
S–Modified MXene as a Catalyst for Accelerated Tetracycline Hydrochloride Electrocatalytic Degradation via ·OH and Active Chlorine Triggering Promotion


Abstract
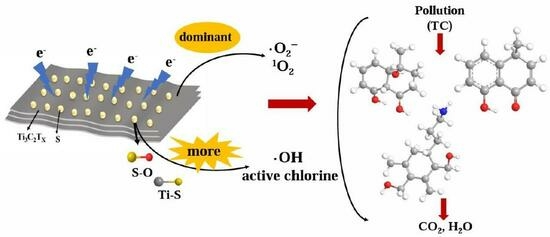
:
1. Introduction
2. Results and Discussion
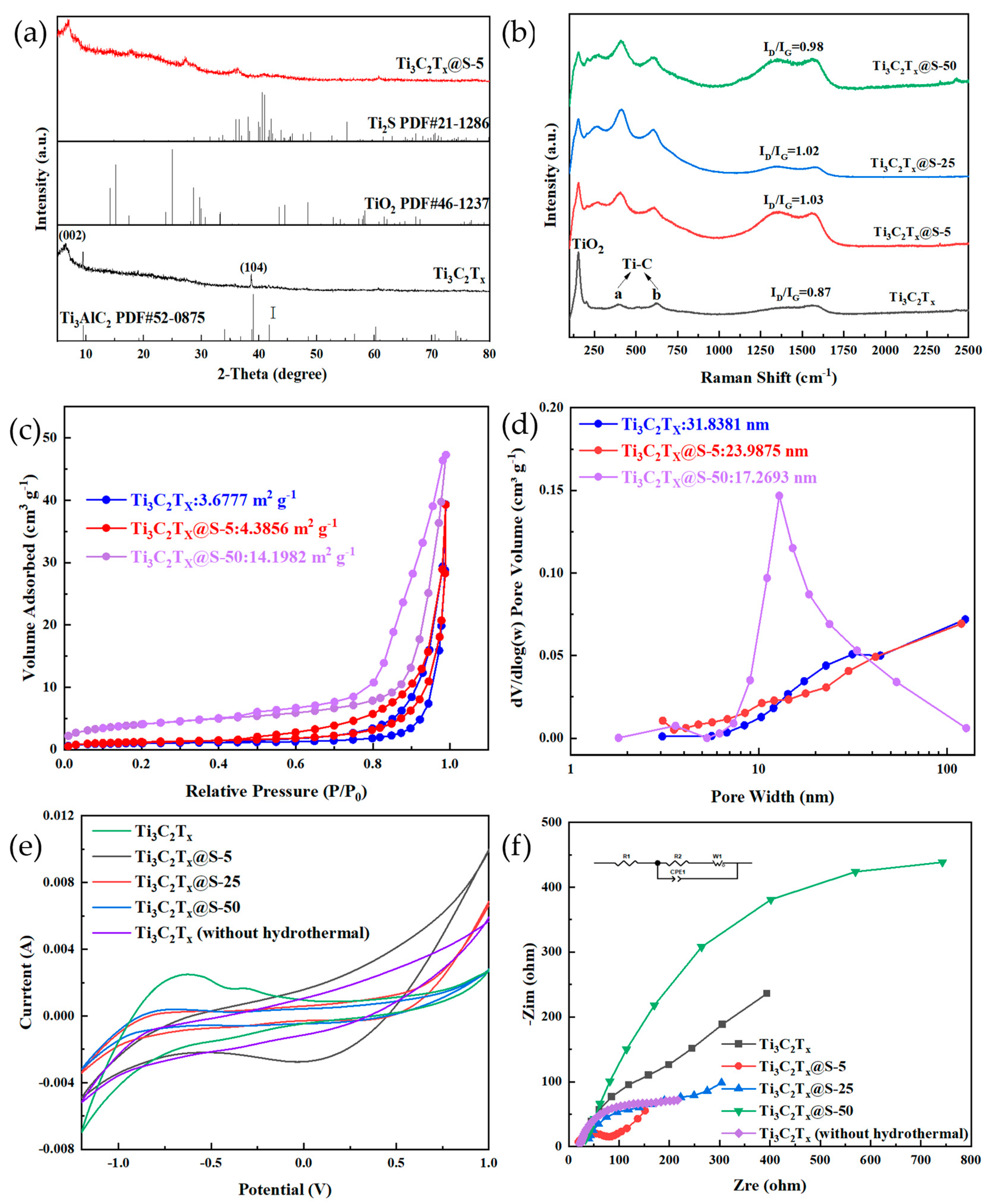
2.1. Characterization Results
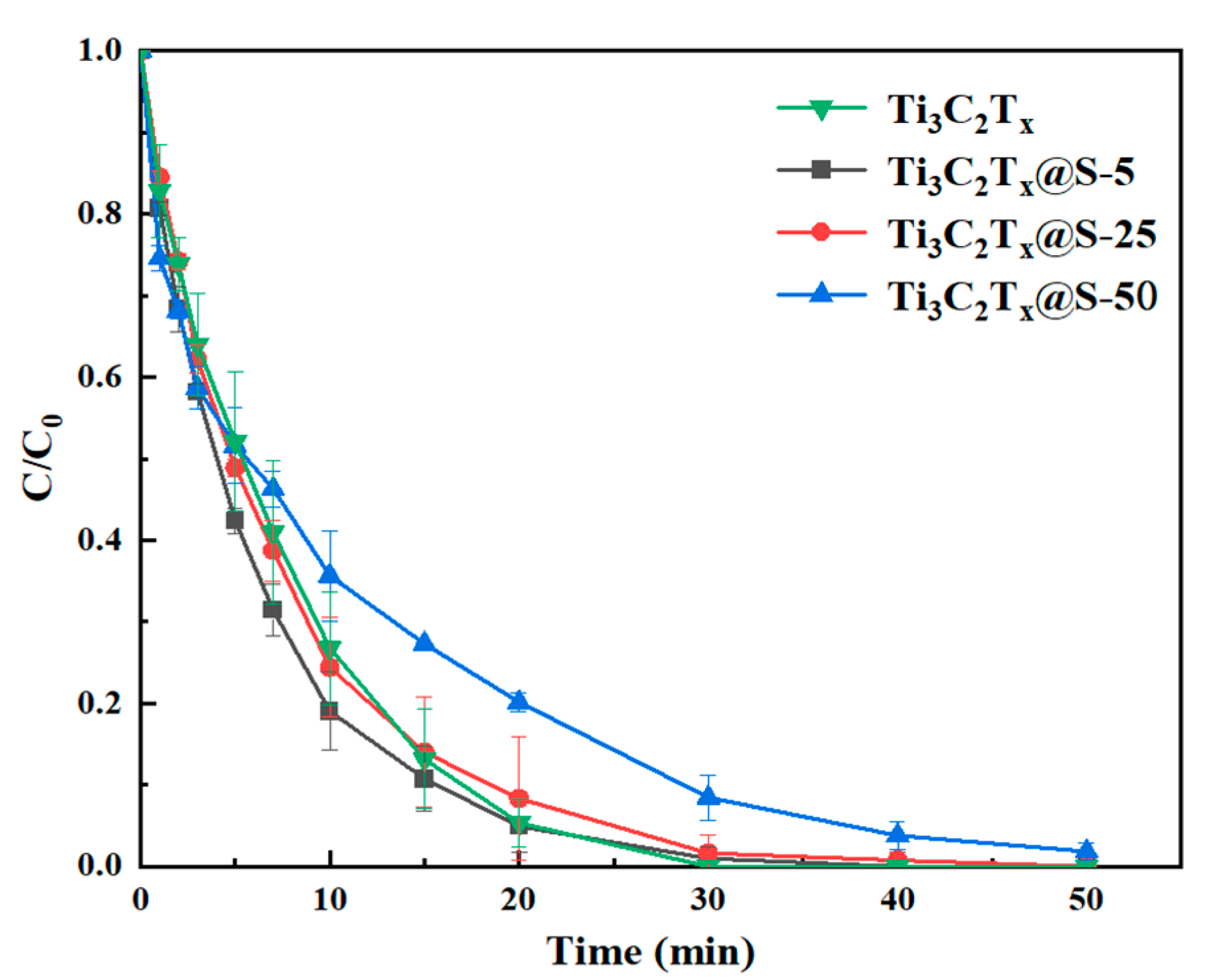
2.2. Catalytic Capacity
2.3. Active Substance Determination
2.4. Identification of the Degradation Pathway
3. Experimental
3.1. Reagents and Materials
3.2. Material Synthesis
3.2.1. Synthesis of Ti3C2Tx MXene without Hydrothermal Nanosheets
3.2.2. Synthesis of Ti3C2Tx@S Nanosheets
3.3. Characterization
3.4. Catalytic Experiment and Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, M.; Zeng, G.M.; Huang, D.L.; Lai, C.; Xu, P.; Zhang, C.; Liu, Y. Hydroxyl radicals based advanced oxidation processes (AOPs) for remediation of soils contaminated with organic compounds: A review. Chem. Eng. J. 2016, 284, 582–598. [Google Scholar] [CrossRef]
- Wei, C.L.; Tao, Y.; An, Y.L.; Tian, Y.; Zhang, Y.C.; Feng, J.K.; Qian, Y.T. Recent Advances of Emerging 2D MXene for Stable and Dendrite-Free Metal Anodes. Adv. Funct. Mater. 2020, 30, 2004613. [Google Scholar] [CrossRef]
- Liu, H.J.; Dong, B. Recent advances and prospects of MXene-based materials for electrocatalysis and energy storage. Mater. Today Phys. 2021, 20, 100469. [Google Scholar] [CrossRef]
- Naguib, M.; Mochalin, V.N.; Barsoum, M.W.; Gogotsi, Y. 25th Anniversary Article: MXenes: A New Family of Two-Dimensional Materials. Adv. Mater. 2014, 26, 992–1005. [Google Scholar] [CrossRef]
- Morales-Garcia, A.; Calle-Vallejo, F.; Illas, F. MXenes: New Horizons in Catalysis. ACS Catal. 2020, 10, 13487–13503. [Google Scholar] [CrossRef]
- Cao, F.C.; Zhang, Y.; Wang, H.Q.; Khan, K.; Tareen, A.K.; Qian, W.J.; Zhang, H.; Agren, H. Recent Advances in Oxidation Stable Chemistry of 2D MXenes. Adv. Mater. 2022, 34, 29. [Google Scholar] [CrossRef]
- Tang, Q.; Zhou, Z.; Shen, P.W. Are MXenes Promising Anode Materials for Li Ion Batteries? Computational Studies on Electronic Properties and Li Storage Capability of Ti3C2 and Ti3C2X2 (X = F, OH) Monolayer. J. Am. Chem. Soc. 2012, 134, 16909–16916. [Google Scholar] [CrossRef]
- Zheng, X.; Wang, Z.L.; Li, J.J.; Wei, L.M. Binder-free S@Ti3C2Tx sandwich structure film as a high-capacity cathode for a stable aluminum-sulfur battery. Sci. China-Mater. 2022, 65, 1463–1475. [Google Scholar] [CrossRef]
