Figure 1.
DFT(B3LYP) optimized structures, after simulated annealing at DFTB level, of the different nanospheres considered in this work. For each one, the stoichiometry, the approximate diameter and the position of the Ti atoms with different coordination are reported (Ti4c in red, Ti5c in green, Ti6c in black, Ti3c(OH) in magenta and Ti4c(OH) in cyan).
Figure 1.
DFT(B3LYP) optimized structures, after simulated annealing at DFTB level, of the different nanospheres considered in this work. For each one, the stoichiometry, the approximate diameter and the position of the Ti atoms with different coordination are reported (Ti4c in red, Ti5c in green, Ti6c in black, Ti3c(OH) in magenta and Ti4c(OH) in cyan).
Figure 2.
Distances distribution (simulated EXAFS) computed with DFT(B3LYP) and DFTB for bulk anatase (a,b), computed with DFT(B3LYP) for the: 1.5 nm (c); 2.2 nm (e); 3.0 nm (g); and 4.4 nm (i) nanospheres; and computed with DFTB for the: 1.5 nm (d); 2.2 nm (f); 3.0 nm (h); and 4.4 nm (j) nanospheres.
Figure 2.
Distances distribution (simulated EXAFS) computed with DFT(B3LYP) and DFTB for bulk anatase (a,b), computed with DFT(B3LYP) for the: 1.5 nm (c); 2.2 nm (e); 3.0 nm (g); and 4.4 nm (i) nanospheres; and computed with DFTB for the: 1.5 nm (d); 2.2 nm (f); 3.0 nm (h); and 4.4 nm (j) nanospheres.
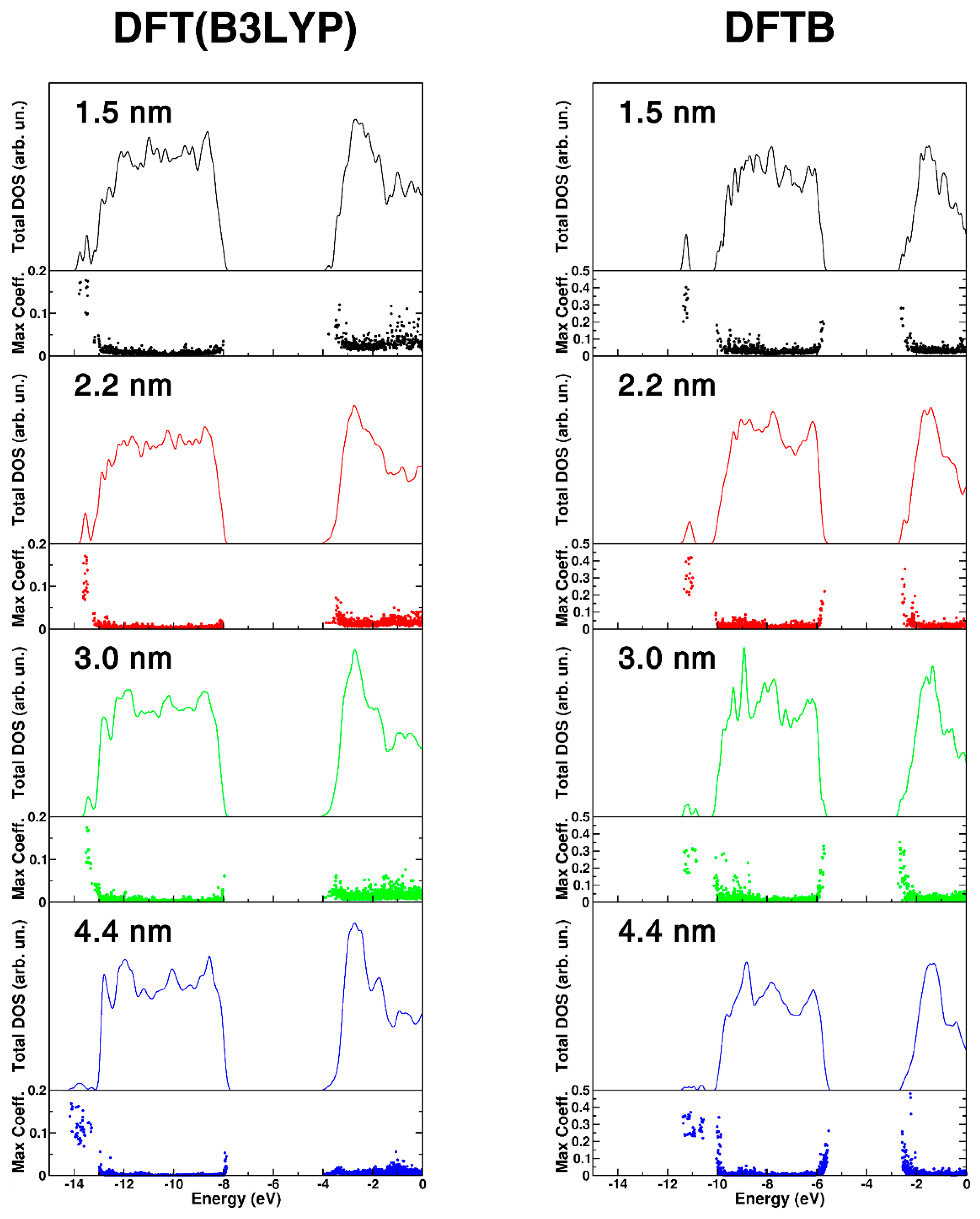
Figure 3.
DFT(B3LYP) and DFTB total (DOS) density of states for different size nanosphere, 1.5 nm (black), 2.2 nm (red), 3.0 nm (green), and 4.4 nm (blue). For each nanosphere, the DOS has been normalized to the number of TiO2 units to have comparable DOS intensities. The maximum atomic orbital coefficient (maxc) of each eigenstate is also reported. High values of maxc correspond to localized states, while low values correspond to delocalized states.
