
Reactor Design for CO2 Photo-Hydrogenation toward Solar Fuels under Ambient Temperature and Pressure


Abstract
:
1. Introduction
2. Results and Discussion
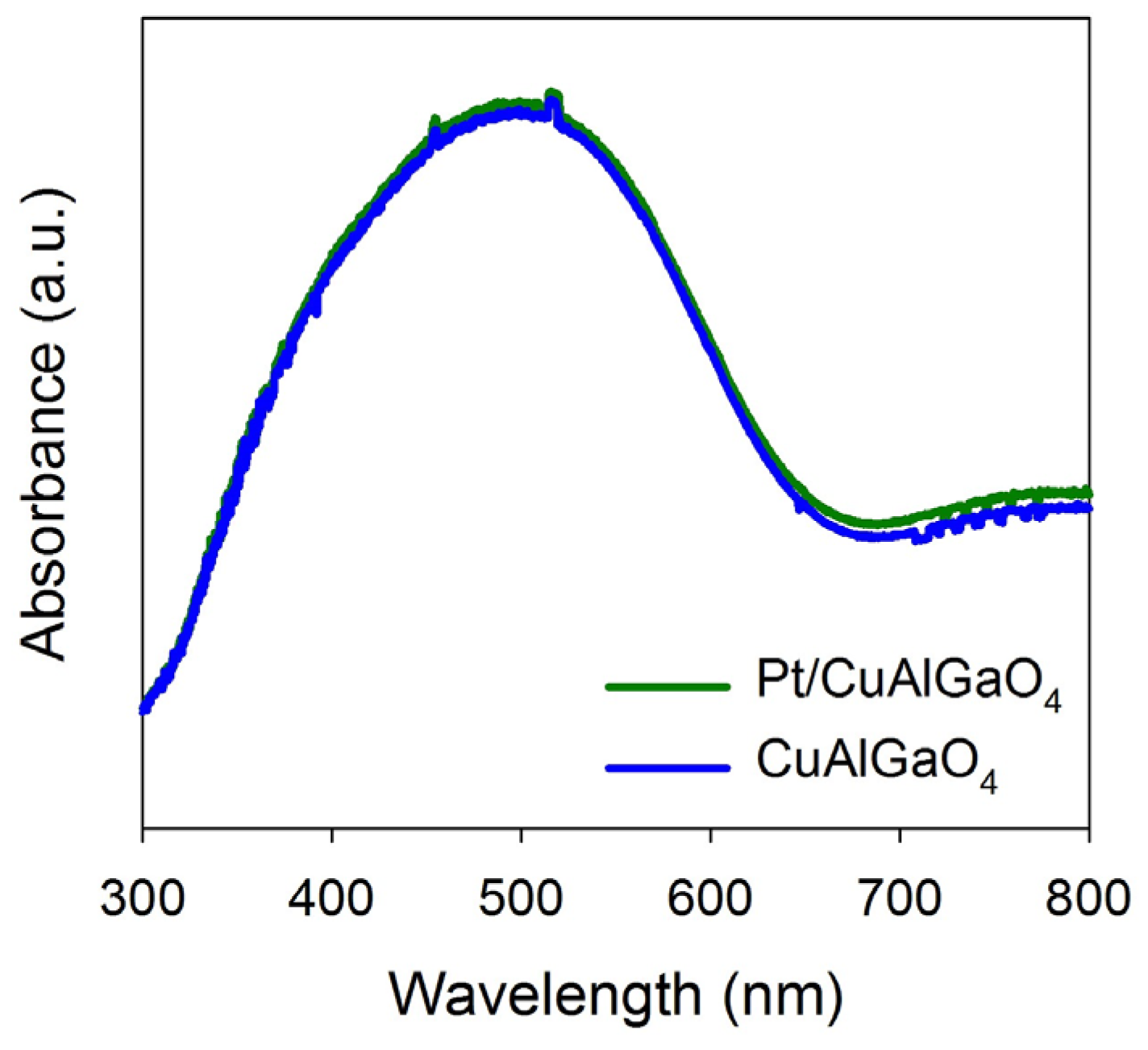
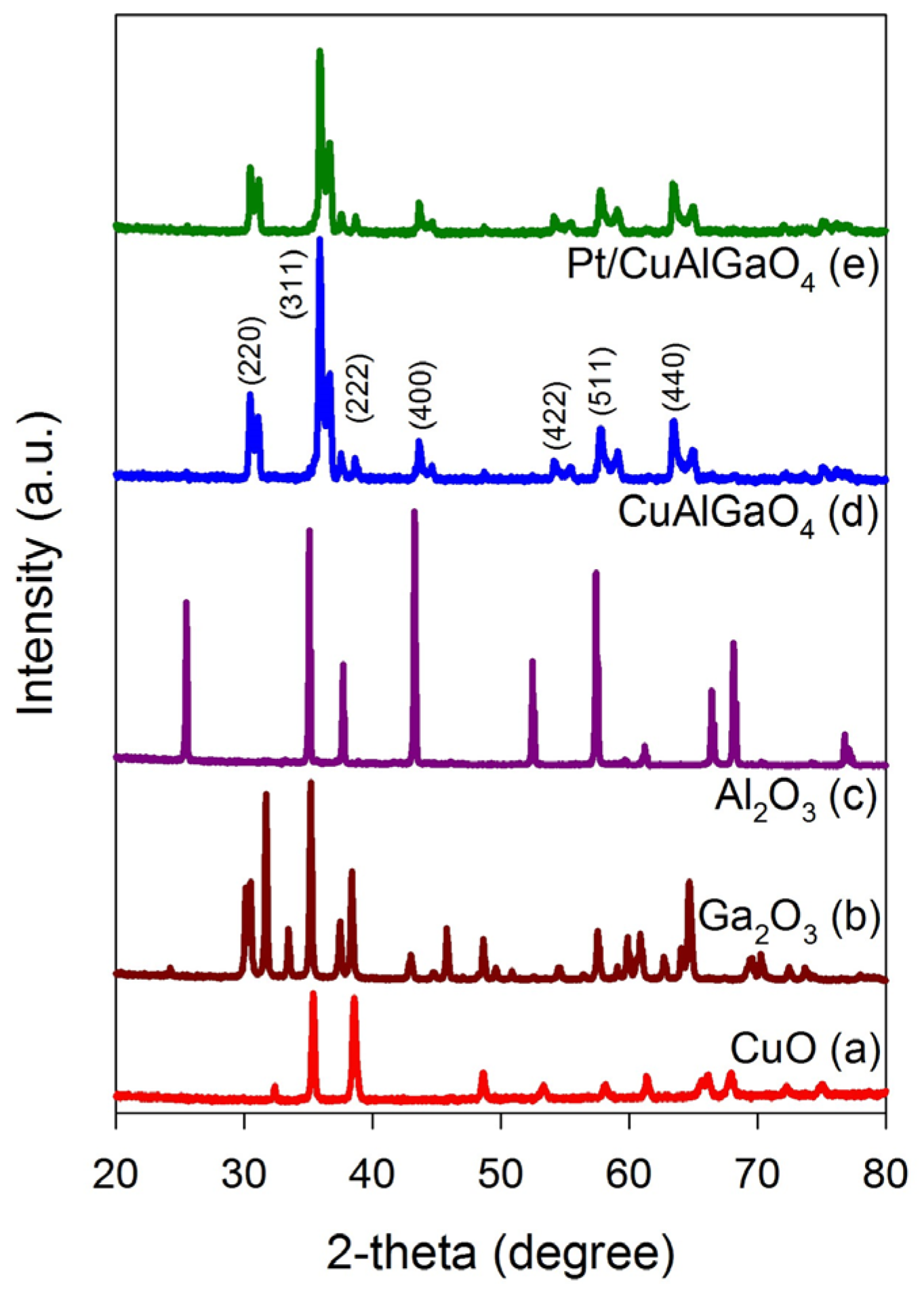
2.1. Characterization of Photocatalysts
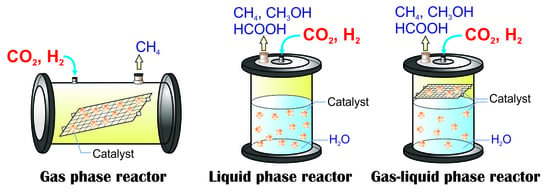
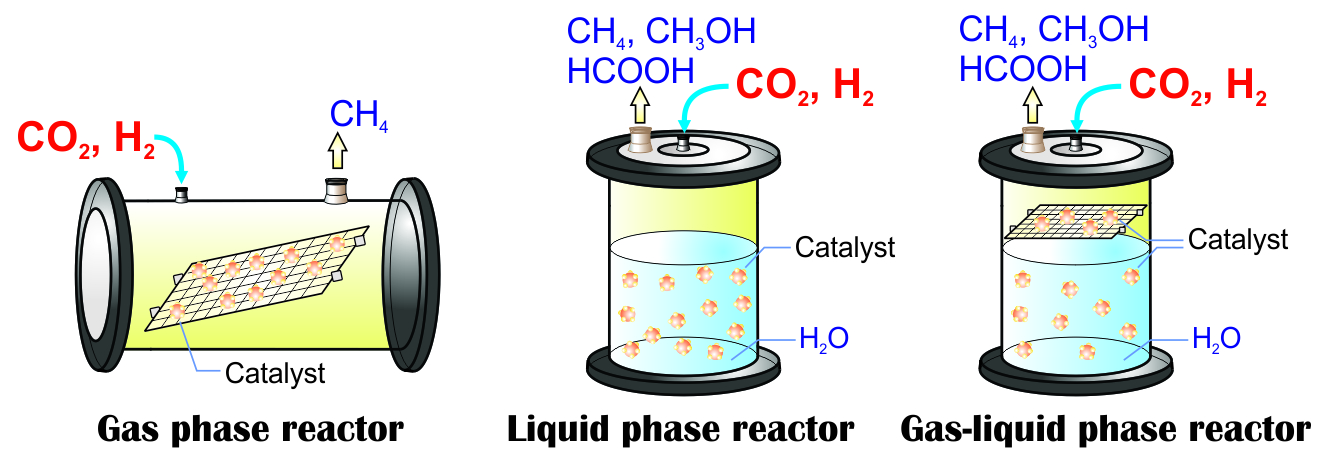
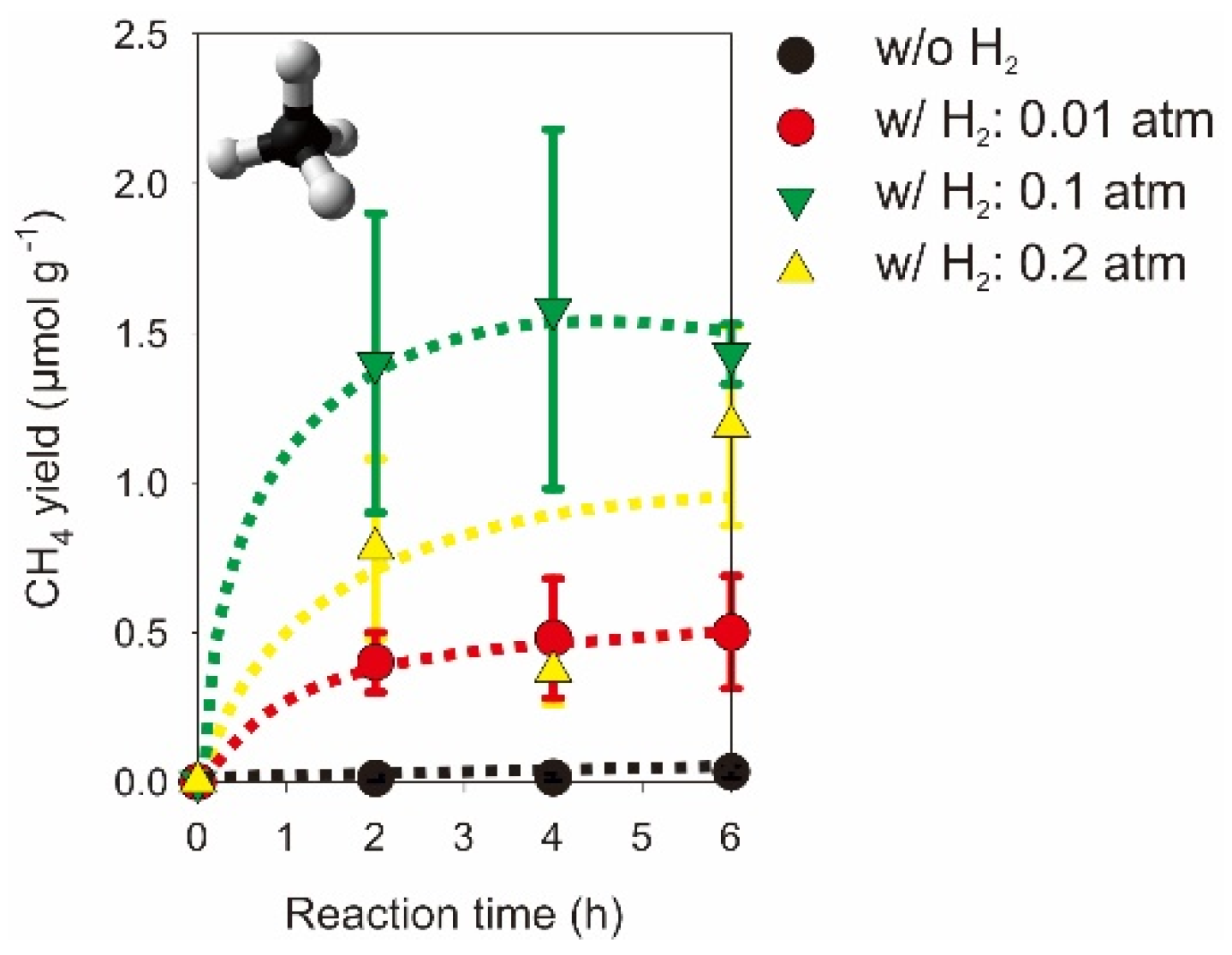
2.2. Photocatalytic CO2 Reduction with Gas, Liquid, Gas-Liquid Phase Reactors
∆H0 = −259.9 kJ/mol; ∆G0 = −132.4 kJ/mol
∆H0 = −137.8 kJ/mol; ∆G0 = −10.7 kJ/mol
∆H0 = −31.0 kJ/mol; ∆G0 = +34.3 kJ/mol
∆H0 = −247.5 kJ/mol; ∆G0 = +170.8 kJ/mol
∆H0 = −131.6 kJ/mol; ∆G0 = −29.9 kJ/mol
∆H0 = −24.8 kJ/mol; ∆G0 = +15.1 kJ/mol
3. Materials and Methods
3.1. Preparation of Photocatalysts
3.2. Characterization of Photocatalysts
3.3. Photo-Hydrogenation of CO2 Reaction
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yang, H.; Xu, Z.; Fan, M.; Gupta, R.; Slimane, R.B.; Bland, A.E.; Wright, I. Progress in carbon dioxide separation and capture: A review. J. Environ. Sci. 2008, 20, 14–27. [Google Scholar] [CrossRef]
- Roy, S.C.; Varghese, O.K.; Paulose, M.; Grimes, C.A. Toward solar fuels: Photocatalytic conversion of carbon dioxide to hydrocarbons. ACS Nano 2010, 4, 1259–1278. [Google Scholar] [CrossRef] [PubMed]
- Lunde, P.J.; Kester, F.L. Rates of methane formation from carbon dioxide and hydrogen over a ruthenium catalyst. J. Catal. 1973, 30, 423–429. [Google Scholar] [CrossRef]
