In Situ Regeneration of Alumina-Supported Cobalt–Iron Catalysts for Hydrogen Production by Catalytic Methane Decomposition

 ,
, 
 and
and 
Abstract
:1. Introduction
2. Results and Discussion
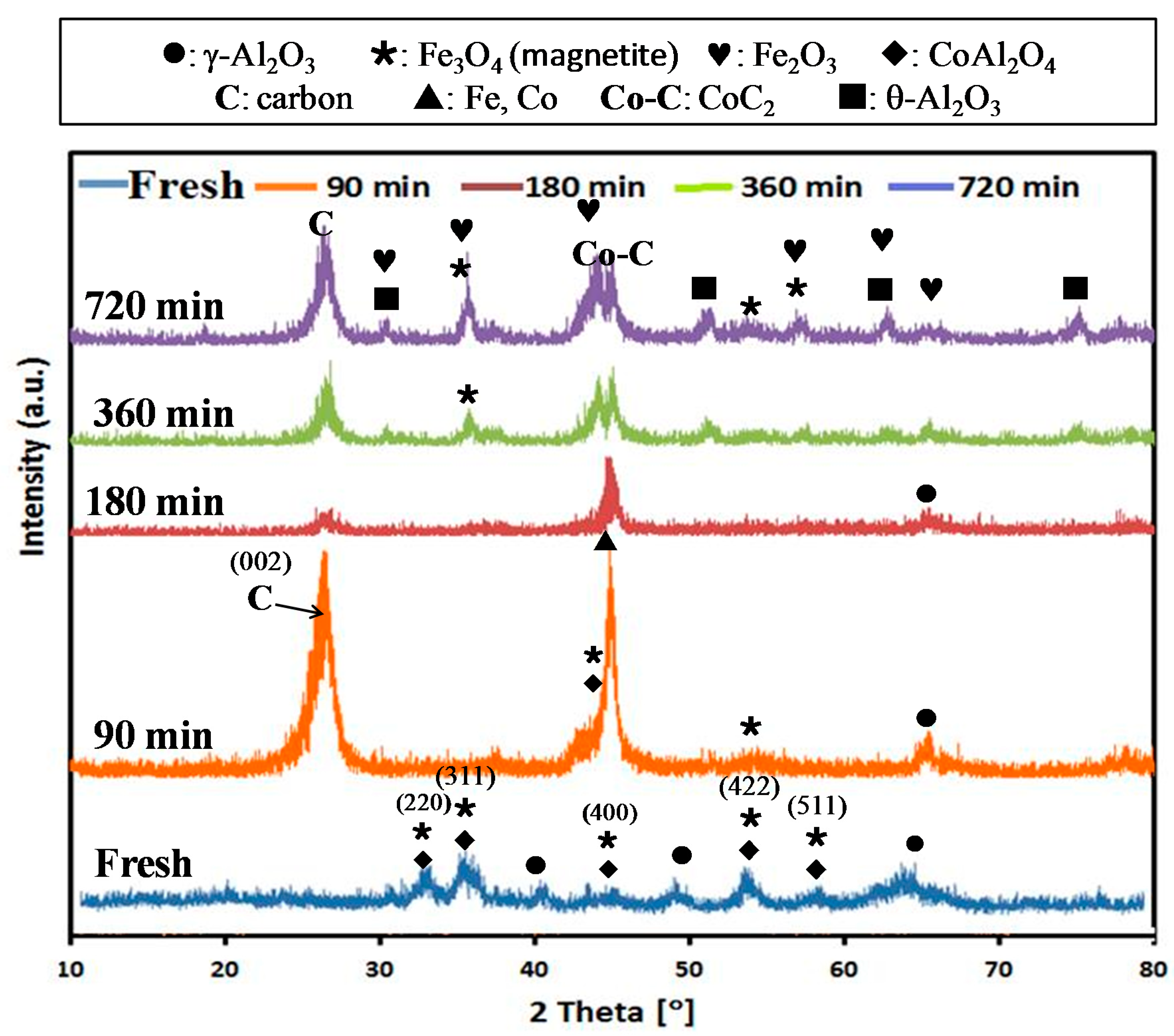
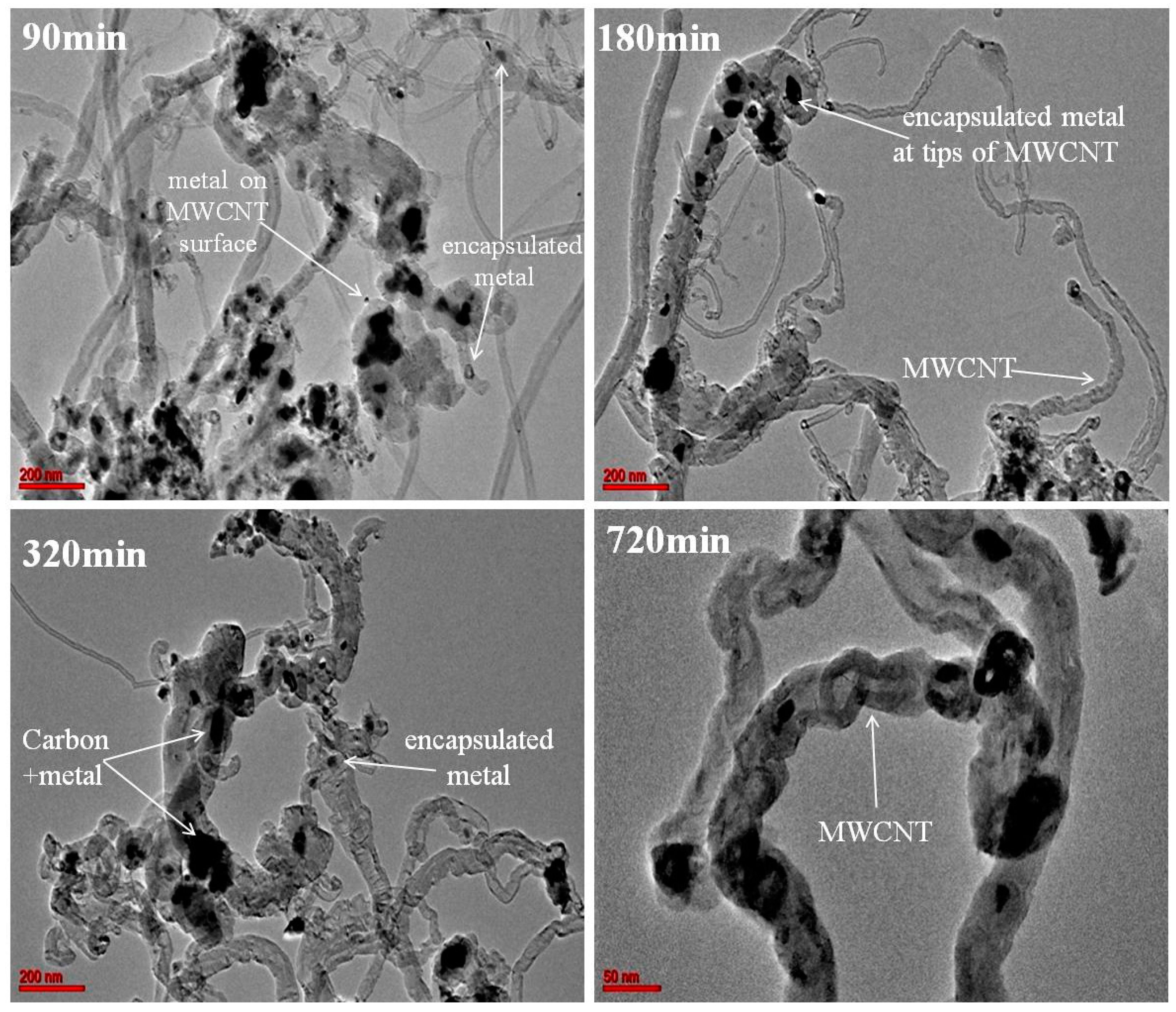
2.1. Structure and Morphology
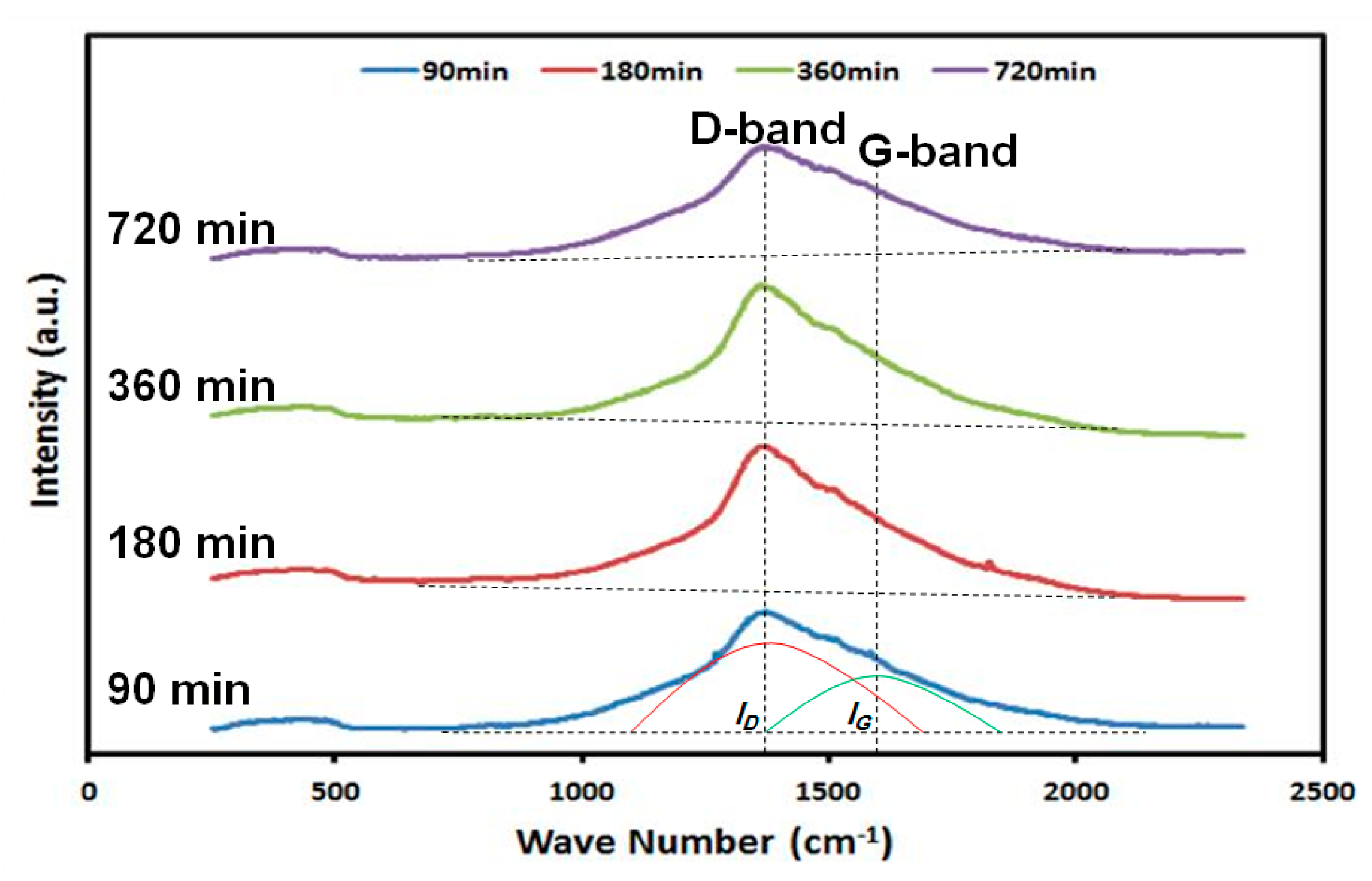
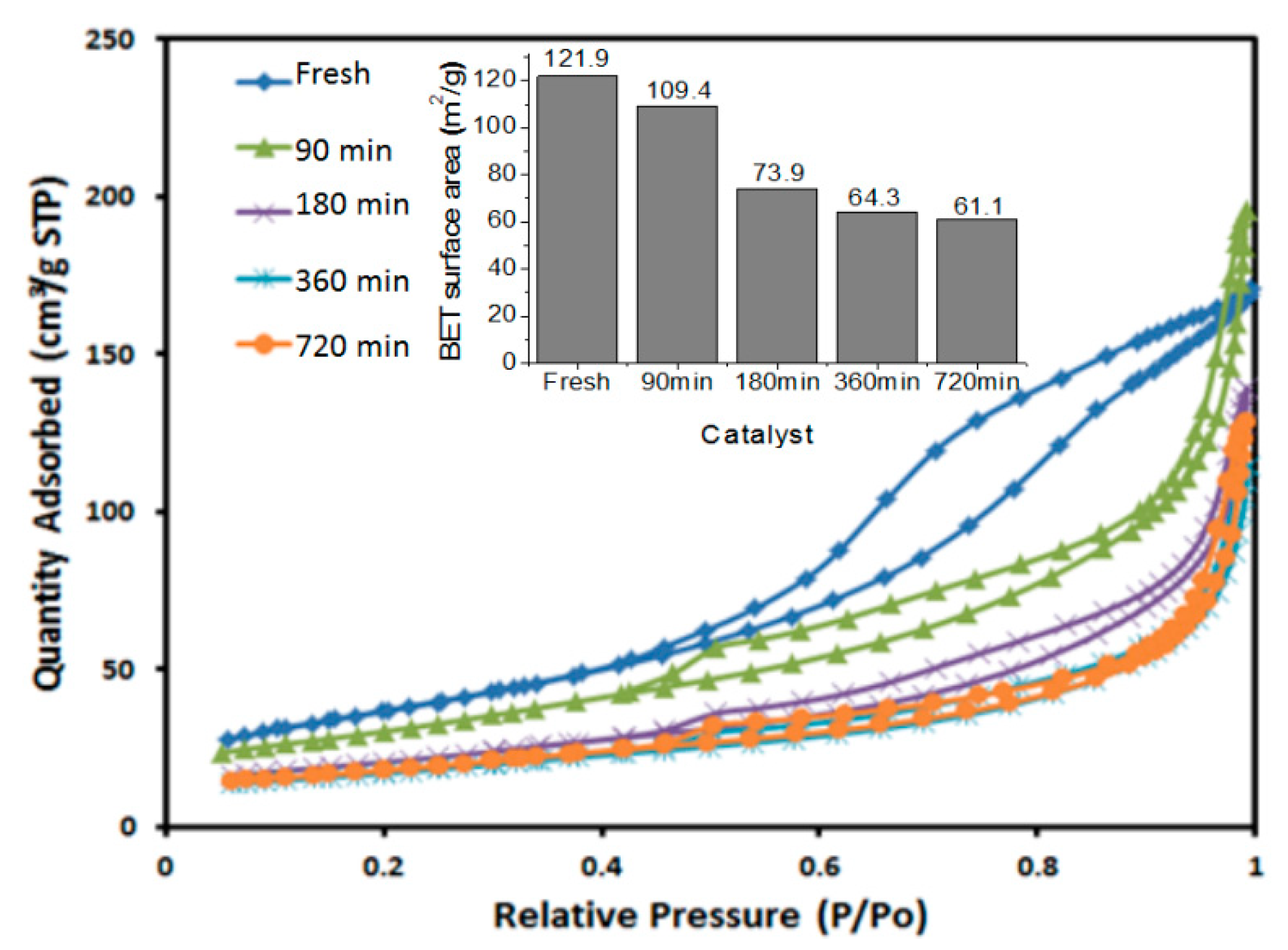
2.2. Surface Characterization
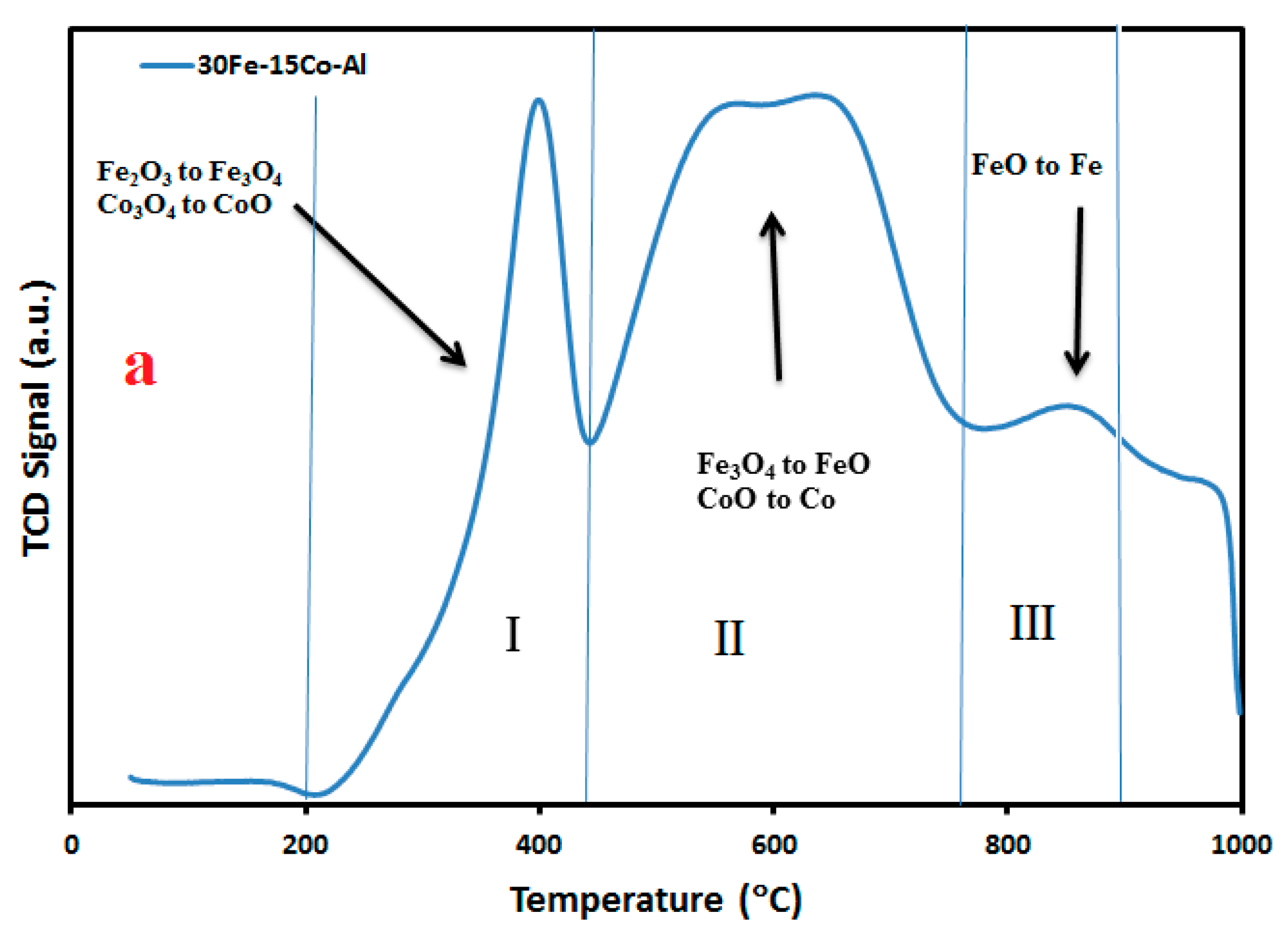