Figure 3.
DFT(B3LYP) and DFTB total (DOS) density of states for different size nanosphere, 1.5 nm (black), 2.2 nm (red), 3.0 nm (green), and 4.4 nm (blue). For each nanosphere, the DOS has been normalized to the number of TiO2 units to have comparable DOS intensities. The maximum atomic orbital coefficient (maxc) of each eigenstate is also reported. High values of maxc correspond to localized states, while low values correspond to delocalized states.
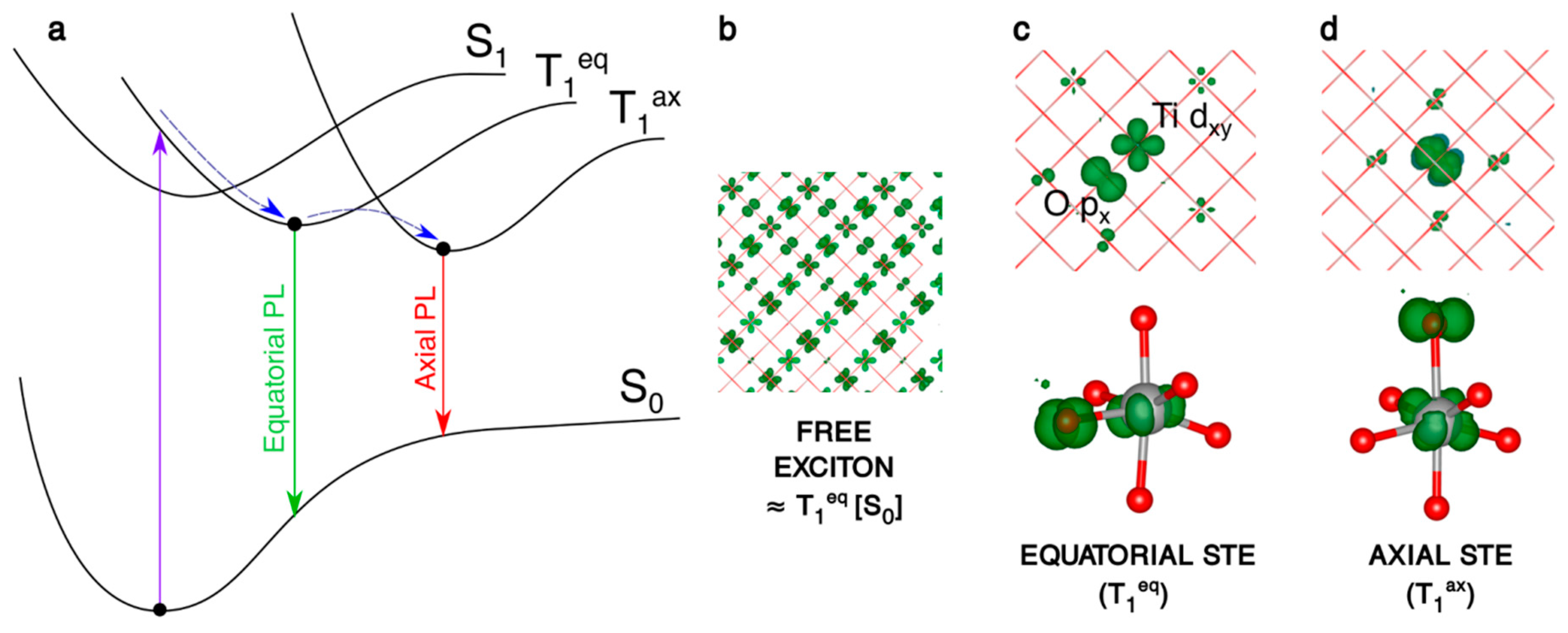
Figure 4.
(a) Schematic representation of the processes involving the electron/hole couple: the vertical excitation S0 → T1, the self-trapping relaxation in the bulk structure and the two different photoemissions T1eq → S0 and T1ax → S0. (b–d) 3D spin density plots of the anatase bulk supercell, as obtained with the B3LYP functional, for the vertical triplet state (b), trapped triplet equatorial (c) and axial (d) exciton in the bulk. The spin density isovalue is 0.01 a.u. (0.0005 a.u. for the vertical triplet).
Figure 4.
(a) Schematic representation of the processes involving the electron/hole couple: the vertical excitation S0 → T1, the self-trapping relaxation in the bulk structure and the two different photoemissions T1eq → S0 and T1ax → S0. (b–d) 3D spin density plots of the anatase bulk supercell, as obtained with the B3LYP functional, for the vertical triplet state (b), trapped triplet equatorial (c) and axial (d) exciton in the bulk. The spin density isovalue is 0.01 a.u. (0.0005 a.u. for the vertical triplet).
