

Mechanistic and Compositional Aspects of Industrial Catalysts for Selective CO2 Hydrogenation Processes



Abstract
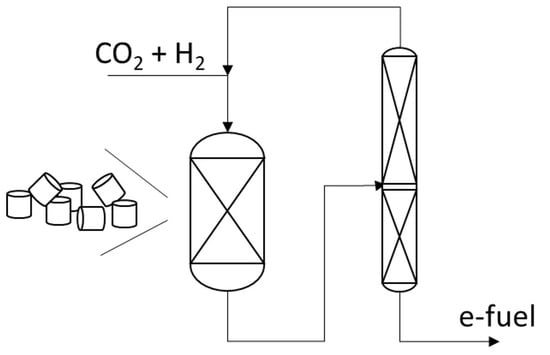
:
1. Introduction
2. The Water-Gas Shift and Reverse Water-Gas Shift Equilibrium and the Catalysts
2.1. High-Temperature Water-Gas Shift (HTWGS) Catalysts
2.2. Low-Temperature Water-Gas Shift (LTWGS) Catalysts
2.3. Medium-Temperature Water-Gas Shift (MTWGS) Catalysts
2.4. Other Industrial Water-Gas Shift Catalysts
2.5. The Reverse Water-Gas Shift Process and Catalysts
3. The Methanol Synthesis and the Catalysts
3.1. Conventional Methanol Synthesis
3.2. Methanol Synthesis from CO2 through Previous Reverse Water-Gas Shift
3.3. Direct Methanol Synthesis from CO2
4. The Methanation Processes and the Catalysts
4.1. Low-Temperature Methanation
4.2. High-Temperature Methanation for Substitute Natural Gas Synthesis
4.3. CO2 Methanation
5. The Production of Higher Hydrocarbons
5.1. Conventional Low-Temperature Fischer-Tropsch (LTFT) Processes
5.2. Conventional High-Temperature Fischer-Tropsch (HTFT) and Medium-Temperature Fischer-Tropsch (MTFT) Processes
5.3. Fischer-Tropsch Processes Using Electrolytic Hydrogen and Captured CO2
6. Mechanistic Aspects of CO2 Hydrogenations
6.1. The Adsorption and Activation of Hydrogen
6.2. The Adsorption and Activation of CO
6.3. The Adsorption and Activation of CO2
7. Mechanistic Aspects and the Role of the Different Catalyst’s Components
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nemmour, A.; Inayat, A.; Janajreh, I.; Ghenai, C. Green hydrogen-based E-fuels (E-methane, E-methanol, E-ammonia) to support clean energy transition: A literature review. Int. J. Hydrogen Energy 2023, 48, 29011–29033. [Google Scholar] [CrossRef]
- Cui, L.; Liu, C.; Yao, B.; Edwards, P.P.; Xiao, T.; Cao, F. A review of catalytic hydrogenation of carbon dioxide: From waste to hydrocarbons. Front. Chem. 2022, 10, 1037997. [Google Scholar] [CrossRef] [PubMed]
- Aasberg-Petersen, K.; Dybkjær, I.; Ovesen, C.V.; Schjødt, N.C.; Sehested, J.; Thomsen, S.G. Natural gas to synthesis gas—Catalysts and catalytic processes. J. Nat. Gas Sci. Eng. 2011, 3, 423–459. [Google Scholar] [CrossRef]
- Boretti, A.; Banik, B.K. Advances in Hydrogen Production from Natural Gas Reforming. Adv. Energy Sustain. Res. 2021, 2, 2100097. [Google Scholar] [CrossRef]
- Dai, F.; Zhang, S.; Luo, Y.; Wang, K.; Liu, Y.; Ji, X. Recent Progress on Hydrogen-Rich Syngas Production from Coal Gasification. Processes 2023, 11, 1765. [Google Scholar] [CrossRef]
- José Juan Bolívar Caballero, J.J.; Zaini, I.N.; Yan, W. Reforming processes for syngas production: A mini-review on the current status, challenges, and prospects for biomass conversion to fuels. Appl. Energy Comb. Sci. 2022, 10, 100064. [Google Scholar] [CrossRef]
- Busca, G. Metal catalysts in hydrogenation and dehydrogenation reactions. In Heterogeneous Catalytic Materials; Busca, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 297–343. [Google Scholar]
- Gao, J.; Liu, Q.; Gu, F.; Liu, B.; Zhong, Z.; Su, F. Recent advances in methanation catalysts for the production of synthetic natural gas. RSC Adv. 2015, 5, 22759–22776. [Google Scholar] [CrossRef]
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on methanation—From fundamentals to current projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Available online: https://www.grandviewresearch.com/industry-analysis/catalyst-market (accessed on 30 October 2023).
- Newsome, D.S. The Water-Gas Shift Reaction. Catal. Rev. 1980, 21, 275–318. [Google Scholar] [CrossRef]
- Baraj, E.; Ciahotný, K.; Hlinčík, T. The water gas shift reaction: Catalysts and reaction mechanism. Fuel 2021, 288, 119817. [Google Scholar] [CrossRef]
- Pattabathula, V.; Richardson, J. Introduction to Ammonia Production. Chem. Eng. Prog. 2016, 112, 69–75. [Google Scholar]
- ThyssenKrupp: Hydrogen Key for Any Refinery. Available online: https://d2zo35mdb530wx.cloudfront.net/_binary/UCPthyssenkruppUhde/6e7007a8-2bd4-47c1-b215-8a4d268c80ed/brochure-hydrogen_scr.pdf (accessed on 30 October 2023).
- Hydrogen, Linde. Available online: https://www.linde-engineering.com/en/images/H2_1_1_e_12_150dpi_NB_tcm19-4258.pdf (accessed on 30 October 2023).
- Ratnasamy, C.; Wagner, J.P. Water Gas Shift Catalysis. Catal. Rev. 2009, 51, 325–440. [Google Scholar] [CrossRef]
- Clariant, Catalysts and Adsorbents for Syngas. Available online: https://www.clariant.com/en/Business-Units/Catalysts/Syngas-Catalysts (accessed on 30 October 2023).
- Lukashuk, L.; van de Water, L.G.A.; van Dijk, H.A.J.; Cobden, P.D.; Dodds, D.L.; Hyde, T.I.; Watson, M.J. A new application of the commercial high temperature water gas shift catalyst for reduction of CO2 emissions in the iron and steel industry: Lab-scale catalyst evaluation. Int. J. Hydrogen Energy 2021, 46, 39023–39035. [Google Scholar] [CrossRef]
- Lee, D.W.; Lee, M.S.; Lee, J.Y.; Kim, S.; Eom, H.-J.; Moon, D.Y.; Lee, K.Y. The review of Cr-free Fe-based catalysts for high-temperature water-gas shift reactions. Catal. Today 2013, 210, 2–9. [Google Scholar] [CrossRef]
- Sourav, S.; Wachs, I.E. Cr-Free, Cu Promoted Fe Oxide-Based Catalysts for High-Temperature Water-Gas Shift (HT-WGS) Reaction. Catalysts 2020, 10, 305. [Google Scholar] [CrossRef]
- Available online: https://www.topsoe.com/our-resources/knowledge/our-products/catalysts/sk-501-flextm (accessed on 30 October 2023).
- Pal, D.B.; Chand, R.; Upadhyay, S.N.; Mishra, P.K. Performance of water gas shift reaction catalysts: A review. Renew. Sustain. Energy Rev. 2018, 93, 549–556. [Google Scholar] [CrossRef]
- Amadeo, N.E.; Laborde, M.A. Hydrogen production from the low-temperature water-gas shift reaction: Kinetics and simulation of the industrial reactor. Int. J. Hydrogen Energy 1995, 20, 949–956. [Google Scholar] [CrossRef]
- Available online: https://www.clariant.com/en/Solutions/Products/2019/02/15/09/47/ShiftMax-217 (accessed on 30 October 2023).
- Süd Chemie, General Catalyst Catalogue. Available online: www.sudchemie.com (accessed on 12 December 2011).
- Available online: https://www.topsoe.com/our-resources/knowledge/our-products/catalysts/lk-823 (accessed on 30 October 2023).
- Available online: https://www.topsoe.com/our-resources/knowledge/our-products/catalysts/lk-853-fencetm (accessed on 30 October 2023).
- Haldor Topsøe, Brochure “Leading Edge Performance LTS Catalysts”. Available online: http://www.topsoe.com (accessed on 12 December 2011).
- KATALKOJM Low Temperature Shift Catalysts. Available online: https://matthey.com/products-and-markets/chemicals/low-temperature-shift-catalysts (accessed on 30 October 2023).