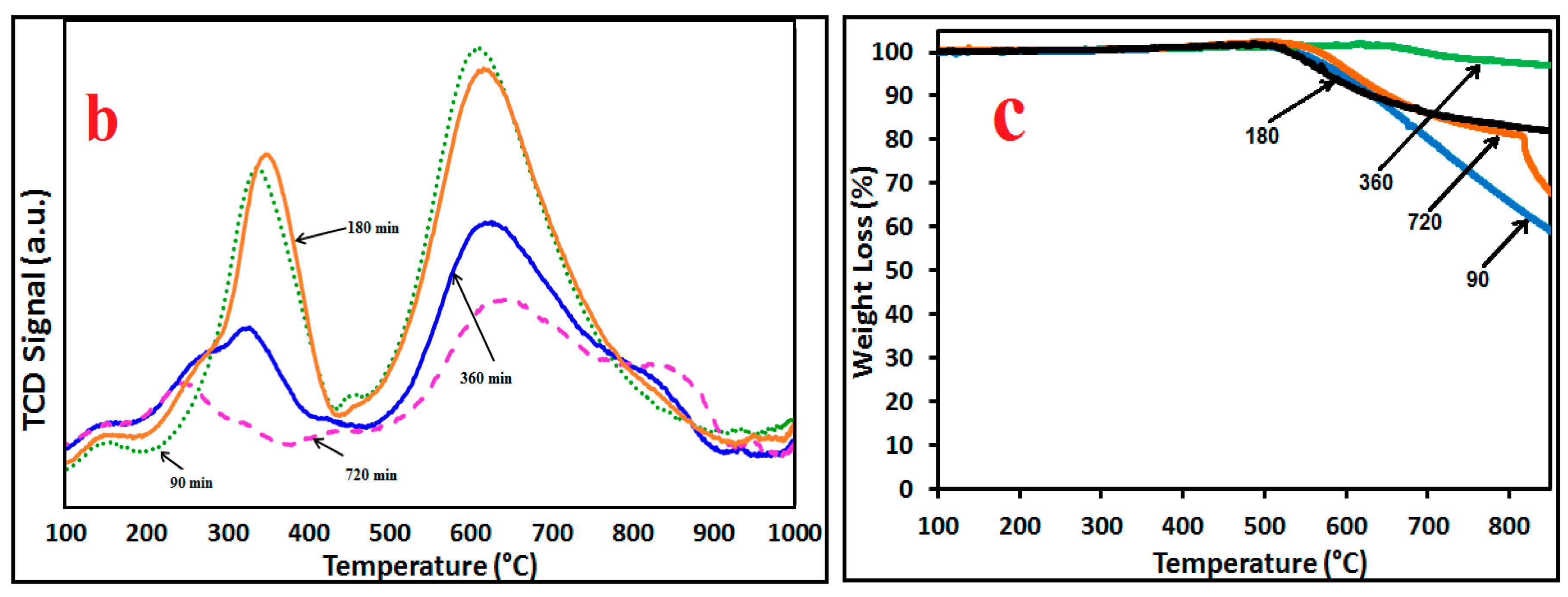
2.3. H2-TPR and TG Analysis
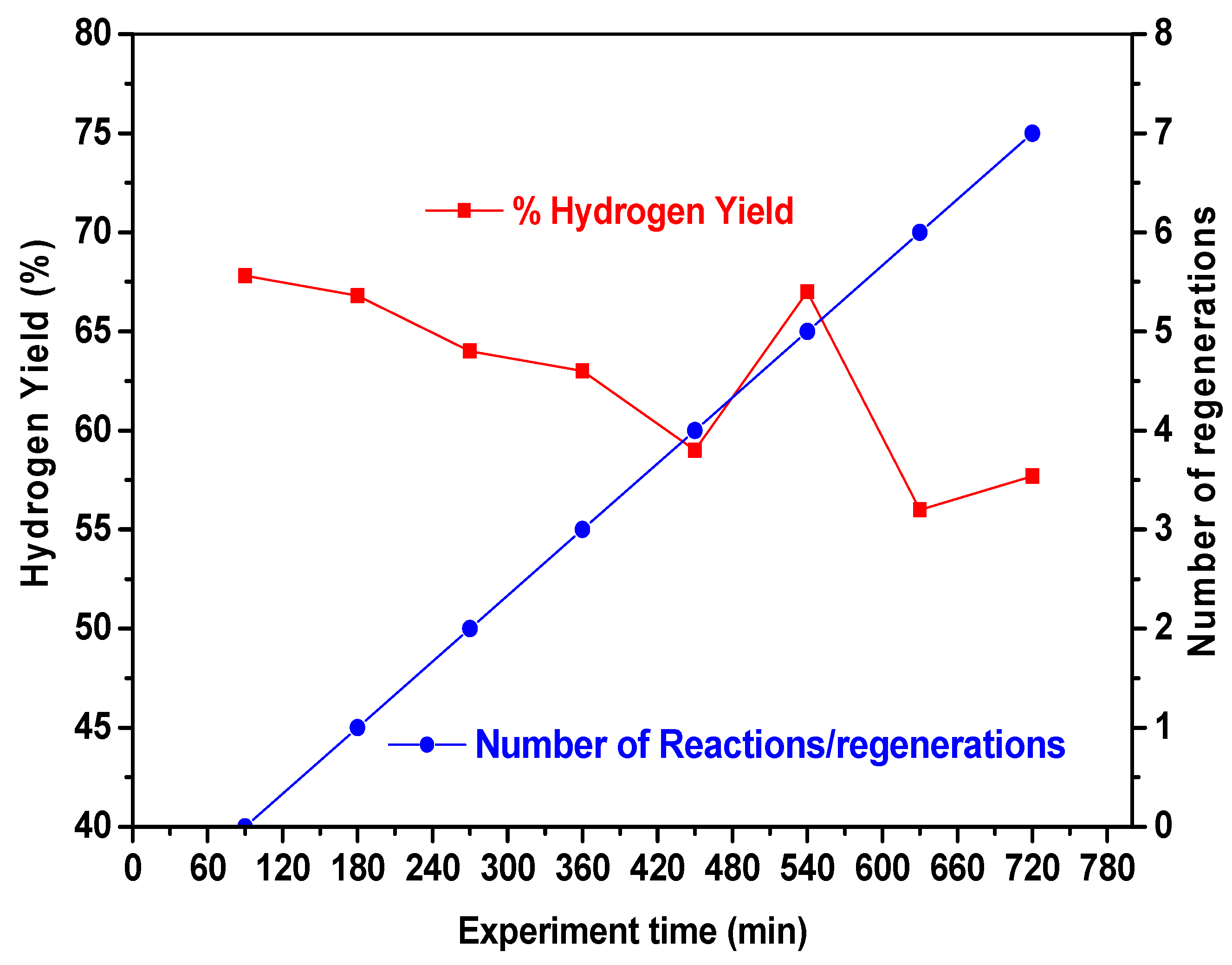
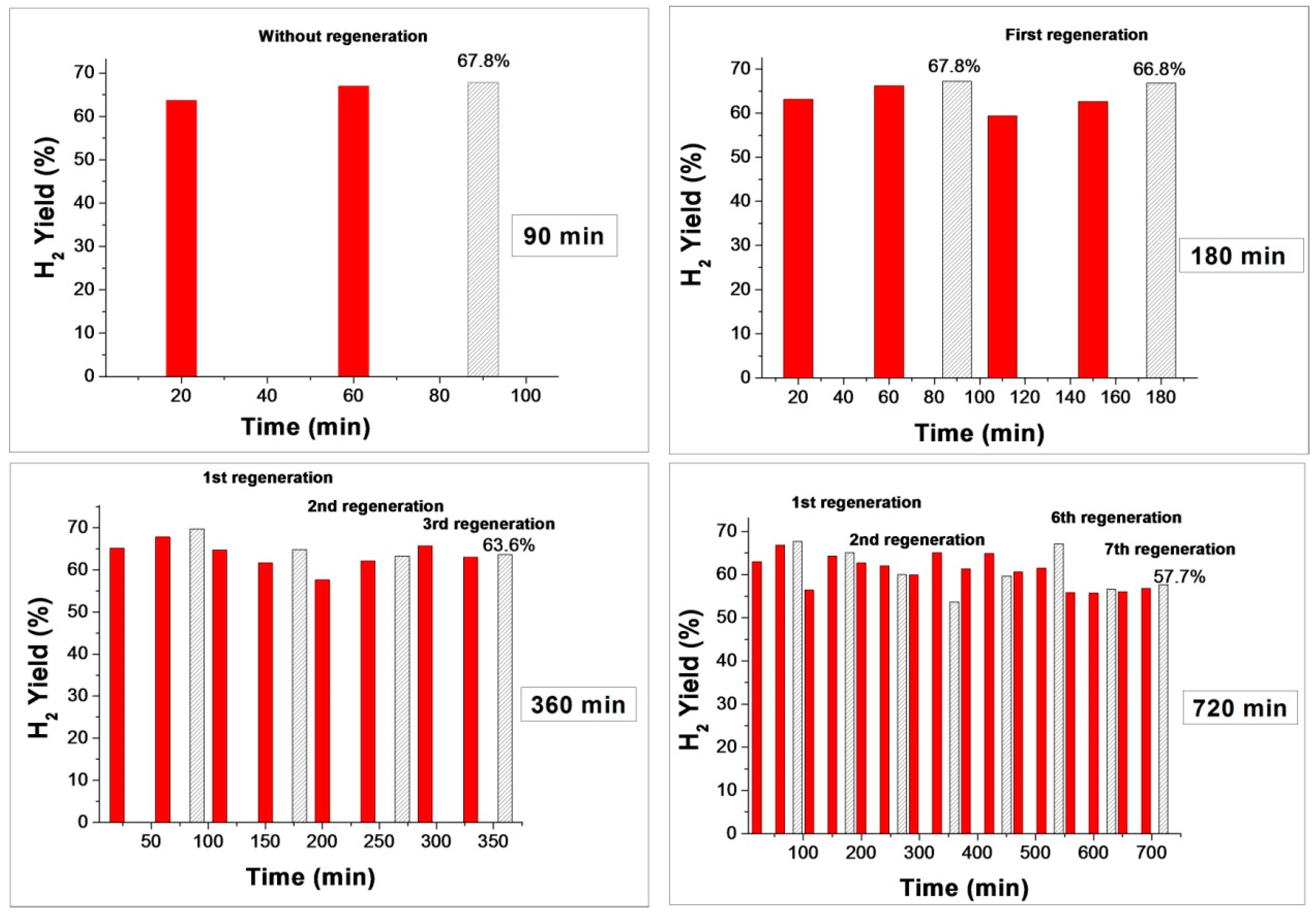
2.4. Catalysts Behavior under CMD-Regeneration Cycles
3. Experimental Section
3.1. Preparation of Fresh Catalyst
3.2. Characterization Methods
3.3. Activity Test (Regeneration Procedure)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dodds, P.E.; Staffell, I.; Hawkes, A.D.; Li, F.; Grunewald, P.; McDowall, W.; Ekins, P.D. Hydrogen