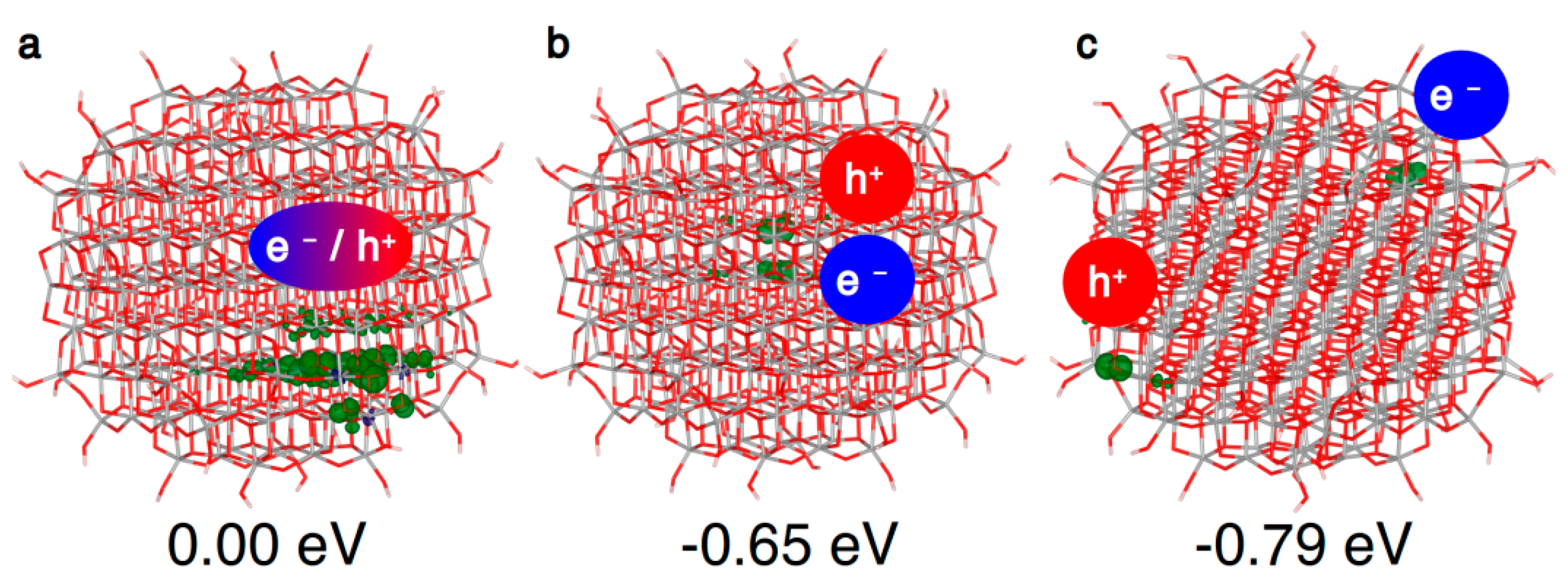
Figure 5.
3D spin density plots of the spherical TiO2 nanoparticle, as obtained with the B3LYP functional, for the vertical triplet state (a), trapped triplet exciton (b) and state (c) with the hole and the electron at the best trapping sites. The spin density isovalue is 0.01 a.u. (0.002 a.u. for the vertical triplet).
Figure 5.
3D spin density plots of the spherical TiO2 nanoparticle, as obtained with the B3LYP functional, for the vertical triplet state (a), trapped triplet exciton (b) and state (c) with the hole and the electron at the best trapping sites. The spin density isovalue is 0.01 a.u. (0.002 a.u. for the vertical triplet).
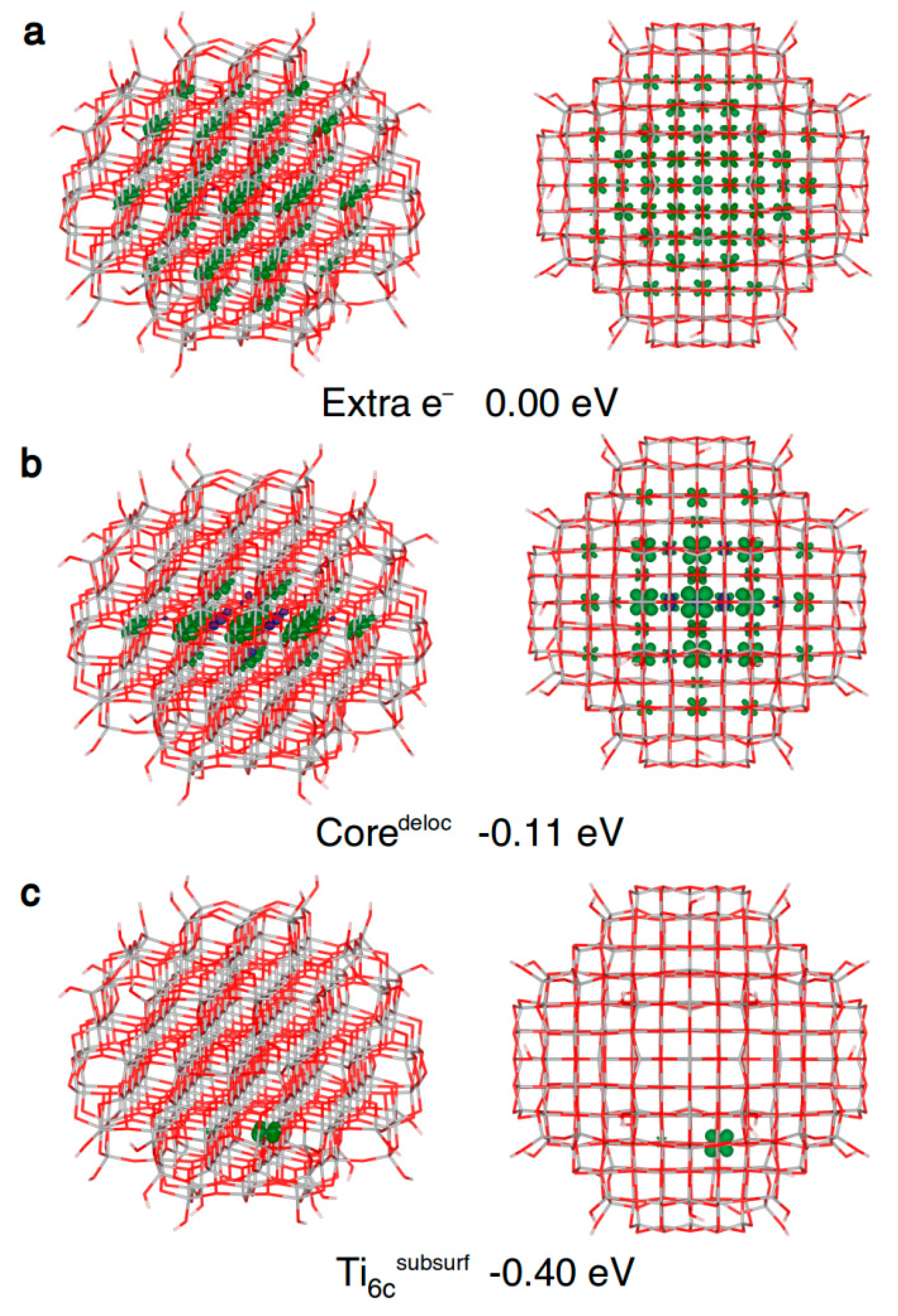
Figure 6.