- Ozcan, O.; Akın, A.N. Methanol steam reforming kinetics using a commercial CuO/ZnO/Al2O3 catalyst: Simulation of a reformer integrated with HT-PEMFC system. Int. J. Hydrogen Energy 2023, 48, 22777–22790. [Google Scholar] [CrossRef]
- Peppley, B.A.; Amphlett, J.C.; Kearns, L.M.; Mann, R.F. Methanol–steam reforming on Cu/ZnO/Al2O3. Part 1: The reaction network. Appl. Catal. A Gen. 1999, 179, 21–29. [Google Scholar] [CrossRef]
- Madon, R.J.; Nagel, P. Low Temperature Water Gas Shift Catalyst. US Patent 0102278, 11 June 2009. [Google Scholar]
- Wilkinson, S.K.; van de Water, L.G.A.; Miller, B.; Simmons, M.J.H.; Stitt, E.H.; Watson, M.J. Understanding the generation of methanol synthesis and water gas shift activity over copper-based catalysts—A spatially resolved experimental kinetic study using steady and non-steady state operation under CO/CO2/H2 feeds. J. Catal. 2016, 337, 208–220. [Google Scholar] [CrossRef]
- Lacerda de Oliveira Campos, B.; Herrera Delgado, K.; Wild, S.; Studt, F.; Pitter, S.; Sauer, J. Surface reaction kinetics of the methanol synthesis and the water gas shift reaction on Cu/ZnO/Al2O3. React. Chem. Eng. 2021, 6, 868–887. [Google Scholar] [CrossRef]
- Sehested, J. Industrial and scientific directions of methanol catalyst development. J. Catal. 2019, 371, 368–375. [Google Scholar] [CrossRef]
- Behrens, M. Meso- and nano-structuring of industrial Cu/ZnO/(Al2O3) catalysts. J. Catal. 2009, 267, 24–29. [Google Scholar] [CrossRef]
- Etim, U.J.; Song, Y.; Zhong, Z. Improving the Cu/ZnO-Based Catalysts for Carbon Dioxide Hydrogenation to Methanol, and the Use of Methanol as a Renewable Energy Storage Media. Front. Energy Res. 2020, 8, 545431. [Google Scholar] [CrossRef]
- Cui, X.; Liu, Y.; Yan, W.; Xue, Y.; Mei, Y.; Li, J.; Gao, X.; Zhang, H.; Zhu, S.; Niu, Y.; et al. Enhancing methanol selectivity of commercial Cu/ZnO/Al2O3 catalyst in CO2 hydrogenation by surface silylation. Appl. Catal. B Environ. 2023, 339, 123099. [Google Scholar] [CrossRef]
- Huang, X.; Beck, A.; Fedorov, A.; Frey, H.; Zhang, B.; Klötzer, B.; vanBokhoven, J.A.; Copéret, C.; Willinger, M.G. Visualizing Structural and Chemical Transformations of an IndustrialCu/ZnO/Al2O3 Pre-catalyst during Activation and CO2 Reduction. ChemCatChem 2022, 14, e202201280. [Google Scholar] [CrossRef]
- Divins, N.J.; Kordus, D.; Timoshenko, J.; Sinev, I.; Zegkinoglou, I.; Bergmann, A.; Chee, S.W.; Widrinna, S.; Karslıoğlu, O.; Mistry, H.; et al. Operando high-pressure investigation of size-controlled CuZn catalysts for the methanol synthesis reaction. Nat. Commun. 2021, 12, 1435. [Google Scholar] [CrossRef] [PubMed]
- Lunkenbein, T.; Schumann, J.; Behrens, M.; Schlögl, R.; Willinger, M.G. Self-assembled catalyst promotion by overgrowth of layered ZnO in industrial Cu/ZnO/Al2O3 catalysts. Angew. Chem. Int. Ed. 2015, 54, 4544–4548. [Google Scholar] [CrossRef]
- Ahn, S.Y.; Kim, K.J.; Kim, B.J.; Shim, J.O.; Jang, W.J.; Roh, H.S. Unravelling the active sites and structure-activity relationship on Cu–ZnO–Al2O3 based catalysts for water-gas shift reaction. Appl. Catal. B Environ. 2023, 325, 122320. [Google Scholar] [CrossRef]
- Montanari, T.; Sisani, M.; Nocchetti, M.; Vivani, R.; Herrera Delgado, M.C.; Ramis, G.; Busca, G.; Costantino, U. Zinc–aluminum hydrotalcites as precursors of basic catalysts: Preparation, characterization and study of the activation of methanol. Catal. Today 2010, 152, 104–109. [Google Scholar] [CrossRef]
- Available online: https://matthey.com/products-and-markets/chemicals/water-gas-shift-catalysts (accessed on 30 October 2023).
- Xu, K.; Ma, C.; Yan, H.; Gu, H.; Wang, W.W.; Li, S.Q.; Meng, Q.L.; Shao, W.P.; Ding, G.H.; Wang, F.R.; et al. Catalytically efficient Ni-NiOx-Y2O3 interface for medium temperature water-gas shift reaction. Nat. Commun. 2022, 13, 2443. [Google Scholar] [CrossRef] [PubMed]
- Antoniak-Jurak, K.; Kowalik, P.; Bicki, R.; Michalska, K.; Prochniak, W.; Wiercioch, P. Cu substituted ZnAl2O4 ex-LDH catalysts for medium-temperature WGS: Effect of Cu/Zn ratio and thermal treatment on catalyst efficiency. Int. J. Hydrogen Energy 2019, 44, 27390–27400. [Google Scholar] [CrossRef]
- Clariant’s ShiftMax® 300 MTS Catalyst Delivers Excellent Performance at BHCC Hydrogen unit in Zhejiang, China, Press Release, 2 June 2020. Available online: https://www.clariant.com/en/Corporate/News/2020/06/Clariantrsquos-ShiftMaxreg-300-MTS-catalyst-delivers-excellent-performance-at-BHCC-hydrogen-unit-in (accessed on 30 October 2023).
- Available online: https://www.topsoe.com/our-resources/knowledge/our-products/catalysts/lk-813 (accessed on 20 October 2023).
- Li, Y.; Kottwitz, M.; Vincent, J.L.; Enright, M.J.; Liu, Z.; Zhang, L.; Huang, J.; Senanayake, S.D.; Yang, W.C.D.; Crozier, P.A.; et al. Dynamic structure of active sites in ceria-supported Pt catalysts for the water gas shift reaction. Nat. Commun. 2021, 12, 914. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.topsoe.com/our-resources/knowledge/our-products/catalysts/ssk-10?hsLang=en (accessed on 20 October 2023).
- Bown, R.M.; Joyce, M.; Zhang, Q.; Rmirez Reina, T.; Duya, M.S. Identifying Commercial Opportunities for the Reverse Water Gas Shift Reaction. Energy Technol. 2021, 9, 2100554. [Google Scholar] [CrossRef]
- Available online: https://ntrs.nasa.gov/citations/20020050609 (accessed on 20 October 2023).
- Stone, F.S.; Waller, D. Cu-ZnO and Cu-ZnO/Al2O3 catalysts for the reverse water-gas shift reaction. The effect of the Cu/Zn ratio on precursor characteristics and on the activity of the derived catalysts. Top. Catal. 2003, 22, 305–318. [Google Scholar] [CrossRef]
- Zhu, M.; Ge, Q.; Zhu, X. Catalytic Reduction of CO2 to CO via Reverse Water Gas Shift Reaction: Recent Advances in the Design of Active and Selective Supported Metal Catalysts. Trans. Tianjin Univ. 2020, 26, 172–187. [Google Scholar] [CrossRef]
- Liu, H.X.; Li, S.Q.; Wang, W.W.; Yu, W.Z.; Zhang, W.J.; Ma, C.; Jia, C.J. Partially sintered copper–ceria as excellent catalyst for the high-temperature reverse water gas shift reaction. Nat. Commun. 2022, 13, 2022. [Google Scholar] [CrossRef] [PubMed]
- Topsoe, eFuels Technology for Converting CO2 and Renewable Electricity to Renewable Synthetic Fuels. Available online: https://info.topsoe.com/en/dp-wp-efuels-technology (accessed on 30 October 2023).
- Adelung, S.; Dietrich, R.U. Impact of the reverse water-gas shift operating conditions on the Power-to-Liquid fuel production cost. Fuel 2022, 317, 123440. [Google Scholar] [CrossRef]
- Available online: https://www.topsoe.com/our-resources/knowledge/our-products/equipment/e-react-fuels (accessed on 30 October 2023).
- Wismann, S.T.; Larsen, K.-E.; Mortensen, P.M. Electrical Reverse Shift: Sustainable CO2 Valorization for Industrial Scale. Angew. Chem. Int. Ed. 2022, 61, e202109696. [Google Scholar] [CrossRef]
- Available online: https://www.hydrocarbonengineering.com/clean-fuels/26052022/ineratec-and-clariant-join-forces-for-a-cleaner-future/ (accessed on 30 October 2023).
- Available online: https://matthey.com/en/news/2022/hycogen (accessed on 30 October 2023).
- Available online: https://matthey.com/products-and-markets/chemicals/fischer-tropsch-technology (accessed on 30 October 2023).
- Available online: https://www.hydrocarbonprocessing.com/news/2023/08/e-fuels-axens-paul-wurth-ifpen-agree-to-co-develop-reverse-water-gas-shift-tech/ (accessed on 30 October 2023).
- Joo, O.-S.; Jung, K.-D.; Moon, I.; Rozovskii, A.Y.; Lin, G.I.; Han, S.-H.; Uhm, S.-J. Carbon dioxide hydrogenation to form methanol via a reverse-water-gas-shift reaction (the CAMERE process). Ind. Eng. Chem. Res. 1999, 38, 1808–1812. [Google Scholar] [CrossRef]
- Skrzypek, J.; Lachowska, M.; Serafin, D. Methanol synthesis from CO2 and H2: Dependence of equilibrium conversions and exit equilibrium concentrations of components on the main process variables. Chem. Eng. Sci. 1990, 45, 89–96. [Google Scholar] [CrossRef]
- Iyer, S.S.; Renganathan, T.; Pushpavanam, S.; Kumar, M.V.; Kaisare, N. Generalized thermodynamic analysis of methanol synthesis: Effect of feed composition. J. CO2 Util. 2015, 10, 95–104. [Google Scholar] [CrossRef]
- Topsoe, The Role of the Methanol-Synthesis Catalyst in the Transition towards Green Methanol. Available online: https://video.topsoe.com/webinar-the-role-of-the (accessed on 30 October 2023).
