
Effects of the Electrodeposition Time in the Synthesis of Carbon-Supported Pt(Cu) and Pt-Ru(Cu) Core-Shell Electrocatalysts for Polymer Electrolye Fuel Cells


Abstract
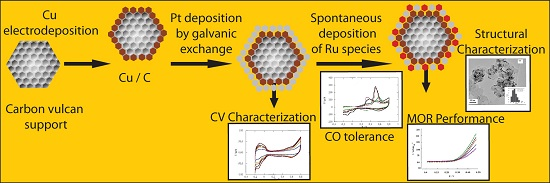
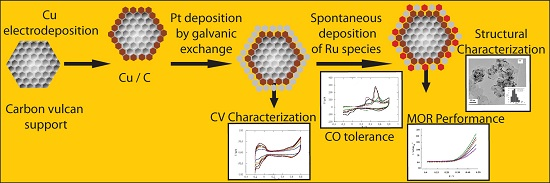
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
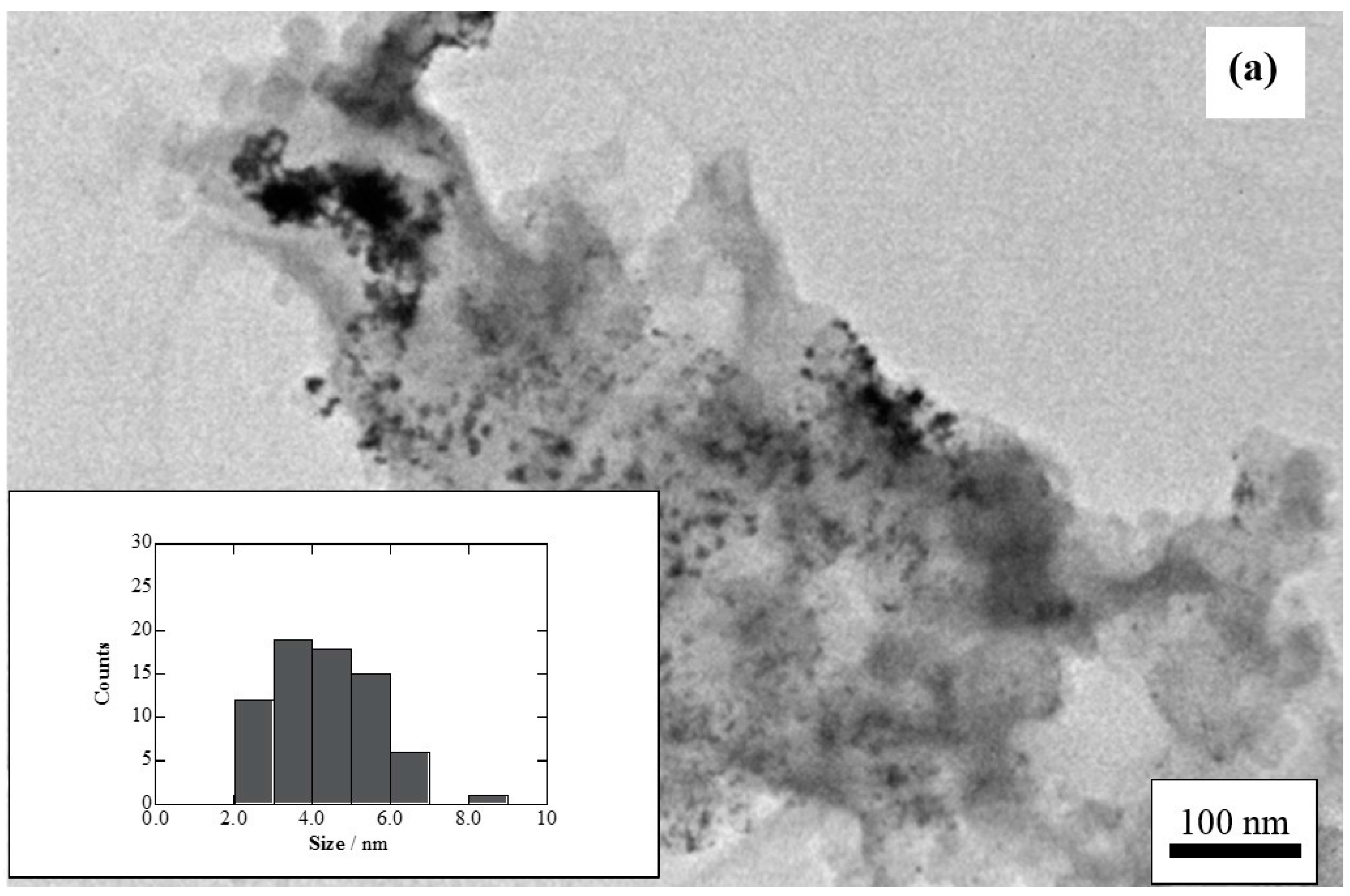
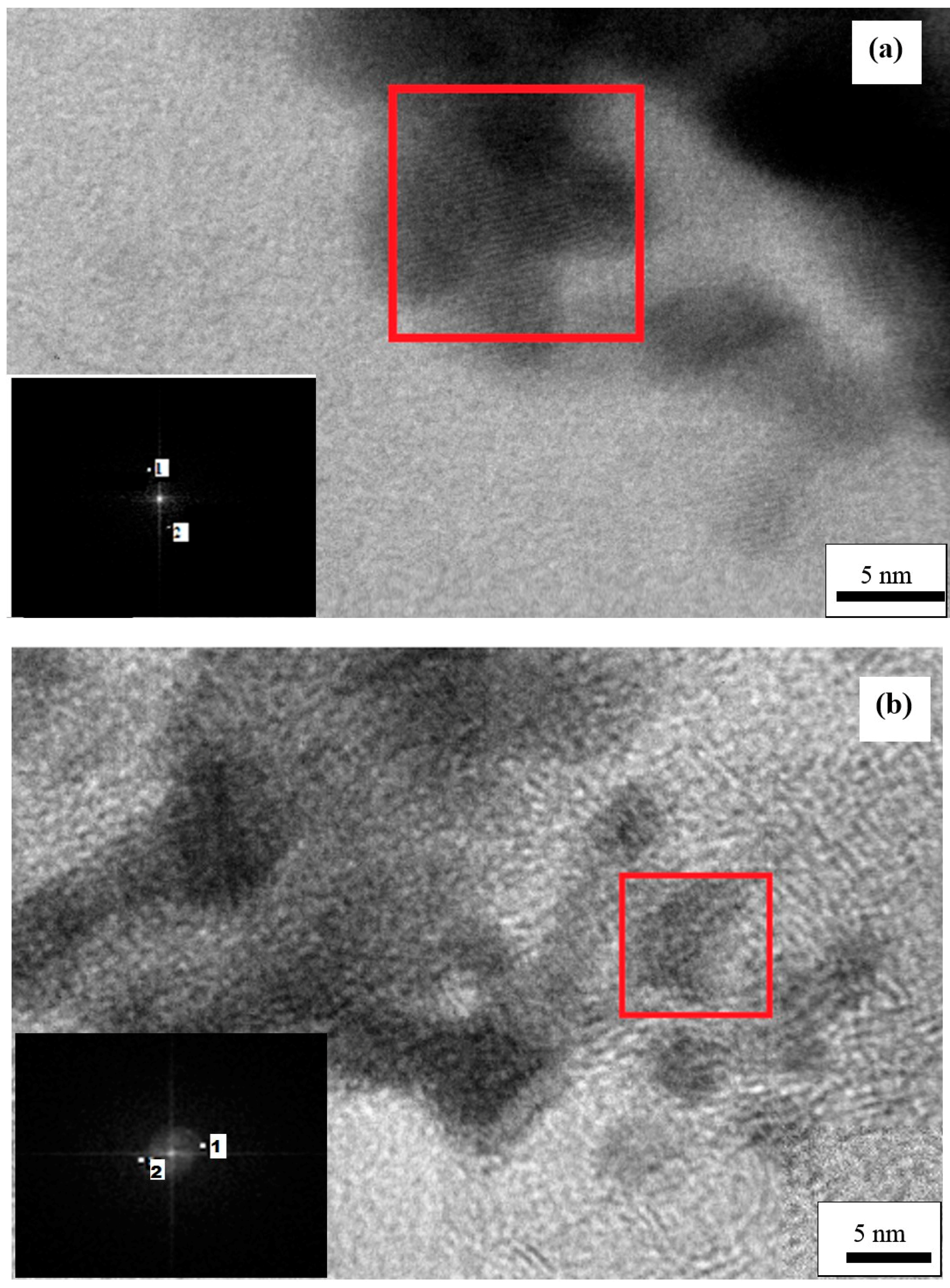
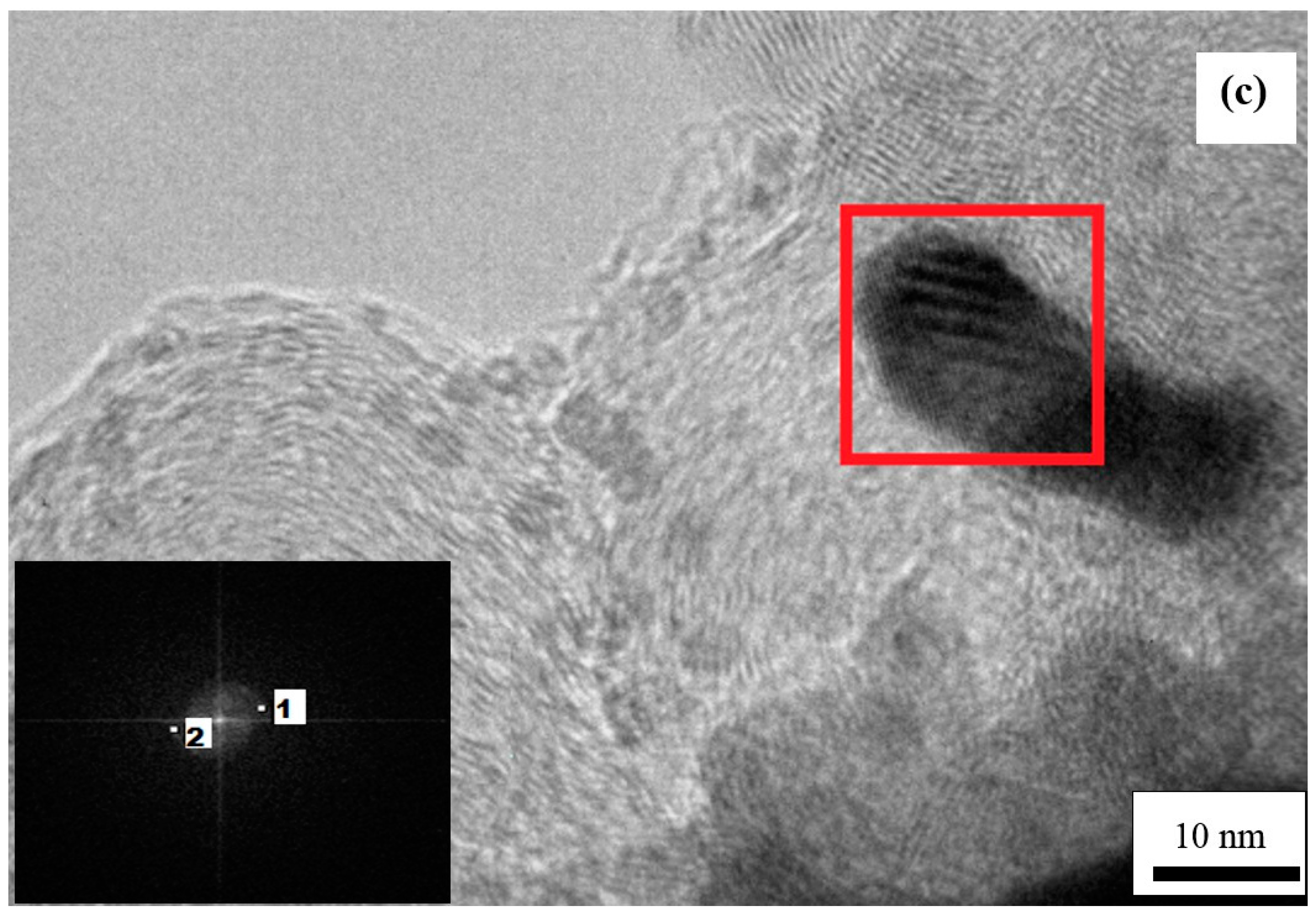
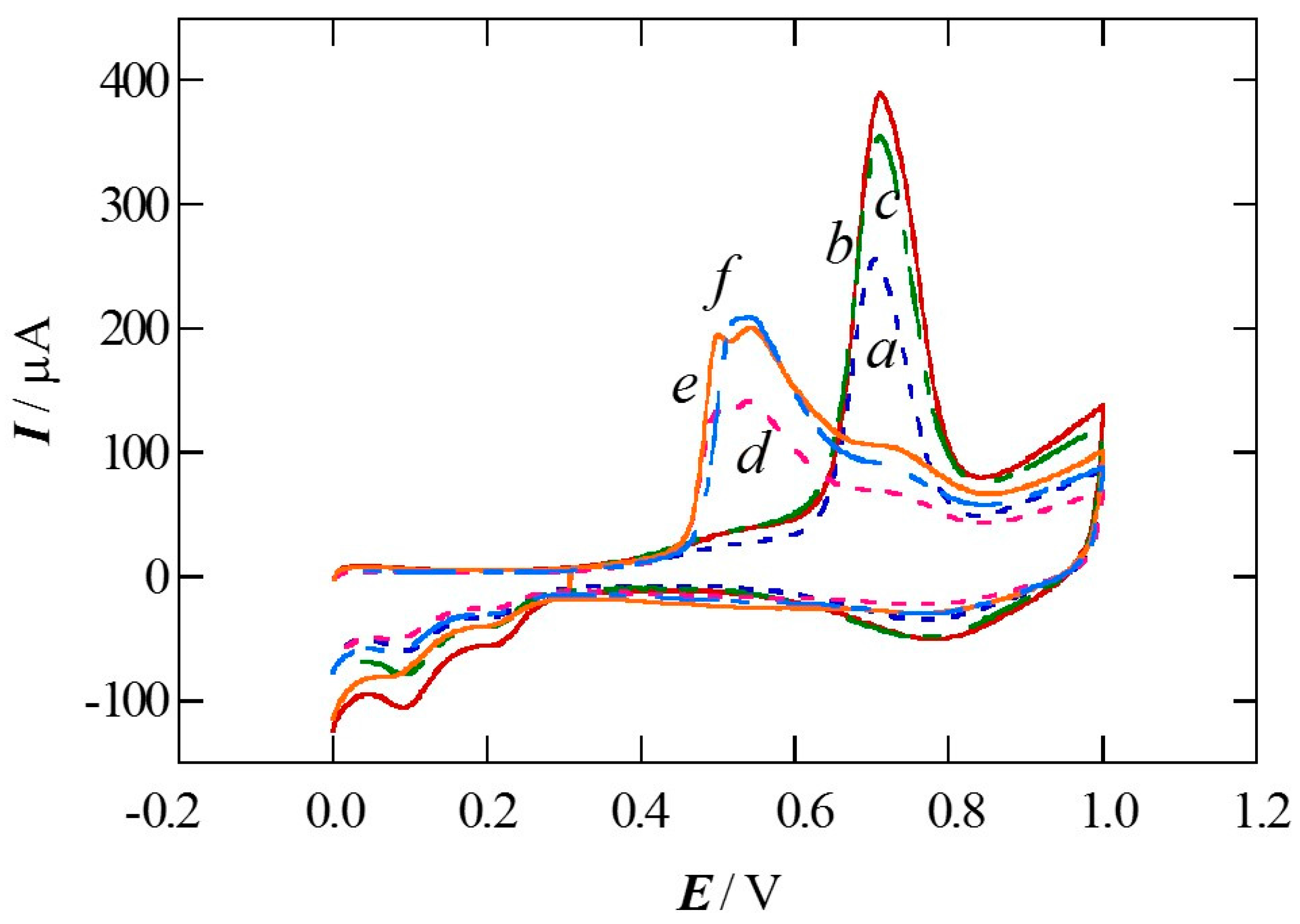
2. Results and Discussion
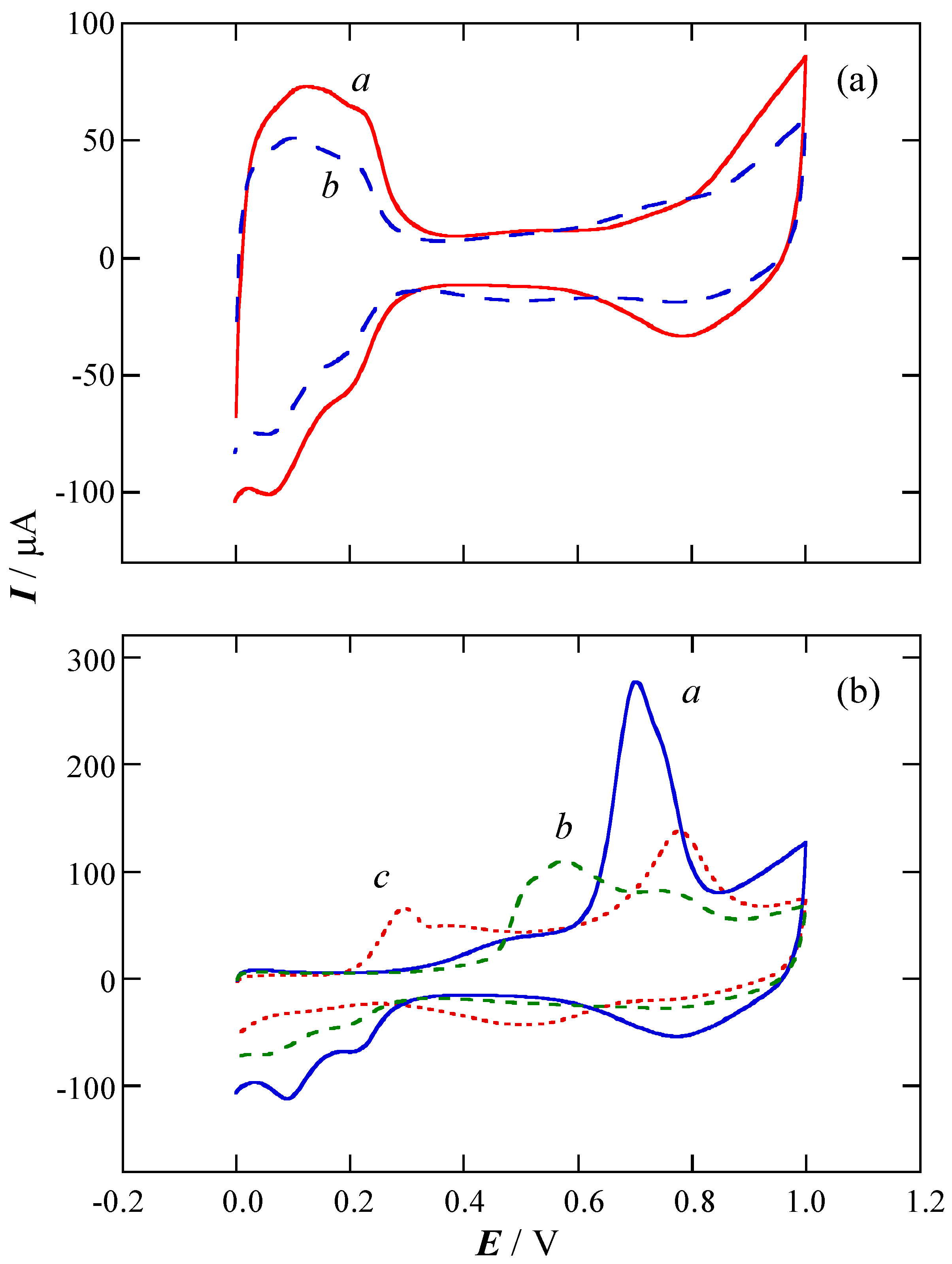
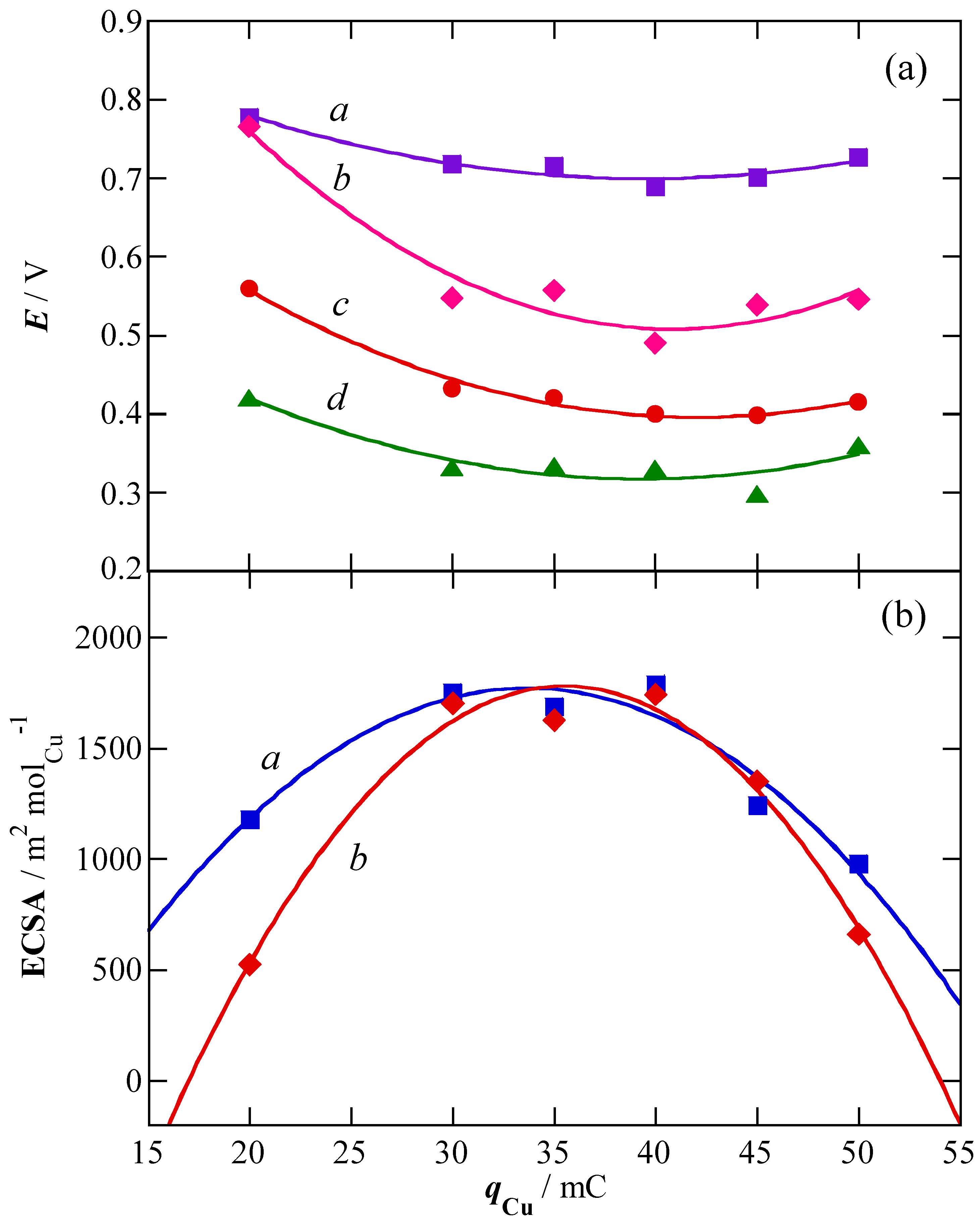
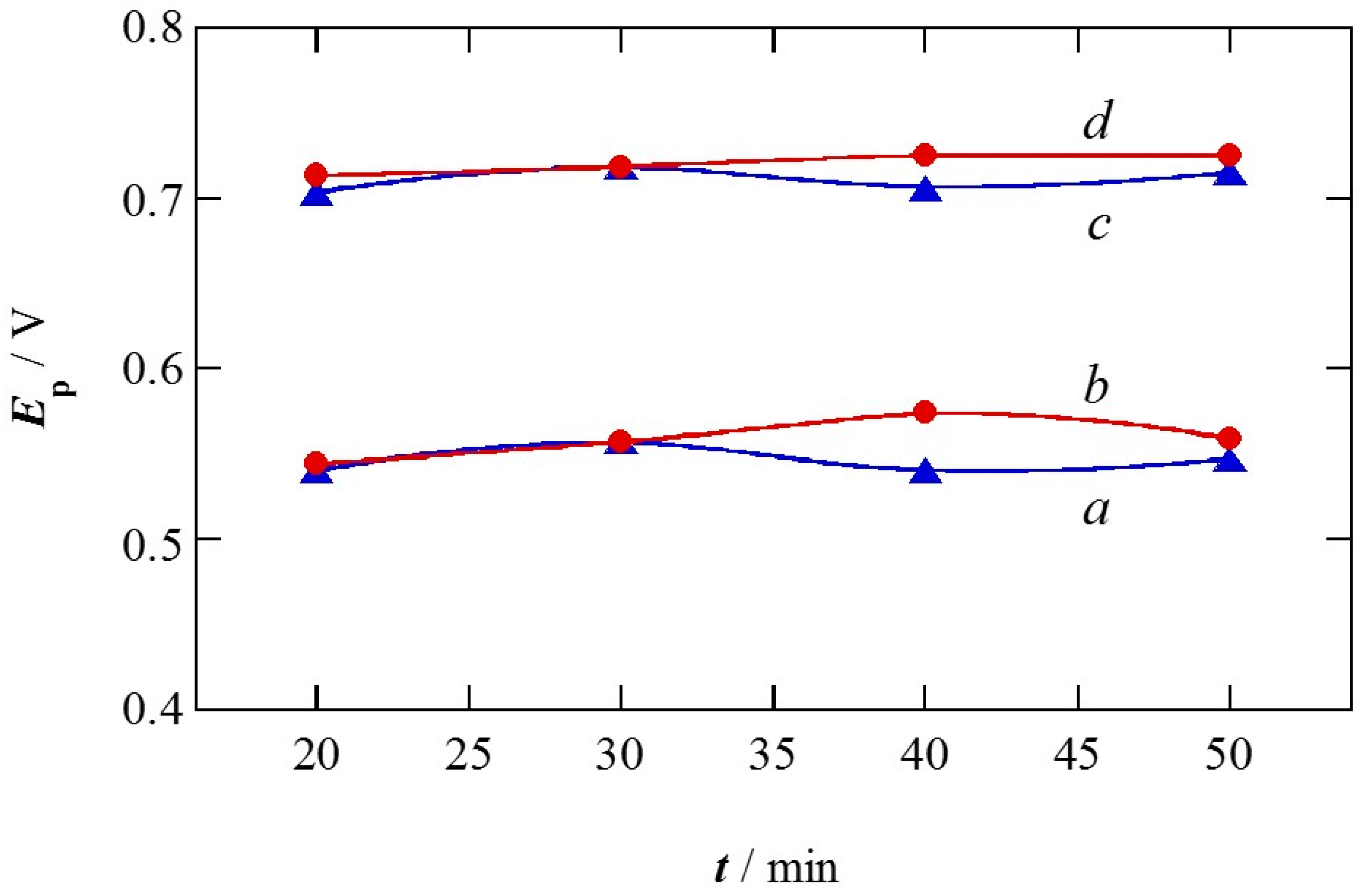
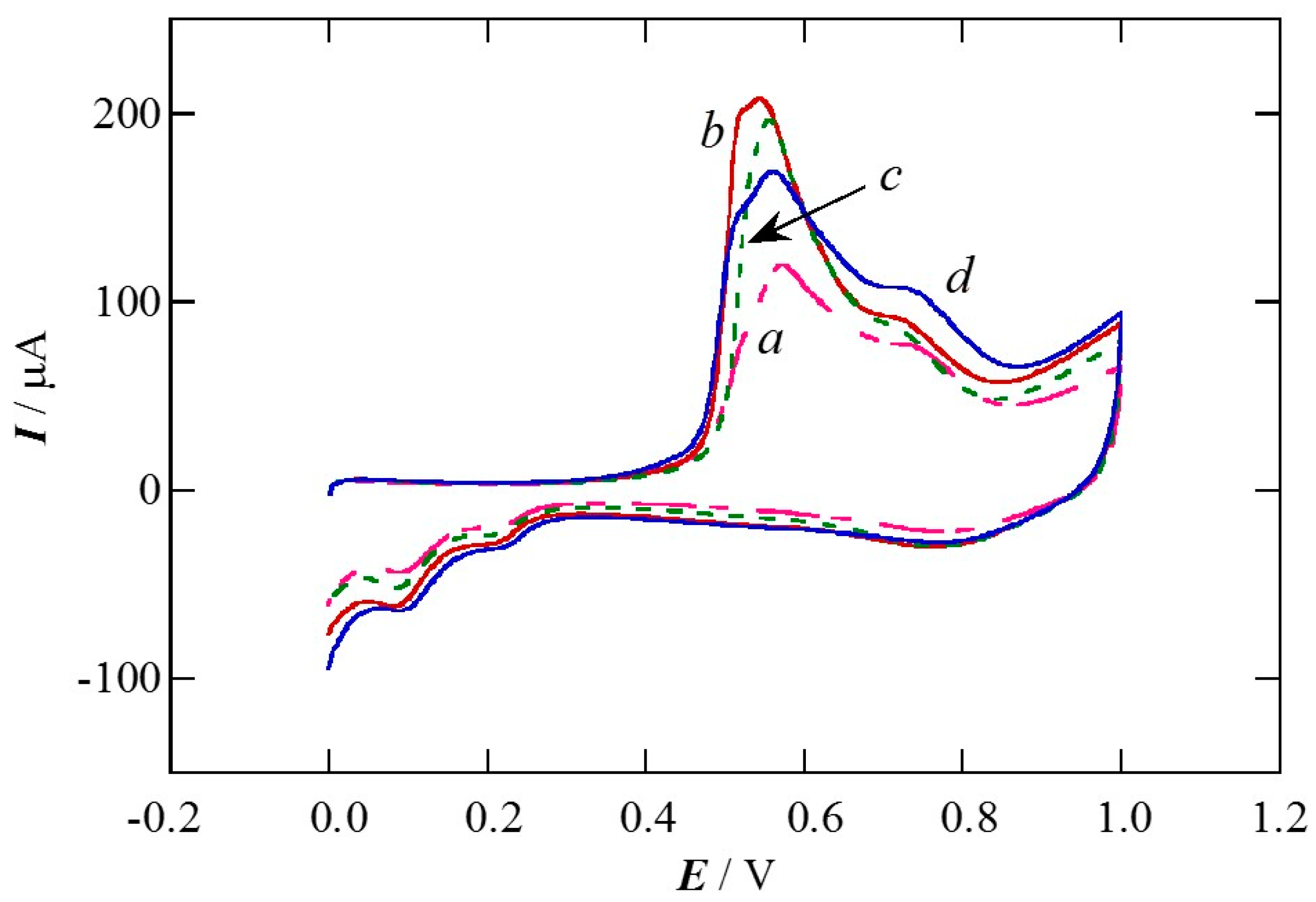
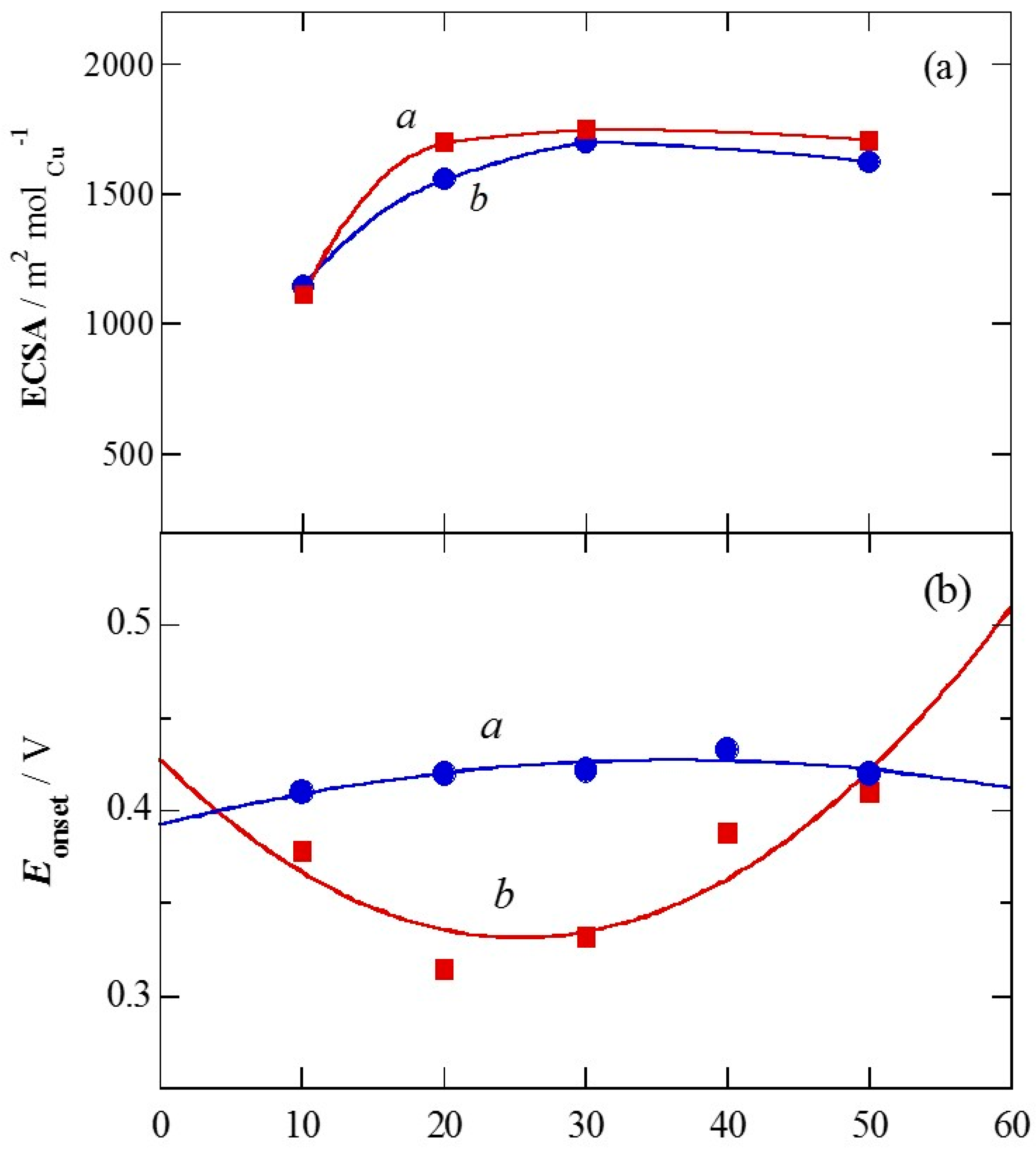
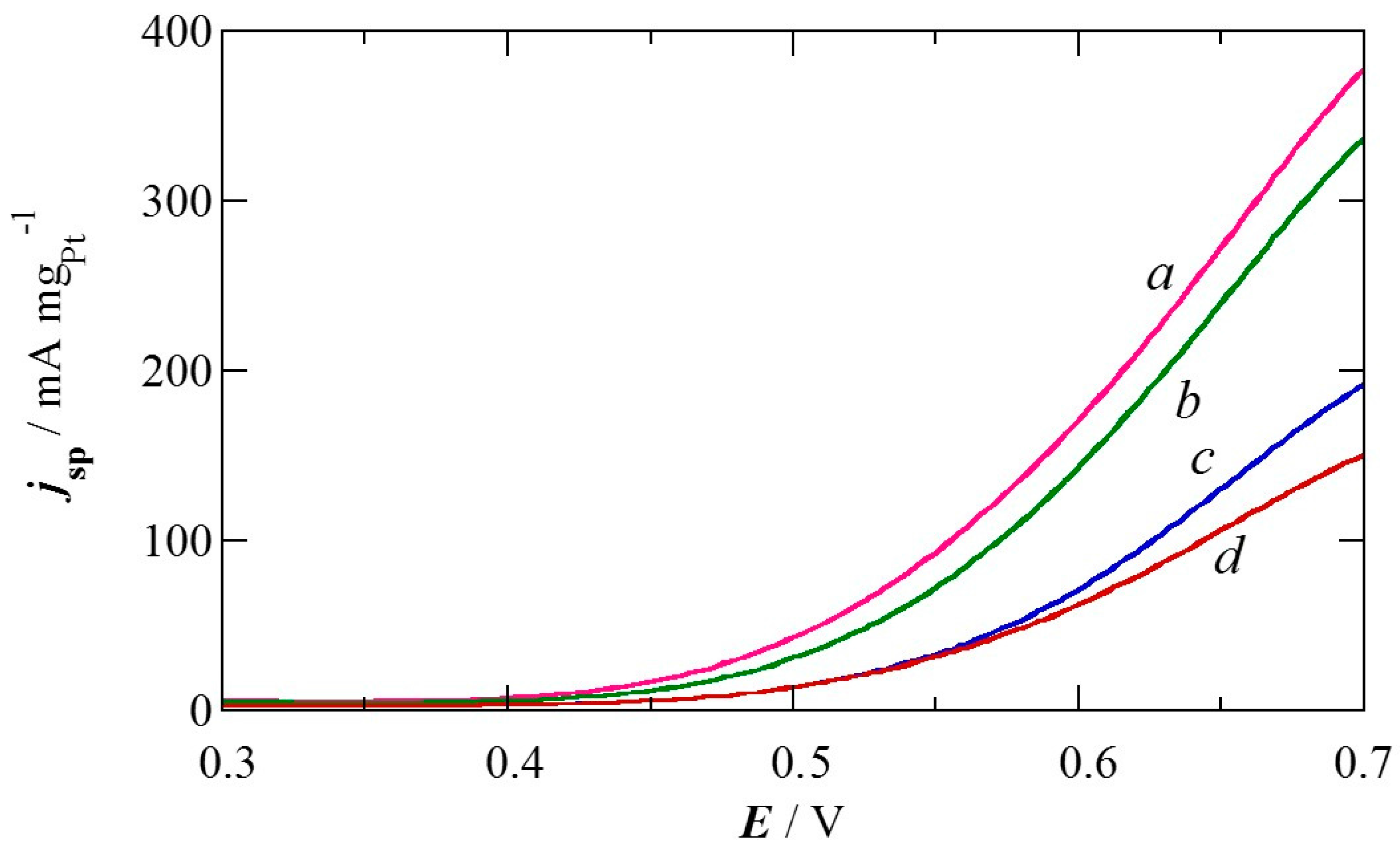
2.1. Charge of Cu Electrodeposition in the Synthesis Sequence i
2.2. Pt Deposition Time by Galvanic Exchange in the Synthesis Sequence ii
2.3. Spontaneous Deposition Time of Ru Species on Pt in the Synthesis Sequence iii
3. Experimental Section
3.1. Materials and Reagents
3.2. Working Electrodes
3.3. Electrochemical Testing
3.4. Structural Characterization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Costamagna, P.; Srinivasan, S. Quantum jumps in the PEMFC science and technology from the 1960s to the year 2000: Part 1. Fundamental scientific aspects. J. Power Sources 2001, 102, 242–252. [Google Scholar] [CrossRef]
- Liu, H.; Song, Ch.; Zhang, L.; Zhang, J.; Wang, H.; Wilkinson, D. A review of anode catalysis in the direct methanol fuel cell. J. Power Sources 2006, 155, 95–110. [Google Scholar] [CrossRef]
- Zainoodin, A.M.; Kamarudin, S.K.; Daub, W.R.W. Electrode in direct methanol fuel cells. Review. Int. J. Hydrogen Energy 2010, 35, 4606–4621. [Google Scholar] [CrossRef]