Front and top view of the 3D plots of spin density of: (a) an extra electron added to the neutral ground state structure (isovalue = 0.001 a.u.); (b) a trapped delocalized electron (isovalue = 0.005 a.u.); and (c) an electron trapped on a subsurface six-fold coordinated titanium atom (isovalue = 0.001 a.u.) in the anatase nanosphere model, as obtained with the B3LYP functional. Below each structure, the energy gain (ΔEtrap) relative to (a) is given.
Figure 6.
Front and top view of the 3D plots of spin density of: (a) an extra electron added to the neutral ground state structure (isovalue = 0.001 a.u.); (b) a trapped delocalized electron (isovalue = 0.005 a.u.); and (c) an electron trapped on a subsurface six-fold coordinated titanium atom (isovalue = 0.001 a.u.) in the anatase nanosphere model, as obtained with the B3LYP functional. Below each structure, the energy gain (ΔEtrap) relative to (a) is given.
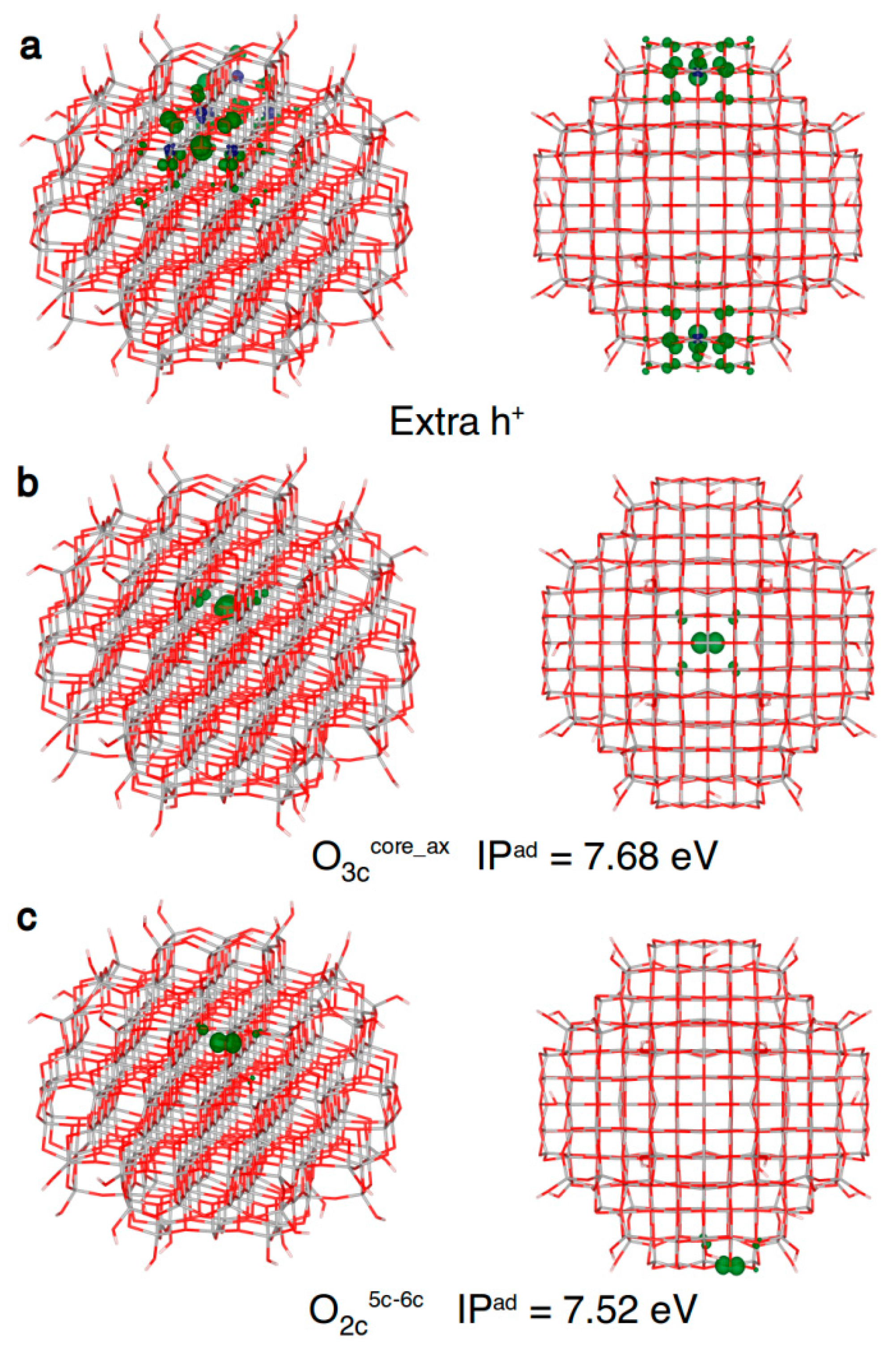
Figure 7.
