ph-H) ppm. 13C NMR (CDCl3): δ 23.15/23.27 (2 × CH2, C-5′ and C-8′), 38.84/39.23 (2 × CH, C-4′ and C-9′), 42.61 (CH2, C-5), 61.88 (CH, C-3), 69.28 (CH2, C-6), 116.46 (CH, 2 × o-Ph-C), 121.28 (CH, p-Ph-C), 126.73/127.06 (2 × CH, C-6 and C-7), 129.19 (CH, 2 × m-Ph-C), 144.70 (Cq, i-Ph-C), 163.99 (Cq, C-2), 179.12/179.19 (2 × Cq, C-1′ and C-3′) ppm. IR (neat): ṽ 3063, 3042, 2971, 2945, 2910, 2853, 1735, 1707, 1600, 1496, 1347, 1274, 1234, 1194, 1165, 1087, 1029, 995, 981, 894, 753, 684 cm−1. Elemental analysis: Calcd for C18H18N2O4·½H2O: C, 64.47; H, 5.71; N, 8.36. Found C, 64.88; H, 5.67; N, 8.49.
- 3-(6-Nitro-1,3-dioxo-indolin-2-yl)-4-phenylmorpholin-2-one (7c): Prepared from N-phenyl morpholine-2-one and 4-nitrophthalimide according to the general procedure, in a reaction performed at 60 °C. Purified by plate chromatography (silica gel) with ethyl acetate/hexane (1:3) as eluent, followed by an elution with dichloromethane/ethyl acetate (8:1). Obtained as pale orange crystals, 12.5 mg, 27% yield, m.p. 192–193 °C. 1H NMR (CDCl3): δ 3.768 (d, 1 H, J 13.4 Hz, C(5)HH), 4.213 (t, 1 H, 11.8 Hz, C(5)HH), 4.65–4.80 (m, 2 H, 6-H), 6.549 (s, H-3), 6.923 (t, 1 H, J 7.1 Hz, p-Ph-H), 6.994 (d, 2 H, 2 × o-Ph-H), 7.23–7.34 (m, 2 H, 2 × m-Ph-H), 8.028 (d, 1 H, J 8.0 Hz, H-8′), 8.597 (d, 1 H, J 8.1 Hz, H-7′), 8.635 (s, 1 H, H-5′) ppm. 13C NMR (CDCl3): δ 42.550 (CH2, C-5), 62.272 (CH, C-3), 69.444 (CH2, C-6), 115.93 (CH, 2 × o-Ph-C), 119.20 (CH, C-5′), 121.58 (CH, p-Ph-C), 125.06 (CH, C-7′), 129.54/129.58 (CH, 2 × m-Ph-C), 132.92 (Cq, C-9′), 135.84 (Cq, C-4′), 144.54 (Cq, i-Ph-C), 151.89 (Cq, C-6′), 163.92 (Cq, C(O)-2), 165.10/165.38 (2 × Cq, C(O)-1′ and C(O)-3′) ppm. IR (neat): ṽ 3100, 2900, 1776, 1722, 1600, 1500, 1338, 1250, 1225, 1200, 1100, 1056, 1013, 962, 932, 856, 744, 716, 687, 641, 524, 500 cm−1. Elemental analysis: Calcd for C18H13N3O6·¼H2O: C, 58.15; H, 3.65; N, 11.30. Found C, 58.01; H, 3.57; N, 11.00.
- 3-(2,5-Dioxopyrrol-1-yl)-4-phenylmorpholin-2-one (7d): Prepared from N-phenyl morpholine-2-one and maleimide according to the general procedure, in a reaction performed at 60 °C. Purified by plate chromatography (silica gel) with ethyl acetate/hexane (1:2) as eluent, followed by an elution with dichloromethane/ethyl acetate (8:1). Obtained as pale yellow crystals, 15.6 mg, 36% yield, m.p. 121 °C. 1H NMR (CDCl3): δ 3.697 (td, 1 H, J 2.6, 13.4 Hz, C(5)HH), 4.104 (overlapp ddd, 1 H, J 3.4, 7.44, 13.7 Hz, C(5)HH), 4.59–4.73 (m, 2 H, 2 × H-6), 6.299 (s, 1 H, H-3), 6.605 (s, 2 H, H-3′ and H-4′), 6.92–6.99 (m, 3 H, o-Ph-H and p-Ph-H), 7.294 (t, 2 H, J 7.8 Hz, 2 × m-Ph-H) ppm. 13C NMR (CDCl3): δ 12.159 (CH2, C-5), 61.651 (CH, C-3), 69.272 (CH2, C-6), 115.92 (2 × CH, o-Ph-C), 121.35 (CH, p-Ph-C), 129.41 (2 × CH, C-3′ and C-4′), 134.41 (2 × CH, m-Ph-C), 144.71 (CH, i-Ph-C), 164.22 (Cq, C-2), 169.71 (2 × Cq, C-2′ and C-5′) ppm. IR (neat): ṽ 3102, 2968, 2909, 2867, 1738, 1704, 1603, 1505, 1462, 1372, 1361, 1341, 1272, 1210, 1145, 1082, 1033, 983, 851, 828, 796, 750, 693, 649, 439 cm−1. Elemental analysis: Calcd for C14H12N2O4·½H2O: C, 59.78; H, 4.66; N, 9.96. Found C, 59.46; H, 4.26; N, 9.39. HRMS (ESI): Calcd for C14H12N2NaO4: 295.0689. Found: 295.08687. Calcd for C14H12KN2O4: 331.0430. Found: 311.0429.