- Dos Santos, L.; Colmati, F.; González, E.R. Preparation and characterization of supported Pt-Ru catalysts with a high Ru content. J. Power Sources 2006, 159, 869–877. [Google Scholar] [CrossRef]
- Antolini, E. Platinum-based ternary catalysts for low temperature fuel cells: Part I. Preparation methods and structural characteristics. Appl. Catal. B Environ. 2007, 74, 324–336. [Google Scholar] [CrossRef]
- Iwasita, T. Methanol and CO electrooxidation. In Handbook of Fuel Cells-Fundamentals, Technology and Applications; Vielstich, W., Gasteiger, H.A., Lamm, A., Eds.; John Wiley & Sons: New York, NY, USA, 2003; Volume 3, pp. 603–622. [Google Scholar]
- Ruth, K.; Vogt, M.; Zuber, R. Development of CO-tolerant catalysts. In Handbook of Fuel Cells-Fundamentals, Technology and Applications; Vielstich, W., Gasteiger, H.A., Lamm, A., Eds.; John Wiley & Sons: New York, NY, USA, 2003; Volume 3, pp. 489–496. [Google Scholar]
- Lamy, C.; Lima, A.; LeRhun, V.; Delime, F.; Coutanceau, C.; Léger, J.M. Recent advances in the development of direct alcohol fuel cells (DAFC). J. Power Sources 2002, 105, 283–296. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Markovic, N.M.; Ross, N.P., Jr.; Cairns, E.J. CO electrooxidation on well-characterized Pt-Ru alloys. J. Phys. Chem. 1994, 98, 617–625. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Markovic, N.M.; Ross, P.N., Jr. H2 and CO electrooxidation on well-characterized Pt, Ru and Pt-Ru Rotating disk electrode studies of the pure gases including temperature effects. J. Phys. Chem. 1995, 99, 8290–8298. [Google Scholar] [CrossRef]