and fuel cell technologies for heating: A review. Int. J. Hydrog. Energy 2015, 40, 2065–2083. [Google Scholar] [CrossRef]
- Deng, J.; Lv, X.; Gao, J.; Pu, A.; Li, M.; Sun, X.; Zhong, J. Facile synthesis of carbon-coated hematite nanostructures for solar water splitting. Energy Environ. Sci. 2013, 6, 1965–1970. [Google Scholar] [CrossRef]
- Lima, S.M.; Cruz, I.O.; Jacobs, G.; Davis, B.H.; Mattos, L.V.; Noronha, F.B. Steam reforming, partial oxidation, and oxidative steam reforming of ethanol over Pt/CeZrO2 catalyst. J. Catal. 2008, 257, 356–368. [Google Scholar]
- Siriwardane, R.; Tian, H.; Fisher, J. Production of pure hydrogen and synthesis gas with Cu-Fe oxygen carriers using combined processes of chemical looping combustion and methane decomposition/reforming. Int. J. Hydrog. Energy 2015, 40, 1698–1708. [Google Scholar] [CrossRef]
- Al-Fatesh, S.A.; Ibrahim, A.A.; Abu-Dahrieh, J.K.; Al-Awadi, A.S.; El-Toni, A.M.; Fakeeha, A.H.; Abasaeed, A.E. Gallium-Promoted Ni Catalyst Supported on MCM-41 for Dry Reforming of Methane. Catalysts 2018, 8, 229. [Google Scholar] [CrossRef]
- Osman, A.I.; Abu-Dahrieh, J.K.; Cherkasov, N.; Fernandez-Garcia, J.; Walker, D.; Walton, R.I.; Rooney, D.W.; Rebrov, E. A highly active and synergistic Pt/Mo2C/Al2O3 catalyst for water-gas shift reaction. Mol. Catal. 2018, 455, 38–47. [Google Scholar] [CrossRef]
