
A Study of Low-Temperature CO Oxidation over Mesoporous CuO-TiO2 Nanotube Catalysts




Abstract
:
1. Introduction
2. Results and Discussion
2.1. Morphology and Elements Distribution
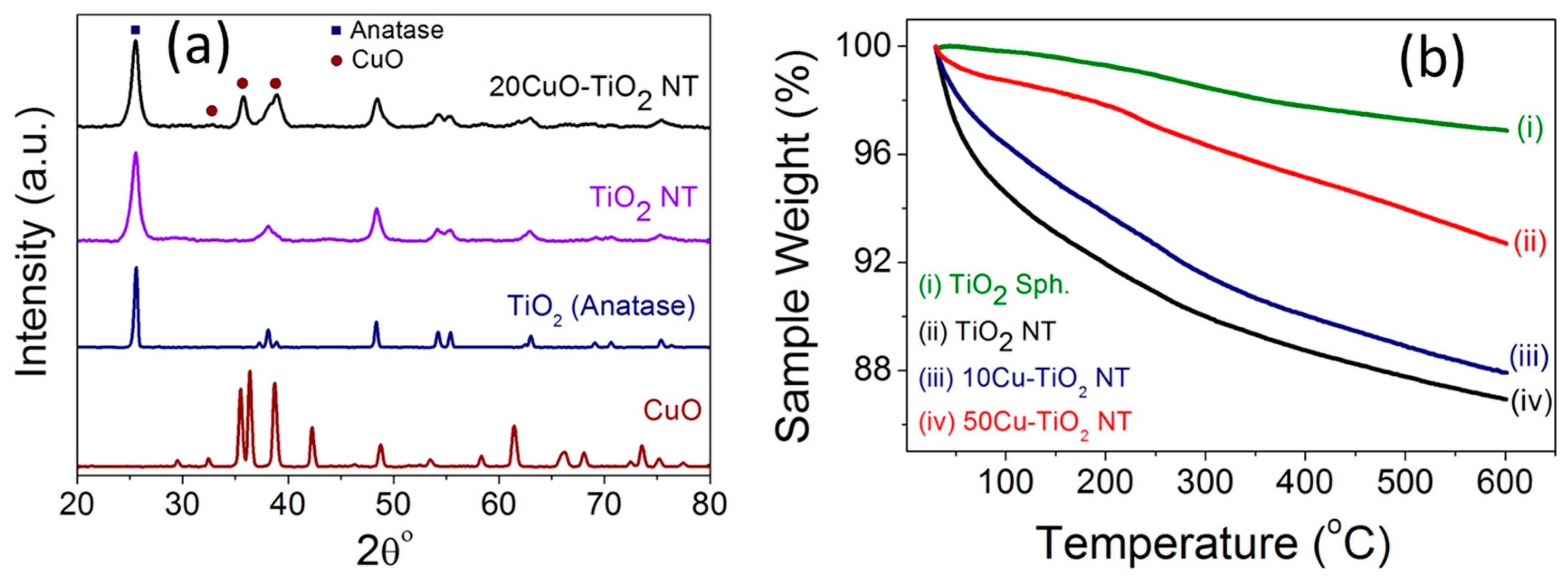
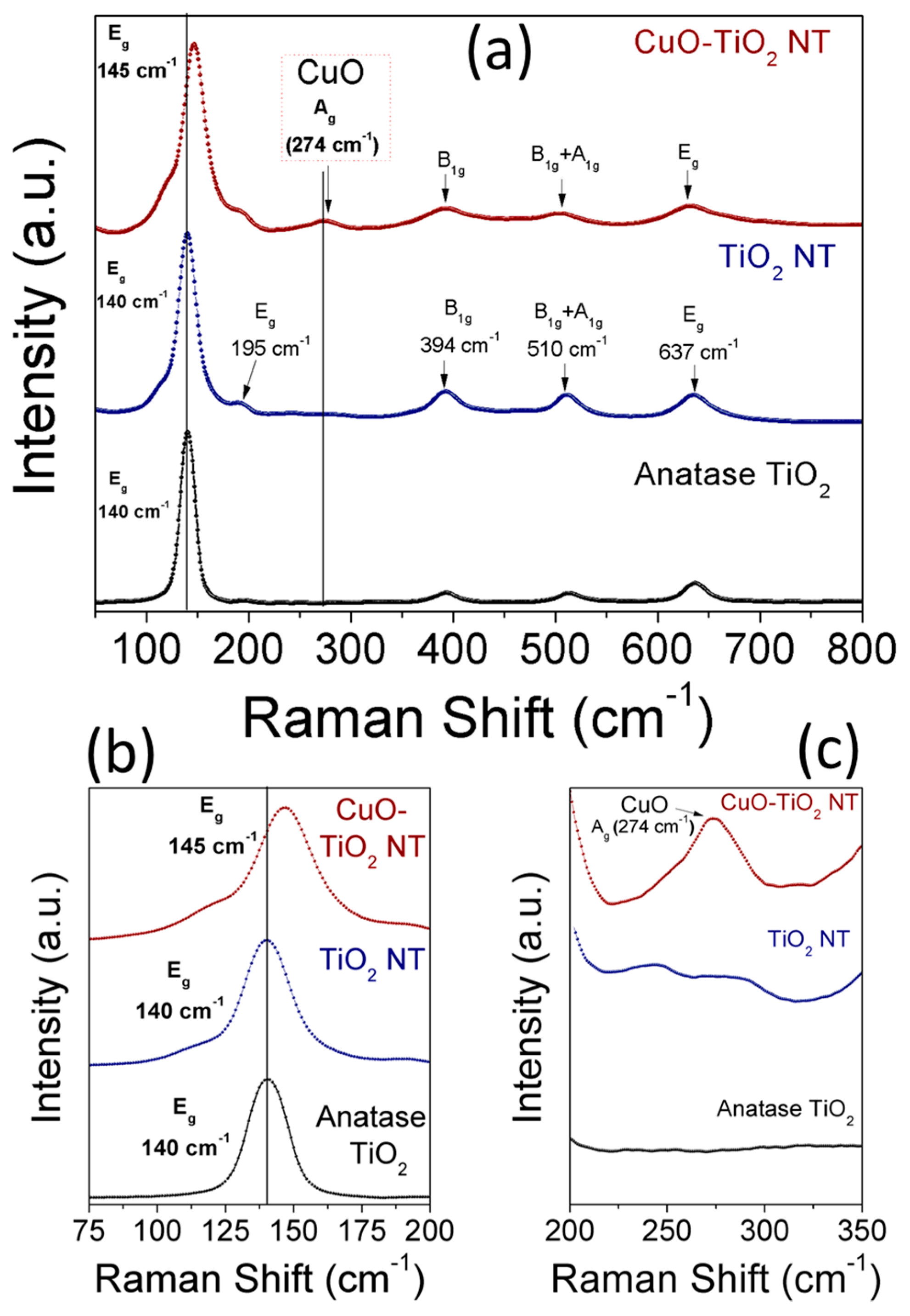
2.2. The Crystal Structure and Thermal Stability
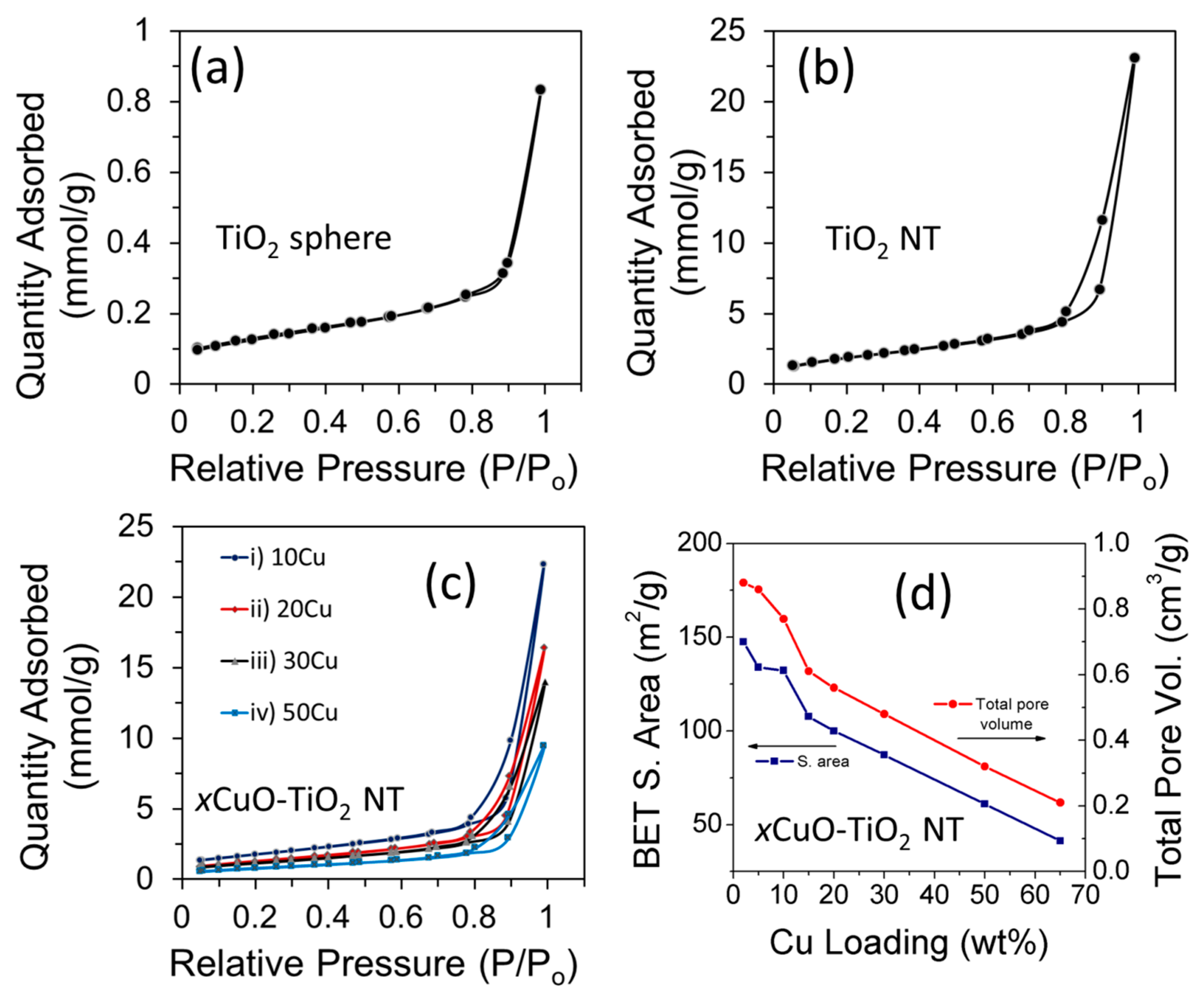
2.3. Surface Properties
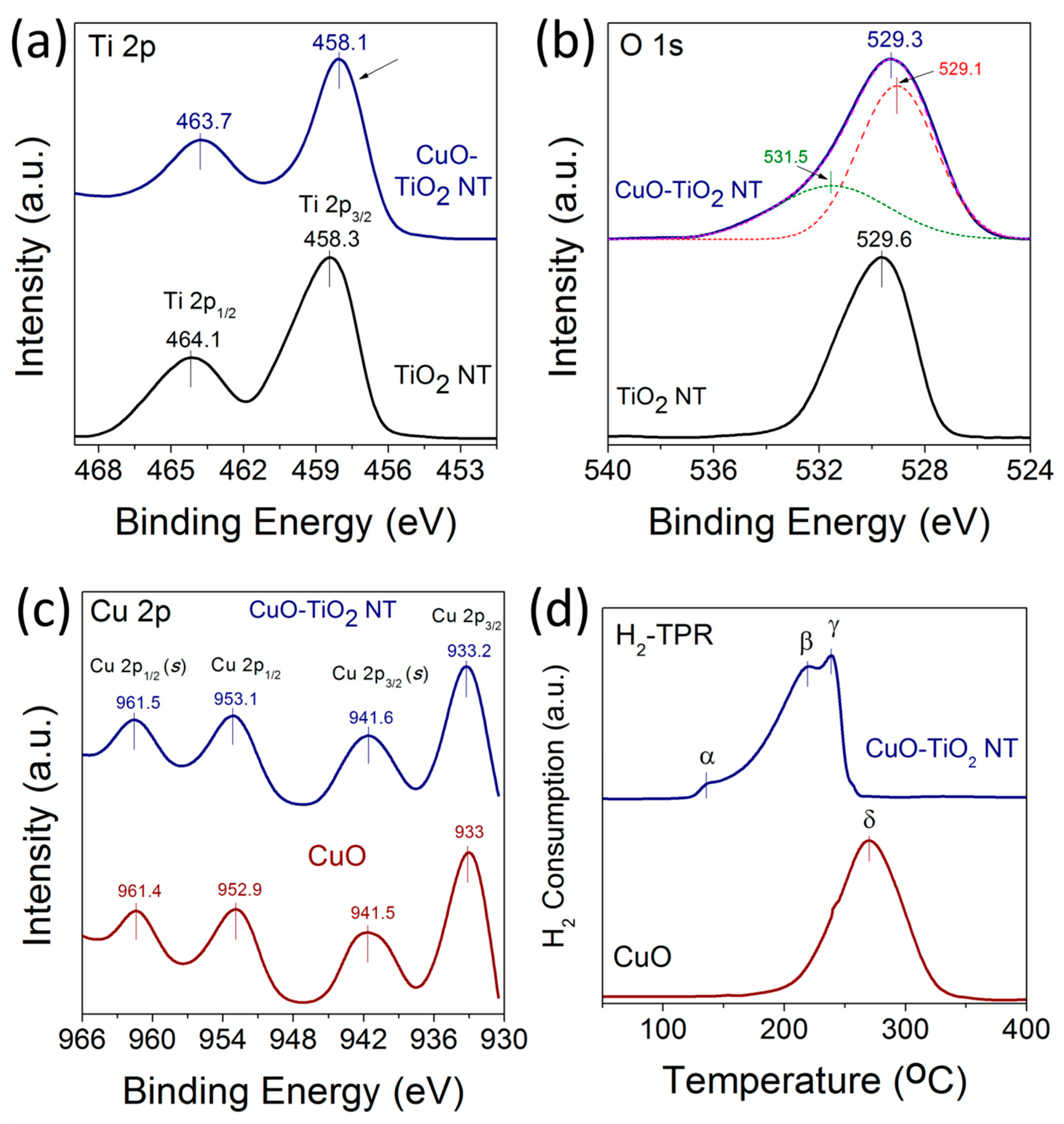
2.4. Valence States
2.5. Metal-Support Interactions
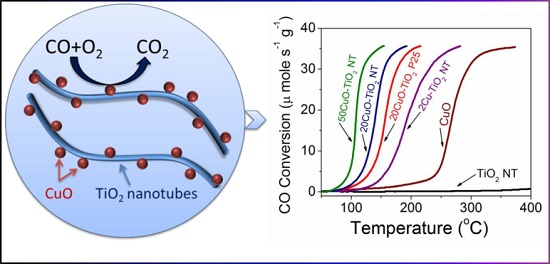
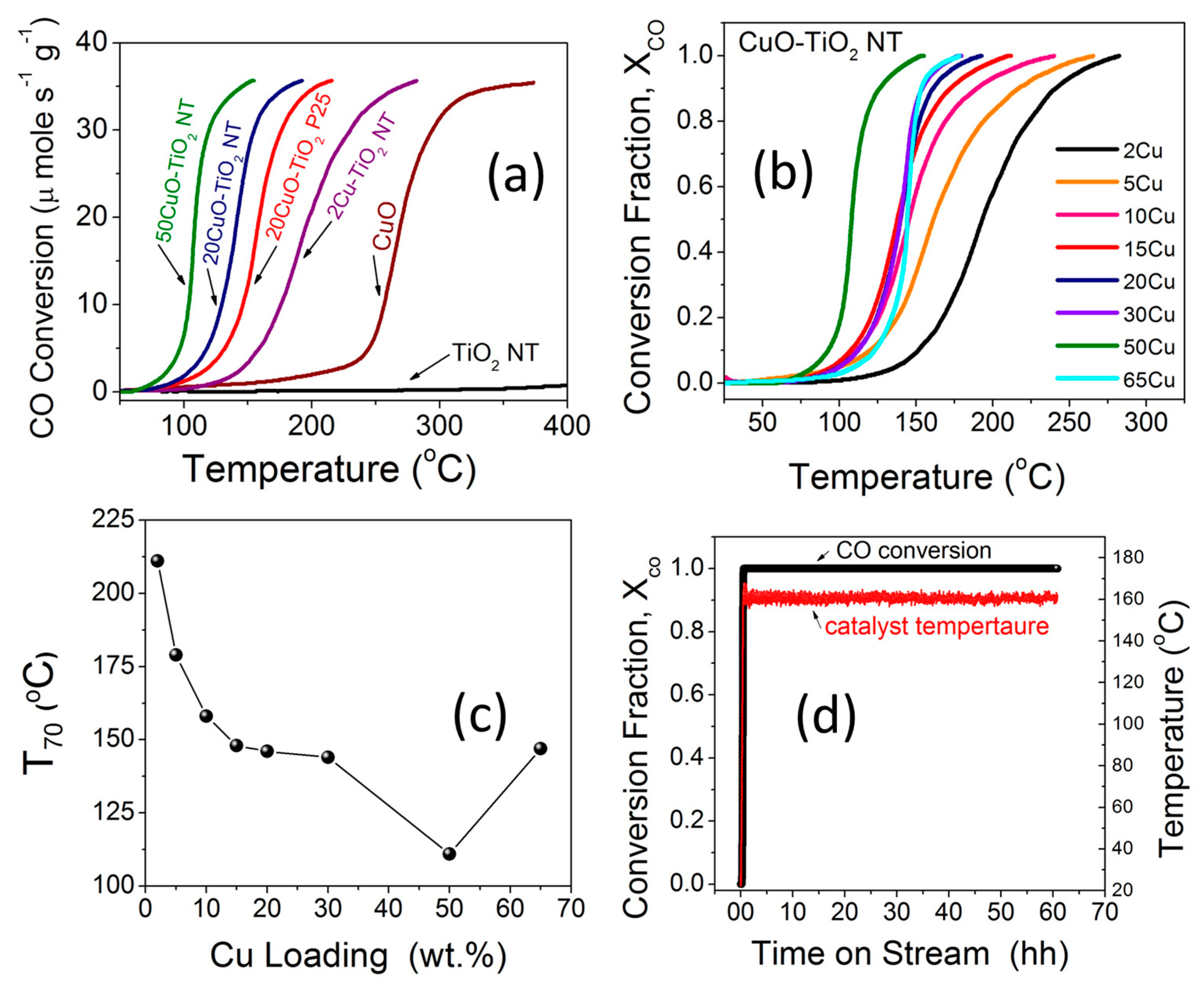
2.6. Catalytic Activity, Stability and Kinetics
2.6.1. Effect of Cu Content on CO Oxidation
2.6.2. Long-Term Stability
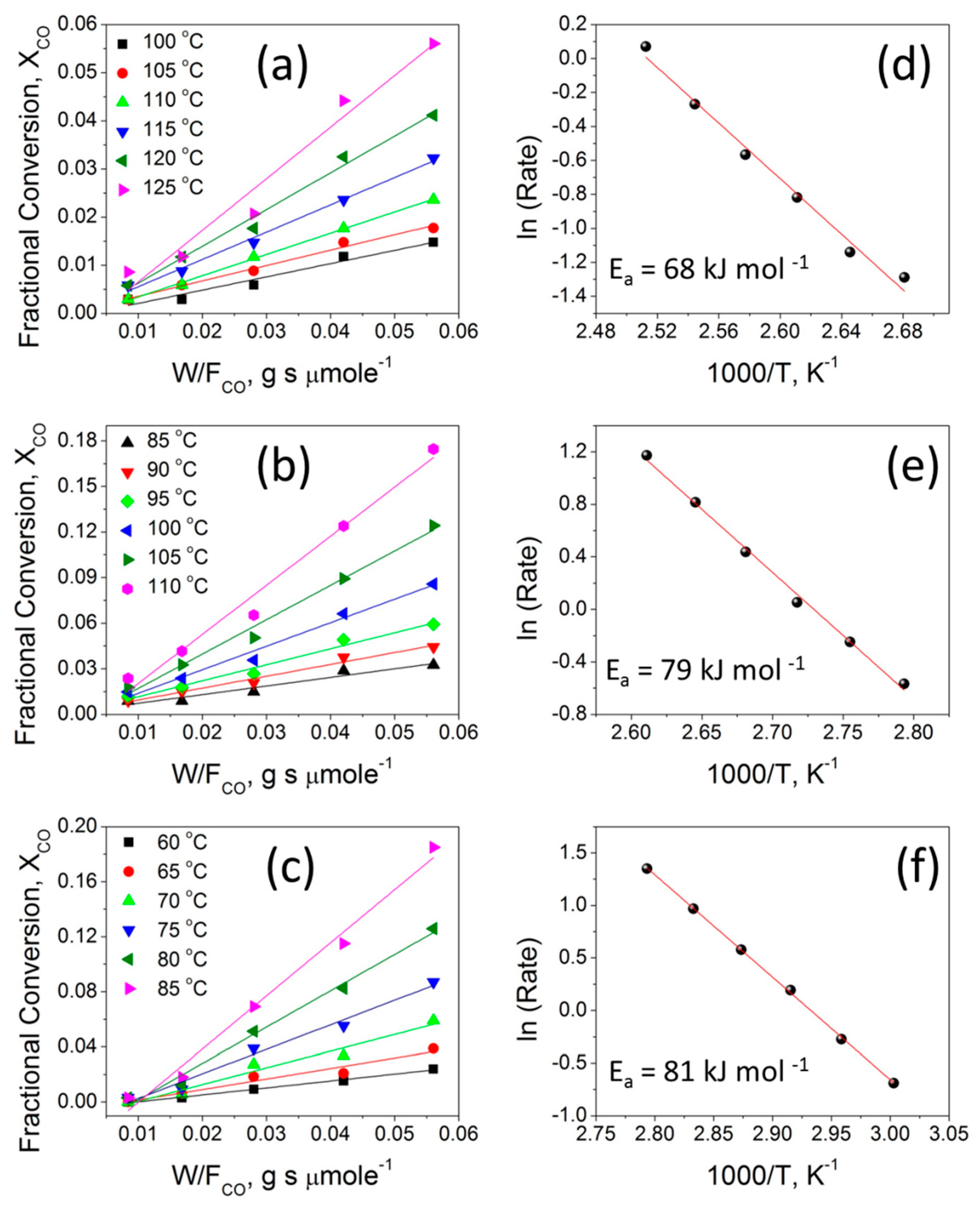
2.6.3. Kinetic Studies
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Synthesis of TiO2 Nanotubes Support
3.2.2. Synthesis of CuO-TiO2 NT Catalysts
3.2.3. Catalysts Characterization
3.2.4. Catalytic Activity, Stability and Kinetics Measurements
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Deng, W.; Flytzani-Stephanopoulos, M. On the issue of the deactivation of Au–Ceria and Pt–Ceria water–Gas shift catalysts in practical fuel-Cell applications. Angew. Chem. Int. Ed. 2006, 45, 2285–2289. [Google Scholar] [CrossRef] [PubMed]
- Shelef, M.; McCabe, R.W. Twenty-Five years after introduction of automotive catalysts: What next? Catal. Today 2000, 62, 35–50. [Google Scholar] [CrossRef]
- Polychronopoulou, K.; Zedan, A.F.; Katsiotis, M.S.; Baker, M.A.; AlKhoori, A.A.; AlQaradawi, S.Y.; Hinder, S.J.; AlHassan, S. Rapid microwave assisted sol-gel synthesis of CeO2 and CexSm1−xO2 nanoparticle catalysts for CO oxidation. Mol. Catal. 2017, 428, 41–55. [Google Scholar] [CrossRef]
- Veith, G.M.; Lupini, A.R.; Rashkeev, S.; Pennycook, S.J.; Mullins, D.R.; Schwartz, V.; Bridges, C.A.; Dudney, N.J. Thermal stability and catalytic activity of gold nanoparticles supported on silica. J. Catal. 2009, 262, 92–101. [Google Scholar] [CrossRef]
- In, S.-I.; Vaughn, D.D.; Schaak, R.E. Hybrid CuO-TiO2−xNx hollow nanocubes for photocatalytic conversion of CO2 into methane under solar irradiation. Angew. Chem. Int. Ed. 2012, 51, 3915–3918. [Google Scholar] [CrossRef] [PubMed]