- Thampi, K.R.; Kiwi, J.; Gratzel, M. Methanation and photo-methanation of carbon dioxide at room temperature and atmospheric pressure. Nature 1987, 327, 506–508. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kent, R.A.; Yung, M.M.; Kuhn, J.N. Carbon dioxide conversion by reverse water-gas shift chemical looping on perovskite-type oxides. Ind. Eng. Chem. Res. 2014, 53, 5828–5837. [Google Scholar] [CrossRef]
- Bustamante, F.; Enick, R.M.; Cugini, A.V.; Killmeyer, R.P.; Howard, B.H.; Rothenberger, K.S.; Ciocco, M.V.; Morreale, B.D.; Chattopadhyay, S.; Shi, S. High-temperature kinetics of the homogeneous reverse water-gas shift reaction. AIChE J. 2004, 50, 1028–1041. [Google Scholar] [CrossRef]
- Noji, T.; Jin, T.; Nango, M.; Kamiya, N.; Amao, Y. CO2 photoreduction by formate dehydrogenase and a Ru-complex in a nanoporous glass reactor. ACS Appl. Mater. Interfaces 2017, 9, 3260–3265. [Google Scholar] [CrossRef]
- Abe, T.; Tanizawa, M.; Watanabe, K.; Taguchi, A. CO2 methanation property of Ru nanoparticle-loaded TiO2 prepared by a polygonal barrel-sputtering method. Energy Environ. Sci. 2009, 2, 315–321. [Google Scholar] [CrossRef]
- Liu, X.; Song, Y.; Geng, W.; Li, H.; Xiao, L.; Wu, W. Cu-Mo2C/MCM-41: An efficient catalyst for the selective synthesis of methanol from CO2. Catalysts 2016, 6, 75. [Google Scholar] [CrossRef]
- Huang, C.; Chen, S.; Fei, X.; Liu, D.; Zhang, Y. Catalytic hydrogenation of CO2 to methanol: Study of synergistic effect on adsorption properties of CO2 and H2 in CuO/ZnO/ZrO2 system. Catalysts 2015, 5, 1846. [Google Scholar] [CrossRef]
- Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G.A.; Prakash, G.K.S. Conversion of CO2 from air into methanol using a polyamine and a homogeneous ruthenium catalyst. J. Am. Chem. Soc. 2016, 138, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Yui, T.; Kan, A.; Saitoh, C.; Koike, K.; Ibusuki, T.; Ishitani, O. Photochemical reduction of CO2 using TiO2: Effects of organic adsorbates on TiO2 and deposition of Pd onto TiO2. ACS Appl. Mater. Interfaces 2011, 3, 2594–2600. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Q.; Zhao, Y.; Wu, W.; Chen, J.; Meng, H. Green synthesis and photo-catalytic performances for ZnO-reduced graphene oxide nanocomposites. J. Colloid Interface Sci. 2013, 411, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.-C.; Sun, X.-Z.; Liu, J.-G. Photo-reduction of CO2 using a rhenium complex covalently supported on a graphene/TiO2 composite. ChemSusChem 2016, 9, 1698–1703. [Google Scholar] [CrossRef] [PubMed]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, B.; Mokhtarianpour, M.; Ensafi, A.A.; Hadadzadeh, H.; Shakeri, J. Electrocatalytic reduction of CO2 using the dinuclear rhenium(i) complex [ReCl(CO)3(μ-tptzH)Re(CO)3]. Polyhedron 2015, 101, 160–164. [Google Scholar] [CrossRef]
- Inoue, T.; Fujishima, A.; Konishi, S.; Honda, K. Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders. Nature 1979, 277, 637–638. [Google Scholar] [CrossRef]
