1. Introduction
Aerogels consisting of metal NPs emerge in the last decade as a very interesting class of electrocatalysts due to their 3D highly porous structure which facilitates the mass transfer and increases the concentration of active sites available per mass of catalyst [
1,
2,
3]. Among all aerogels [
4], cryoaerogels are especially interesting for electrocatalytic applications due to (i) the easy, fast, and versatile fabrication method (without chemical selectivity), (ii) the possibility of controlling the macroscopic shape of the gel, and (iii) the possibility of obtaining thin and homogeneous films of the material directly attached to the desired substrate/electrode [
5,
6]. By simply freezing and freeze-drying aqueous colloidal solutions of nanoparticles, cryoaerogels can be obtained, regardless of their surface chemistry, shape, or composition. The resulting self-supported porous nanocrystal-based cryogel thin films obtained by cryogelation showcase high specific surface areas and open porous structure, which convert them into better catalysts in comparison to the non-gelated nanoparticle colloid [
7,
8].
In the present work, we will focus on the study of the catalytic activity of cryogels from metallic NPs toward the ethanol oxidation reaction in alkaline media. Several works have reported outstanding electrocatalytic activities by using metal aerogels as catalysts for the ethanol oxidation reaction [
9,
10,
11]. This is especially relevant for the development of direct alcohol fuel cells (DAFCs) as an alternative to hydrogen fuel cells. Alcohols are easier to store and transport compared to hydrogen and they showcase higher energy densities. Methanol and ethanol are the most studied alcohols for the development of DAFCs and some prototypes are already commercially available. Ethanol is considered a more renewable alcohol compared to methanol, has a higher energy density (8030 Wh kg
−1 vs. 4820 Wh kg
−1), and is less toxic [
12,
13]. Also, ethanol is inexpensive and can be obtained from biomass through environmentally friendly methods, making DEFCs very promising as an alternative for the production of electrical energy.
Some of the challenges encountered in the development of DEFCs are (i) the high cost of the catalyst, which is normally relying on noble metals, (ii) the high catalyst loading needed, and (iii) the difficulties to completely oxidize the ethanol to CO
2, which requires breaking the C–C bond. Typical noble metals used for the EOR are Pt, Pd, Au, and Rh [
14]. However, due to their high price and easy poisoning by the reaction intermediates, it is necessary to further investigate other materials [
15]. The aim is to obtain less expensive electrocatalysts which can assist with the complete oxidation of ethanol reducing the overpotential. In addition, it has been reported that a good electrocatalyst for the EOR should be also able to adsorb OH species, preventing the electrode surface passivation caused by poisonous intermediates in the reaction. For that, one promising solution is the use of metal oxides as co-catalysts or supports for the noble metal, which can give rise to catalysts with higher catalytic activities with the additional advantage of reducing considerably the cost of the material [
16,
17,
18,
19]. The enhanced activity is normally explained through the occurrence of electronic and bifunctional mechanisms over the noble metal, which leads to an improvement in the electron transfer kinetics and higher tolerances against poisoning [
20,
21].
In particular, iron oxide is an inexpensive material and is largely used in many industrial processes such as the synthesis of ammonia and dehydrogenation of organic substrates, while there is a lot of active research dedicated to the study of iron oxide nanocatalysts for the degradation of organic dyes [
22], among other environmental applications [
23]. Their magnetic properties allow catalyst recovery by magnetic separation, interesting for example in the field of wastewater treatment, and can be selectively heated upon application of alternating magnetic fields, which can improve the yield of different reactions [
24]. For the ethanol oxidation reaction, some works have reported higher performances of noble metal catalysts mixed with iron oxide nanoparticles in comparison to the noble metal alone, either when the iron oxide is functioning as a co-catalysts or supports for the noble metal [
18,
19,
21,
25,
26]. All these studies point out that the catalysts made out of noble metals mixed with iron oxide nanoparticles could be superior materials for their use in direct ethanol fuel cells.
The aim of this work is to investigate alternative catalysts with better performances towards the ethanol oxidation reaction in alkaline media while reducing their overall cost. Mixtures of γ-Fe2O3 NPs with very few amount of Pt and Pd (1 wt%) are converted into cryogels with high surface areas and high porosity and deposited as thin films on modified ITO substrates. Then, the catalytic activity of these multicomponent (noble metal-metal oxide) cryogels towards the EOR is tested by means of cyclic voltammetry.