- Aricó, A.S.; Baglio, V.; Di Blasi, A.; Modica, E.; Antonucci, P.L.; Antonucci, V. Analysis of the high-temperature methanol oxidation behavior at carbon-supported Pt-Ru catalysts. J. Electroanal. Chem. 2003, 557, 167–176. [Google Scholar] [CrossRef]
- Tegou, A.; Papadimitriou, S.; Pavlidou, E.; Kokkinidis, G.; Sotiropoulos, S. Oxygen reduction at platinum- and gold-coated copper deposits on glassy carbon substrates. J. Electroanal. Chem. 2007, 608, 67–77. [Google Scholar] [CrossRef]
- Papadimitriou, S.; Tegou, A.; Pavlidou, E.; Armyanov, S.; Valova, E.; Kokkinidis, G.; Sotiropoulos, S. Preparation and characterization of platinum- and gold-coated copper, iron, cobalt and nickel deposits on glassy carbon substrates. Electrochim. Acta 2008, 53, 6559–6567. [Google Scholar] [CrossRef]
- Podlovchenko, B.I.; Gladysheva, T.D.; Filatov, Y.; Yashina, L.V. The use of galvanic displacement in synthesizing Pt(Cu) catalysts with the core-sell structure. Russ. J. Electrochem. 2010, 46, 1189–1197. [Google Scholar] [CrossRef]
- Kuznetsov, V.V.; Podlovchenko, B.I.; Kavyrshina, K.V.; Maksimov, Y.M. Oxidation of methanol on Pt(Mo) electrodes obtained using galvanic displacement method. Russ. J. Electrochem. 2010, 46, 1353–1359. [Google Scholar] [CrossRef]
- Podlovchenko, B.I.; Krivchenko, V.A.; Maksimov, Y.M.; Gladysheva, T.D.; Yashina, L.V.; Evlashin, S.A.; Pilevsky, A.A. Specific features of the formation on Pt(Cu) catalysts by galvanic displacement with carbon nanowalls used as support. Electrochim. Acta 2012, 76, 137–144. [Google Scholar] [CrossRef]