- Sastre, F.; Puga, A.V.; Liu, L.; Corma, A.; García, H. Complete photocatalytic reduction of CO2 to methane by H2 under solar light irradiation. J. Am. Chem. Soc. 2014, 136, 6798–6801. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Baeg, J.-O.; Kale, B.B.; Yadav, R.K.; Moon, S.-J.; Kong, K.-j.; So, W.-W. An efficient visible-light active photocatalyst CuAlGaO4 for solar hydrogen production. Catal. Commun. 2011, 12, 651–654. [Google Scholar] [CrossRef]
- Lee, W.-H.; Liao, C.-H.; Tsai, M.-F.; Huang, C.-W.; Wu, J.C.S. A novel twin reactor for CO2 photoreduction to mimic artificial photosynthesis. Appl. Catal. B 2013, 132–133, 445–451. [Google Scholar] [CrossRef]
- Li, K.; Peng, B.; Peng, T. Recent advances in heterogeneous photocatalytic CO2 conversion to solar fuels. ACS Catal. 2016, 6, 7485–7527. [Google Scholar] [CrossRef]
- Lo, C.-C.; Hung, C.-H.; Yuan, C.-S.; Wu, J.-F. Photoreduction of carbon dioxide with H2 and H2O over TiO2 and ZrO2 in a circulated photocatalytic reactor. Sol. Energy Mater. Sol. Cells 2007, 91, 1765–1774. [Google Scholar] [CrossRef]
- Nguyen, T.-V.; Wu, J.C.S.; Chiou, C.-H. Photoreduction of CO2 over ruthenium dye-sensitized TiO2-based catalysts under concentrated natural sunlight. Catal. Commun. 2008, 9, 2073–2076. [Google Scholar] [CrossRef]
- Abbott, M.M.; Smith, J.M.; Van Ness, H.C. Introduction to Chemical Engineering Thermodynamics; McGraw-Hill Book Co. InC.: Boston, MA, USA, 1949; pp. 619–626. [Google Scholar]
- Cheng, Y.-H.; Nguyen, V.-H.; Chan, H.-Y.; Wu, J.C.S.; Wang, W.-H. Photo-enhanced hydrogenation of CO2 to mimic photosynthesis by CO co-feed in a novel twin reactor. Appl. Energy 2015, 147, 318–324. [Google Scholar] [CrossRef]