- Carrettin, S.; Hao, Y.; Aguilar-Guerrero, V.; Gates, B.C.; Trasobares, S.; Calvino, J.J.; Corma, A. Increasing the number of oxygen vacancies on TiO2 by doping with iron increases the activity of supported gold for co oxidation. Chem. Eur. J. 2007, 13, 7771–7779. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, S.; Zhao, Y.; Wang, X.; Wang, S.; Wu, S.; Zhang, S.; Huang, W. Synthesis and characterization of CuO/TiO2 catalysts for low-Temperature CO oxidation. Catal. Commun. 2006, 7, 1029–1034. [Google Scholar] [CrossRef]
- Hafez, A.M.; Zedan, A.F.; AlQaradawi, S.Y.; Salem, N.M.; Allam, N.K. Computational study on oxynitride perovskites for CO2 photoreduction. Energy Convers. Manag. 2016, 122, 207–214. [Google Scholar] [CrossRef]
- Anil, C.; Madras, G. Kinetics of CO oxidation over Cu doped Mn3O4. J. Mol. Catal. Chem. 2016, 424, 106–114. [Google Scholar] [CrossRef]
- Hornes, A.; Hungria, A.B.; Bera, P.; Camara, A.L.; Fernandez-Garcia, M.; Martinez-Arias, A.; Barrio, L.; Estrella, M.; Zhou, G.; Fonseca, J.J.; et al. Inverse CeO2/CuO catalyst as an alternative to classical direct configurations for preferential oxidation of CO in hydrogen-Rich stream. J. Am. Chem. Soc. 2009, 132, 34–35. [Google Scholar] [CrossRef] [PubMed]
- Caputo, T.; Lisi, L.; Pirone, R.; Russo, G. Kinetics of the preferential oxidation of CO over CuO/CeO2 catalysts in H2-Rich gases. Ind. Eng. Chem. Res. 2007, 46, 6793–6800. [Google Scholar] [CrossRef]
- Royer, S.; Duprez, D. Catalytic oxidation of carbon monoxide over transition metal oxides. Chem. Cat. Chem. 2011, 3, 24–65. [Google Scholar] [CrossRef]
- Kim, H.Y.; Liu, P. Complex catalytic behaviors of cutiox mixed-Oxide during CO oxidation. J. Phys. Chem. C 2015, 119, 22985–22991. [Google Scholar] [CrossRef]
- Chen, G.; Xu, Q.; Yang, Y.; Li, C.; Huang, T.; Sun, G.; Zhang, S.; Ma, D.; Li, X. Facile and mild strategy to construct mesoporous CeO2–CuO nanorods with enhanced catalytic activity toward CO oxidation. ACS Appl. Mater. Interfaces 2015, 7, 23538–23544. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Chen, T.C.; Chen, C.C.; Lai, Y.T.; You, J.H.; Chou, T.M.; Chen, C.H.; Lee, J.-F. Effect of Ti3+ on TiO2-Supported Cu catalysts used for CO oxidation. Langmuir 2012, 28, 9996–10006. [Google Scholar] [CrossRef] [PubMed]