Front and top view of the 3D plots of spin density of: (a) an excess hole resulting from a vertical ionization of the model (isovalue = 0.001 a.u.); (b) a trapped hole in the core (isovalue = 0.01 a.u.); and (c) a hole trapped on a two-fold coordinated oxygen atom (isovalue = 0.01 a.u.) in the anatase nanosphere model, as obtained with the B3LYP functional. Below the structures with a trapped hole the adiabatic ionization potential (IPad) as a measure of the hole trapping ability is given.
Figure 7.
Front and top view of the 3D plots of spin density of: (a) an excess hole resulting from a vertical ionization of the model (isovalue = 0.001 a.u.); (b) a trapped hole in the core (isovalue = 0.01 a.u.); and (c) a hole trapped on a two-fold coordinated oxygen atom (isovalue = 0.01 a.u.) in the anatase nanosphere model, as obtained with the B3LYP functional. Below the structures with a trapped hole the adiabatic ionization potential (IPad) as a measure of the hole trapping ability is given.
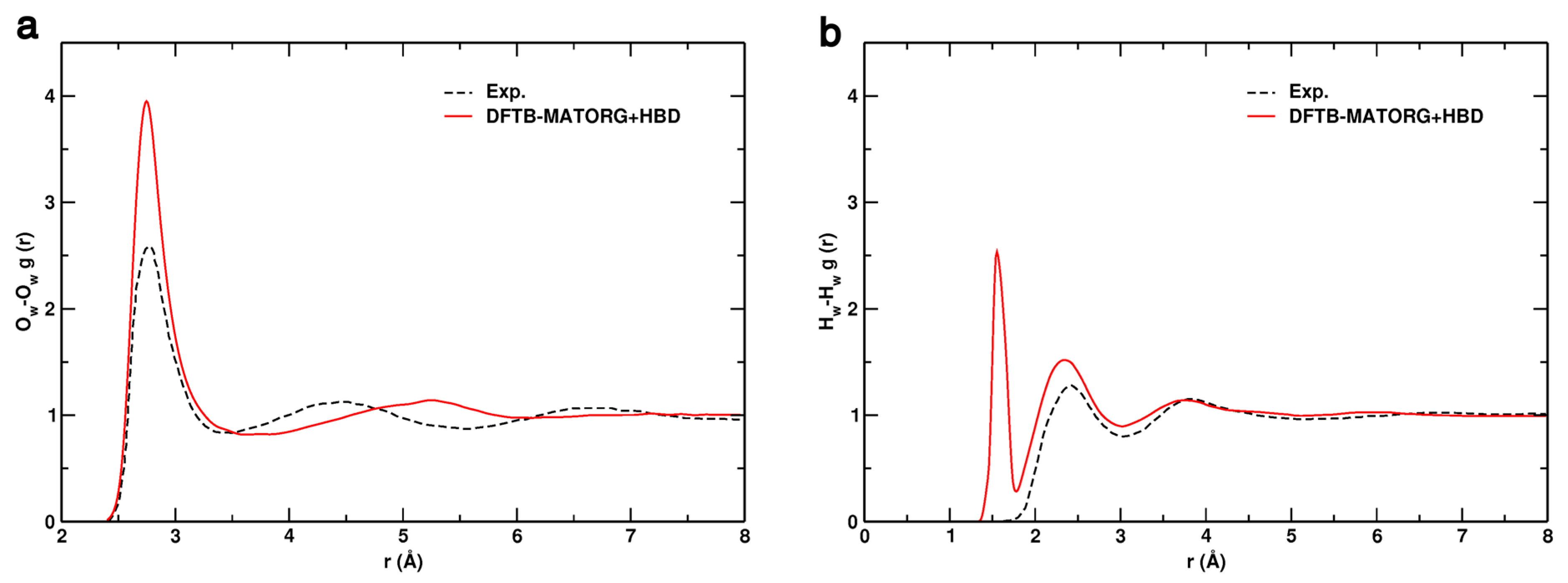
Figure 8.
Comparison of the: oxygen–oxygen (Ow-Ow) (a); and hydrogen–hydrogen (Hw-Hw) (b) radial distribution functions (RDF) obtained experimentally (dashed black line) and calculated with the DFTB-MATORG+HBD method.
Figure 8.
Comparison of the: oxygen–oxygen (Ow-Ow) (a); and hydrogen–hydrogen (Hw-Hw) (b) radial distribution functions (RDF) obtained experimentally (dashed black line) and calculated with the DFTB-MATORG+HBD method.