- Wender, I. Reactions of synthesis gas. Fuel Proc. Technol. 1996, 48, 189–297. [Google Scholar] [CrossRef]
- Lange, J.P. Methanol synthesis: A short review of technology improvement. Catal. Today 2001, 64, 3–8. [Google Scholar] [CrossRef]
- Sheldon, D. Methanol Production—A Technical History. Johns. Matthey Technol. Rev. 2017, 61, 172–182. [Google Scholar] [CrossRef]
- Available online: https://engineering.airliquide.com/technologies/methanol (accessed on 30 October 2023).
- Available online: https://www.topsoe.com/processes/methanol (accessed on 30 October 2023).
- Xu, X.; Liu, Y.; Zhang, F.; Di, W.; Zhang, Y. Clean coal technologies in China based on methanol platform. Catal. Today 2017, 298, 61–66. [Google Scholar] [CrossRef]
- Dieterich, V.; Buttler, A.; Hanel, A.; Spliethoff, H.; Fendt, S. Power-to-liquid via synthesis of methanol, DME or Fischer-Tropsch-fuels: A review. Energy Environ. Sci. 2020, 13, 3207–3252. [Google Scholar] [CrossRef]
- Xiao, K.; Wang, Q.; Qi, X.; Zhong, L. For Better Industrial Cu/ZnO/Al2O3 Methanol Synthesis Catalyst: A Compositional Study. Catal. Lett. 2017, 147, 1581–1591. [Google Scholar] [CrossRef]
- Behrens, M.; Girgsdies, F.; Trunschke, A.; Schlögl, R. Minerals as Model Compounds for Cu/ZnO Catalyst Precursors: Structural and Thermal Properties and IR Spectra of Mineral and Synthetic (Zincian) Malachite, Rosasite and Aurichalcite and a Catalyst Precursor Mixture. Eur. J. Inorg. Chem. 2009, 2009, 1347–1357. [Google Scholar] [CrossRef]
- Liu, X.M.; Lu, G.Q.; Yan, Z.F.; Beltramini, J. Recent Advances in Catalysts for Methanol Synthesis via Hydrogenation of CO and CO2. Ind. Eng. Chem. Res. 2003, 42, 6518–6530. [Google Scholar] [CrossRef]
- Waugh, K.C. Methanol synthesis. Catal. Lett. 2012, 142, 1153–1166. [Google Scholar] [CrossRef]
- Topsoe, High Activity Methanol Synthesis Catalyst. 2014. Available online: https://www.topsoe.com/our-resources/knowledge/our-products/process-licensing/syncor-methanoltm?hsLang=en (accessed on 30 October 2023).
- Kuld, S.; Thorhauge, M.; Falsig, H.; Elkjær, C.F.; Helveg, S.; Chorkendorff, I.; Sehested, J. Quantifying the promotion of Cu catalysts by ZnO for methanol synthesis. Science 2016, 352, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Osborne, S.; Schwartz, H.; Ringe, N.; Reitmeier, S. A flexible tool for methanol symthesis. Nitrogen Syngas 2021, 373, 44–50. [Google Scholar]
- Available online: https://www.clariant.com/en/Business-Units/Catalysts/Syngas-Catalysts/Methanol (accessed on 30 October 2023).
- Johnson Matthey, Latest Catalyst Provides More Methanol for Longer, Nitrogen & Syngas September/October 2021. Available online: https://matthey.com/documents/161599/440829/Reprint+-+N%2BS+-+Latest+catalyst+provides+more+methanol+for+longer.pdf/b8751743-9309-5b50-1259-8428455aba9d?t=1668002379563 (accessed on 30 October 2023).
- Available online: https://matthey.com/documents/161599/162780/An-update-on-KATALCO-51-102-synthesis-catalyst.pdf/cccb1b34-d35e-e62f-d481-9fe2d8f1568f (accessed on 30 October 2023).
- Nielsen, N.D.; Jensen, A.D.; Christensen, J.M. The roles of CO and CO2 in high pressure methanol synthesis over Cu-based catalysts. J. Catal. 2021, 393, 324–334. [Google Scholar] [CrossRef]
- Available online: https://www.basf.com/global/en/media/news-releases/2019/05/p-19-218.html (accessed on 30 October 2023).
- Ruland, H.; Song, H.; Laudenschleger, D.; Stürmer, S.; Schmidt, S.; He, J.; Kähler, K.; Muhler, M.; Schlög, R. CO2 Hydrogenation with Cu/ZnO/Al2O3: A Benchmark Study. ChemCatChem 2020, 12, 3216–3222. [Google Scholar] [CrossRef]
- Available online: https://info.topsoe.com/hyoctane-leaflet-dlp (accessed on 30 October 2023).
- Gao, J.; Wang, Y.; Ping, Y.; Hu, D.; Xu, G.; Gu, F.; Su, F. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RSC Adv. 2012, 2, 2358–2368. [Google Scholar] [CrossRef]
- Stangeland, K.; Kalai, D.; Li, H.; Yu, Z. CO2 methanation: The effect of catalysts and reaction conditions. Energy Procedia 2017, 105, 2022–2027. [Google Scholar] [CrossRef]
- Katla, D.; Węcel, D.; Jurczyk, M.; Skorek-Osikowska, A. Preliminary experimental study of a methanation reactor for conversion of H2 and CO2 into synthetic natural gas (SNG). Energy 2023, 263, 125881. [Google Scholar] [CrossRef]
- Torrente-Murciano, L.; Mattia, D.; Jones, M.D.; Plucinski, P.K. Formation of hydrocarbons via CO2 hydrogenation—A thermodynamic study. J. CO2 Util. 2014, 6, 34–39. [Google Scholar] [CrossRef]
- Lim, J.Y.; McGregor, J.; Sederman, A.J.; Dennis, J.S. The role of the Boudouard and water-gas shift reactions in the methanation of CO or CO2 over Ni/γ-Al2O3 catalyst. Chem. Eng. Sci. 2016, 152, 754–766. [Google Scholar] [CrossRef]
- Pearce, B.B.; Twigg, M.V.; Woodward, C. Methanation. In Catalyst Handbook, 2nd ed.; Twigg, M.V., Ed.; Wolfe Pub.: London, UK, 1989; pp. 340–383. [Google Scholar]
- Golosman, E.Z.; Efremov, V.N. Industrial Catalysts for the Hydrogenation of Carbon Oxides. Catal. Ind. 2012, 4, 267–283. [Google Scholar] [CrossRef]
- Miguel, C.V.; Mendes, A.; Madeira, L.M. Intrinsic kinetics of CO2 methanation over an industrial nickel-based catalyst. J. CO2 Util. 2018, 25, 128–136. [Google Scholar] [CrossRef]
- Available online: http://www.topsoe.com/business_areas/ammonia/processes/methanation.aspx (accessed on 30 October 2023).
- Johnson Matthey Methanation Catalysts. Available online: https://matthey.com/documents/161599/440146/JM+Methanation+product+brochure+%28c2020%29.pdf/56eaef18-4782-776f-ed2a-c332f5f16659?t=1653488109779 (accessed on 30 October 2023).
- Fujita, S.I.; Takezawa, N. Difference in the selectivity of CO and CO2 methanation reactions. Chem. Eng. J. 1997, 68, 63–68. [Google Scholar] [CrossRef]
- Schmider, D.; Maier, L.; Deutschmann, O. Reaction Kinetics of CO and CO2 Methanation over Nickel. Ind. Eng. Chem. Res. 2021, 60, 5792–5805. [Google Scholar] [CrossRef]
- Burger, T.; Donaubauera, P.; Hinrichsen, O. On the kinetics of the co-methanation of CO and CO2 on a co-precipitated Ni-Al catalyst. Appl. Catal. B Environ. 2021, 282, 119408. [Google Scholar] [CrossRef]
- Schumacher, J.; Meyer, D.; Friedland, J.; Güttel, R. Methanation of CO/CO2 mixtures: Evaluation of pellet size effect on methane formation rate and reactant selectivity. Chem. Eng. J. 2023, 463, 142451. [Google Scholar] [CrossRef]
- Dagle, R.A.; Wang, Y.; Xia, G.G.; Strohm, J.J.; Holladay, J.; Palo, D.R. Selective CO methanation catalysts for fuel processing applications. Appl. Catal. A Gen. 2007, 326, 213–218. [Google Scholar] [CrossRef]
- Kopyscinski, J.; Schildhauer, T.J.; Biollaz, S.M.A. Production of synthetic natural gas (SNG) from coal and dry biomass e a technology review from 1950 to 2009. Fuel 2010, 89, 1763–1783. [Google Scholar] [CrossRef]
- Røstrup-Nielsen, J.R.; Pedersen, K.; Sehested, J. High temperature methanation. Sintering and structure sensitivity. Appl Catal. A Gen 2007, 330, 134–138. [Google Scholar] [CrossRef]
- Bolt, A.; Dincer, I.; Agelin-Chaab, M. A critical review of synthetic natural gas production techniques and technologies. J. Nat. Gas Sci. Eng. 2020, 84, 103670. [Google Scholar] [CrossRef]
- Schaaf, T.; Grünig, J.; Schuster, M.R.; Rothenfluh, T.; Orth, A. Methanation of CO2—Storage of renewable energy in a gas distribution system. Energy Sustain. Soc. 2014, 4, 2. [Google Scholar] [CrossRef]
- Li, J.; Tian, Y.; Yan, X.; Yang, J.; Wang, Y.; Xu, W.; Xie, K. Approach and potential of replacing oil and natural gas with coal in China. Front. Energy 2020, 14, 419–431. [Google Scholar] [CrossRef]
- Available online: https://www.netl.doe.gov/sites/default/files/netl-file/tremp-2009.pdf (accessed on 30 October 2023).
- Available online: https://www.topsoe.com/processes/rng (accessed on 30 October 2023).