- Bonanni, S.; Aït-Mansour, K.; Harbich, W.; Brune, H. Effect of the TiO2 reduction state on the catalytic CO oxidation on deposited size-Selected pt clusters. J. Am. Chem. Soc. 2012, 134, 3445–3450. [Google Scholar] [CrossRef] [PubMed]
- Fahim, N.F.; Sekino, T. A novel method for synthesis of titania nanotube powders using rapid breakdown anodization. Chem. Mater. 2009, 21, 1967–1979. [Google Scholar] [CrossRef]
- Yuan, L.; Meng, S.; Zhou, Y.; Yue, Z. Controlled synthesis of anatase TiO2 nanotube and nanowire arrays via aao template-Based hydrolysis. J. Mater. Chem. A 2013, 1, 2552–2557. [Google Scholar] [CrossRef]
- Kubo, T.; Nakahira, A. Local structure of TiO2-derived nanotubes prepared by the hydrothermal process. J. Phys. Chem. C 2008, 112, 1658–1662. [Google Scholar] [CrossRef]
- Khan, M.A.; Jung, H.-T.; Yang, O.B. Synthesis and characterization of ultrahigh crystalline TiO2 nanotubes. J. Phys. Chem. B 2006, 110, 6626–6630. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, A.; Kubo, T.; Yamasaki, Y. Microstructural control of mesoporous bulk composed of TiO2-derived titanate nanotubes. ACS Appl. Mater. Interfaces 2010, 2, 1136–1140. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, A.; Kubo, T.; Numako, C. TiO2-derived titanate nanotubes by hydrothermal process with acid treatments and their microstructural evaluation. ACS Appl. Mater. Interfaces 2010, 2, 2611–2616. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Cao, Y.; Huang, Q.; He, H.; Liu, Y.; Guo, W.; Hong, M. High-Temperature formation of titanate nanotubes and the transformation mechanism of nanotubes into nanowires. Cryst. Growth Des. 2009, 9, 3632–3637. [Google Scholar] [CrossRef]