- Wagner, C.D.; Muilenberg, G.E. Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Data for Use in X-ray Photoelectron Spectroscopy; Perkin-Elmer Corp., Physical Electronics Division: Eden Prairie, MN, USA, 1979; p. 190. [Google Scholar]
- Nash, T. The colorimetric estimation of formaldehyde by means of the hantzsch reaction. Biochem. J. 1953, 55, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Zhang, W.; Ren, J.; Xu, R. Photocatalytic reduction of CO2: A brief review on product analysis and systematic methods. Anal. Methods 2013, 5, 1086–1097. [Google Scholar] [CrossRef]
- Yang, C.-C.; Vernimmen, J.; Meynen, V.; Cool, P.; Mul, G. Mechanistic study of hydrocarbon formation in photocatalytic CO2 reduction over Ti-SBA-15. J. Catal. 2011, 284, 1–8. [Google Scholar] [CrossRef]
- Mei, B.; Pougin, A.; Strunk, J. Influence of photodeposited gold nanoparticles on the photocatalytic activity of titanate species in the reduction of CO2 to hydrocarbons. J. Catal. 2013, 306, 184–189. [Google Scholar] [CrossRef]
- Xu, X.; Moulijn, J.A. Mitigation of CO2 by chemical conversion: Plausible chemical reactions and promising products. Energy Fuels 1996, 10, 305–325. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conversion Process | Main Products | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|
| Biological | HCOOH: conversion efficiency of (22 ± 9) × 10−2% | Biological capability to synthesize liquid fuels | Complicated and cumbersome biological processes | [7] |
| Catalytic (Heterogeneous catalysis) | CH4: 3.8 × 104 μmol·h−1·g−1 catalyst | High efficiency | High temperature requirement | [8] |