- Torcida, M.F.; Curto, D.; Martín, M. Design and optimization of CO2 hydrogenation multibed reactors. Chem. Eng. Res. Des. 2022, 181, 89–100. [Google Scholar] [CrossRef]
- Available online: http://www.topsoe.com/products/CatalystPortfolio.aspx (accessed on 30 October 2023).
- Nguyen, T.T.M.; Wissing, L.; Skjøth-Rasmussen, M.S. High temperature methanation: Catalyst considerations. Catal. Today 2013, 215, 233–238. [Google Scholar] [CrossRef]
- Available online: http://www.oilngasprocess.com/petrochemical/substitute-natural-gas-sng-process-by-davy-process-technology-uk.html (accessed on 30 October 2023).
- Ross, J.R.H. Nickel Catalysts for C1 Reactions: Recollections from a Career in Heterogeneous Catalysis. Top. Catal. 2021, 64, 896–906. [Google Scholar] [CrossRef]
- Romano, L.; Ruggeri, F. Methane from syngas—Status of Amec Foster Wheeler VESTA technology development. Energy Procedia 2015, 81, 249–254. [Google Scholar] [CrossRef]
- Depetri, V.; Collodi, G.; Mancuso, L.; Ruggeri, F. VESTA Methanation Applications for Small Scale, Multipurpose, Green SNG Production. Chem. Eng. Trans. 2018, 65, 415–420. [Google Scholar]
- Available online: https://www.clariant.com/en/Corporate/News/2016/07/VESTA-Oncethrough-Methanation-New-Technology-with-Wide-H2CO-Flexibility-Successfully-Passes-Pilot-Te (accessed on 30 October 2023).
- Götz, M.; Lefebvre, J.; Mörs, F.; Koch, A.M.; Graf, F.; Bajohr, S.; Reimert, R.; Kolb, T. Renewable Power-to-Gas: A Technological and Economic Review. Renew. Energy 2016, 85, 1371–1390. [Google Scholar] [CrossRef]
- Vega Puga, E.; Moumin, G.; Neumann, N.C.; Roeb, M.; Ardone, A.; Sattler, C. Holistic View on Synthetic Natural Gas Production: A Technical, Economic and Environmental Analysis. Energies 2022, 15, 1608. [Google Scholar] [CrossRef]
- Thema, M.; Bauer, F.; Sterner, M. Sterner Power-to-Gas: Electrolysis and methanation status review. Renew. Sustain. Energy Rev. 2019, 112, 775–778. [Google Scholar] [CrossRef]
- Pintér, G. The development of global power-to-methane potentials between 2000 and 2020: A comparative overview of international projects. Appl. Energy 2024, 353, 122094. [Google Scholar] [CrossRef]
- Tripodi, A.; Conte, F.; Rossetti, I. Carbon Dioxide Methanation: Design of a Fully Integrated Plant. Energy Fuels 2020, 34, 7242–7256. [Google Scholar] [CrossRef]
- Uddin, Z.; Bor-Yih Yu, B.Y.; Lee, H.Y. Evaluation of alternative processes of CO2 methanation: Design, optimization, control, techno-economic and environmental analysis. J. CO2 Util. 2022, 60, 101974. [Google Scholar] [CrossRef]
- Lee, W.J.; Li, C.; Prajitno, H.; Yoo, J.; Patel, J.; Yang, Y.; Lim, S. Recent trend in thermal catalytic low temperature CO2 methanation: A critical review. Catal. Today 2021, 368, 2–19. [Google Scholar] [CrossRef]
- Mebrahtu, C.; Nohl, M.; Dittrich, L.; Foit, S.R.; de Haart, L.G.L.; Eichel, R.A.; Palkovits, R. Integrated Co-Electrolysis and Syngas Methanation for the Direct Production of Synthetic Natural Gas from CO2 and H2O. ChemSusChem 2021, 14, 2295–2302. [Google Scholar] [CrossRef] [PubMed]
- Schlereth, D.; Hinrichsen, O. A fixed-bed reactor modeling study on the methanation of CO2. Chem. Eng. Res. Des. 2014, 92, 702–712. [Google Scholar] [CrossRef]
- Available online: https://www.clariant.com/en/Business-Units/Catalysts/Energy-Transition/Carbon-Capture-and-Utilization (accessed on 30 October 2023).
- Mohd Ridzuan, N.D.; Shaharun, M.S.; Anawar, M.A.; Ud-Din, I. Ni-Based Catalyst for Carbon Dioxide Methanation: A Review on Performance and Progress. Catalysts 2022, 12, 469. [Google Scholar] [CrossRef]
- Busca, G.; Spennati, E.; Riani, P.; Garbarino, G. Looking for an Optimal Composition of Nickel-Based Catalysts for CO2 Methanation. Energies 2023, 16, 5304. [Google Scholar] [CrossRef]
- Stenger, H.G., Jr.; Askonas, C.F. Thermodynamic Product Distributions for the Fischer-Tropsch Synthesis. Ind. Eng. Chem. Fundamen. 1986, 25, 410–413. [Google Scholar] [CrossRef]
- Chen, J.; Yang, C. Thermodynamic Equilibrium Analysis of Product Distribution in the Fischer−Tropsch Process Under Different Operating Conditions. ACS Omega 2019, 4, 22237–22244. [Google Scholar] [CrossRef]
- Leckel, D. Diesel Production from Fischer−Tropsch: The Past, the Present, and New Concepts. Energy Fuels 2009, 23, 2342–2358. [Google Scholar] [CrossRef]
- Zennaro, R.; Hugues, F.; Caprani, E. The Eni—IFP/Axens GTL Technology: From R&D to a Successful Scale-Up. In Proceedings of the DGMK/SCI-Conference “Synthesis Gas Chemistry”, Dresden, Germany, 4–6 October 2006; Available online: https://www.osti.gov/etdeweb/servlets/purl/20840759 (accessed on 30 October 2023).
- Martinelli, M.; Gnanamani, M.K.; LeViness, S.; Jacobs, G.; Shafer, W.D. An overview of Fischer-Tropsch Synthesis: XtL processes, catalysts and reactors. Appl. Catal. A Gen. 2020, 608, 11774. [Google Scholar] [CrossRef]
- Niu, C.; Xia, M.; Chen, C.; Ma, Z.; Jia, L.; Hou, B.; Li, D. Effect of process conditions on the product distribution of Fischer-Tropsch synthesis over an industrial cobalt-based catalyst using a fixed-bed reactor. Appl. Catal. A Gen. 2020, 601, 117630. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Development of Novel Cobalt Fischer−Tropsch Catalysts. Chem. Rev. 2007, 107, 1692–1744. [Google Scholar] [CrossRef] [PubMed]
- Cornaro, U.; Rossini, S.; Montanari, T.; Finocchio, E.; Busca, G. K-doping of Co/Al2O3 low temperature Fischer-Tropsch catalysts. Catal. Today 2012, 197, 101–108. [Google Scholar] [CrossRef]
- Spennati, E.; Garbarino, G.; Riani, P.; Busca, G. Alumina-supported cobalt catalysts in the hydrogenation of CO2 at atmospheric pressure. Int. J. Hydrogen Energy 2023, 48, 25006–25015. [Google Scholar] [CrossRef]
- Diehl, F.; Khodakov, A.Y. Promotion of Cobalt Fischer-Tropsch Catalysts with Noble Metals: A Review. Oil Gas Sci. Technol. Rev. IFP 2009, 64, 11–24. [Google Scholar] [CrossRef]
- Claeys, M.; Dry, M.E.; van Steen, E.; du Plessis, E.; van Berge, P.J.; Saib, A.M.; Moodley, D.J. In situ magnetometer study on the formation and stability of cobalt carbide in Fischer-Tropsch synthesis. J. Catal. 2014, 318, 193–202. [Google Scholar] [CrossRef]
- Hazemann, P.; Decottignies, D.; Maury, S.; Humbert, S.; Meunier, F.C.; Schuurman, Y. Selectivity loss in Fischer-Tropsch synthesis: The effect of cobalt carbide formation. J. Catal. 2021, 397, 1–12. [Google Scholar] [CrossRef]
- Shiba, N.C.; Liu, X.; Yao, Y. Advances in lower olefin production over cobalt-based catalysts via the Fischer-Tropsch process. Fuel Proc. Technol. 2022, 238, 107489. [Google Scholar] [CrossRef]
- Peacock, M.; Paterson, J.; Reed, L.; Davies, S.; Carter, S.; Coe, A.; Clarkson, J. Innovation in Fischer-Tropsch: Developing Fundamental Understanding to Support Commercial Opportunities. Top. Catal. 2020, 63, 328–339. [Google Scholar] [CrossRef]