- Wajid Shah, M.; Zhu, Y.; Fan, X.; Zhao, J.; Li, Y.; Asim, S.; Wang, C. Facile synthesis of defective TiO2−x nanocrystals with high surface area and tailoring bandgap for visible-Light photocatalysis. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Fang, B.; Xing, Y.; Bonakdarpour, A.; Zhang, S.; Wilkinson, D.P. Hierarchical CuO–TiO2 hollow microspheres for highly efficient photodriven reduction of CO2 to CH4. ACS Sustain. Chem. Eng. 2015, 3, 2381–2388. [Google Scholar] [CrossRef]
- Thi Hiep, N.; Thu Loan, N.; Thi Dieu Thuy, U.; Quang Liem, N. Synthesis and characterization of nano-CuO and CuO/TiO2 photocatalysts. Adv. Nat. Sci. Nanosci. Nanotechnol. 2013, 4. [Google Scholar] [CrossRef]
- Senthilkumar, V.; Kim, Y.S.; Chandrasekaran, S.; Rajagopalan, B.; Kim, E.J.; Chung, J.S. Comparative supercapacitance performance of CuO nanostructures for energy storage device applications. RSC Adv. 2015, 5, 20545–20553. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, C. Improved photocatalytic activity of nano CuO-Incorporated TiO2 granules prepared by spray drying. Prog. Nat. Sci. Mater. Int. 2015, 25, 334–341. [Google Scholar] [CrossRef]
- Kochkar, H.; Lakhdhar, N.; Berhault, G.; Bausach, M.; Ghorbel, A. Optimization of the alkaline hydrothermal route to titanate nanotubes by a doehlert matrix experience design. J. Phys. Chem. C 2009, 113, 1672–1679. [Google Scholar] [CrossRef]
- Jang, J.S.; Choi, S.H.; Kim, D.H.; Jang, J.W.; Lee, K.S.; Lee, J.S. Enhanced photocatalytic hydrogen production from water−Methanol solution by nickel intercalated into titanate nanotube. J. Phys. Chem. C 2009, 113, 8990–8996. [Google Scholar] [CrossRef]
- Ganesh, I.; Kumar, P.P.; Annapoorna, I.; Sumliner, J.M.; Ramakrishna, M.; Hebalkar, N.Y.; Padmanabham, G.; Sundararajan, G. Preparation and characterization of Cu-Doped TiO2 materials for electrochemical, photoelectrochemical, and photocatalytic applications. Appl. Surf. Sci. 2014, 293, 229–247. [Google Scholar] [CrossRef]
- Ren, R.; Wen, Z.; Cui, S.; Hou, Y.; Guo, X.; Chen, J. Controllable synthesis and tunable photocatalytic properties of Ti3+-Doped TiO2. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Duan, W.; Liu, B.; Chen, X.; Yang, F.; Guo, J. The effects of doping copper and mesoporous structure on photocatalytic properties of TiO2. J. Nanomater. 2014. [Google Scholar] [CrossRef]