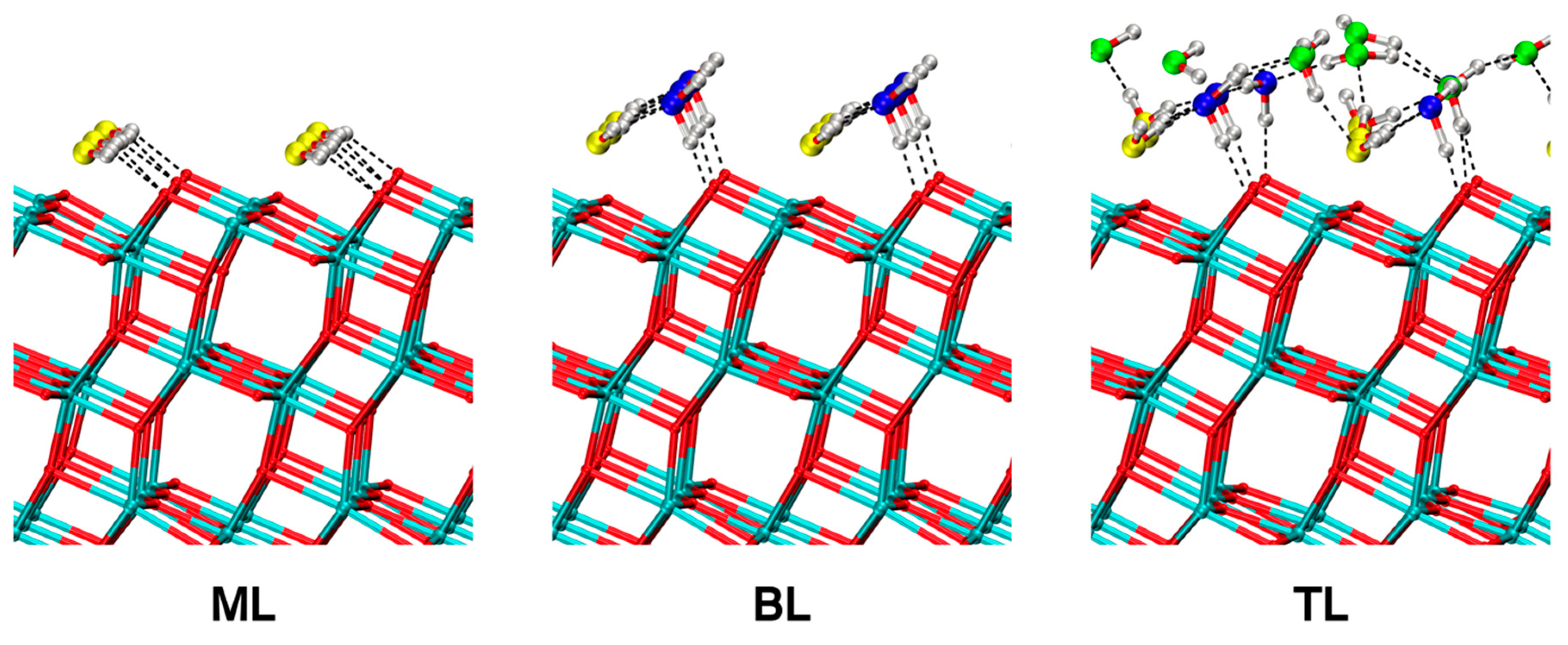
Figure 9.
DFTB-MATORG+HBD structures of a monolayer (ML), a bilayer (BL) and a trilayer (TL) of water on the (101) TiO2 anatase surface. Dashed lines correspond to H-bonds.
Figure 9.
DFTB-MATORG+HBD structures of a monolayer (ML), a bilayer (BL) and a trilayer (TL) of water on the (101) TiO2 anatase surface. Dashed lines correspond to H-bonds.
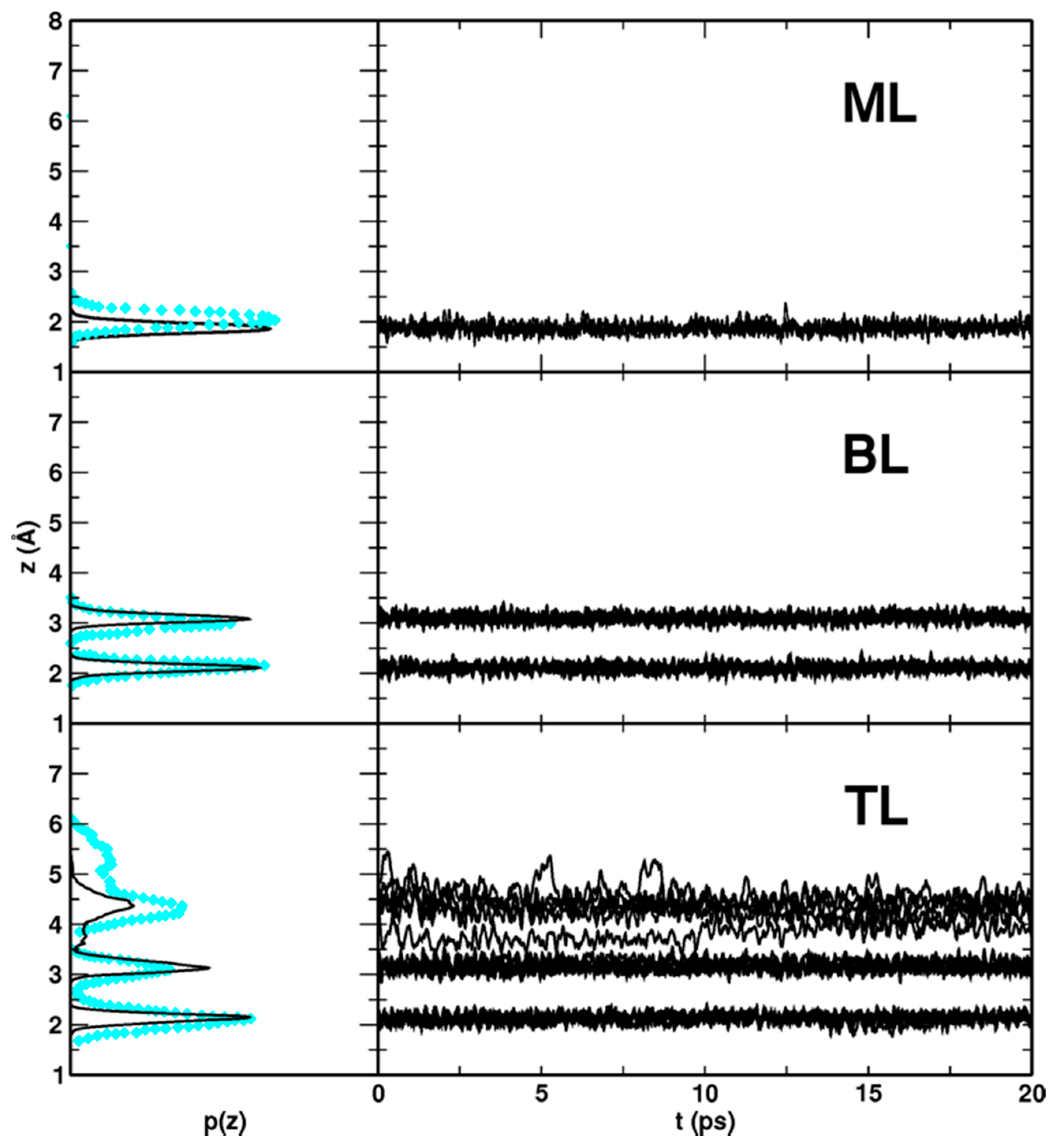
Figure 10.