- Tegou, A.; Armyanov, S.; Valova, E.; Steenhaut, O.; Hubin, A.; Kokkinidis, G.; Sotiropoulus, S. Mixed platinum-gold electrocatalysts for borohydride oxidation prepared by the galvanic replacement of nickel deposits. J. Electroanal. Chem. 2009, 634, 104–110. [Google Scholar] [CrossRef]
- Caballero-Manrique, G.; Velázquez-Palenzuela, A.; Centellas, F.; Garrido, J.A.; Arias, C.; Rodríguez, R.M.; Brillas, E.; Cabot, P.L. Electrochemical synthesis and characterization of carbon-supported Pt and Pt-Ru nanoparticles with Cu cores for CO and methanol oxidation in polymer electrolyte fuel cells. Int. J. Hydrogen Energy 2014, 39, 12859–12869. [Google Scholar] [CrossRef]
- Mohl, M.; Kumar, A.; Reddy, A.L.M.; Kukovecz, A.; Konya, Z.; Kiricsi, I.; Vajtai, R.; Ajayan, P.M. Synthesis of catalytic porous metallic nanorods by galvanic exchange reaction. J. Phys. Chem. C 2010, 114, 389–393. [Google Scholar] [CrossRef]
- Mohl, M.; Dobo, D.; Kukovecz, A.; Konya, Z.; Kordas, K.; Wei, J.; Vajtai, R.; Ajayan, P.M. Formation of CuPd and CuPt bimetallic nanotubes by galvanic replacement reaction. J. Phys. Chem. C 2011, 115, 9403–9409. [Google Scholar] [CrossRef]
- Cheng, B.; Cheng, D.; Zhu, J. Synthesis of PtCu nanowires in nonoaqueous solvent with enhanced activity and stability for oxygen reduction reaction. J. Power Sources 2014, 267, 380–387. [Google Scholar] [CrossRef]
- Kokkinidis, G.; Papoutsis, A.; Stoychev, D.; Milchev, A. Electroless deposition of Pt on Ti—Catalytic activity for the hydrogen evolution reaction. J. Electroanal. Chem. 2000, 486, 48–55. [Google Scholar] [CrossRef]