- Zhao, X.Q.; Liu, M.; Chen, Y.; Hou, B.; Zhang, N.; Chen, B.B.; Yang, N.; Chen, K.; Li, J.L.; An, L.A. Fabrication of layered Ti3C2 with an accordion-like structure as a potential cathode material for high performance lithium-sulfur batteries. J. Mater. Chem. A 2015, 3, 7870–7876. [Google Scholar] [CrossRef]
- Zhang, F.; Zhou, Y.; Zhang, Y.; Li, D.C.; Huang, Z.C. Facile synthesis of sulfur@titanium carbide Mxene as high performance cathode for lithium-sulfur batteries. Nanophotonics 2020, 9, 2025–2032. [Google Scholar] [CrossRef]
- Borchert, H. X-ray Diffraction. In Solar Cells Based on Colloidal Nanocrystals; Springer International Publishing: Cham, Switzerland, 2014; pp. 79–94. [Google Scholar]
- Du, C.; Wu, J.; Yang, P.; Li, S.Y.; Xu, J.M.; Song, K.X. Embedding S@TiO2 nanospheres into MXene layers as high rate cyclability cathodes for lithium-sulfur batteries. Electrochim. Acta 2019, 295, 1067–1074. [Google Scholar] [CrossRef]
- Liang, C.H.; Meng, G.W.; Wang, G.Z.; Zhang, L.D.; Zhang, S.Y. Catalytic synthesis of Ti2S nanofibers. Chem. Mat. 2001, 13, 2150–2153. [Google Scholar] [CrossRef]
- Bao, W.Z.; Xie, X.Q.; Xu, J.; Guo, X.; Song, J.J.; Wu, W.J.; Su, D.W.; Wang, G.X. Confined Sulfur in 3D MXene/Reduced Graphene Oxide Hybrid Nanosheets for Lithium-Sulfur Battery. Chem. Eur. J. 2017, 23, 12613–12619. [Google Scholar] [CrossRef] [PubMed]
- Adomavičiūtė-Grabusovė, S.; Ramanavičius, S.; Popov, A.; Šablinskas, V.; Gogotsi, O.; Ramanavičius, A. Selective Enhancement of SERS Spectral Bands of Salicylic Acid Adsorbate on 2D Ti3C2Tx-Based MXene Film. Chemosensors 2021, 9, 223. [Google Scholar] [CrossRef]
- Balaji, S.S.; Karnan, M.; Anandhaganesh, P.; Tauquir, S.M.; Sathish, M. Performance evaluation of B-doped graphene prepared via two different methods in symmetric supercapacitor using various electrolytes. Appl. Surf. Sci. 2019, 491, 560–569. [Google Scholar] [CrossRef]
- Fang, Y.Z.; Hu, R.; Liu, B.Y.; Zhang, Y.Y.; Zhu, K.; Yan, J.; Ye, K.; Cheng, K.; Wang, G.L.; Cao, D.X. MXene-derived TiO2/reduced graphene oxide composite with an enhanced capacitive capacity for Li-ion and K-ion batteries. J. Mater. Chem. A 2019, 7, 5363–5372. [Google Scholar] [CrossRef]