- Espinoza, R.L.; Steynberg, A.P.; Jager, B.; Vosloo, A.C. Low temperature Fischer-Tropsch synthesis from a Sasol perspective. Appl. Catal. A Gen. 1999, 186, 13–26. [Google Scholar] [CrossRef]
- Weststrate, C.J.; Sharma, D.; Garcia Rodriguez, D.; Gleeson, M.A.; Fredriksson, H.O.A.; Niemantsverdriet, J.M. Mechanistic insight into carbon-carbon bond formation on cobalt under simulated FischerTropsch synthesis conditions. Nat. Commun. 2020, 11, 750. [Google Scholar] [CrossRef] [PubMed]
- Perego, C.; Bortolo, R.; Zennaro, R. Gas to liquids technologies for natural gas reserves valorization: The Eni experience. Catal. Today 2009, 142, 9–16. [Google Scholar] [CrossRef]
- Ma, W.; Jacobs, G.; Sparks, D.E.; Todic, B.; Bukur, D.B.; Davis, B.H. Quantitative comparison of iron and cobalt based catalysts for the Fischer-Tropsch synthesis under clean and poisoning conditions. Catal. Today 2020, 343, 125–136. [Google Scholar] [CrossRef]
- Steynberg, A.P.; Espinoza, R.L.; Jager, B.; Vosloo, A.C. High temperature Fischer-Tropsch synthesis in commercial practice. Appl. Catal. A Gen. 1999, 186, 41–54. [Google Scholar] [CrossRef]
- Xu, J.; Yang, Y.; Li, Y. Fischer-Tropsch synthesis process development: Steps from fundamentals to industrial processes. Curr. Opin. Chem. Eng. 2013, 2, 354–363. [Google Scholar] [CrossRef]
- Xu, J.; Yang, Y.; Li, Y.W. Recent development in converting coal to clean fuels in China. Fuel 2015, 152, 122–130. [Google Scholar] [CrossRef]
- Zhang, J.; Abbas, M.; Chen, J. The Evolution of Fe Phases of a Fused Iron Catalyst during Reduction and Fischer-Tropsch Synthesis. Catal. Sci. Technol. 2017, 7, 3626–3636. [Google Scholar] [CrossRef]
- Lyu, S.; Wang, L.; Li, Z.; Yin, S.; Chen, J.; Zhang, Y.; Li, J.; Wang, Y. Stabilization of ε-iron carbide as high-temperature catalyst under realistic Fischer-Tropsch synthesis conditions. Nat. Commun. 2020, 11, 6219. [Google Scholar] [CrossRef]
- Luo, M.; Hamdeh, H.; Davis, B.H. Fischer-Tropsch Synthesis: Catalyst activation of low alpha iron catalyst. Catal. Today 2009, 140, 127–134. [Google Scholar] [CrossRef]
- Gaube, J.; Klein, H.F. The promoter effect of alkali in Fischer-Tropsch iron and cobalt catalysts. Appl. Catal. A Gen. 2008, 350, 126–132. [Google Scholar] [CrossRef]
- De Smit, E.; Cinquini, F.; Beale, A.M.; Safonova, O.V.; van Beek, W.; Sautet, P.; Weckhuysen, B.M. Stability and Reactivity of ϵ-χ-θ Iron Carbide Catalyst Phases in Fischer-Tropsch Synthesis: Controlling μC. J. Am. Chem. Soc. 2010, 132, 14928–14941. [Google Scholar] [CrossRef]
- Lu, F.; Chen, X.; Lei, Z.; Wen, L.; Zhang, Y. Revealing the activity of different iron carbides for Fischer-Tropsch synthesis. Appl. Catal. B Environ. 2021, 281, 119521. [Google Scholar] [CrossRef]
- Ordomsky, V.V.; Legras, B.; Cheng, K.; Paul, S.; Khodakov, A.Y. The role of carbon atoms of supported iron carbides in Fischer-Tropsch synthesis. Catal. Sci. Technol. 2015, 5, 1433–1437. [Google Scholar] [CrossRef]
- Pham, T.H.; Qi, Y.; Yang, J.; Duan, X.; Qian, G.; Zhou, X.; Chen, D.; Yuan, W. Insights into Hägg Iron-Carbide-Catalyzed, D. Fischer−Tropsch Synthesis: Suppression of CH4 Formation and Enhancement of C−C Coupling on χ-Fe5C2(510). ACS Catal. 2015, 5, 2203–2208. [Google Scholar] [CrossRef]
- Atsonios, K.; Li, J.; Inglezakis, V.J. Process analysis and comparative assessment of advanced thermochemical pathways for e-kerosene production. Energy 2023, 278, 127868. [Google Scholar] [CrossRef]
- Visconti, C.G.; Martinelli, M.; Falbo, L.; Fratalocchi, L.; Lietti, L. CO2 hydrogenation to hydrocarbons over Co and Fe-based Fischer-Tropsch catalysts. Catal. Today 2016, 277, 161–170. [Google Scholar] [CrossRef]
- Choi, Y.H.; Jang, Y.J.; Park, H.; Kim, W.Y.; Lee, Y.H.; Choi, S.H.; Lee, J.S. Carbon dioxide Fischer-Tropsch synthesis: A new path to carbon-neutral fuels. Appl. Catal. B Environ. 2017, 202, 605–610. [Google Scholar] [CrossRef]
- Wei, J.; Ge, Q.; Yao, R.; Wen, Z.; Fang, C.; Guo, L.; Xu, H.; Sun, J. Directly converting CO2 into a gasoline fuel. Nat. Commun. 2017, 8, 15174. [Google Scholar] [CrossRef]
- Cheng, Y.; Chen, Y.; Zhang, S.; Wu, X.; Chen, C.; Shi, X.; Qing, M.; Li, J.; Liu, C.L.; Dong, W.S. High-yield production of aromatics over CuFeO2/hierarchical HZSM-5 via CO2 Fischer-Tropsch synthesis. Green Chem. 2023, 25, 3570–3584. [Google Scholar] [CrossRef]
- Zhang, L.; Dang, Y.; Zhou, X.; Gao, P.; van Bavel, A.P.; Wang, H.; Li, S.; Shi, L.; Yang, Y.; Vovk, E.I.; et al. Direct conversion of CO2 to a jet fuel over CoFe alloy catalysts. Innovation 2021, 2, 100170. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, F. Science Alert, 27 April 2015. Available online: https://www.sciencealert.com/audi-have-successfully-made-diesel-fuel-from-air-and-water (accessed on 30 October 2023).
- Busca, G. The surface acidity of solid oxides and its characterization by IR spectroscopic methods. An attempt at systematization. Phys. Chem. Chem. Phys. 1999, 1, 723–736. [Google Scholar] [CrossRef]
- Busca, G. Bases and basic materials in industrial and environmental chemistry. Liquid versus solid basicity. Chem. Rev. 2010, 110, 2217–2249. [Google Scholar] [CrossRef] [PubMed]
- Helali, Z.; Jedidi, A.; Syzgantseva, O.A.; Calatayud, M.; Minot, C. Scaling reducibility of metal oxides. Theor. Chem. Acc. 2017, 136, 100. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Why gold is the noblest of all the metals. Nature 1995, 376, 238. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Theoretical surface science and catalysis—Calculations and concepts. Adv. Catal. 2000, 45, 71–129. [Google Scholar]
- Nørskov, J.K.; Abild-Pedersen, F.; Studt, F.; Bligaard, T. Density functional theory in surface chemistry and catalysis. Proc. Natl. Acad. Sci. USA 2011, 108, 937–943. [Google Scholar] [CrossRef]
- Takigawa, I.; Shimizu, K.; Tsuda, K.; Takakusa, S. Machine-learning prediction of the d-band center for metals and bimetals. RSC Adv. 2016, 6, 52587. [Google Scholar] [CrossRef]
- Kristinsdóttir, L.; Skúlason, E. A systematic DFT study of hydrogen diffusion on transition metal surfaces. Surf. Sci. 2012, 606, 1400–1404. [Google Scholar] [CrossRef]
- Vázquez-Parga, D.; Jurado, A.; Roldan, A.; Viñes, F. A computational map of the probe CO molecule adsorption and dissociation on transition 672 metal low Miller indices surfaces. Appl. Surf. Sci. 2023, 618, 156581. [Google Scholar] [CrossRef]
- Jin, W.; Wang, Y.; Liu, T.; Ding, C.; Guo, H. CO2 chemisorption and dissociation on flat and stepped transition metal surfaces. Appl. Surf. Sci. 2022, 599, 154024. [Google Scholar] [CrossRef]
- Available online: www.dailymetalprice.com (accessed on 14 November 2023).