- Kokkinidis, G.; Lazarov, V.; Papoutsis, A.; Stoychev, D.; Milchev, A. Electroless deposition of Pt on Ti: Part II. Catalytic activity for oxygen reduction. J. Electroanal. Chem. 2001, 511, 20–30. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, Y.; Xie, Y.; Yang, Z.; Huang, H.; Zhou, Q. Preparation of highly dispersed CuPt nanoparticles on ionic-liquid-assisted grapheme sheets for direct methanol fuel cell. Chem. Eng. J. 2012, 197, 80–87. [Google Scholar] [CrossRef]
- Jia, Y.; Jiang, Y.; Zhang, J.; Zhang, L.; Chen, Q.; Xie, Z.; Zheng, L. Unique excavated rhombic dodecahedral PtCu3 alloy nanocrystals constructed with ultrathin nanosheets of high-energy (110) facets. J. Am. Chem. Soc. 2014, 136, 3748–3751. [Google Scholar] [CrossRef] [PubMed]
- Geboes, B.; Mintsouli, I.; Wouters, B.; Georgieva, J.; Kakaroglou, A.; Sotiropoulos, S.; Valova, E.; Armyanov, S.; Hubin, A.; Breugelmans, T. Surface and electrochemical characterization of a Pt-Cu/C nano-structured electrocatalysts, prepared by galvanic displacement. Appl. Catal. B: Environ. 2014, 150–151, 249–256. [Google Scholar] [CrossRef]
- Ammam, M.; Easton, E.B. PtCu/C and Pt(Cu)/C catalysts: Synthesis, characterization and catalytic activity towards ethanol electrooxidation. J. Power Sources 2013, 222, 79–87. [Google Scholar] [CrossRef]
- Mintsouli, I.; Georgieva, J.; Armyanov, S.; Valova, E.; Avdeev, G.; Hubin, A.; Steenhaut, O.; Dille, J.; Tsiplakides, D.; Balomenou, S.; et al. Pt-Cu electrocatalysts for methanol oxidation prepared by partial galvanic replacement of Cu/carbon powder precursors. Appl. Catal. B: Environ. 2013, 136–137, 160–167. [Google Scholar] [CrossRef]
- Qiu, H.J.; Xu, H.T.; Li, X.; Wang, J.Q.; Wang, Y. Core-shell-structured nanoporous PtCu with high C content and enhanced catalytic performance. J. Mater. Chem. A 2015, 3, 7939–7944. [Google Scholar] [CrossRef]