2. Results and Discussion
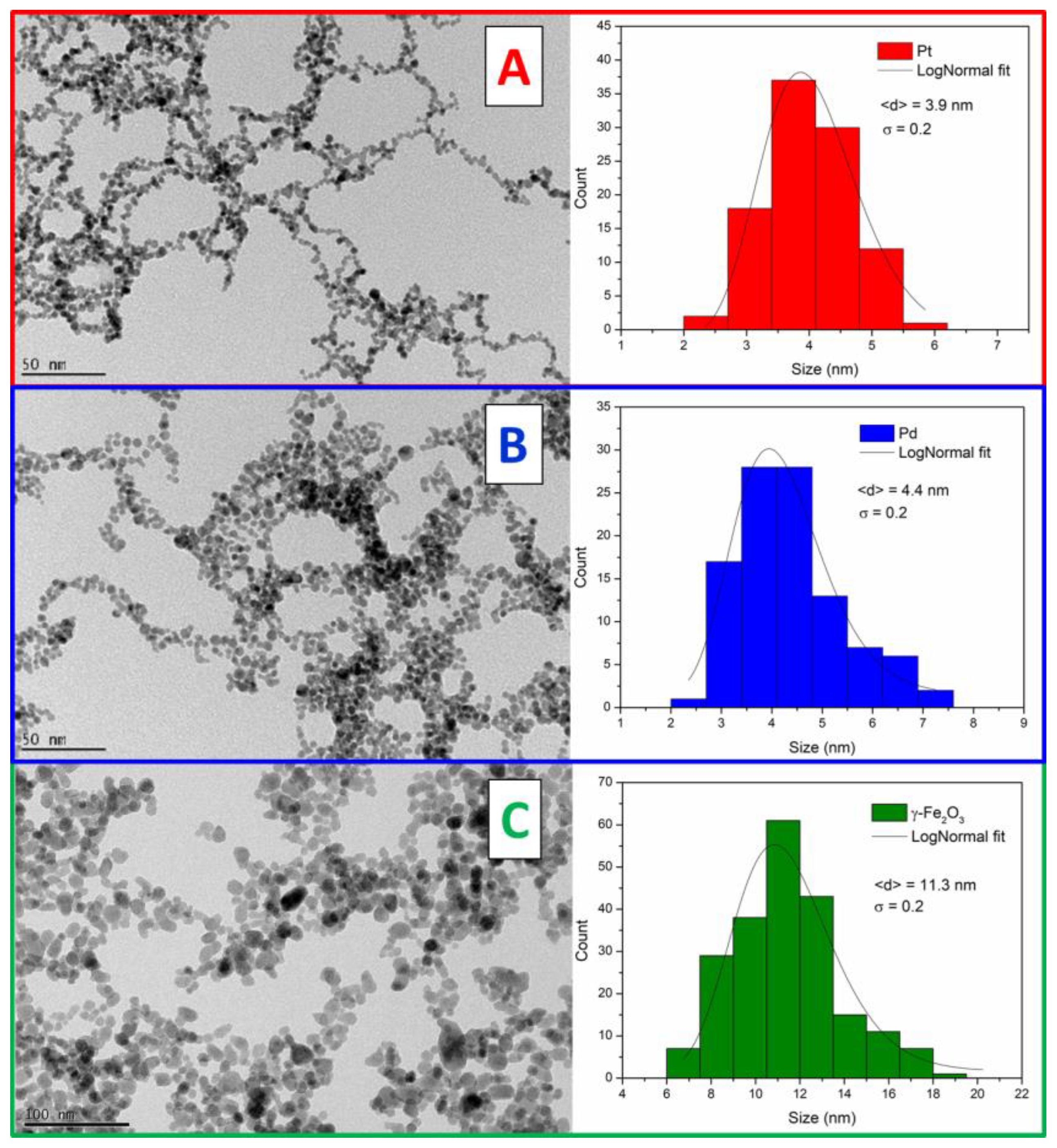
The morphology and size distribution of the synthesized Pt, Pd, and γ-Fe
2O
3 NPs are shown in
Figure 1. All samples have quasi-spherical shape, with sizes of 3.9, 4.4, and 11.3 nm, for the Pt, Pd and γ-Fe
2O
3 NPs, respectively. For the size distribution, more than 100 nanoparticles are measured and fitted to log-normal distribution. The arrangement of the iron oxide and the noble metal NPs in the cryogel structure can affect the catalytic activity, for example, by modifying the accessible catalytic active sites.
In that regard, it has been reported previously that the surface charge of the nanoparticles plays a significant role in the final homogeneity of the multicomponent cryogels upon freezing and subsequent freeze-drying [
7]. In particular, the cryogelation of nanoparticles with different sizes and with similar surface charges leads to local aggregation, while opposite charges lead to homogeneous distributions of the different materials within the cryogel [
7].
In
Table 1, DLS measurements of the different samples, pure Pt, Pd and γ-Fe
2O
3 NPs as well as the 1 wt% Pt or Pd/γ-Fe
2O
3 NPs mixtures are shown. γ-Fe
2O
3 has a hydrodynamic size of around 36 nm, in comparison to 6 nm for Pt and 9 nm for Pd (
Table 1,
Figure S1), being the most aggregated sample. Nevertheless, when 1 wt% Pt or Pd is mixed with the iron oxide, the hydrodynamic size remains unchanged, so no further aggregation is observed upon mixing. The zeta potential show that the surface charge of the γ-Fe
2O
3 nanoparticles (acidic conditions pH = 3) is positive while the surface charge of the noble metals is negative (acidic conditions pH = 5) due to the citrate surface ligands. Interestingly, upon mixing, the resulting zeta potential is still negative but has lower value (absolute value), suggesting that the Pt and Pd nanoparticles are arranging around the iron oxide nanoparticle surface. Therefore, a good homogeneous distribution of the Pt and Pd with the iron oxide is expected due to the opposite surface charges. TEM is performed for the mixed colloids (
Figure 2), confirming that there is no segregation of different components or big aggregates, better visible in the case of the Pt due to the higher difference in sizes compared to γ-Fe
2O
3 NPs. Also, it can be clearly seen that several Pt NPs arrange around one γ-Fe
2O
3 nanoparticle, the arrangement favored by the different surface charges, which in addition can prevent the further aggregation of the iron oxide nanoparticles. In the case of the Pd, a similar distribution of the nanoparticles as in the case of only iron oxide (
Figure 1C) is obtained.