DFTB-MATORG+HBD distribution p(z) and time evolution z(t) of the distances between the water molecules (O atoms) of the monolayer (ML), bilayer (BL) and trilayer (TL) and the titania surface (Ti5c atoms). In cyan diamonds, values calculated with DFT(PBE) Car–Parrinello simulations are shown.
Figure 10.
DFTB-MATORG+HBD distribution p(z) and time evolution z(t) of the distances between the water molecules (O atoms) of the monolayer (ML), bilayer (BL) and trilayer (TL) and the titania surface (Ti5c atoms). In cyan diamonds, values calculated with DFT(PBE) Car–Parrinello simulations are shown.
Table 1.
Amount of Ti atoms with different coordination for the variously sized nanospheres, as optimized at DFTB level, in terms of their number and percentage with respect to the total number of Ti atoms, is reported. Corresponding values for the optimized nanosphere at the DFT(B3LYP) level are in parenthesis, only when different from DFTB.
Table 1.
Amount of Ti atoms with different coordination for the variously sized nanospheres, as optimized at DFTB level, in terms of their number and percentage with respect to the total number of Ti atoms, is reported. Corresponding values for the optimized nanosphere at the DFT(B3LYP) level are in parenthesis, only when different from DFTB.
| DFTB [DFT(B3LYP)] | Number | % | Number | % | Number | % | Number | % |
|---|
| Ti Site | 1.5 nm | 2.2 nm | 3.0 nm | 4.4 nm |
|---|
| Ti4c | 20 | 19.8 | 36 | 16.1 | 53 | 13.3 | 106 | 8.4 |
| (19) | (18.8) |
| Ti5c | 20 | 19.8 | 43 | 19.2 | 69 | 17.3 | 159 | 12.6 |
| (21) | (20.8) | (49) | (22.0) | (65) | (16.3) |
| Ti6c_sup | 20 | 19.8 | 28 | 12.6 | 72 | 18 | 157 | 12.4 |
| (24) | (10.8) | (75) | (18.8) |
| Ti6c | 29 | 28.7 | 96 | 43.1 | 181 | 43.4 | 791 | 62.5 |
| (94] | (42.1) | (182) | (45.6) |
| Ti3c(OH) | 8 | 7.9 | 8 | 3.6 | 16 | 4 | 20 | 1.6 |
| Ti4c(OH) | 4 | 4 | 12 | 5.4 | 8 | 2 | 32 | 2.5 |
Table 2.
HOMO-LUMO electronic gap () and electronic BAND gap (expressed in eV) calculated for nanospheres of different size and for bulk anatase with both DFT(B3LYP) and DFTB methods.
Table 2.
HOMO-LUMO electronic gap () and electronic BAND gap (expressed in eV) calculated for nanospheres of different size and for bulk anatase with both DFT(B3LYP) and DFTB methods.
| MODEL | | |
|---|
| Nanospheres | DFT(B3LYP) | DFTB | DFT(B3LYP) | DFTB |
|---|
| 1.5 nm | 4.23 | 3.12 | 4.81 | 3.62 |
| 2.2 nm | 4.13 | 3.11 | 4.31 | 3.55 |
| 3.0 nm | 4.00 | 2.95 | 4.13 | 3.42 |
| 4.4 nm | 3.92 | 2.95 | 3.96 | 3.33 |
| BULK | - | - | 3.81 | 3.22 |
Table 3.
Trapping Energy (ΔEtrap) of the exciton in the triplet state and the corresponding photoluminescence (PL) in the axial and equatorial configuration for bulk anatase and for the nanosphere. All energies are in eV.
Table 3.
Trapping Energy (ΔEtrap) of the exciton in the triplet state and the corresponding photoluminescence (PL) in the axial and equatorial configuration for bulk anatase and for the nanosphere. All energies are in eV.
| | | Bulk | Nanosphere |
|---|
| Ti3+-Oax− | ΔEtrap | −0.59 | −0.65 |
| PL | 1.99 | 1.96 |
| Ti3+-Oeq− | ΔEtrap | −0.49 | |
| PL | 2.35 | |
Table 4.
Trapping energy (ΔEtrap) for electrons at different sites in the spherical anatase nanoparticles, as obtained with B3LYP functional. The reference zero for ΔEtrap is obtained by adding one electron to the nanosphere in its neutral ground state geometry, with no atomic relaxation. The charge localization (%electron) is also given. The sites nomenclature is defined in the text.
Table 4.
Trapping energy (ΔEtrap) for electrons at different sites in the spherical anatase nanoparticles, as obtained with B3LYP functional. The reference zero for ΔEtrap is obtained by adding one electron to the nanosphere in its neutral ground state geometry, with no atomic relaxation. The charge localization (%electron) is also given. The sites nomenclature is defined in the text.
| Position | ΔEtrap (eV) | %electron |
|---|
| Ti4cequator | −0.09 | 88% |
| Ti4c(OH) | No trapping | |
| Ti5c | No trapping | |
| Ti6csubsurf | −0.40 | 85% |
| Ti6ccore | −0.13 | 62% |
| Coredeloc | −0.11 | 19% |
Table 5.
Adiabatic ionization potential (IPad) for holes at different sites of the spherical anatase nanoparticle, as obtained with B3LYP functional. The charge localization (%hole) is also reported. The sites nomenclature is defined in the text.
Table 5.