- Huang, Y.; Zhao, T.; Zeng, L.; Tan, P.; Xu, J. A facile approach for preparation of highly dispersed platinum-copper/carbon nanocatalyst toward formic acid electro-oxidation. Electrochim. Acta 2016, 190, 956–963. [Google Scholar] [CrossRef]
- Solla-Gullón, J.; Vidal-Iglesias, F.J.; Herrero, E.; Feliu, J.M.; Aldaz, A. CO monolayer oxidation on semi-spherical and preferentially oriented (100) and (111) platinum nanoparticles. Electrochem. Commun. 2006, 8, 189–194. [Google Scholar] [CrossRef]
- Velázquez-Palenzuela, A.; Brillas, E.; Arias, C.; Centellas, F.; Garrido, J.A.; Rodríguez, R.M.; Cabot, P.L. Structural characterization of Ru-modified carbon-supported Pt nanoparticles using spontaneous deposition with CO oxidation activity. J. Phys. Chem. C 2012, 116, 18469–18478. [Google Scholar]
- MacDonald, J.P.; Gualtieri, B.; Runga, N.; Teliz, E.; Zinola, C.F. Modification of platinum surfaces by spontanoeous deposition: methanol oxidation electrocatalysis. Int. J. Hydrogen Energy 2008, 33, 7048–7061. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Markovic, N.; Ross, P.N., Jr.; Cairns, E.J. Temperature-dependent methanol electro-oxidation on well-characterized Pt-Ru alloys. J. Electrochem. Soc. 1994, 141, 1795–1803. [Google Scholar] [CrossRef]
- Sugimoto, W.; Yokoshima, K.; Murakami, Y.; Takasu, Y. Charge storage mechanism of nanostructured anhydrous and hydrous ruthenium-based oxides. Electrochim. Acta 2006, 52, 1742–1748. [Google Scholar] [CrossRef]