Iron oxide superparamagnetic nanoparticles are desirable in this work because their net magnetization in the absence of an applied field is zero, so no further aggregation of the nanoparticles will occur and thus a better mixing with the noble metal is possible. For the magnetic characterization of the γ-Fe
2O
3 NPs, SQUID magnetometry is performed. As can be seen in
Figure S2, the saturation magnetization of the γ-Fe
2O
3 NPs is M
s (300 K) = 75 emu/g, value expected for bulk maghemite γ-Fe
2O
3 [
27]. In addition, the coercive field at 300 K is negligible (<20 Oe) and the blocking temperature of the sample is around 210 K, so at room temperature the sample used is in the superparamagnetic regime.
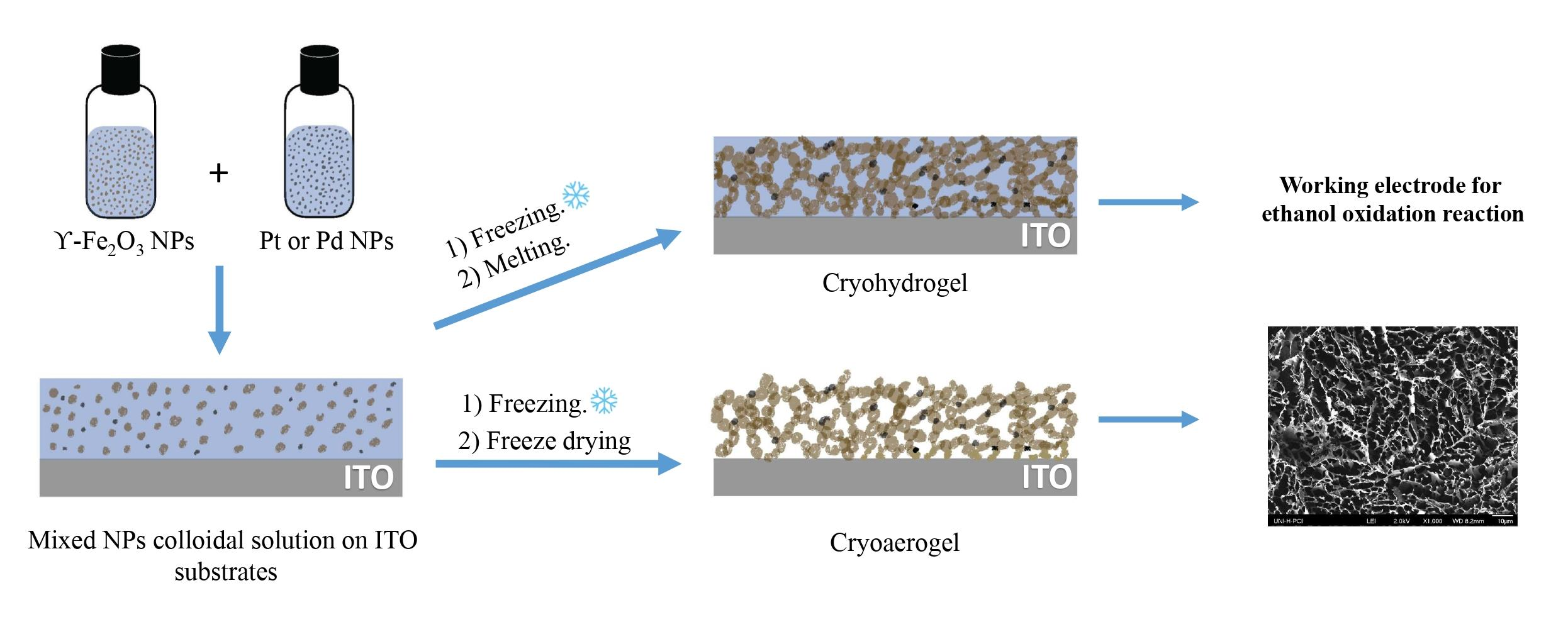
Cryogels are prepared by flash-freezing the colloidal NPs aqueous solution at cryogenic temperatures. On one hand, subsequent freeze-drying results in the formation of cryoaerogel structure. On the other hand, cryohydrogels are obtained by thawing the flash-frozen sample. It is important to highlight that to produce cryogels from colloidal NPs, a high concentration of the NPs in aqueous media is required (at least 0.1 vol%). For this, we synthesized Pt, Pd, and γ-Fe
2O
3 NPs in water. Afterward, Pt or Pd NPs were mixed with γ-Fe
2O
3 NPs to obtain a final concentration of 1 wt% of the noble metal. In the flash-freezing step, the NPs are pushed to fill the spaces in between the ice crystallites. In the freeze-drying step, upon sublimation, the frozen ice crystallites are turned into vapor state resulting in a highly porous self-supported cryoaerogel structure. Cryohydrogels obtained by gentle thawing have been found to retain the morphological characteristics of cryoaerogels [
8]. Here, we prepare cryogel thin films by spreading 3 µL of the mixed noble metal/γ-Fe
2O
3 colloidal NPs aqueous solution over an area of 5 × 5 mm
2 on the ITO/substrate surface, followed by dipping the substrate in isopentane/Liq. N
2 as freezing media. The thickness of the cryogel thin film is around 100 μm corresponding to the volume of NPs solution applied on the substrate. A top view of the cryoaerogel thin film structure of Pt/γ-Fe
2O
3 and Pd/γ-Fe
2O
3 can be seen in the SEM images (
Figure 3). Highly porous connected networks of mixed Pt/γ-Fe
2O
3 or Pd/γ-Fe
2O
3 are observed. The nanoparticles assemble together forming sheet-like structures, which are further connected to form dendritic porous self-supported networks.