- Wu, Q.; Li, J.; Zhang, W.; Qian, H.; She, W.; Pan, H.; Wen, J.; Zhang, X.; Liu, X.; Jiang, X. Antibacterial property, angiogenic and osteogenic activity of Cu-Incorporated TiO2 coating. J. Mater. Chem. B 2014, 2, 6738–6748. [Google Scholar] [CrossRef]
- Deng, C.; Li, B.; Dong, L.; Zhang, F.; Fan, M.; Jin, G.; Gao, J.; Gao, L.; Zhang, F.; Zhou, X. No reduction by co over CuO supported on CeO2-Doped TiO2: The effect of the amount of a few CeO2. Phys. Chem. Chem. Phys. 2015, 17, 16092–16109. [Google Scholar] [CrossRef] [PubMed]
- Ola, O.; Mercedes Maroto-Valer, M. Copper based TiO2 honeycomb monoliths for CO2 photoreduction. Catal. Sci. Technol. 2014, 4, 1631–1637. [Google Scholar] [CrossRef]
- Tsai, C.-Y.; Hsi, H.-C.; Kuo, T.-H.; Chang, Y.-M.; Liou, J.-H. Preparation of cu-Doped TiO2 photocatalyst with thermal plasma torch for low-Concentration mercury removal. Aerosol Air Qual. Res. 2013, 13. [Google Scholar] [CrossRef]
- Li, W.; Liang, R.; Hu, A.; Huang, Z.; Zhou, Y.N. Generation of oxygen vacancies in visible light activated one-Dimensional iodine TiO2 photocatalysts. RSC Adv. 2014, 4, 36959–36966. [Google Scholar] [CrossRef]
- Shan, Z.; Archana, P.S.; Shen, G.; Gupta, A.; Bakker, M.G.; Pan, S. Nanocot: Low-Cost nanostructured electrode containing carbon, oxygen, and titanium for efficient oxygen evolution reaction. J. Am. Chem. Soc. 2015, 137, 11996–12005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-G.; Ma, L.-L.; Li, J.-L.; Yu, Y. In situ fenton reagent generated from TiO2/Cu2O composite film: A new way to utilize TiO2 under visible light irradiation. Environ. Sci. Technol. 2007, 41, 6264–6269. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, B.; Fan, Y.; Feng, N.; Qiu, A.; He, J.M.J.; Yang, H.; Chen, Y. The effect of titania polymorph on the strong metal-Support interaction of Pd/TiO2 catalysts and their application in the liquid phase selective hydrogenation of long chain alkadienes. J. Mol. Catal. A Chem. 2004, 216, 107–114. [Google Scholar] [CrossRef]