- 3-(2,5-Dioxopyrrolidin-1-yl)-4-phenylmorpholin-2-one (7e): Prepared from N-phenyl morpholine-2-one and succinimide according to the general procedure, in a reaction performed at 60 °C. Purified by plate chromatography (silica gel) with dichloromethane/ethyl acetate (8:1) as eluent. Obtained as buff-colored crystals, 30.6 mg, 83% yield, m.p. 157 °C. 1H NMR (CDCl3): δ 2.663 (s, 4 H, 2 × H-3′ and 2 × H-4′), 3.700 (d, 1 H, J 13.4 Hz, C(5)HH), 4.15 (t, 1 H, J 13.5 Hz, C(5)HH), 4.55–4.71 (m, 2 H, 2 × H-6), 6.340 (s, 1 H, H-3), 6.89–6.98 (m, 3 H, 2 × o-Ph-H + p-Ph-H), 7.290 (t, 1 H, J 7.7 Hz, 2 × m-Ph-H) ppm. 13C NMR (CDCl3): δ 27.98 (2 × CH2, C-3′ and C-4′), 42.55 (CH2, C-5), 61.873 (CH2, C-3), 69.30 (CH2, C-6), 115.59 (2 × CH, o-Ph-C), 121.10 (CH, p-Ph-C), 129.43 (2 × CH, m-Ph-C), 144.64 (CH, i-Ph-C), 164.03 (Cq, C-2), 176.19 (2 × Cq, C-2′ and C-5′) ppm. IR (neat): ṽ 2988, 2937, 2906, 2865, 1741, 1706, 1597, 1496, 1375, 1269, 1208, 1177, 1084, 984, 753, 698 cm−1. Elemental analysis: Calcd for C14H14N2O4·¼H2O: C, 60.31; H, 5.24; N, 10.04. Found C, 60.26; H, 5.22; N, 9.64.
- 3-(3-Ethyl-3-methyl-2,5-dioxopyrrolidin-1-yl)-4-phenylmorpholin-2-one (7f): Prepared N-morpholine-2-one and (R)-ethosuximide according to the general procedure in a reaction performed at 60 °C. Purified by plate chromatography (silica gel) with dichloromethane/ethyl acetate (8:1) as eluent to afford a 1:1 mixture of two inseparable diastereoisomers. Obtained as buff-colored crystals, 29.1 mg, 71% yield, m.p. 78 °C. 1H NMR (CDCl3): δ (mixture of two diastereoisomers) 0.629 and 0.682 (t, 3 H, J 7.4 Hz, CH3-8′), 1.121 (s, 3 H, CH3-6′), 1.36–1.49 (m, 1 H, CHH-7′), 1.52–1.66 (m, 1 H, CHH-7′), 2.319 (2 × overlapp. d, 1 H, CHH-4′), 2.542 (2 × overlapp. d, 1 H, CHH-4′), 3.56–3.66 (m, 1 H, CHH-5), 4.160 (appar. t, 1 H, CHH-5), 4.57–4.71 (m, 2 H, H-6), 6.334 and 6.348 (overlapp. s, 1 H, H-3), 6.87–6.97 (m, 3 H, 2 × o-Ph-H and p-Ph-H), 7.22–7.33 (m, 2 × m-Ph-H) ppm. 13C NMR (CDCl3): δ (mixture of two diastereoisomers) 8.142 and 8.244 (CH3, C-8′), 23.638 and 23.844 (CH3, C-6′), 30.483 and 30.720 (CH2, C-7′), 39.985 and 40.045 (CH2, C-4′), 42.672 and 42.713 (CH2, C-5), 44.166 and 44.336 (Cq, C-3), 61.762 and 61.939 (CH, C-3), 69.234 and 69.293 (CH2, C-6), 116.11 and 116.54 (CH, 2 × o-Ph-C), 121.26 and 121.45 (CH, p-Ph-C), 129.23 (CH, 2 × m-Ph-C), 144.72 and 144.79 (Cq, i-Ph-C), 164.06 (Cq, C(O)-2), 175.12 and 175.22 (Cq, C(O)-2′ and C(O)-5′) ppm. IR (neat): ṽ 3012, 2940, 2900, 2858, 1760, 1712, 1600, 1500, 1383, 1267, 1208, 1146, 1092, 983, 742, 692, 667 cm−1. Elemental analysis: Calcd for C17H20N2O4·½H2O: C, 62.76; H, 6.50; N, 8.61. Found C, 62.84; H, 6.70; N, 8.48.