As we have shown in our previous study [
8], the electrocatalytic performance of Pt, Pd, and Au towards EOR is significantly improved in the case of cryogel coatings compared to simply drop-casted NPs. The higher electrochemical active surface area (ECSA) and improved electrocatalytic activity can be attributed to the high porosity and connectivity of the structure. Moreover, controlling the freezing speed by freezing in isopentane at its melting point further enhances the electrocatalytic activity, by changing the thin film superstructure from lamellar shape (in case of liq. N
2) to cellular/dendritic shape (in case of isopentane). As well, in the previous study [
8], it was shown that the cryohydrogels exhibit the same electrocatalytic performance no matter if they were obtained by just thawing the cryogels or by freeze-drying the cryogels and rewetting of the obtained cryoaerogels. Additionally, cryohydrogels obtained from simple thawing have been found to exhibit the same morphological characteristics as freeze-dried cryoaerogels. As a result, the thawing procedure was adapted since a less complicated and faster steps are required for their application in the electrocatalysis of EOR.
In the present work, the electrocatalytic performance of cryohydrogels from pure Pt, Pd, and 1 wt% Pt/γ-Fe2O3 or Pd/γ-Fe2O3 towards EOR under alkaline conditions was studied and compared by means of cyclic voltammetry. The mechanism of the overall ethanol oxidation reaction on the surface of noble metals is usually explained by two main pathways as follows:
In alkaline media, the reaction starts first with a chemisorption step to form CH
3CH
2OH
ad on the noble metal surface followed by a dehydrogenation step to form CH
3CHO
ad/CH
3CO
ad. The reaction will then undergo one of the following two pathways. C1 pathway, where complete oxidation of ethanol takes place, involves C–C bond cleavage, formation of CO intermediates, and finally formation of carbonates and/or CO
2 as end products (with 12 e
− transfer). On the other hand, partial oxidation takes place when CH
3CHO
ad/CH
3CO
ad is directly oxidized into acetaldehyde (with 2 e
− transfer) or acetate (with 4 e
− transfer) through C2 pathway. The challenge here is to facilitate the C1 pathway which allows efficient conversion of the ethanol chemical energy into electricity. The CO adsorbed intermediates are commonly described as surface poisons, efficient removal of these poisons facilitates C1 pathway [
28].