- Yang, Y.L.; Huang, Z.; Liu, Y.Y.; Guo, D.; Zhang, Q.; Hong, J.M. Mechanism exploration of highly conductive Ni-metal organic frameworks/reduced graphene oxide heterostructure for electrocatalytic degradation of paracetamol: Functions of metal sites, organic ligands, and rGO basement. J. Colloid Interface Sci. 2023, 629, 667–682. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.M.; Zheng, J.H.; Nai, J.W.; Jin, C.B.; Yuan, H.D.; Sheng, O.W.; Liu, Y.J.; Fang, R.Y.; Zhang, W.K.; Huang, H.; et al. Atomic Sulfur Covalently Engineered Interlayers of Ti3C2 MXene for Ultra-Fast Sodium-Ion Storage by Enhanced Pseudocapacitance. Adv. Funct. Mater. 2019, 29, 10. [Google Scholar] [CrossRef]
- Riyanto; Sahroni, I.; Bindumadhavan, K.; Chang, P.Y.; Doong, R.A. Boron Doped Graphene Quantum Structure and MoS2 Nanohybrid as Anode Materials for Highly Reversible Lithium Storage. Front. Chem. 2019, 7, 12. [Google Scholar] [CrossRef]
- Ni, J.F.; Fu, S.D.; Wu, C.; Maier, J.; Yu, Y.; Li, L. Self-Supported Nanotube Arrays of Sulfur-Doped TiO2 Enabling Ultrastable and Robust Sodium Storage. Adv. Mater. 2016, 28, 2259–2265. [Google Scholar] [CrossRef]
- Wu, S.S.; Feng, Y.F.; Wu, K.D.; Jiang, W.Q.; Xue, Z.F.; Xiong, D.P.; Chen, L.; Feng, Z.Y.; Wen, K.H.; Li, Z.Y.; et al. Mxene Ti3C2 generated TiO2 nanoparticles in situ and uniformly embedded in rGO sheets as high stable anodes for potassium ion batteries. J. Alloy. Compd. 2023, 930, 12. [Google Scholar] [CrossRef]
- Li, L.; Jiang, G.X.; An, C.H.; Xie, Z.J.; Wang, Y.J.; Jiao, L.F.; Yuan, H.T. Hierarchical Ti3C2@TiO2 MXene hybrids with tunable interlayer distance for highly durable lithium-ion batteries. Nanoscale 2020, 12, 10369–10379. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.G.; Liu, Y.Y.; Li, R.R.; Li, J.L. Two-dimensional Ti3C2Tx@S as cathode for room temperature sodium-sulfur batteries. Ionics 2019, 25, 5373–5382. [Google Scholar] [CrossRef]
- Tang, X.H.; Li, D.Y. Sulfur-doped highly ordered TiO2 nanotubular arrays with visible light response. J. Phys. Chem. C 2008, 112, 5405–5409. [Google Scholar] [CrossRef]
- Huo, X.G.; Wang, X.X.; Li, Z.Y.; Liu, J.; Li, J.L. Two-dimensional composite of D-Ti3C2Tx@S@TiO2 (MXene) as the cathode material for aluminum-ion batteries. Nanoscale 2020, 12, 3387–3399. [Google Scholar] [CrossRef]
- Li, J.B.; Yan, D.; Hou, S.J.; Li, Y.Q.; Lu, T.; Yao, Y.F.; Pan, L.K. Improved sodium-ion storage performance of Ti3C2Tx MXenes by sulfur doping. J. Mater. Chem. A 2018, 6, 1234–1243. [Google Scholar] [CrossRef]