- 3-(2,6-Dioxopiperidin-1-yl)-4-phenylmorpholin-2-one (7h): Prepared from N-phenyl morpholine-2-one and glutarimide according to the general procedure, but omitting the addition of acetic acid, in a reaction performed at 80 °C. Purified by plate chromatography (silica gel) with hexane/ethyl acetate (2:1) as eluent. Obtained as buff-colored crystals, 16.0 mg, 36% yield, m.p. 165 °C. 1H NMR (CDCl3): δ 1.723 (br s, 2 H, 2 × H-4′), 2.574 (t, J 11.1 Hz, 4 H, 2 × H-3′ and 2 × H-5′), 3.568 (dd, 1 H, J 1.8, 12.7 Hz, C(5)HH), 4.104 (m, 1 H, C(5)HH), 4.50–4.69 (m, 2 H, 2 × H-6), 6.866 (d, 2 H, J 7.5 Hz, 2 × o-Ph-H), 6.923 (t, 1 H, J 7.3 Hz, p-Ph-H), 6.905 (s, 1 H, H-3), 7.261 (t, 2 H, J 7.3. Hz, m-Ph-H) ppm. 13C NMR (CDCl3): δ 16.416 (CH2, C-4′), 32.456 (2 × CH2, C-3′ and C-5′), 43.367 (CH2, C-5), 61.424 (CH2, C-3), 68.907 (CH2, C-6), 116.48 (CH, 2 × o-Ph-C), 120.96 (CH, p-Ph-C), 129.14 (CH, 2 × m-Ph-C), 145.16 (Cq, i-Ph-C), 165.25 (Cq, C(O)-2), 172.23 (Cq, 2 × C(O), C-2′ and C-6′) ppm. IR (neat): ṽ 2967, 2932, 2906, 2862, 1749, 1722, 1671, 1595, 1494, 1368, 1344,1310, 1271, 1247, 1213, 1171, 1134, 1086, 1012, 988, 750, 701, 438 cm−1. Elemental analysis: Calcd for C15H16N2O4·¼H2O: C, 61.68; H, 5.84; N, 9.25. Found C, 61.30; H, 5.76; N, 9.74.
- 3-(1,3-Dioxoindolin-2-yl)-4-(p-tolyl)-morpholin-2-one (7k): Prepared from N-tolylmorpholine-2-one and phthalimide according to the general procedure in a reaction performed at 60 °C. Purified by plate chromatography (silica gel) with dichloromethane/ethyl acetate/hexane (8:1:3) as eluent. Obtained as buff-colored crystals, 33.4 mg, 75% yield. 1H NMR (CDCl3): δ 2.240 (s, 3 H, CH3), 3.669 (d, J 13.3 Hz, C(5)HH), 4.179 (overlapp. ddd, J 7.6, 13.5 Hz, C(5)HH), 4.61–4.79 (m, 2 × H-6), 6.457 (s, 1 H, H-3), 6.924 (d, 2 H, J 8.5 Hz, 2 × o-Ph-H), 7.071 (d, 2 H, J 8.5 Hz, 2 × m-Ph-H), 7.719 (dd, J 3.1, 5.4 Hz, C-6′ and C-7′), 7.824 (dd, J 3.1, 5.5 Hz, H-5′ and H-8′) ppm. 13C NMR (CDCl3): δ 20.402 (CH3), 42.688 (CH2, C-5), 62.113 (CH, C-3), 69.359 (CH2, C-6), 116.46 (CH, 2 × o-Ph-C), 123.76 (CH, C-5′ and C-8′), 129.90 (CH, 2 × m-Ph-C), 130.94 (Cq, p-Ph-C), 131.55 (Cq, C-4′ and C-9′), 134.37 (CH, C-6′ and C-7′), 142.62 (Cq, i-Ph-C), 164.53 (Cq, C(O)-2), 167.44 (Cq, C(O)-1′ and C(O)-3′) ppm. IR (neat): ṽ 3037, 2960, 2922, 2861, 1756, 1712, 1617, 1519, 1470, 1379, 1269, 1204, 1084, 986, 896, 808, 716, 644, 516 cm−1. Elemental analysis: Calcd for C19H16N2O4·¼H2O: C, 63.12; H, 5.24; N, 7.74. Found C, 63.33; H, 4.74; N, 7.37.