- Chen, C.-A.; Chen, Y.-M.; Huang, Y.-S.; Tsai, D.-S.; Tiong, K.-K.; Liao, P.-C. Synthesis and characterization of well-Aligned anatase TiO2 nanocrystals on fused silicavia metal-Organic vapor deposition. CrystEngComm 2009, 11, 2313–2318. [Google Scholar] [CrossRef]
- Tian, F.; Zhang, Y.; Zhang, J.; Pan, C. Raman spectroscopy: A new approach to measure the percentage of anatase TiO2 exposed (001) facets. J. Phys. Chem. C 2012, 116, 7515–7519. [Google Scholar] [CrossRef]
- Sahoo, S.; Arora, A.K.; Sridharan, V. Raman line shapes of optical phonons of different symmetries in anatase TiO2 nanocrystals. J. Phys. Chem. C 2009, 113, 16927–16933. [Google Scholar] [CrossRef]
- Santara, B.; Giri, P.K.; Imakita, K.; Fujii, M. Evidence for ti interstitial induced extended visible absorption and near infrared photoluminescence from undoped TiO2 nanoribbons: An in situ photoluminescence study. J. Phys. Chem. C 2013, 117, 23402–23411. [Google Scholar] [CrossRef]
- Choudhury, B.; Dey, M.; Choudhury, A. Defect generation, d-d transition, and band gap reduction in Cu-Doped TiO2 nanoparticles. Int. Nano Lett. 2013, 3, 1–8. [Google Scholar] [CrossRef]
- Choudhury, B.; Choudhury, A. Dopant induced changes in structural and optical properties of Cr3+ doped TiO2 nanoparticles. Mater. Chem. Phys. 2012, 132, 1112–1118. [Google Scholar] [CrossRef]
- Navas, J.; Sanchez-Coronilla, A.; Aguilar, T.; Hernandez, N.C.; de los Santos, D.M.; Sanchez-Marquez, J.; Zorrilla, D.; Fernandez-Lorenzo, C.; Alcantara, R.; Martin-Calleja, J. Experimental and theoretical study of the electronic properties of Cu-Doped anatase TiO2. Phys. Chem. Chem. Phys. 2014, 16, 3835–3845. [Google Scholar] [CrossRef] [PubMed]
- Vaseem, M.; Hong, A.R.; Kim, R.-T.; Hahn, Y.-B. Copper oxide quantum dot ink for inkjet-Driven digitally controlled high mobility field effect transistors. J. Mater. Chem. C 2013, 1, 2112–2120. [Google Scholar] [CrossRef]
- Kung, M.C.; Davis, R.J.; Kung, H.H. Understanding Au-Catalyzed low-Temperature CO oxidation. J. Phys. Chem. C 2007, 111, 11767–11775. [Google Scholar] [CrossRef]
- Haruta, M. Catalysis of gold nanoparticles deposited on metal oxides. CATTECH 2002, 6, 102–115. [Google Scholar] [CrossRef]