| CH3OH: 8.8% of CO2 conversion with the corresponded selectivity of 63% | [9] | |||
| CH3OH: 193.9 g·kgcat−1·h−1 | [10] | |||
| Catalytic (Homogeneous catalysis) | CH3OH: 79% yield | [11] | ||
| Photocatalytic (Heterogeneous catalysis) | CH4: 0.56 μmol h−1·g−1 catalyst | Storage of solar energy | Low efficiency | [12] |
| CH3OH: 4.6 μmol h−1·g−1 catalyst | [13] | |||
| Photocatalytic (Homogeneous catalysis) | CO: 12.66 h−1 of TOF | [14] | ||
| Electrocatalytic | Liquid fuels | Converting CO2 directly to liquid fuels (long-chain molecule) | High energy barrier needs overcoming | [15,16] |
| Element | Atomic Ratio (%) | |
|---|---|---|
| CuAlGaO4 | Pt/CuAlGaO4 | |
| O | 61.9 | 61.6 |
| Al | 23.0 | 22.0 |
| Cu | 12.8 | 13.0 |
| Ga | 2.3 | 3.2 |
| Pt | N/D 1 | 0.2 |
| No. | Type of Reactors | Experimental Conditions | Yield of Products (µmol·g−1) | |||
|---|---|---|---|---|---|---|
| CO2 | H2 (0.01 atm) | Photocatalyst (1 wt % Pt/CuAlGaO4) | Light Source | |||
| 1 | Gas phase reactor | X 1 | O 2 | O | O | BDL 3 |
| 2 | O | O | X | O | BDL | |
| 3 | O | O | O | X | BDL | |
| 4 | Liquid phase reactor | X | O | O | O | BDL |
| 5 | O | O | X | O | BDL | |
| 6 | O | O | O | X | BDL | |
| 7 | Gas-liquid phase reactor | X | O | O | O | BDL |
| 8 | O | O | X | O | BDL | |
| 9 | O | O | O | X | BDL | |
| Entry | Phase | H2 Partial Pressure (atm) | Product Yields (µmol·g−1) | |||
|---|---|---|---|---|---|---|
| CH4 | CH3OH | HCOOH | Total HCs 1 | |||
| 1 | Gas | 0.00 | 0.012 ± 0.010 | BDL 2 | BDL | 0.012 |