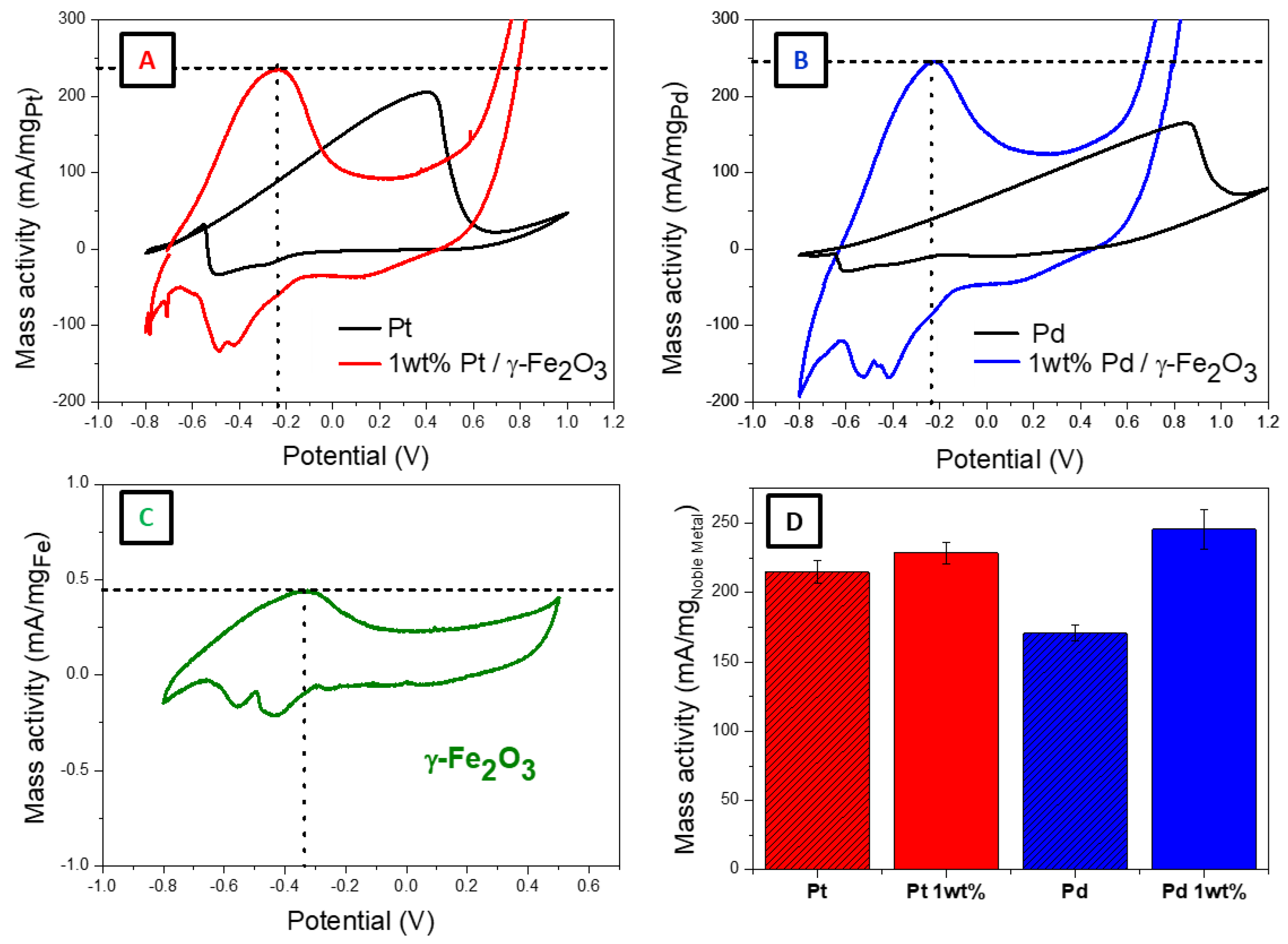
Cyclic voltammetry measurements are done for the cryohydrogels of pure noble metal and the mixtures with γ-Fe
2O
3 (1 wt% of the noble metal).
Figure 4 displays the cyclic voltammograms obtained in the presence of 0.25 M EtOH/1 M KOH at a scan rate of 50 mV/s. For all cryohydrogels, a strong anodic peak corresponding to EtOH oxidation on the surface is observed. A less intense peak is observed in the backward scan corresponding to the removal of the CO poisonous species which are produced during the reaction. The onset and the peak potentials for the electrocatalytic reaction give insights into the reaction kinetics, as they shift to more negative values means better reaction kinetics. Although for EOR the theoretical thermodynamic potential is relatively low (less than 0.1 V) [
29], practically, higher oxidation potentials are required to reach the peak current specially in the case of Pt and Pd based-catalysts. This overpotential can be attributed to the low oxophilicity of Pt and Pd which hinders the first chemisorption step of ethanol on the catalyst surface. So, lowering the overpotentials is a crucial step to obtain efficient electrocatalysis of EOR [
30].
The onset potentials, defined as the potential value at mass activity of 100 mA/mg
metal [
31], was significantly decreased. Pure Pt and Pd cryoaerogels showed onset potentials of −0.18 and +0.26 V, respectively, however for Pt/γ-Fe
2O
3 and Pd/γ-Fe
2O
3 the values are −0.52 and −0.50 V, respectively. The peak potential of Pt and Pd cryoaerogels are 0.40 and 0.85 V respectively. However, Pt/γ-Fe
2O
3 and Pd/γ-Fe
2O
3 showed much lower values of peak potentials, −0.24 and −0.23 V respectively. This means that much better reaction kinetics for ethanol oxidation was observed in the case of mixing the noble metals with γ-Fe
2O
3. This can be explained by the bifunctional mechanism of γ-Fe
2O
3. On one hand, the more oxophilic behavior of γ-Fe
2O
3 than Pt and Pd noble metals facilitates the dehydrogenation step of the adsorbed ethanol on the noble metal surface. As a result, the peak potential is greatly decreased in the case of mixed noble metal/iron oxide structures compared to pure noble metals. On the other hand, the electronic structure of the noble metal NPs is modified when they are in close contact with iron oxide NPs [
32] which weakens the bond between the noble metal and the CO intermediates. Also, the oxophilic iron oxide NPs increase the tolerance against poisonous CO intermediates by facilitating their oxidation driving the reaction into C1 pathway [
19].