- Zou, X.; Xu, J.; Qi, S.; Suo, Z.; An, L.; Li, F. Effects of preparation conditions of Au/FeOx/Al2O3 catalysts prepared by a modified two-Step method on the stability for CO oxidation. J. Nat. Gas Chem. 2011, 20, 41–47. [Google Scholar] [CrossRef]
- Kang, M.Y.; Yun, H.J.; Yu, S.; Kim, W.; Kim, N.D.; Yi, J. Effect of TiO2 crystalline phase on CO oxidation over CuO catalysts supported on TiO2. J. Mol. Catal. A Chem. 2013, 368–369, 72–77. [Google Scholar] [CrossRef]
- Carabineiro, S.A.C.; Bogdanchikova, N.; Avalos-Borja, M.; Pestryakov, A.; Tavares, P.B.; Figueiredo, J.L. Gold supported on metal oxides for carbon monoxide oxidation. Nano Res. 2011, 4, 180–193. [Google Scholar] [CrossRef]
- Satsuma, A.; Yanagihara, M.; Osaki, K.; Saeki, Y.; Liu, H.; Yamamoto, Y.; Arai, S.; Ohyama, J. Promotion of low-temperature oxidation of co over pd supported on titania-Coated ceria. RSC Adv. 2014, 4, 54187–54193. [Google Scholar] [CrossRef]
- Vedyagin, A.A.; Volodin, A.M.; Kenzhin, R.M.; Chesnokov, V.V.; Mishakov, I.V. CO oxidation over Pd/ZrO2 catalysts: Role of support’s donor sites. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Ayastuy, J.L.; Gurbani, A.; González-Marcos, M.P.; Gutiérrez-Ortiz, M.A. Kinetics of carbon monoxide oxidation over CuO supported on nanosized CeO2. Ind. Eng. Chem. Res. 2009, 48, 5633–5641. [Google Scholar] [CrossRef]
- Duprat, F. Light-Off curve of catalytic reaction and kinetics. Chem. Eng. Sci. 2002, 57, 901–911. [Google Scholar] [CrossRef]
- Lu, J.-Q.; Sun, C.-X.; Li, N.; Jia, A.-P.; Luo, M.-F. Kinetic study of co oxidation over CuO/MO2 (m = si, ti and ce) catalysts. Appl. Surf. Sci. 2013, 287, 124–134. [Google Scholar] [CrossRef]
- Roduner, E. Understanding catalysis. Chem. Soc. Rev. 2014, 43, 8226–8239. [Google Scholar] [CrossRef] [PubMed]
- Jia, A.-P.; Deng, Y.; Hu, G.-S.; Luo, M.-F.; Lu, J.-Q. Kinetic and activity study of CO oxidation over CuO–MnOx–CeO2 catalysts. React. Kinet. Mech. Catal. 2016, 117, 503–520. [Google Scholar] [CrossRef]