Adiabatic ionization potential (IPad) for holes at different sites of the spherical anatase nanoparticle, as obtained with B3LYP functional. The charge localization (%hole) is also reported. The sites nomenclature is defined in the text.
| Position | IPad (eV) | %hole |
|---|
| O2c4c–6c | 7.74 | 89%/11% |
| O2c5c–6c | 7.52 | 92%/8% |
| O3ccore_ax | 7.68 | 90% |
| Ti3c(OH) | No trapping |
| Ti4c(OH) | No trapping |
| Ti5c(OH) | 7.81 | 85%/15% |
Table 6.
Lattice parameters a and c and their c/a ratio for bulk TiO2 anatase. Values computed with the DFTB and DFT methods are reported and compared with the experimental values. In parenthesis, the absolute errors referred to the experimental data are shown.
Table 6.
Lattice parameters a and c and their c/a ratio for bulk TiO2 anatase. Values computed with the DFTB and DFT methods are reported and compared with the experimental values. In parenthesis, the absolute errors referred to the experimental data are shown.
| Method | Reference | (Å) | (Å) | (Å) |
|---|
| DFTB-MATORG+HBD | This work | 3.796 (+0.014) | 9.790 (+0.288) | 2.579 (+0.067) |
| DFT(PBE) | This work | 3.789 (+0.007) | 9.612 (+0.110) | 2.537 (+0.025) |
| DFT(PBE) | Ref. [81] | 3.786 (+0.004) | 9.737 (+0.235) | 2.572 (+0.060) |
| DFT(B3LYP) | Ref. [82] | 3.783 (+0.001) | 9.805 (+0.303) | 2.592 (+0.080) |
| DFT(B3LYP) | Ref. [17] | 3.789 (+0.007) | 9.777 (+0.275) | 2.580 (+0.068) |
| DFT(HSE06) | Ref. [17] | 3.766 (−0.016) | 9.663 (+0.161) | 2.566 (+0.054) |
| Exp. | Ref. [47] | 3.782 | 9.502 | 2.512 |
Table 7.
Water dimer binding energy () and oxygen–oxygen distance (RO-O). Values obtained with DFTB (MATORG+HBD), higher-level methods (CCSD, PBE and B3LYP) and experiments are reported. In parenthesis, the absolute errors referred to the experimental data are shown.
Table 7.
Water dimer binding energy () and oxygen–oxygen distance (RO-O). Values obtained with DFTB (MATORG+HBD), higher-level methods (CCSD, PBE and B3LYP) and experiments are reported. In parenthesis, the absolute errors referred to the experimental data are shown.
| Method | Reference | (eV) | RO-O (Å) |
|---|
| DFTB-MATORG+HBD | This work | 0.199 (–0.037) | 2.815 (−0.157) |
| DFT(PBE) | Ref. [83] | 0.222 | 2.889 |
| DFT(B3LYP) | Ref. [83] | 0.198 | 2.926 |
| CCSD | Ref. [84] | 0.218 | 2.912 |
| Exp. | Refs. [85,86] | 0.236 | 2.972 |
Table 8.
Values calculated with DFT and DFTB methods of the adsorption energies per molecule () of water on the TiO2 (101) anatase slab in the molecular (H2O) and dissociated (OH, H) state. Different coverages are considered (low, θ = 0.25 and full, θ = 1). The experimental adsorption energy of the water monolayer on the (101) surface is also reported. The absolute errors (in parenthesis) reported for DFTB are calculated with respect to the PBE values from this work.
Table 8.
Values calculated with DFT and DFTB methods of the adsorption energies per molecule () of water on the TiO2 (101) anatase slab in the molecular (H2O) and dissociated (OH, H) state. Different coverages are considered (low, θ = 0.25 and full, θ = 1). The experimental adsorption energy of the water monolayer on the (101) surface is also reported. The absolute errors (in parenthesis) reported for DFTB are calculated with respect to the PBE values from this work.
| Method | Reference | Coverage, θ | , H2O (eV) | , OH, H (eV) |
|---|
| DFTB-MATORG | This work | 0.25 | –1.08 (+0.41) | –0.54 (+0.22) |
| 1 | –0.96 (+0.34) | –0.58 (+0.15) |
| DFTB-MATORG+HBD | This work | 0.25 | –0.80 (+0.13) | –0.31 (–0.01) |
| 1 | –0.71 (+0.09) | –0.40 (–0.03) |
| DFT(PBE) | This work | 0.25 | –0.67 | –0.32 |
| 1 | –0.62 | –0.43 |
| DFT(PBE) | Ref. [88] | 0.25 | –0.74 | –0.23 |
| 1 | –0.72 | –0.44 |
| Exp. | Refs. [89,90] | 1 | –0.5/–0.7 | |
Table 9.
Values calculated with DFT and DFTB methods of the binding energy per molecule ( in eV) of the water monolayer (ML), bilayer (BL) and trilayer (TL) on the (101) TiO2 anatase surface after an optimization run from the last snapshot of the MD simulation. The binding energy ( is defined as the difference between the total energy of the titania/water interface equilibrium structure and the sum of the total energy of six isolated water molecules plus the total energy of the optimized slab with one water layer less.
Table 9.
Values calculated with DFT and DFTB methods of the binding energy per molecule ( in eV) of the water monolayer (ML), bilayer (BL) and trilayer (TL) on the (101) TiO2 anatase surface after an optimization run from the last snapshot of the MD simulation. The binding energy ( is defined as the difference between the total energy of the titania/water interface equilibrium structure and the sum of the total energy of six isolated water molecules plus the total energy of the optimized slab with one water layer less.
| Water Configuration | | (eV) | |
|---|
| DFTB-MATORG+HBD | DFT(PBE) a | DFT(PBE) b |
|---|
| ML | –0.70 | –0.62 | –0.69 |
| BL | –0.73 | –0.67 | –0.65 |
| TL | –0.53 | –0.53 | –0.56 |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}