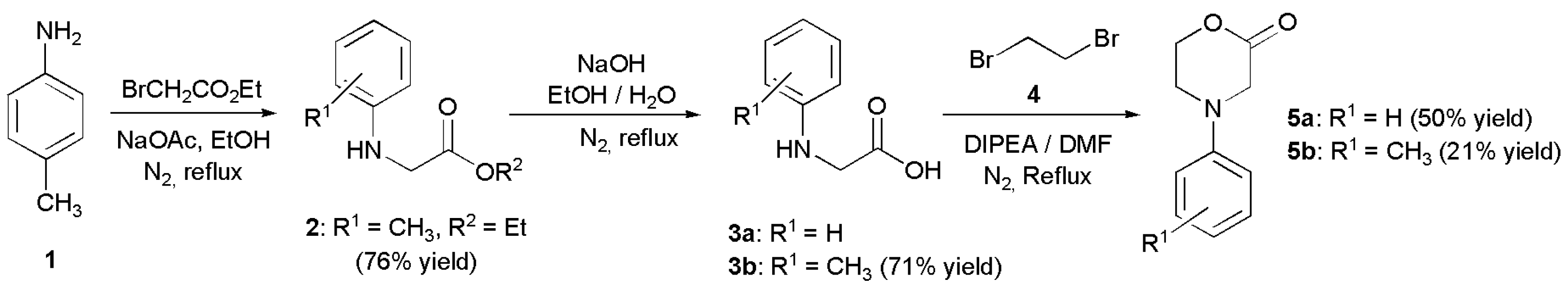
- 4-(p-Tolyl)-morpholin-2-one (5b): Prepared as 5a. Purified by column chromatography (silica gel) with ethyl acetate/hexane (1:2) as eluent. Obtained as buff-coloured crystals, 0.188 mg, 21% yield, m.p. 72 °C. 1H NMR (CDCl3): 2.313 (s, 3 H, CH3), 3.469 (m, 2 × H-5), 4.089 (s, 2 × H-3), 4.575 (m, 2 × H-6),6.76 (d, 2 H, J 7.4 Hz, 2 × o-CH ), 7.15 (d, 2 H, J 7.4 Hz, 2 × m-CH) ppm. 13C NMR (CDCl3): δ 20.355 (CH3), 44.757, (CH2, C-5), 50.868, (CH2, C-3), 67.793, (CH2, C-6), 114, 39 (CH, 2 × o-Ph-C), 129.73 (Cq, p-C), 130.03 (CH, 2 × m-Ph-C), 145.80, (Cq, i-Ph-C), 167.49 (Cq, C(O)-2) ppm. IR (neat): ṽ 3037, 2999, 2960, 2916, 2852, 1718, 1617, 1514, 1462, 1381, 1275, 1234, 1079, 978, 938, 814, 796, 520 cm−1. Elemental analysis: Calcd for C11H13NO2·¼H2O: C, 67.50; H, 6.95; N, 7.16. Found C, 67.12; H, 6.83; N, 7.12.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tian, T.; Li, Z.; Li, C.-J. Cross-dehydrogenative coupling: A sustainable reaction for C–C bond formations. Green Chem. 2021, 23, 6789–6862. [Google Scholar] [CrossRef]
- Faisca Phillips, A.M.; Guedes da Silva, M.d.F.C.; Pombeiro, A.J.L. New trends in enantioselective cross-dehydrogenative coupling. Catalysts 2020, 10, 529. [Google Scholar] [CrossRef]
- Huang, C.Y.; Kang, H.; Li, J.; Li, C.J. En route to intermolecular cross-dehydrogenative coupling Reactions. J. Org. Chem. 2019, 84, 12705–12721. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S. Catalytic enantioselective cross dehydrogenative coupling of sp3 C–H of heterocycles. Org. Biomol. Chem. 2019, 17, 9683–9692. [Google Scholar] [CrossRef]
- Phillips, A.M.F.; Pombeiro, A.J.L. Recent developments in transition metal-catalyzed cross-dehydrogenative coupling reactions of ethers and thioethers. ChemCatChem 2018, 10, 3354–3383. [Google Scholar] [CrossRef]
- Varun, B.V.; Dhineshkumar, J.; Bettadapur, K.R.; Siddaraju, Y.; Alagiri, K.; Prabhu, K.R. Recent advancements in dehydrogenative cross coupling reactions for C–C bond formation. Tetrahedron Lett. 2017, 58, 803–824. [Google Scholar] [CrossRef]
- Kozlowski, M.C. Oxidative coupling in complexity building transforms. Acc. Chem. Res. 2017, 50, 638–643. [Google Scholar] [CrossRef]
- Liu, C.; Yuan, J.; Gao, M.; Tang, S.; Li, W.; Shi, R.; Lei, A. Oxidative coupling between two hydrocarbons: An update of recent C-H functionalizations. Chem. Rev. 2015, 115, 12138–12204. [Google Scholar] [CrossRef]
- Yeung, C.S.; Dong, V.M. Catalytic dehydrogenative cross-coupling: Forming carbon-carbon bonds by oxidizing two carbon-hydrogen bonds. Chem. Rev. 2011, 111, 1215–1292. [Google Scholar] [CrossRef]
- Phillips, A.M.F.; Pombeiro, A.J.L. The functionalization of amino acids, peptides, and derivatives by cross-dehydrogenative coupling. In Handbook of CH-Functionalization; Maiti, D., Ed.; Wiley-VCH: Weinheim, Germany, 2022. [Google Scholar] [CrossRef]
- San Segundo, M.; Correa, A. Cross-dehydrogenative coupling reactions for the functionalization of α-amino acid derivatives and peptides. Synthesis 2018, 50, 2853–2866. [Google Scholar] [CrossRef]
- Noisier, A.F.M.; Brimble, M.A. C–H functionalization in the synthesis of amino acids and peptides. Chem. Rev. 2014, 114, 8775–8806. [Google Scholar] [CrossRef] [PubMed]
- San Segundo, M.; Guerrero, I.; Correa, A. Co-catalyzed C(sp3)–H oxidative coupling of glycine and peptide derivatives. Org. Lett. 2017, 19, 5288–5291. [Google Scholar] [CrossRef] [PubMed]
- Boutureira, O.; Bernardes, G.J.L. Advances in chemical protein modification. Chem. Rev. 2015, 115, 2174–2195. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.-Y.; Berti, F.; Adamo, R. Towards the next generation of biomedicines by site-selective conjugation. Chem. Soc. Rev. 2016, 45, 1691–1719. [Google Scholar] [CrossRef]
- Qvit, N.; Rubin, S.J.S.; Urban, T.J.; Mochly-Rosen, D.; Gross, E.C.R. Peptidomimetic therapeutics: Scientific approaches and opportunities. Drug Discov. Today 2017, 22, 454–462. [Google Scholar] [CrossRef]