- Wang, A.N.; Chen, Y.X.; Liu, L.; Liu, X.; Wang, Z.L.; Zhang, Y. Sulfur nanoparticles/Ti3C2Tx MXene with an optimum sulfur content as a cathode for highly stable lithium-sulfur batteries. Dalton Trans. 2021, 50, 5574–5581. [Google Scholar] [CrossRef]
- Sires, I.; Brillas, E. Remediation of water pollution caused by pharmaceutical residues based on electrochemical separation and degradation technologies: A review. Environ. Int. 2012, 40, 212–229. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhou, Y.L.; Yu, Y.B.; Chen, B.Y.; Hong, J.M. Exploring catalytic performance of boron-doped graphene electrode for electrochemical degradation of acetaminophen. Appl. Surf. Sci. 2020, 508, 13. [Google Scholar] [CrossRef]
- Wang, L.Q.; Li, R.Y.; Zhang, Y.M.; Gao, Y.X.; Xiao, X.; Zhang, Z.W.; Chen, T.; Zhao, Y. Tetracycline degradation mechanism of peroxymonosulfate activated by oxygen-doped carbon nitride. RSC Adv. 2023, 13, 6368–6377. [Google Scholar] [CrossRef]
- Zhi, D.; Qin, J.L.; Zhou, H.; Wang, J.B.; Yang, S.X. Removal of tetracycline by electrochemical oxidation using a Ti/SnO2-Sb anode: Characterization, kinetics, and degradation pathway. J. Appl. Electrochem. 2017, 47, 1313–1322. [Google Scholar] [CrossRef]
- Eftekhari, A. Electrocatalysts for hydrogen evolution reaction. Int. J. Hydrog. Energy 2017, 42, 11053–11077. [Google Scholar] [CrossRef]
- Kadeer, K.; Reheman, A.; Maimaitizi, H.; Talifu, D.; Tursun, Y.; Abulizi, A. Preparation of rGO/AgCl QDs and its enhanced photoelectrocatalytic performance for the degradation of Tetracycline. J. Am. Ceram. Soc. 2019, 102, 5342–5352. [Google Scholar] [CrossRef]
- Tang, S.F.; Zhao, M.Z.; Yuan, D.L.; Li, X.; Zhang, X.Y.; Wang, Z.B.; Jiao, T.F.; Wang, K. MnFe2O4 nanoparticles promoted electrochemical oxidation coupling with persulfate activation for tetracycline degradation. Sep. Purif. Technol. 2021, 255, 8. [Google Scholar] [CrossRef]
- Liu, C.Y.; Fu, D.F.; Li, H.H. Behaviour of multi-component mixtures of tetracyclines when degraded by photoelectrocatalytic and electrocatalytic technologies. Environ. Technol. 2012, 33, 791–799. [Google Scholar] [CrossRef]