- Busca, G. Catalysts for hydrogenations, dehydrogenations and methathesis: Sulphides and oxides. In Heterogeneous Catalytic Materials; Busca, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 345–374. [Google Scholar]
- Ghiotti, G.; Chiorino, A.; Boccuzzi, F. Surface chemistry and electronic effects of H2 (D2) on two different microcrystalline ZnO powders. Surf. Sci. 1993, 287–288, 228–234. [Google Scholar] [CrossRef]
- French, S.A.; Sokol, A.A.; Bromley, S.T.; Catlow, C.R.A.; Rogers, S.C.; Sherwood, P. Assignment of the Complex Vibrational Spectra of the Hydrogenated ZnO Polar Surfaces Using QM/MM Embedding. J. Chem. Phys. 2003, 118, 317–320. [Google Scholar] [CrossRef]
- Kiss, J.; Witt, A.; Meyer, B.; Marx, D. Methanol synthesis on ZnO(0001): I. Hydrogen coverage, charge state of oxygen vacancies, and chemical reactivity. J. Chem. Phys. 2009, 130, 184706. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, M.; Strunk, J.; Hinrichsen, O.; Muhler, M.; Fink, K.; Meyer, B.; Wöll, C. Active sites on oxide surfaces: ZnO-catalyzed synthesis of methanol from CO and H2. Angew. Chem. 2005, 44, 2790–2794. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, C.; Saussey, J.; Lavalley, J.C.; Djega-Mariadassou, G. Definition of polycrystalline ZnO catalytic sites and their role in CO hydrogenation. Appl. Catal. 1986, 25, 59–68. [Google Scholar] [CrossRef]
- Busca, G. Fourier transform-infrared spectroscopic study of the adsorption of hydrogen on chromia and on some metal chromites. J. Catal. 1989, 120, 303–313. [Google Scholar] [CrossRef]
- Collins, S.E.; Baltanás, M.A.; Bonivardi, A.L. Hydrogen chemisorption on gallium oxide polymorphs. Langmuir 2005, 21, 962–970. [Google Scholar] [CrossRef]
- Kondo, J.N.; Sakata, Y.; Domen, K.; Maruya, K.; Onishi, T. Infrared study of hydrogen adsorbed on ZrO2. J. Chem. Soc. Farad. Trans. 1990, 86, 397–401. [Google Scholar] [CrossRef]
- Hofmann, A.; Clark, S.J.; Oppel, M.; Hahndorf, I. Hydrogen adsorption on the tetragonal ZrO2(101) surface: A theoretical study of an important catalytic reactant. Phys. Chem. Chem. Phys. 2002, 4, 3500–3508. [Google Scholar] [CrossRef]
- Syzgantseva, O.; Calatayud, M.; Minot, C. Revealing the Surface Reactivity of Zirconia by Periodic DFT Calculations. J. Phys. Chem. C 2010, 114, 11918–11923. [Google Scholar] [CrossRef]
- Cavalleri, M.; Pelmenschikov, A.; Morosi, G.; Gamba, A.; Coluccia, S.; Martra, G. Dissociative adsorption of H2 on defect sites of MgO: A combined IR spectroscopic and quantum chemical study. Stud. Surf. Sci. Catal. 2001, 140, 131–139. [Google Scholar]
- Binet, C.; Daturi, M.; Lavalley, J.C. IR study of polycrystalline ceria properties in oxidised and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar] [CrossRef]
- Vilé, G.; Bridier, B.; Wichert, J.; Pérez-Ramírez, J. Ceria in hydrogenation catalysis: High selectivity in the conversion of alkynes to olefins. Angew. Chem. 2012, 51, 8620–8623. [Google Scholar] [CrossRef]
- Doornkamp, C.; Ponec, V. The universal character of the Mars and Van Krevelen mechanism. J. Mol. Catal. A Chem. 2000, 162, 19–32. [Google Scholar] [CrossRef]
- Chen, H.T.; Choi, Y.M.; Liu, M.; Lin, M.C. A Theoretical Study of Surface Reduction Mechanisms of CeO2(111) and (110) by H2. ChemPhysChem 2007, 8, 849–855. [Google Scholar] [CrossRef]
- Li, Z.; Werner, K.; Chen, L.; Jia, A.; Qian, K.; Zhong, J.Q.; You, R.; Wu, L.; Zhang, L.; Pan, H.; et al. Interaction of Hydrogen with Ceria: Hydroxylation, Reduction, and Hydride Formation on the Surface and in the Bulk. Chem. Eur. J. 2021, 27, 5268–5276. [Google Scholar] [CrossRef]
- Wei, B.; Calatayud, M. Hydrogen activation on Anatase TiO2: Effect of surface termination. Catal. Today 2022, 397–399, 113–120. [Google Scholar] [CrossRef]
- Yua, X.; Zhang, X.; Wang, S. High coverage hydrogen adsorption on the Fe3O4(1 1 0) surface. Appl. Surf. Sci. 2015, 353, 973–978. [Google Scholar] [CrossRef]
- Bielz, T.; Lorenz, H.; Jochum, W.; Kaindl, R.; Klauser, F.; Klötzer, B.; Penner, S. Hydrogen on In2O3: Reducibility, Bonding, Defect Formation, and Reactivity. J. Phys. Chem. C 2010, 114, 9022–9029. [Google Scholar] [CrossRef]
- Posada-Borbón, A.; Grönbeck, H. Hydrogen adsorption on In2O3(111) and In2O3(110). Phys. Chem. Chem. Phys. 2020, 22, 16193–16202. [Google Scholar] [CrossRef] [PubMed]
- Rukini, A.; Rhamdhani, M.A.; Brooks, G.A.; Van den Bulck, A. Metals Production and Metal Oxides Reduction Using Hydrogen: A Review. J. Sustain. Metall. 2022, 8, 1–24. [Google Scholar] [CrossRef]
- Sheppard, D.A.; Buckley, C.E. Hydrogen adsorption on porous silica. Int. J. Hydrogen Energy 2008, 33, 1688–1692. [Google Scholar] [CrossRef]
- Amenomiya, Y. Adsorption of Hydrogen and H2-D2 Exchange Reaction on Alumina. J. Catal. 1971, 22, 109–121. [Google Scholar] [CrossRef]
- Weller, S.W.; Montagna, A.A. Studies of Alumina I. Reaction with Hydrogen at Elevated Temperatures. J. Catal. 1971, 21, 303–311. [Google Scholar] [CrossRef]
- Kramer, R.; Andre, M. Adsorption of Atomic Hydrogen on Alumina by Hydrogen Spillover. J. Catal. 1979, 58, 287–295. [Google Scholar] [CrossRef]
- Digne, M.; Sautet, P.; Raybaud, P.; Euzen, P.; Toulhoat, H. Use of DFT to achieve a rational understanding of acid–basic properties of γ-alumina surfaces. J. Catal. 2004, 226, 54–68. [Google Scholar] [CrossRef]
- Wischert, R.; Laurent, P.; Copéret, C.; Delbecq, F.; Sautet, P. γ-Alumina: The essential and unexpected role of water for the structure, stability, and reactivity of “defect” sites. J. Am. Chem. Soc. 2012, 134, 14430–14449. [Google Scholar] [CrossRef]
- Karim, W.; Spreafico, C.; Kleibert, A.; Gobrecht, J.; VandeVondele, J.; Ekinci, Y.; van Bokhoven, J.A. Catalyst support effects on hydrogen spillover. Nature 2017, 541, 68–71. [Google Scholar] [CrossRef]
- Garbarino, G.; Travi, I.; Pani, M.; Carnasciali, M.M.; Busca, G. Pure vs ultra-pure γ-alumina: A spectroscopic study and catalysis of ethanol conversion. Catal. Commun. 2015, 70, 77–81. [Google Scholar] [CrossRef]
- Shen, H.; Li, H.; Yang, Z.; Li, C. Magic of hydrogen spillover: Understanding and application. Green Energy Environ. 2022, 7, 1161–1198. [Google Scholar] [CrossRef]
- Beck, A.; Rzepka, P.; Marshall, K.P.; Stoian, D.; Willinger, M.G.; van Bokhoven, J.A. Hydrogen Interaction with Oxide Supports in the Presence and Absence of Platinum. J. Phys. Chem. C 2022, 126, 17589–17597. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, M.; Wang, A.; Zhang, T. Selective Hydrogenation over Supported Metal Catalysts: From Nanoparticles to Single Atoms. Chem. Rev. 2020, 120, 683–733. [Google Scholar] [CrossRef] [PubMed]
- Arena, F.; Italiano, G.; Barbera, K.; Bordiga, S.; Bonura, G.; Spadaro, L.; Frusteri, F. Solid-state interactions, adsorption sites and functionality of Cu-ZnO/ZrO2 catalysts in the CO2 hydrogenation to CH3OH. Appl. Catal. A Gen. 2008, 350, 16–23. [Google Scholar] [CrossRef]
- Paul, J.F.; Sautet, P. Comparison of the nature of the hydrogen-metal bond on Pd(111) and Ni(111) by a periodic density functional method. Surf. Sci. 1996, 356, L403–L409. [Google Scholar] [CrossRef]
- Huda, M.N.; Kleinman, L. Hydrogen adsorption and dissociation on small platinum clusters: An electronic structure density functional study. Phys. Rev. B 2006, 74, 195407. [Google Scholar] [CrossRef]
- Ferrin, P.; Kandoi, S.; Nilekar, A.U.; Mavrikakis, M. Hydrogen adsorption, absorption and diffusion on and in transition metal surfaces: A DFT study. Surf. Sci. 2012, 606, 679–689. [Google Scholar] [CrossRef]
- Greeley, J.; Marikakis, M. Surface and subsurface hydrogen: Adsorption properties on transition metals and near-surface alloys. J. Phys. Chem. B 2005, 109, 3460–3471. [Google Scholar] [CrossRef]
- Chizallet, C.; Bonnard, C.; Krebs, E.; Bisson, L.; Thomazeau, C.; Raybaud, P. Thermodynamic Stability of Buta-1,3-diene and But-1-ene on Pd(111) and (100) Surfaces under H2 Pressure: A DFT Study. J. Phys. Chem. C 2011, 115, 12135–12149. [Google Scholar] [CrossRef]