- Blaskovich, M.A.T. Unusual amino acids in medicinal chemistry. J. Med. Chem. 2016, 59, 10807–10836. [Google Scholar] [CrossRef]
- Elander, R.P. Industrial production of β-lactam antibiotics. Appl. Microbiol. Biotechnol. 2003, 61, 385–392. [Google Scholar] [CrossRef]
- Ballard, A.; Narduolo, S.; Ahmad, H.O.; Keymer, N.I.; Asaad, N.; Cosgrove, D.A.; Buurma, N.J.; Leach, A.G. Racemisation in Chemistry and Biology. Chem. Eur. J. 2020, 26, 3661–3687. [Google Scholar] [CrossRef]
- Shirakawa, S.; Maruoka, K. Recent developments in asymmetric phase-transfer reactions. Angew. Chem. Int. Ed. 2013, 52, 4312–4348. [Google Scholar] [CrossRef]
- Hashimoto, T.; Maruoka, K. Recent development and application of chiral phase-transfer catalysts. Chem. Rev. 2007, 107, 5656–5682. [Google Scholar] [CrossRef]
- Bada, J.L. Racemization of Amino Acids. In Chemistry and Biochemistry of the Amino Acids; Barrett, G.C., Ed.; Springer: Dordrecht, Germany, 1985; pp. 399–414. [Google Scholar]
- King, T.A.; Mandrup Kandemir, J.; Walsh, S.J.; Spring, D.R. Photocatalytic methods for amino acid modification. Chem. Soc. Rev. 2021, 50, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Ohata, J.; Martin, S.C.; Ball, Z.T. Metal-Mediated Functionalization of Natural peptides and proteins: Panning for bioconjugation gold. Angew. Chem. Int. Ed. 2019, 58, 6176–6199. [Google Scholar] [CrossRef] [PubMed]
- de Gruyter, J.N.; Malins, L.R.; Baran, P.S. Residue-specific peptide modification: A chemist’s guide. Biochemistry 2017, 56, 3863–3873. [Google Scholar] [CrossRef] [PubMed]
- Faraggi, T.M.; Rouget-Virbel, C.; Rincón, J.A.; Barberis, M.; Mateos, C.; García-Cerrada, S.; Agejas, J.; de Frutos, O.; MacMillan, D.W.C. Synthesis of enantiopure unnatural amino acids by metallaphotoredox catalysis. Org. Process Res. Dev. 2021, 25, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Mani, V.S.; Narayanasamy, R. Conformationally constrained amino acids in peptide design. SSRN 2016. [Google Scholar] [CrossRef]
- Perez, J.J. Designing peptidomimetics. Curr. Top. Med. Chem. 2018, 18, 566–590. [Google Scholar] [CrossRef]
- Lenci, E.; Trabocchi, A. Peptidomimetic toolbox for drug discovery. Chem. Soc. Rev. 2020, 49, 3262–3277. [Google Scholar] [CrossRef]
- Robertson, N.S.; Spring, D.R. Using peptidomimetics and constrained peptides as valuable tools for inhibiting protein–protein interactions. Molecules 2018, 23, 959. [Google Scholar] [CrossRef]
- Morrison, C. Constrained peptides’ time to shine? Nat. Rev. Drug Discov. 2018, 17, 531–533. [Google Scholar] [CrossRef]
- Chowdhury, R. Eosin-Y/Cu(OAc)2-catalyzed aerobic oxidative coupling reactions of glycine esters in the dark. Org. Biomol. Chem. 2022, 20, 5387–5392. [Google Scholar] [CrossRef]
- Daggupati, R.V.; Malapaka, C. Cu(i)-Catalyzed amidation/imidation of N-arylglycine ester derivatives via C–N coupling under mild conditions. Org. Chem. Front. 2018, 5, 788–792. [Google Scholar] [CrossRef]
- Sun, B.; Wang, Y.; Li, D.; Jin, C.; Su, W. A copper/O2-mediated direct sp3 C–H/N–H cross-dehydrogen coupling reaction of acylated amines and N-aryl glycine esters. Org. Biomol. Chem. 2018, 16, 2902–2909. [Google Scholar] [CrossRef]
- Xiao, L.-J.; Zhu, Z.-Q.; Guo, D.; Xie, Z.-B.; Lu, Y.; Le, Z.-G. Copper-catalyzed cross-dehydrogenative-coupling reaction of N-arylglycine esters with imides or amides for synthesis of α-substituted α-amino acid esters. Synlett 2018, 29, 1659–1663. [Google Scholar] [CrossRef]
- Huo, C.; Dong, J.; Su, Y.; Tang, J.; Chen, F. Iron-catalyzed oxidative sp3 carbon-hydrogen bond functionalization of 3,4-dihydro-1,4-benzoxazin-2-ones. Chem. Commun. 2016, 52, 13341–13344. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, A.; Moazami, M.; Abaee, M.S.; Mirzaei, M. Ionic liquid-catalyzed synthesis of (1,4-benzoxazin-3-yl) malonate derivatives via cross-dehydrogenative-coupling reactions. Heterocycl. Commun. 2022, 28, 51–57. [Google Scholar] [CrossRef]