The peak current in the forward and backward scan can be denoted as I
f and I
b respectively. As the ratio of I
f/I
b increases, a higher tolerance of the electrocatalyst against poisoning by the reaction intermediates is obtained. For example, the I
f/I
b ratio in the case of pure Pt is found to be 6, which is considered a significantly high value compared to Pt and Pd NPs based catalysts found in the literature for the same reaction conditions [
33]. As can be seen in
Figure 4A,B the I
f/I
b ratio is high in the case of pure and mixed cryohydrogels. The ratio is even higher in the case of mixed samples in comparison to the pure ones. These observations mean that the materials that we present here exhibit much higher tolerance against surface poisoning with the reaction intermediates, products, and by-products than other materials in literature [
33]. One reason can be attributed to the cryohydrogel structure itself providing a very high surface area available for the reaction. Another reason is the bifunctional effect of mixing γ-Fe
2O
3 with the noble metal.
The peak current intensity at the peak potential was normalized to the noble metal mass in order to calculate the mass activity. The mass activities of different noble metal-based structures are presented in
Figure 4D and
Table 2. The Pt/γ-Fe
2O
3 mixture exhibits nearly the same mass activity as pure Pd. An increase in the mass activity of around 44% in the case of Pd/γ-Fe
2O
3 mixture is observed compared to the pure Pd cryohydrogel.
The electrochemical surface area (ECSA) is a key parameter in analyzing the electrocatalytic activity for the oxidation of different alcohols, which can be determined by integrating the hydrogen adsorption peak from the backward scan of the CV curves following Equation (1) [
8]:
where
is the amount of charge exchange during the adsorption of hydrogen atoms on the metal, Q is the charge required to oxidize a monolayer of hydrogen on the metal surface, usually assumed constant,
is the scan rate (50 mV/s) and
is the noble metal loading on the measured electrode [
19,
34]. For the calculation, typical values of Q are assumed for Pt and Pd, which are 420 μC/cm
2 and 405 μC/cm
2 [
8,
29]. The estimated values can be found in
Table 2. The mass activity is obtained dividing the current by the Pt or Pd mass in each case. The specific activity is derived from the ECSA and the mass activity and represent the intrinsic activity of the catalyst [
35].
To shed light on the stability of the cryogel catalysts, we have performed TEM measurements for the 1 wt% Pt/γ-Fe
2O
3 and Pd/γ-Fe
2O
3 samples before and after the CV measurements (
Figure S3). We observed no major changes in the cryogel structure, the NPs keep their origin arrangement within the cryogel thin sheet with no sign of size or shape changes. In addition, in
Figure S4 we show CV measurements of the samples for 10 cycles. We observed very small differences in the mass activities between the different cycles.
Mixing noble metals with γ-Fe
2O
3 in the cryogel structure for ethanol oxidation has several advantages. First, the bifunctional mechanism, in which OH species are adsorbed on the surface of the oxophilic γ-Fe
2O
3. This adsorbed OH species facilitates the dehydrogenation of CH
3CH
2OH
ad [
19]. Moreover, γ-Fe
2O
3 facilitates the removal of poisonous CO intermediates from the surface of the noble metal by the same mechanism which increases selectivity towards C1 pathway [
19]. Due to the fact that noble metals are not highly oxophilic as iron, pure noble metals can’t facilitate this mechanism effectively [
29]. Second, the electronic structure of the noble metal is modified when the γ-Fe
2O
3 is in close contact, a charge transfer from the noble metal to the iron occurs resulting in less electronic density on the noble metal [
32]. As a result, the noble metal bond to the CO intermediates is weakened which facilitates the dehydrogenation step. Finally, as a result of the mentioned effects, the overall electrocatalytic performance represented by the mass activity, the electrochemically active surface area (ECSA) and the oxidation potential are greatly enhanced (
Table 2).
Summarizing, the mechanism of the EtOH oxidation reaction on the surface of noble metal/γ-Fe2O3 cryohydrogels can be explained by a first step of adsorption of the CO species via O on the noble metal surface active sites followed by facilitated dehydrogenation step by the OH species adsorbed on the γ-Fe2O3 surface. Afterwards, the reaction will go through C1 or C2 pathway as explained earlier. The more favorable pathway for the reaction on the surface of our new materials is not confirmed yet in this study, however C1 pathway is highly expected due to the observed better reaction kinetics, efficient removal of poisonous CO intermediates and the high ECSA achieved by the porous cryohydrogel structure.