- Shao, C.; Zhang, J.; Liu, Y.; Jiang, Y.; Jia, Y.; Li, G.; Sun, Z. Effective degradation of tetracycline by Pd/AG/ITO electrode: Electrode preparation, characterization, kinetics, degradation mechanism and toxicity assessment. J. Environ. Chem. Eng. 2023, 11, 110344. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Pang, D.; Song, H.; Zhang, S. Simultaneous electrochemical degradation of tetracycline and metronidazole through a high-efficiency and low-energy-consumption advanced oxidation process. Chemosphere 2022, 292, 133469. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, Y. Spinel CuxCo1−xMn2O4 electrode for effectively cleaning organic wastewater via electrocatalytic oxidation. Sep. Purif. Technol. 2021, 258, 118024. [Google Scholar] [CrossRef]
- Sun, W.; Sun, Y.; Shah, K.J.; Chiang, P.-C.; Zheng, H. Electrocatalytic oxidation of tetracycline by Bi-Sn-Sb/γ-Al2O3 three-dimensional particle electrode. J. Hazard. Mater. 2019, 370, 24–32. [Google Scholar] [CrossRef]
- Neta, P.; Huie, R.E.; Ross, A.B. Rate Constants for Reactions of Inorganic Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. [Google Scholar] [CrossRef]
- Wang, N.; Ma, W.J.; Ren, Z.Q.; Du, Y.C.; Xu, P.; Han, X.J. Prussian blue analogues derived porous nitrogen-doped carbon microspheres as high-performance metal-free peroxymonosulfate activators for non-radical-dominated degradation of organic pollutants. J. Mater. Chem. A 2018, 6, 884–895. [Google Scholar] [CrossRef]
- Zhang, S.W.; Gao, H.H.; Xu, X.T.; Cao, R.Y.; Yang, H.C.; Xu, X.J.; Li, J.X. MOF-derived CoN/N-C@SiO2 yolk-shell nanoreactor with dual active sites for highly efficient catalytic advanced oxidation processes. Chem. Eng. J. 2020, 381, 12. [Google Scholar] [CrossRef]
- Sun, P.; Liu, H.; Feng, M.B.; Guo, L.; Zhai, Z.C.; Fang, Y.S.; Zhang, X.S.; Sharma, V.K. Nitrogen-sulfur co-doped industrial graphene as an efficient peroxymonosulfate activator: Singlet oxygen-dominated catalytic degradation of organic contaminants. Appl. Catal. B-Environ. 2019, 251, 335–345. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, Q.H.; Zhang, J.; Xiang, M.H.; Zhou, Y.H.; Chen, Z.Y.; Chen, Y.B.; Yao, T.T. Broad spectrum driven Y doped BiO2−x for enhanced degradation of tetracycline: Synergy between singlet oxygen and free radicals. Appl. Surf. Sci. 2023, 607, 14. [Google Scholar] [CrossRef]
- Chen, S.; Hu, T.F.; Zhang, Q.; Hong, J.M. UV-annealing synthesis of sulfur-doped graphene for bisphenol A electrocatalytic degradation. Appl. Surf. Sci. 2021, 569, 11. [Google Scholar] [CrossRef]