- Ashwood, M.S.; Cottrell, I.F.; Davies, A.J. Stereoselective synthesis of 2-(S)-(3,5-bis(trifluoromethyl)benzyloxy)-3-(S)-phenyl-1,4-oxazine. Tetrahedron Asymmetry 1997, 8, 957–963. [Google Scholar] [CrossRef]
- Trstenjak, U.; Ilaš, J.; Kikelj, D. Advances in the synthesis of morpholin-3-ones and morpholin-2-ones. Synthesis 2012, 44, 3551–3578. [Google Scholar] [CrossRef]
- Nelson, T.D. Synthesis of aprepitant. In Strategies and Tactics in Organic Synthesis; Harmata, M., Ed.; Academic Press: Amsterdam, The Netherlands, 2005; Volume 6, pp. 321–351. [Google Scholar]
- Ku, I.W.; Cho, S.; Doddareddy, M.R.; Jang, M.S.; Keum, G.; Lee, J.H.; Chung, B.Y.; Kim, Y.; Rhim, H.; Kang, S.B. Morpholin-2-one derivatives as novel selective T-type Ca2+ channel blockers. Bioorg. Med. Chem. Lett. 2006, 16, 5244–5248. [Google Scholar] [CrossRef]
- Gonzalez, A.Z.; Eksterowicz, J.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Duquette, J.; Fox, B.M.; Fu, J.; et al. Selective and potent morpholinone inhibitors of the MDM2-p53 protein-protein interaction. J. Med. Chem. 2014, 57, 2472–2488. [Google Scholar] [CrossRef]
- Mock, J.N.; Taliaferro, J.P.; Lu, X.; Patel, S.K.; Cummings, B.S.; Long, T.E. Haloenol pyranones and morpholinones as antineoplastic agents of prostate cancer. Bioorg. Med. Chem. Lett. 2012, 22, 4854–4858. [Google Scholar] [CrossRef]
- Bardiot, D.; Thevissen, K.; De Brucker, K.; Peeters, A.; Cos, P.; Taborda, C.P.; McNaughton, M.; Maes, L.; Chaltin, P.; Cammue, B.P.; et al. 2-(2-oxo-morpholin-3-yl)-acetamide derivatives as broad-spectrum antifungal agents. J. Med. Chem. 2015, 58, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Blake, T.R.; Waymouth, R.M. Organocatalytic ring-opening polymerization of morpholinones: New strategies to functionalized polyesters. J. Am. Chem. Soc. 2014, 136, 9252–9255. [Google Scholar] [CrossRef] [PubMed]
- Kashima, C.; Harada, K. Nucleophilic ring-opening reactions of morpholin-2-ones. A resolution of dl-(secondary-alkyl)amines. J. Org. Chem. 1989, 54, 789–792. [Google Scholar] [CrossRef]
- Basu, A.; Kunduru, K.R.; Katzhendler, J.; Domb, A.J. Poly(α-hydroxy acid)s and poly(α-hydroxy acid-co-α-amino acid)s derived from amino acid. Adv. Drug Deliv. Rev. 2016, 107, 82–96. [Google Scholar] [CrossRef]
- Cordeiro, R.A.; Serra, A.; Coelho, J.F.J.; Faneca, H. Poly(β-amino ester)-based gene delivery systems: From discovery to therapeutic applications. J. Control. Release 2019, 310, 155–187. [Google Scholar] [CrossRef]
- Iqbal, S.; Qu, Y.; Dong, Z.; Zhao, J.; Khan, A.R.; Rehman, S.; Zhao, Z. Poly(β-amino esters) based potential drug delivery and targeting polymer; an overview and perspectives (review). Eur. Polym. J. 2020, 141, 110097. [Google Scholar] [CrossRef]
- Zheng, Y.-N.; Zheng, H.; Li, T.; Wei, W.-T. Recent advances in copper-catalyzed C–N bond formation involving N-centered radicals. ChemSusChem 2021, 14, 5340–5358. [Google Scholar] [CrossRef]
- Park, Y.; Kim, Y.; Chang, S. Transition metal-catalyzed C–H amination: Scope, mechanism, and applications. Chem. Rev. 2017, 117, 9247–9301. [Google Scholar] [CrossRef]
- Bariwal, J.; Van der Eycken, E. C–N bond forming cross-coupling reactions: An overview. Chem. Soc. Rev. 2013, 42, 9283–9303. [Google Scholar] [CrossRef]
- Lamberth, C. Synthesis and applications of cyclic imides in agrochemistry. In Imides: Medicinal, Agricultural, Synthetic Applications and Natural Products Chemistry; Luzzio, F.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 335–352. [Google Scholar] [CrossRef]
- Li, J.J. Imide-containing synthetic drugs. In Imides: Medicinal, Agricultural, Synthetic Applications and Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2019; pp. 353–366. [Google Scholar] [CrossRef]
- Luzzio, F.A. Thalidomide and analogues. In Imides: Medicinal, Agricultural, Synthetic Applications and Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2019; pp. 367–429. [Google Scholar] [CrossRef]
- Zerilli, T.; Ocheretyaner, E. Apremilast (Otezla): A new oral treatment for adults with psoriasis and psoriatic arthritis. Pharm. Ther. 2015, 40, 495–500. [Google Scholar]