3. Materials and Methods
3.1. Materials
Dihydrogen hexachloroplatinate (IV) hexahydrate and iron(II) chloride nonahydrate (99%) were purchased from Alfa Aesar, Kandel, Germany. Isopropanol (ACS reagent, ≥99.5%), iron(III) chloride hexahydrate (99.0%), hydrogen peroxide (35%), ammonium hydroxide (ACS reagent, 28–30%), toluene (ACS reagent, ≥99.5%), potassium hydroxide (ACS reagent, ≥85%), ethanol (for spectroscopy Uvasol®), hydrochloric acid (37 wt% in water, 99.99%), 2-methylbutane (isopentane, ReagentPlus, ≥99.5%), sodium borohydride (ReagentPlus, 99%), 3-aminopropyltriethoxysilane (APTES, ≥98%) and palladium(II)chloride (99%)were purchased from Sigma-Aldrich, Darmstadt, Germany. Trisodium citrate dihydrate (99%) was purchased from ABCR, Karlsruhe, Baden-Württemberg, Germany. All chemicals were used as received. Millipore water system from Sartorius (Göttingen, Germany) was used to obtain highly pure Milli-Q water with resistivity value of 18 MΩ.cm. Indium tin oxide (ITO) glass substrates (ITO surface resistance: 12 Ω/square, thickness: 1.1 mm) was purchased from VisionTek Systems, Ltd., Cheshire, UK.
3.2. Synthesis of Pt NPs
Citrate capped Pt NPs of particle size around 5 nm was prepared following the synthesis procedure by Bigall et al. [
36]. Briefly, to 464 mL Milli-Q water, 36.2 mL of 0.2 wt% dihydrogen hexachloroplatinate (IV) hexahydrate was added and brought to boil. Upon boiling, 11.6 mL of 1 wt% trisodium citrate dihydrate was added followed by quick injection of 5.5 mL of 0.076 wt% freshly prepared sodium borohydride after 30 s. The color of the solution changes from yellow to dark brown. The reaction was stopped after 1 min (by simply stopping the stirring of the solution) and the resulted colloidal solution was stored under ambient conditions.
3.3. Synthesis of Pd NPs
According to the same previous procedure [
36], in 50 mL 0.06 M HCl, 0.2565 g palladium(II)chloride was dissolved. To 500 mL Milli-Q water, 10 mL of the PdCl
2 stock solution was added. The prepared solution was heated to 80 °C followed by addition of 11.6 mL of 1 wt% trisodium citrate dehydrate. After 30 s, 5.5 mL of 0.076 wt% freshly prepared sodium borohydride was quickly injected. The color of the solution changes from yellow to dark brown. The reaction was stopped after 1 min and the resulted colloidal solution was stored under ambient conditions.
3.4. Concentration of Pt and Pd NPs Solutions
The volume of each colloidal solution (Pt and Pd NPs) was reduced from around 500 to 50 mL using solvent-resistance ultrafiltration cell (Merck Millipore, Darmstadt, Germany) provided with 10 kDa polyethersulfon membrane (Sartorius, Göttingen, Germany). The colloidal solutions were filled into the ultrafiltration cell and filtered under continuous stirring and pressure of 5 bar until the final volume is around 50 mL. Additionally, the obtained 50 mL of the colloidal NPs solution of Pt and Pd were further concentrated to 1 mL using Amicon® Ultra centrifuge filters from Sigma-Aldrich, Darmstadt, Germany. The 50 mL solution was distributed equally into 4 centrifuge tube filters and centrifuged for 4–6 min at 3000 rcf. The volume was reduced to 1 mL in each tube, then the 4 mL were collected all in one tube and diluted with 0.01 wt% citrate solution to 10 mL. The 10 mL colloidal NPs solution was concentered again to 1 mL by centrifugation for 3 min at 3000 rcf. The last step was repeated 3 times to wash the NPs with citrate solution.
3.5. Synthesis of Iron Oxide γ-Fe2O3 NPs
Maghemite NPs are synthesized by the modified Massart coprecipitation method of a mixture of Fe(II) and Fe(III) salts in alkaline medium [
37,
38]. Briefly, magnetite (Fe
3O
4) NPs are prepared by dissolving 24.4 g of iron(III) chloride hexahydrate (FeCl
3·6H
2O, Sigma Aldrich, Darmstadt, Germany) in 43 mL of Milli-Q water under magnetic stirring and 10.8 g of iron(II) chloride nonahydrate (FeCl
2·9H
2O, Alfa Aesar, Kandel, Germany) in 45 mL of milliQ water. After that, both solutions are mixed and 400 mL of milliQ water are added, yielding a total volume of around 500 mL. 75 mL of an alkaline medium consisting of NH
4OH 28% (Sigma Aldrich, Darmstadt, Germany) is placed into a 1 L glass container and under magnetic stirring the mixture of the chlorides is added, obtaining a black solution. To obtain NPs of around 11 nm, a slow addition (0.2 mL/s) of the aqueous solution into the base is necessary. The solution is heated up to 90 °C and kept at that temperature during 2 h, and after this time, the solution is left to cool down. The black product is washed three times with distilled water using a permanent magnet to remove the supernatant. Maghemite (γ-Fe
2O
3) NPs are obtained by oxidizing the magnetite (Fe
3O
4) NPs previously synthesised.