- Maki-Arvela, P.; Murzin, D.Y. Effect of metal particle shape on hydrogen assisted reactions. Appl. Catal. A Gen. 2021, 618, 118140. [Google Scholar] [CrossRef]
- Oudenhuijzen, M.K.; van Bokhoven, J.A.; Miller, J.T.; Ramaker, D.E.; Koningsberger, D.C. Three-site model for hydrogen adsorption on supported platinum particles: Influence of support ionicity and particle size on the hydrogen coverage. J. Am. Chem. Soc. 2005, 127, 1530–1540. [Google Scholar] [CrossRef] [PubMed]
- Znak, L.; Zieliński, J. Interaction of hydrogen with unsupported and supported nickel. Langmuir 2006, 22, 8758. [Google Scholar] [CrossRef] [PubMed]
- Znak, L.; Zieliński, J. Effects of support on hydrogen adsorption/desorption on nickel. Appl. Catal. A Gen. 2008, 334, 268–276. [Google Scholar] [CrossRef]
- Wang, Y.; Winter, L.R.; Chen, J.G.; Yan, B.Y. CO2 hydrogenation over heterogeneous catalysts at atmospheric pressure: From electronic properties to product selectivity. Green Chem. 2021, 23, 249–267. [Google Scholar] [CrossRef]
- Zabilskiy, M.; Sushkevich, V.L.; Palagin, D.; Newton, M.A.; Krumeich, F.; van Bokhoven, J.A. The unique interplay between copper and zinc during catalytic carbon dioxide hydrogenation to methanol. Nat. Commun. 2020, 11, 2409. [Google Scholar] [CrossRef] [PubMed]
- Silveri, F.; Quesne, M.G.; Roldan, A.; de Leeuw, N.H.; Catlow, C.R.A. Hydrogen adsorption on transition metal carbides: A DFT study. Phys. Chem. Chem. Phys. 2019, 21, 5335–5343. [Google Scholar] [CrossRef]
- Bian, G.; Oonuki, A.; Kobayashi, Y.; Koizumi, N.; Yamada, M. Syngas adsorption on precipitated iron catalysts reduced by H2, syngas or CO and on those used for high pressure FT synthesis by in situ diffuse reflectance FTIR spectroscopy. Appl. Catal. A 2001, 219, 13–24. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, J.; Wang, T.; Song, J.F.; Yang, Y.; Li, Y.F.; Wen, X. Theoretical insight into the interaction between hydrogen and Hägg carbide (χ-Fe5C2) surfaces. Appl. Surf. Sci. 2022, 583, 152538. [Google Scholar] [CrossRef]
- Blyholder, G. Molecular orbital view of chemisorbed carbon monoxide. J. Phys. Chem. 1964, 68, 2772–2777. [Google Scholar] [CrossRef]
- Lupinetti, A.J.; Fau, S.; Frenking, G.; Strauss, S.H. Theoretical Analysis of the Bonding between CO and Positively Charged Atoms. J. Phys. Chem. A 1997, 101, 9551–9559. [Google Scholar] [CrossRef]
- Sthorozhev, P.Y.; Yanko, V.S.; Tsyganenko, A.A.; Rodriguez-Delgado, G.; Otero Areán, C. Isomeric states of polar molecules on ionic surfaces:electrostatic model and FTIR studies. Appl. Surf. Sci. 2004, 238, 390–394. [Google Scholar] [CrossRef]
- Hadjiivanov, K.; Vayssilov, G. Characterization of Oxide Surfaces and Zeolites by Carbon Monoxide as an IR Probe Molecule. Adv. Catal. 2002, 47, 307–511. [Google Scholar]
- Busca, G. Infrared (IR) Spectroscopy. In Springer Handbook of Advanced Catalyst Characterization; Bañares, M.A., Wachs., I., Eds.; Springer Nature: Cham, Switzerland, 2023; pp. 1–30. [Google Scholar]
- Blomqvist, J.; Lehman, L.; Salo, P. CO adsorption on metal-oxide surfaces doped with transition-metal adatoms. Phys. Status Solid. 2012, 249, 1046–1057. [Google Scholar] [CrossRef]
- Gopal, P.G.; Schneider, R.L.; Watters, K.L. Evidence for Production of Surface Formate upon Direct Reaction of CO with Alumina and Magnesia. J. Catal. 1987, 105, 366–372. [Google Scholar] [CrossRef]
- Shido, T.; Asakura, K.; Iwasawa, Y. Reactant-promoted reaction mechanism for catalytic water-gas shift reaction on MgO. J. Catal. 1990, 122, 55–67. [Google Scholar] [CrossRef]
- Kogler, M.; Köck, E.M.; Klötzer, B.; Schachinger, T.; Wallisch, W.; Henn, R.; Huck, C.W.; Hejny, C.; Penner, S. High-Temperature Carbon Deposition on Oxide Surfaces by CO Disproportionation. J. Phys. Chem. C 2016, 120, 1795–1807. [Google Scholar] [CrossRef]
- Dang, J.; Chou, K. A Model for the Reduction of Metal Oxides by Carbon Monoxide. ISIJ Int. 2018, 58, 585–593. [Google Scholar] [CrossRef]
- Abild-Pedersen, F.; Andersson, M.P. CO adsorption energies on metals with correction for high coordination adsorption sites—A density functional study. Surf. Sci. 2007, 601, 1747–1753. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Sheppard, N. Advances in Infrared and Raman Spectroscopy; Hester, R.E., Clark, R.H.J., Eds.; Heyden: London, UK, 1982; Volume 5, pp. 86–95. [Google Scholar]
- Fielicke, A.; Gruene, P.; Meijer, G.; Rayner, D.M. The adsorption of CO on transition metal clusters: A case study of cluster surface chemistry. Surf. Sci. 2009, 603, 1427–1433. [Google Scholar] [CrossRef]
- Lemire, C.; Meyer, R.; Shaikhutdinov, S.K.; Freund, H.-J. CO adsorption on oxide supported gold: From small clusters to monolayer islands and three-dimensional nanoparticles. Surf. Sci. 2004, 552, 27–34. [Google Scholar] [CrossRef]
- Wadayama, T.; Sasaki, Y.; Shiomitsu, K.; Hatta, A. Infrared reflection absorption study of carbon monoxide adsorption on Cu(100)-(2 · 2)p4g-Pd ordered alloy surface. Surf. Sci. 2005, 592, 72–82. [Google Scholar] [CrossRef]
- Sanchez-Escribano, V.; Arrighi, L.; Riani, P.; Marazza, R.; Busca, G. Characterization of Pd-Cu Alloy Nanoparticles on γ-Al2O3-Supported Catalysts. Langmuir 2006, 22, 9214–9219. [Google Scholar] [CrossRef] [PubMed]
- Aghamiria, S.; Ghobeity, A. Nickel carbonyl formation in a fluidized bedreactor: Experimental investigation and modeling. J. Chem. Technol. Biotechnol. 2020, 95, 2921–2929. [Google Scholar] [CrossRef]
- Garbarino, G.; Campodonico, S.; Romero Perez, A.; Carnasciali, M.M.; Riani, P.; Finocchio, E.; Busca, G. Spectroscopic characterization of Ni/Al2O3 catalytic materials for the steam reforming of renewables. Appl. Catal. A Gen. 2013, 452, 163–173. [Google Scholar] [CrossRef]
- Liu, Z.P.; Hu, P. General trends in CO dissociation on transition metal surfaces. J. Chem. Phys. 2001, 114, 8244–8247. [Google Scholar] [CrossRef]
- Chai, J.; Pestman, R.; Chen, W.; Dugulan, A.I.; Feng, B.; Men, Z.; Wang, P.; Hensen, E.J.M. The role of H2 in Fe carburization by CO in Fischer-Tropsch catalysts. J. Catal. 2021, 400, 93–102. [Google Scholar] [CrossRef]
- Gong, J.; Cao, C.; Sun, R.; Cui, L.; Gao, r.; Hao, H. A DFT Insight into the Tuning Effect of Potassium Promoter on the Formation of Carbon Atoms via Carburization Gases Dissociation on Iron-Based Catalysts. Catalysts 2020, 10, 527. [Google Scholar] [CrossRef]
- Hofer, L.J.E.; Peebles, W.C. Preparation and X-ray Diffraction Studies of a New Cobalt Carbide. J. Am. Chem. Soc. 1947, 69, 893–899. [Google Scholar] [CrossRef]
- Struis, R.P.W.J.; Bachelin, D.; Ludwig, C.; Wokaun, A. Studying the Formation of Ni3C from CO and Metallic Ni at T = 265 °C in Situ Using Ni K-Edge X-ray Absorption Spectroscopy. J. Phys. Chem. C 2009, 113, 2443–2451. [Google Scholar] [CrossRef]
- Busca, G.; Lorenzelli, V. Infrared spectroscopic identification of species arising from reactive adsorption of carbon oxides on metal oxide surfaces. Mat. Chem. 1982, 7, 89–126. [Google Scholar] [CrossRef]
- Ramis, G.; Busca, G.; Lorenzelli, V. Low-temperature CO2 adsorption on metal oxides: Spectroscopic characterization of some weakly adsorbed species. Mat. Chem. Phys. 1991, 29, 425–435. [Google Scholar] [CrossRef]
- Solymosi, F. The bonding, structure and reactions of CO2 adsorbed on clean and promoted metal surfaces. J. Mol. Catal. 1991, 65, 337–358. [Google Scholar] [CrossRef]
- Freund, H.-J.; Roberts, M.W. Surface chemistry of carbon dioxide. Surf. Sci. Rep. 1996, 25, 225–273. [Google Scholar] [CrossRef]
- Taifan, W.; Boily, J.F.; Baltrusaitis, J. Surface chemistry of carbon dioxide revisited. Surf. Sci. Rep. 2016, 71, 595–671. [Google Scholar] [CrossRef]
- Liu, C.; Cundari, T.R.; Wilson, A.K. CO2 Reduction on Transition Metal (Fe, Co, Ni, and Cu) Surfaces: In Comparison with Homogeneous Catalysis. J. Phys. Chem. C 2012, 116, 5681–5688. [Google Scholar] [CrossRef]