- In Top 200 Pharmaceuticals by Retail Sales in 2021, compiled and produced by M. H. Qureshi from the Njarðarson group (University of Arizona) as described by McGrath, N.A.; Brichacek, M.; Njardarson, J.T. A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Ed. 2010, 87, 1348–1349. Available online: https://njardarson.lab.arizona.edu/sites/njardarson.lab.arizona.edu/files/Top%20200%20Pharmaceuticals%202021V2.pdf (accessed on 8 June 2023). [CrossRef]
- In Top 200 Small Molecule Pharmaceuticals by Retail Sales in 2021, compiled and produced by M. H. Qureshi from the Njarðarson group (University of Arizona) as described by McGrath, N.A.; Brichacek, M.; Njardarson, J.T. A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Ed. 2010, 87, 1348–1349. Available online: https://njardarson.lab.arizona.edu/sites/njardarson.lab.arizona.edu/files/Top%20200%20Small%20Molecules%202021V3.pdf (accessed on 8 June 2023). [CrossRef]
- Mullard, A. Targeted protein degraders crowd into the clinic. Nat. Rev. Drug Discov. 2021, 20, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Crews, C.M. PROTACs: Past, present and future. Chem. Soc. Rev. 2022, 51, 5214–5236. [Google Scholar] [CrossRef] [PubMed]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Ishoey, M.; Chorn, S.; Singh, N.; Jaeger, M.G.; Brand, M.; Paulk, J.; Bauer, S.; Erb, M.A.; Parapatics, K.; Müller, A.C.; et al. Translation termination factor GSPT1 is a phenotypically relevant off-target of heterobifunctional phthalimide degraders. ACS Chem. Biol. 2018, 13, 553–560. [Google Scholar] [CrossRef]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef]
- Breugst, M.; Tokuyasu, T.; Mayr, H. Nucleophilic reactivities of imide and amide anions. J. Org. Chem. 2010, 75, 5250–5258. [Google Scholar] [CrossRef]
- Rohlmann, R.; Stopka, T.; Richter, H.; García Mancheño, O. Iron-catalyzed oxidative tandem reactions with TEMPO oxoammonium salts: Synthesis of dihydroquinazolines and quinolines. J. Org. Chem. 2013, 78, 6050–6064. [Google Scholar] [CrossRef]
- Johnston, A.D.; Asmussen, E.; Bowen, R.L. Substitutes for N-phenylglycine in adhesive bonding to dentin. J. Dent. Res. 1989, 68, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Behbehani, H.; Ibrahim, H.M. Synthetic strategy for pyrazolo[1,5-a]pyridine and pyrido[1,2-b]indazole derivatives through AcOH and O2-promoted cross-dehydrogenative coupling reactions between 1,3-dicarbonyl compounds and N-amino-2-iminopyridines. ACS Omega 2019, 4, 15289–15303. [Google Scholar] [CrossRef] [PubMed]
- Forkosh, H.; Vershinin, V.; Reiss, H.; Pappo, D. Stereoselective synthesis of optically pure 2-amino-2′-hydroxy-1,1′-binaphthyls. Org. Lett. 2018, 20, 2459–2463. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Yoshida, K.; Tokuyama, H. Acetic acid promoted metal-free aerobic carbon-carbon bond forming reactions at α-position of tertiary amines. Org. Lett. 2014, 16, 4194–4197. [Google Scholar] [CrossRef]
- Froehr, T.; Sindlinger, C.P.; Kloeckner, U.; Finkbeiner, P.; Nachtsheim, B.J. A metal-free amination of benzoxazoles–The first example of an iodide-catalyzed oxidative amination of heteroarenes. Org. Lett. 2011, 13, 3754–3757. [Google Scholar] [CrossRef]
- Ma, S.; Wu, L.; Liu, M.; Xu, X.; Huang, Y.; Wang, Y. Highly enantioselective aza-Michael addition reactions of 4-nitrophthalimide with α,β-unsaturated ketones. RSC Adv. 2013, 3, 11498–11501. [Google Scholar] [CrossRef]
- Narute, S.; Pappo, D. Iron phosphate catalyzed asymmetric cross-dehydrogenative coupling of 2-naphthols with β-ketoesters. Org. Lett. 2017, 19, 2917–2920. [Google Scholar] [CrossRef]
- Lee, A.; Betori, R.C.; Crane, E.A.; Scheidt, K.A. An enantioselective cross-dehydrogenative coupling catalysis approach to substituted tetrahydropyrans. J. Am. Chem. Soc. 2018, 140, 6212–6216. [Google Scholar] [CrossRef]
- World Health Organization. World Health Organization Model List of Essential Medicines: 22nd List 2019; World Health Organization: Geneva, Switzerland, 2019.