- Velázquez-Palenzuela, A.; Brillas, E.; Arias, C.; Centellas, F.; Garrido, J.A.; Rodríguez, R.M.; Cabot, P.L. Carbon monoxide, methanol and ethanol electro-oxidation on Ru decorated carbon-supported Pt nanoparticles prepared by spontaneous deposition. J. Power Sources 2013, 225, 163–171. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Gasteiger, H.A.; Stäb, G.D.; Urban, P.M.; Kolb, D.M.; Behm, R.J. Characterization of high-surface-area electrocatalysts using a rotating disk electrode configuration. J. Electrochem. Soc. 1998, 145, 2354–2358. [Google Scholar] [CrossRef]
- Gómez de la Fuente, J.L.; Martínez-Huerta, M.V.; Rojas, S.; Hernández-Fernández, P.; Terreros, P.; Fierrro, J.L.G.; Peña, M.A. Tailoring and structure of PtRu nanoparticles supported on functionalized carbon for DMFC applications: New evidence of the hydrous ruthenium oxide phase. Appl. Catal. B Environ. 2009, 88, 505–514. [Google Scholar] [CrossRef]
- Chrzanowski, W.; Wieckowski, A. Ultrathin films of ruthenium on low index platinum single crystal surfaces: An electrochemical study. Langmuir 1997, 13, 5974–5978. [Google Scholar] [CrossRef]
- WWW-MINCRYST. Crystallographic and Crystallochemical Database for Minerals and their Structural Analogues. Institute of Experimental Mineralogy. Russian Academy of Sciences. Available online: http://database.iem.ac.ru/mincryst/index.php (accessed on 7 January 2016).
- Rolison, D.R.; Hagans, P.L.; Swider, K.E.; Long, J.W. Role of hydrous ruthenium oxide in Pt-Ru direct methanol fuel cell anode electrocatalysts. The importance of mixed electron/proton conductivity. Langmuir 1999, 15, 774–779. [Google Scholar] [CrossRef]
- Cabot Corporation. Specialty Chemicals and Performance Materials. Available online: http://www.cabotcorp.com (accessed on 10 October 2015).