- Stan, S.D.; Daeschel, M.A. 5,5-Dimethyl-2-pyrrolidone-N-oxyl formation in electron spin resonance studies of electrolyzed NaCl solution using 5,5-dimethyl-1-pyrroline-N-oxide as a spin trapping agent. J. Agric. Food Chem. 2005, 53, 4906–4910. [Google Scholar] [CrossRef]
- Xiao, C.M.; Zhang, M.; Wang, C.H.; Yan, X.; Zhang, H.; Chen, S.S.; Yao, Y.Y.; Qi, J.W.; Zhang, S.T.; Li, J.S. 2D metal-organic framework derived hollow Co/NC carbon sheets for peroxymonosulfate activation. Chem. Eng. J. 2022, 444, 11. [Google Scholar] [CrossRef]
- Chen, X.Q.; Huang, Z.; Liu, Y.Y.; Zhang, Q.; Hong, J.M. Cu-HAB/GO composite with superior active surface area/sites and conductivity properties for the electrocatalytic degradation of tetracycline hydrochloride. J. Phys. Chem. Solids 2023, 172, 15. [Google Scholar] [CrossRef]
- Martinez-Huitle, C.A.; Brillas, E. Decontamination of wastewaters containing synthetic organic dyes by electrochemical methods: A general review. Appl. Catal. B-Environ. 2009, 87, 105–145. [Google Scholar] [CrossRef]
- Chen, Z.B.; Fu, M.; Yuan, C.X.; Hu, X.L.; Bai, J.W.; Pan, R.; Lu, P.; Tang, M. Study on the degradation of tetracycline in wastewater by micro-nano bubbles activated hydrogen peroxide. Environ. Technol. 2022, 43, 3580–3590. [Google Scholar] [CrossRef]
- Li, X.B.; Fan, S.S.; Jin, C.J.; Gao, M.C.; Zhao, Y.G.; Guo, L.; Ji, J.Y.; She, Z.L. Electrochemical degradation of tetracycline hydrochloride in sulfate solutions on boron-doped diamond electrode: The accumulation and transformation of persulfate. Chemosphere 2022, 305, 10. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Liu, C.; Wu, Y.; Xia, M.Z.; Wang, F.Y. Porous P, Fe-doped g-C3N4 nanostructure with enhanced photo-Fenton activity for removal of tetracycline hydrochloride: Mechanism insight, DFT calculation and degradation pathways. Chemosphere 2022, 291, 11. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.Y.; Huang, Z.; Liu, Y.Y.; Hong, J.M.; Zhang, Q. New electron transfer bridge construction between Ni3(HTTP)2 and graphene oxide layers for enhanced electrocatalytic oxidation of tetracycline hydrochloride. Chem. Eng. J. 2023, 451, 13. [Google Scholar] [CrossRef]