An acid treatment is carried out to oxidize the Fe3O4 to γ-Fe2O3. For that 300 mL of nitric acid (HNO3, 2 M, Sigma Aldrich, Darmstadt, Germany) is added to the washed product and magnetically stirred for 15 min. After that, the supernatant is removed, and 75 mL aqueous solution of iron (II) nitrate 9-hydrate (Fe(NO3)3, 1 M) and 130 mL of distilled water are added. The mixture is heated until boiling temperature while magnetically stirring and kept in these conditions for 30 min. When it cools down, the supernatant is removed by magnetic decantation again, and 300 mL of HNO3 (2 M) is added. After 15 min of magnetic stirring, the supernatant is removed, and the brown product is washed three times with acetone. For that, acetone is added to the solution and mixed. After that, the acetone is discarded by magnetic decantation and more acetone is added. This procedure is repeated three times and the nanoparticles are redispersed in distilled water. Finally, the remaining acetone is removed by means of a rotary evaporator and the sample is concentrated to a total volume of around 50 mL.
3.6. Preparation of the Substrates
To prepare the ITO-coated glass substrates for coating, pieces of 150 × 300 mm were cut and cleaned in isopropanol followed by water for 10 min each. The cleaned substrates were then stirred in a mixture of water: hydrogen peroxide: ammonia in a ratio of 5:1:1 at 70 °C for 2 h. Afterwards, the substrates were washed with water 3–4 times followed by washing with isopropanol and finally dried with a compressed air stream. The substrates were then stirred for another 2 h at the same temperature in a solvent mixture consisting of 1% v/v APTES in toluene. Finally, the substrates were rinsed in toluene to get rid of excess APTES, dried with compressed air stream and then stored at 80 °C until use.
3.7. Preparation of Cryogels
The concentration of the NPs stock solutions used was 30, 57 and 64 g/L for Pt, Pd and γ-Fe2O3, respectively. The NPs colloidal solutions were mixed to obtain 1 wt% Pt or Pd with γ-Fe2O3 NPs. The NPs stock solutions were stored at 4 °C and remained stable at least up to 3 months. To prepare the cryogels, a square area of 5 × 5 mm was defined with a scotch tape (Scotch 3M, Neuss, Germany) on the modified ITO coated side of the substrates. With a micropipette, a volume of 3 µL of the NPs stock solution was applied in the specified area. Isopentane/liq. N2 freezing medium was prepared as follows, around 100 mL of isopentane was placed in a dewar, then liq. N2 was added gradually with gentle stirring with a spatula until isopentane was completely frozen. The mixture was left for few minutes until the isopentane reaches its melting point. The substrate was quickly dipped into isopentane/liq. N2 (−160 °C) and left for 10 min. The substrates were stored at −20 °C overnight and finally freeze-dried at 0.076 mbar overnight using (Christ Alpha 1–2 LD+, Profcontrol, Schönwalde-Glien, Germany) freeze dryer. For the characterization of the materials, cryoaerogels were used, while for the electrochemical measurements the materials were re-wetted and used in form of cryohydrogels.
3.8. Characterization
Transmission electron microscopy (TEM) was performed using a Tecnai G2 F20 TMP (from FEI, Hillsboro, OR, USA) equipped with a 200 kV field emission gun. The samples were prepared on a carbon coated Cu grid (mesh width 300 μm) either by drop casting in case of the colloidal NPs solutions or by touching the surface in case of the cryoaerogels. Image J (1.53 K) software is used to determine the size distribution of the NPs by measuring more than 100 nanoparticles. Scanning electron microscopy (SEM) was performed using a JEOL JSM 6700F electron microscope, Peabody, Massachusetts, US operated at 2 kV. The samples were prepared by sticking the substrate on a 25 mm SEM holder by adhesive carbon tape (plano) and glued by silver ink. The hydrodynamic size of the nanoparticles in suspension and their surface charge were measured by dynamic light scattering (DLS) in a Zetasizer ZSP, Malvern Analytical, Malvern, UK. Disposable cuvettes and disposable folded capillary cells (DTS 1070, Malvern Analytical, Malvern, UK) were used for size and zeta potential measurements respectively. All samples were diluted and measured three times. The concentration of the nanoparticle suspensions was determined by atomic absorption spectroscopy (Varian AA140 Atomic Absorption Spectrometer from Varian, Inc., Palo Alto, CA, USA) for Pt, Pd and Fe.
SQUID magnetometry (Superconducting Quantum Interference Device) from Quantum Design, California, US was used to investigate the magnetic properties of the iron oxide nanoparticles. For that, 20 μL of the sample is dropped in a cotton and placed in a gelatine capsule, stuffing the capsule with more cotton to prevent the sample to move during the measurements. The capsule is then inserted and immobilized in a straw and measured. Hysteresis cycles are measured at 5 K and 300 K from −3 T to 3 T and the thermal dependency of the magnetization is studied by means of ZFC-FC curves, measuring as the temperature increases from 5 K to 300 K and applying a field of 0.01 T (100 Oe).
3.9. Electrocatalytic Measurements
Cyclic voltammetry was carried out using a Metrohm Autolab PGSTA T204 potentiostat from Metrohm, Herisau, Switzerland, in a three-electrode setup consisting of Ag/AgCl reference electrode, platinum wire counter electrode and ITO substrate as the working electrode. CV measurements were performed in a mixture of 0.25 M EtOH/1 M KOH at a scan rate of 50 mV/s and a potential range between −0.8 and 1.0 V.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}