- Maillard, F.; Savinova, E.; Stimming, U. CO monolayer oxidation on Pt nanoparticles: Further insights into the particle size effects. J. Electroanal. Chem. 2007, 599, 221–232. [Google Scholar] [CrossRef]











© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caballero-Manrique, G.; Nadeem, I.M.; Brillas, E.; Centellas, F.; Garrido, J.A.; Rodríguez, R.M.; Cabot, P.-L. Effects of the Electrodeposition Time in the Synthesis of Carbon-Supported Pt(Cu) and Pt-Ru(Cu) Core-Shell Electrocatalysts for Polymer Electrolye Fuel Cells. Catalysts 2016, 6, 125. https://doi.org/10.3390/catal6080125
Caballero-Manrique G, Nadeem IM, Brillas E, Centellas F, Garrido JA, Rodríguez RM, Cabot P-L. Effects of the Electrodeposition Time in the Synthesis of Carbon-Supported Pt(Cu) and Pt-Ru(Cu) Core-Shell Electrocatalysts for Polymer Electrolye Fuel Cells. Catalysts. 2016; 6(8):125. https://doi.org/10.3390/catal6080125
Chicago/Turabian StyleCaballero-Manrique, Griselda, Immad Muhammed Nadeem, Enric Brillas, Francesc Centellas, José Antonio Garrido, Rosa María Rodríguez, and Pere-Lluís Cabot. 2016. "Effects of the Electrodeposition Time in the Synthesis of Carbon-Supported Pt(Cu) and Pt-Ru(Cu) Core-Shell Electrocatalysts for Polymer Electrolye Fuel Cells" Catalysts 6, no. 8: 125. https://doi.org/10.3390/catal6080125
APA StyleCaballero-Manrique, G., Nadeem, I. M., Brillas, E., Centellas, F., Garrido, J. A., Rodríguez, R. M., & Cabot, P. -L. (2016). Effects of the Electrodeposition Time in the Synthesis of Carbon-Supported Pt(Cu) and Pt-Ru(Cu) Core-Shell Electrocatalysts for Polymer Electrolye Fuel Cells. Catalysts, 6(8), 125. https://doi.org/10.3390/catal6080125