| 2 | 0.01 | 0.400 ± 0.100 | BDL | BDL | 0.400 | |
| 3 | 0.20 | 0.780 ± 0.300 | BDL | BDL | 0.780 | |
| 4 | Liquid | 0.00 | 0.010 ± 0.010 | 0.235 ± 0.100 | BDL | 0.245 |
| 5 | 0.01 | 0.149 ± 0.050 | BDL | 0.094 ± 0.045 | 0.243 | |
| 6 | 0.20 | 0.112 ± 0.030 | 0.340 ± 0.200 | BDL | 0.452 | |
| 7 | Gas-Liquid | 0.00 | 0.014 ± 0.010 | 0.285 ± 0.100 | BDL | 0.299 |
| 8 | 0.01 | 0.480 ± 0.200 | 7.352 ± 2.100 | 0.470 ± 0.100 | 8.302 | |
| 9 | 0.20 | 0.666 ± 0.120 | 0.445 ± 0.110 | 0.145 ± 0.010 | 1.255 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-Y.; Yu, J.C.-C.; Nguyen, V.-H.; Wu, J.C.-S.; Wang, W.-H.; Kočí, K. Reactor Design for CO2 Photo-Hydrogenation toward Solar Fuels under Ambient Temperature and Pressure. Catalysts 2017, 7, 63. https://doi.org/10.3390/catal7020063
Chen C-Y, Yu JC-C, Nguyen V-H, Wu JC-S, Wang W-H, Kočí K. Reactor Design for CO2 Photo-Hydrogenation toward Solar Fuels under Ambient Temperature and Pressure. Catalysts. 2017; 7(2):63. https://doi.org/10.3390/catal7020063
Chicago/Turabian StyleChen, Chun-Ying, Joseph Che-Chin Yu, Van-Huy Nguyen, Jeffrey Chi-Sheng Wu, Wei-Hon Wang, and Kamila Kočí. 2017. "Reactor Design for CO2 Photo-Hydrogenation toward Solar Fuels under Ambient Temperature and Pressure" Catalysts 7, no. 2: 63. https://doi.org/10.3390/catal7020063
APA StyleChen, C. -Y., Yu, J. C. -C., Nguyen, V. -H., Wu, J. C. -S., Wang, W. -H., & Kočí, K. (2017). Reactor Design for CO2 Photo-Hydrogenation toward Solar Fuels under Ambient Temperature and Pressure. Catalysts, 7(2), 63. https://doi.org/10.3390/catal7020063