- Liu, X.; Sun, L.; Deng, W.Q. Theoretical Investigation of CO2 Adsorption and Dissociation on Low Index Surfaces of Transition Metals. J. Phys. Chem. C 2018, 122, 8306–8314. [Google Scholar] [CrossRef]
- Ernst, K.-H.; Schlatterbeck, D.; Christmann, K. Adsorption of carbon dioxide on Cu(110) and on hydrogen and oxygen covered Cu(110) surfaces. Phys. Chem. Chem. Phys. 1999, 1, 4105–4112. [Google Scholar] [CrossRef]
- Bonicke, I.A.; Kirstein, W.; Thieme, F. A study on CO2 dissociation on a stepped (332) copper surface. Surf. Sci. 1994, 307–309, 177. [Google Scholar] [CrossRef]
- Cai, J.; Han, Y.; Chen, S.; Crumlin, E.J.; Yang, B.; Li, Y.; Liu, Z. CO2 Activation on Ni(111) and Ni(100) Surfaces in the Presence of H2O: An Ambient-Pressure X-ray Photoelectron Spectroscopy Study. J. Phys. Chem. C 2019, 123, 12176–12182. [Google Scholar] [CrossRef]
- Cong, V.Y.; Son, N.V.; Diem, D.Q.; Pham, S.Q.T. A comparison of water-gas shift reaction on ZnO (1010) surface and 6Cu cluster deposited over ZnO (1010) surface using density functional theory studies. J. Mol. Model. 2022, 28, 84. [Google Scholar] [CrossRef] [PubMed]
- Grabow, L.C.; Mavrikakis, M. Mechanism of Methanol Synthesis on Cu through CO2 and CO Hydrogenation. ACS Catal. 2011, 1, 365–384. [Google Scholar] [CrossRef]
- Palomino, R.M.; Ramírez, P.J.; Liu, Z.; Hamlyn, R.; Waluy, I.; Mahapatra, M.; Orozco, I.; Hunt, A.; Simonovis, J.P.; Senanayake, S.D.; et al. Hydrogenation of CO2 on ZnO/Cu(100) and ZnO/Cu(111) Catalysts: Role of Copper Structure and Metal–Oxide Interface in Methanol Synthesis. J. Phys. Chem. B 2018, 122, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Saussey, J.; Lavalley, J.C.; Lamotte, J.; Rais, T. I.r. spectroscopic evidence of formyl species formed by CO and H2 Co-adsorption on ZnO and Cu–ZnO. J. Chem. Soc. Chem. Commun. 1982, 278–279. [Google Scholar] [CrossRef]
- Enger, B.C.; Holmen, A. Nickel and Fischer-Tropsch Synthesis. Catal. Rev. 2012, 54, 437–488. [Google Scholar] [CrossRef]
- Sanchez-Escribano, V.; Larrubia Vargas, M.A.; Finocchio, E.; Busca, G. On the mechanisms and the selectivity determining steps in syngas conversion over supported metal catalysts: An IR study. Appl. Catal. A Gen. 2007, 316, 68–74. [Google Scholar] [CrossRef]
- Heine, C.; Lechner, B.A.J.; Bluhm, H.; Salmeron, M. Recycling of CO2: Probing the Chemical State of The Ni(111) Surface During the Methanation Reaction with Ambient-pressure X-ray Photoelectron Spectroscopy. J. Am. Chem. Soc. 2016, 138, 13246–13252. [Google Scholar] [CrossRef]
- Huang, M.-X.; Liu, F.; He, C.-C.; Yang, S.-Q.; Chen, W.Y.; Ouyang, L.; Zhao, L.; Yu, J. Interface promoted CO2 methanation: A theoretical study of Ni/La2O3. Chem. Phys. Lett. 2021, 768, 138396. [Google Scholar] [CrossRef]
- Montanari, T.; Castoldi, L.; Lietti, L.; Busca, G. Basic catalysis and catalysis assisted by basicity: FT-IR and TPD characterization of potassium-doped alumina. Appl. Catal. A Gen. 2011, 400, 61–69. [Google Scholar] [CrossRef]
- Fasolini, A.; Spennati, E.; Atakoohi, S.E.; Percivale, M.; Busca, G.; Basile, F.; Garbarino, G. A study of CO2 hydrogenation over Ni-MgAlOx catalysts derived from hydrotalcite precursors. Catal. Today 2003, 423, 114271. [Google Scholar] [CrossRef]
- Garbarino, G.; Wang, C.; Valsamakis, I.; Chitsazan, S.; Riani, P.; Finocchio, E.; Flytzani-Stephanopoulos, M.; Busca, G. Acido-basicity of lanthana/alumina catalysts and their activity in ethanol conversion. Appl. Catal. B Environ. 2017, 200, 458–468. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process | Main Component | Other Components | P Range | T Range | Feed Composition | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CO | H2 | CO2 | H2O | Other | |||||||||
| Phase | wt% | Comp. | Role | wt% | Bar | K | mol% | mol% | mol% | mol% | mol% | ||
| WGS | HT, high SR a | Fe3O4 | 70–90 | Cr2O3, | stabilizer | <10 | 30–50 | 650–720 | 5–10 | 30–40 | 3–8 | 30–40 | N2 < 15 |
| Cu | promoter | <3 | CH4 < 0.5 | ||||||||||
| MgO | promoter | <1 | Ar < 0.5 | ||||||||||
| HT, low SR a | ZnO-ZnAl2O4 | ~100 | 6–12 | 45–50 | 4–10 | 15–20 | N2 < 18 | ||||||
| CH4 < 0.6 | |||||||||||||
| Ar < 0.6 | |||||||||||||
| LT | Cu | >40 | ZnO | Activity promoter, stabilizer | 50–30 | 30–50 | 450–573 | 2–2.5 | 35–50 | 9–15 | 20–40 | N2 12–16 | |
| Al2O3 (in ZnO) | stabilizer | <20 | CH4 < 0.5 | ||||||||||
| Cs2O, Na2O | Selectivity promoter | <1 | Ar < 0.5 | ||||||||||
| Methanol synthesis | Cu | >50 | ZnO | Activity promoter, stabilizer | ~40 | 50–150 | 473–523 | 10–35 | 40–75 | 1–13 | <2 | CH4 < 15 | |
| Al2O3 (in ZnO) | stabilizer | <10 | |||||||||||
| MgO | promoter | ~2 | |||||||||||
| SiO2 | stabilizer | ||||||||||||
| Methanation | LT a | Ru | 0.3 | γ-Al2O3 | support | 99 | 30–50 | 440–550 | 0.5 | 75 | 0.2 | --- | N2 24 |
| Ni | 20–50 | γ-Al2O3 | support | <80 | 30–50 | 470–620 | CH4 0.2 | ||||||
| MgO, CaO, La2O3 | stabilizer | <20 | Ar 0.3 | ||||||||||
| HT | Ni | >20 | MgAl2O4 La2O3-Al2O3 CaO-Al2O3 | support, stabilizer | <80 | >30 | 500–970 | 25–35 | 35–75 | 1–30 | --- | CH4 < 10 | |
| FTS | LT | Co | 15–30 | γ-Al2O3 | support | <80 | 20–30 | 473–523 | 25–45 | 45–70 | 0–5 | <1 | N2 < 10 CH4 < 10 |
| Ru, Rh, Pt or Pd | activator | <0.1 | |||||||||||
| ZrO2, CeO2, La2O3 | promoter, stabilizer | <10 | |||||||||||
| CoC2 | Inert b | <6 | |||||||||||
| HT | Fe | 90 | SiO2 | promoter | <5 | 20–40 | 590–630 | 35–60 | 35–60 | 0–5 | <1 | N2 < 10 CH4 < 10 | |
| Cu | promoter | <5 | |||||||||||
| K2O | promoter | <5 | |||||||||||
| Fe carbides | active phases b | ||||||||||||
| Fe3O4 | |||||||||||||
| d-Band Center | Hydrogen Adsorption | Strongest CO Adsorption | Fastest CO Dissociation | Strongest CO2 Adsorption | Price | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔEFCC | ΔEontop | Eads | Face | Geometry | Eads | Eatt | Face | Eads | Face | Site | ||||
| eV | eV | eV | eV | USD/lb | ||||||||||
| Cu | Fcc | −2.67 | 0.07 | 0.62 | −0.68 | 011 | short bridge | 1.81 | 2.68 | 011 | 0.25 | 332 | terrace | 3.67 |
| Ni | Fcc | −1.29 | −0.37 | 0.19 | −1.84 | 111 | threefold | −0.07 | 1.64 | 001 | −0.38 | 332 | terrace | 7.79 |
| Co | Hcp | −1.17 | −0.31 | 0.28 | −1.58 | 11–20 | short bridge | 0.40 | 1.33 | 11–20 | −0.59 | 015 | terrace | 15.16 |
| Fe | Bcc | −0.92 | −0.54 | 0.23 | −2.20 | 011 | threefold | −1.38 | 1.08 | 011 | −1.23 | 321 | step | <<1 |
| Pd | Fcc | −1.83 | −0.42 | 0.11 | −1.77 | 111 | threefold | 1.40 | 2.83 | 001 | −0.10 | 332 | terrace | 16,289.28 |
| Ru | Hcp | −1.41 | −0.41 | −0.10 | −1.95 | 10–10 | terminal | 0.18 | 1.38 | 11–20 | −0.90 | 015 | step | 7440.00 |
| Pt | Fcc | −2.25 | −0.37 | −0.38 | −1.81 | 001 | bridge | 2.27 | 4.09 | 111 | −0.07 | 332 | terrace | 14,206.72 |
| Ref. | [173] | [174] | [174] | [175] | [175] | [175] | [175] | [175] | [175] | [176] | [176] | [176] | [177] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Busca, G.; Spennati, E.; Riani, P.; Garbarino, G. Mechanistic and Compositional Aspects of Industrial Catalysts for Selective CO2 Hydrogenation Processes. Catalysts 2024, 14, 95. https://doi.org/10.3390/catal14020095
Busca G, Spennati E, Riani P, Garbarino G. Mechanistic and Compositional Aspects of Industrial Catalysts for Selective CO2 Hydrogenation Processes. Catalysts. 2024; 14(2):95. https://doi.org/10.3390/catal14020095
Chicago/Turabian StyleBusca, Guido, Elena Spennati, Paola Riani, and Gabriella Garbarino. 2024. "Mechanistic and Compositional Aspects of Industrial Catalysts for Selective CO2 Hydrogenation Processes" Catalysts 14, no. 2: 95. https://doi.org/10.3390/catal14020095
APA StyleBusca, G., Spennati, E., Riani, P., & Garbarino, G. (2024). Mechanistic and Compositional Aspects of Industrial Catalysts for Selective CO2 Hydrogenation Processes. Catalysts, 14(2), 95. https://doi.org/10.3390/catal14020095