- Sun, K.C.; Qadir, M.B.; Jeong, S.H. Hydrothermal synthesis of TiO2 nanotubes and their application as an over-Layer for dye-Sensitized solar cells. RSC Adv. 2014, 4, 23223–23230. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | BET Surface Area (m²/g) | Total Pore Volume (cm³/g) | Average Pore Width (nm) |
|---|---|---|---|
| CuO | 33.5 | 0.30 | 35.8 |
| TiO2 P25 | 44.8 | 0.15 | 12.2 |
| TiO2 (Anatase) | 9.1 | 0.02 | 12.6 |
| TiO2 NT | 183.2 | 0.76 | 16.7 |
| 2CuO-TiO2 NT | 147.5 | 0.88 | 23.9 |
| 5CuO-TiO2 NT | 133.9 | 0.86 | 25.7 |
| 10CuO-TiO2 NT | 132.2 | 0.77 | 23.4 |
| 15CuO-TiO2 NT | 107.6 | 0.61 | 22.7 |
| 20CuO-TiO2 NT | 99.9 | 0.56 | 22.7 |
| 30CuO-TiO2 NT | 87.2 | 0.48 | 22.2 |
| 50CuO-TiO2 NT | 60.95 | 0.32 | 21.4 |
| 65CuO-TiO2 NT | 41.3 | 0.21 | 21.2 |
| Catalyst | Reaction Rate (µmole s−1 g−1) 1 | CO Conversion (%) at T = 155 °C | Temperatures | Ea (kJ mol−1) 2 | |
|---|---|---|---|---|---|
| T50 | T70 | ||||
| 2CuO-TiO2 NT | 4.3 | 12 | 193 | 211 | 68.0 ± 3.8 |
| 5CuO-TiO2 NT | 14 | 40 | 161 | 179 | - |
| 10CuO-TiO2 NT | 23 | 66 | 145 | 158 | - |
| 15CuO-TiO2 NT | 28 | 78 | 137 | 148 | - |
| 20CuO-TiO2 NT | 30 | 84 | 139 | 146 | - |
| 20CuO- TiO2 P25 | 17 | 46 | 156 | 166 | 79.8 ± 2.4 |
| 30CuO-TiO2 NT | 32 | 91 | 139 | 144 | - |
| 50CuO-TiO2 NT | 36 | 100 | 108 | 111 | 81.0 ±0.7 |
| 65CuO-TiO2 NT | 33 | 33 | 144 | 147 | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zedan, A.F.; Allam, N.K.; AlQaradawi, S.Y. A Study of Low-Temperature CO Oxidation over Mesoporous CuO-TiO2 Nanotube Catalysts. Catalysts 2017, 7, 129. https://doi.org/10.3390/catal7050129
Zedan AF, Allam NK, AlQaradawi SY. A Study of Low-Temperature CO Oxidation over Mesoporous CuO-TiO2 Nanotube Catalysts. Catalysts. 2017; 7(5):129. https://doi.org/10.3390/catal7050129
Chicago/Turabian StyleZedan, Abdallah F., Nageh K. Allam, and Siham Y. AlQaradawi. 2017. "A Study of Low-Temperature CO Oxidation over Mesoporous CuO-TiO2 Nanotube Catalysts" Catalysts 7, no. 5: 129. https://doi.org/10.3390/catal7050129
APA StyleZedan, A. F., Allam, N. K., & AlQaradawi, S. Y. (2017). A Study of Low-Temperature CO Oxidation over Mesoporous CuO-TiO2 Nanotube Catalysts. Catalysts, 7(5), 129. https://doi.org/10.3390/catal7050129