- Wang, H.L.; Chen, T.H.; Chen, D.; Zou, X.H.; Li, M.X.; Huang, F.J.; Sun, F.W.; Wang, C.; Shu, D.B.; Liu, H.B. Sulfurized oolitic hematite as a heterogeneous Fenton-like catalyst for tetracycline antibiotic degradation. Appl. Catal. B-Environ. 2020, 260, 13. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Ti3C2Tx (At%) | Ti3C2Tx@S–5 (At%) |
|---|---|---|
| F | 49.44 | 30.90 |
| Al | 4.68 | 1.94 |
| S | 0.00 | 0.99 |
| Cl | 1.82 | 2.06 |
| Ti | 44.06 | 64.11 |
| Total | 100.00 | 100.00 |
| Materials | Reaction Conditions | Initial Concentration of TC | Efficiency | Ref. |
|---|---|---|---|---|
| rGO/AgCl QDs | 0.1 mol/L Na2SO4 | 20 mg L−1 | 20.73%@120 min | [34] |
| EO/PS–MnFe2O4 | J = 20 mA cm−2, pH = 4.5, 2 mmol L−1 PS | 25 mg L−1 | 86.23%@60 min | [35] |
| TiO2 Manotubes | V = 1.0 V, air aeration | 15 mg L−1 | 50%@180 min | [36] |
| Pd/AG/ITO | I = 8 mA, 0.2 mol L−1 NaCl | 12 mg L−1 | 85.21%@120 min | [37] |
| TiO2−x–10 | J = 10 mA cm−2, 0.075 mol L−1 Na2SO4 | 50 mg L−1 | 92.6%@60 min | [38] |
| Spinel CuxCo1−xMn2O4 | J = 20 mA cm−2, pH = 3, 0.05 mol·L−1 Na2SO4 | 20 mg L−1 | 91.3%@120 min | [39] |
| Bi–Sn–Sb/γ–Al2O3 | J = 0.1 A·cm−2, pH = 5.9 | 100 mg L−1 | 85.9%@180 min | [40] |
| Ti3C2Tx@S–5 | J = 20 mA cm−2, pH = 6, 17 mmol L−1 NaCl | 10 mg L−1 | 100%@30 min | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, F.; Huang, Z.; Liu, Y.-Y.; Zhang, Q.; Chang, C.-T. S–Modified MXene as a Catalyst for Accelerated Tetracycline Hydrochloride Electrocatalytic Degradation via ·OH and Active Chlorine Triggering Promotion. Catalysts 2023, 13, 1237. https://doi.org/10.3390/catal13091237
Zhang F, Huang Z, Liu Y-Y, Zhang Q, Chang C-T. S–Modified MXene as a Catalyst for Accelerated Tetracycline Hydrochloride Electrocatalytic Degradation via ·OH and Active Chlorine Triggering Promotion. Catalysts. 2023; 13(9):1237. https://doi.org/10.3390/catal13091237
Chicago/Turabian StyleZhang, Fan, Zhi Huang, Yan-Ying Liu, Qian Zhang, and Chang-Tang Chang. 2023. "S–Modified MXene as a Catalyst for Accelerated Tetracycline Hydrochloride Electrocatalytic Degradation via ·OH and Active Chlorine Triggering Promotion" Catalysts 13, no. 9: 1237. https://doi.org/10.3390/catal13091237
APA StyleZhang, F., Huang, Z., Liu, Y. -Y., Zhang, Q., & Chang, C. -T. (2023). S–Modified MXene as a Catalyst for Accelerated Tetracycline Hydrochloride Electrocatalytic Degradation via ·OH and Active Chlorine Triggering Promotion. Catalysts, 13(9), 1237. https://doi.org/10.3390/catal13091237