- Funes-Ardoiz, I.; Maseras, F. Oxidative coupling mechanisms: Current state of understanding. ACS Catal. 2018, 8, 1161–1172. [Google Scholar] [CrossRef]
- Allen, S.E.; Walvoord, R.R.; Padilla-Salinas, R.; Kozlowski, M.C. Aerobic copper-catalyzed organic reactions. Chem. Rev. 2013, 113, 6234–6458. [Google Scholar] [CrossRef]
- Wendlandt, A.E.; Suess, A.M.; Stahl, S.S. Copper-catalyzed aerobic oxidative C–H functionalizations: Trends and mechanistic insights. Angew. Chem. Int. Ed. 2011, 50, 11062–11087. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.-J.; Song, L.-J.; Yang, Y.-F.; Zhang, X.; Wiest, O.; Wu, Y.-D. Computational studies on the mechanism of the copper-catalyzed sp3-C–H cross-dehydrogenative coupling reaction. ChemPlusChem 2013, 78, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Young, J.G.; Onyebuagu, W. Synthesis and characterization of di-disubstituted phthalocyanines. J. Org. Chem. 1990, 55, 2155–2159. [Google Scholar] [CrossRef]
- Howell, B.A.; Dangalle, H.; Al-Omari, M. Thermal characteristics of precursors to a difunctional imide monomer. J. Therm. Anal. Calorim. 2011, 106, 129–137. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Ent. | Catalyst (mol%) | Solvent | Oxidant | [Phth] (M) | Additive | Yield 2 (%) |
|---|---|---|---|---|---|---|
| 1 | Cu(I)Cl | DCE | O2 | 0.1 | None | 23 |
| 2 | Cu(I)Cl | DMSO | air | 0.1 | None | Traces |
| 3 | Cu(I)Cl (10) | MeCN | air | 0.2 | None | 37 |
| 4 | Cu(I)Cl (10) | MeCN | O2 | 0.2 | None | 54 |
| 5 3 | Cu(I)Cl (10) | MeCN | O2 | 0.2 | None | 61 |
| 6 | Cu(I)Cl | MeCN | O2 | 0.1 | None | 70 |
| 7 4 | Cu(I)Cl (10) | MeCN | O2 | 0.1 | None | 70 |
| 8 | Cu(I)Cl | MeCN | O2 | 0.3 | None | 60 |
| 9 | Cu(I)Cl (30) | MeCN | O2 | 0.1 | None | 65 |
| 10 | Cu(II)Cl2 | MeCN | O2 | 0.1 | None | Traces |
| 11 | Cu(I)Br | MeCN | O2 | 0.1 | None | 49 |
| 12 | Fe(II)Cl2 | MeCN | O2 | 0.1 | None | 3 |
| 13 | Cu(II)(OAc)2 | MeCN | O2 | 0.1 | None | 42 |
| 14 5 | Cu(I)Cl (10) | MeCN | DTBP (2 equiv)/N2 | 0.1 | None | 40 |
| 15 5 | Cu(I)Cl (10) | MeCN | DTBP (3 equiv)/N2 | 0.1 | None | 55 |
| 16 | Cu(I)Cl | MeCN | DTBP (1 equiv)/N2 | 0.1 | None | 80 |
| 17 | Cu(I)Cl | MeCN | O2 | 0.1 | Et3N (1.0 equiv) | 0 |
| 18 | Cu(I)Cl | MeCN | O2 | 0.1 | Mol. sieves | Traces |
| 19 | Cu(I)Cl | MeCN | O2 | 0.1 | Pyridine (1.0 equiv) | 31 |
| 20 | Cu(I)Cl | MeCN | O2 | 0.1 | AcOH (1.0 equiv) | 87 |
| 21 | Cu(I)Cl | MeCN | O2 | 0.1 | AcOH (1.5 equiv) | 93 |
| 22 | None | MeCN | air | 0.1 | AcOH (1.5 equiv) | 80 |
| 23 | None | MeCN | N2 | 0.1 | AcOH (1.5 equiv) | 53 |
| 24 | none | MeCN | air | 0.1 | None | 8 |

| Entry | Morpholinone | Imide | Product (M) | Yield (%) | |
|---|---|---|---|---|---|
| 1 |  |  | 7a |  | 68 (93) |
| 2 | |  | 7b |  | 93 2 |
| 3 | |  | 7c |  | 27 |
| 4 | |  | 7d |  | 36 (85) |
| 5 | |  | 7e |  | 83 3 |
| 6 | |  | 7f |  | 71 (dr = 1:1) |
| 7 | |  | 7g |  | ND |
| 8 | |  | 7h |  | 36 (75) (at 80 °C) 4 |
| 9 | |  | 7i |  | ND |
| 10 | |  | 7j |  | ND |
| 11 |  |  | 7k |  | 75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faisca Phillips, A.M.; Pombeiro, A.J.L. A Mild and Sustainable Procedure for the Functionalization of Morpholin-2-Ones by Oxidative Imidation Reactions. Catalysts 2023, 13, 1072. https://doi.org/10.3390/catal13071072
Faisca Phillips AM, Pombeiro AJL. A Mild and Sustainable Procedure for the Functionalization of Morpholin-2-Ones by Oxidative Imidation Reactions. Catalysts. 2023; 13(7):1072. https://doi.org/10.3390/catal13071072
Chicago/Turabian StyleFaisca Phillips, Ana Maria, and Armando J. L. Pombeiro. 2023. "A Mild and Sustainable Procedure for the Functionalization of Morpholin-2-Ones by Oxidative Imidation Reactions" Catalysts 13, no. 7: 1072. https://doi.org/10.3390/catal13071072
APA StyleFaisca Phillips, A. M., & Pombeiro, A. J. L. (2023). A Mild and Sustainable Procedure for the Functionalization of Morpholin-2-Ones by Oxidative Imidation Reactions. Catalysts, 13(7), 1072. https://doi.org/10.3390/catal13071072