- Osman, A.I.; Meudal, J.; Laffir, F.; Thompson, J.; Rooney, D. Enhanced catalytic activity of Ni on η-Al2O3 and ZSM-5 on addition of ceria zirconia for the partial oxidation of methane. App. Catal. B Environ. 2017, 212, 68–79. [Google Scholar] [CrossRef]
- Politano, A.; Cattelan, M.; Boukhvalov, D.W.; Campi, D.; Cupolillo, A.; Gnoli, S.; Apostol, N.G.; Lacovig, P.; Lizzit, S.; Far, D.; et al. Unveiling the Mechanisms Leading to H2 Production Promoted by Water Decomposition on Epitaxial Graphene at Room Temperature. ACS Nano 2016, 10, 4543–4549. [Google Scholar] [CrossRef] [PubMed]
- Abbas, H.F.; Daud, W.M.A. Hydrogen production by methane decomposition: A review. Int. J. Hydrog. Energy 2010, 35, 1160–1190. [Google Scholar] [CrossRef]
- Diehm, C.; Deutschmann, O. Hydrogen production by catalytic partial oxidation of methane over staged Pd/Rh coated monoliths: Spatially resolved concentration and temperature profiles. Int. J. Hydrog. Energy 2014, 39, 17998–18004. [Google Scholar] [CrossRef]
- Vigneault, A.; Grace, J.R. Hydrogen production in multi-Channel membrane reactor via steam methane reforming and methane catalytic combustion. Int. J. Hydrog. Energy 2015, 40, 233–243. [Google Scholar] [CrossRef]
- Wang, Y.; Peng, J.; Zhou, C.; Lim, Z.-Y.; Wu, C.; Ye, S.; Wang, G. Effect of Pr addition on the properties of Ni/Al2O3 catalysts with an application in the autothermal reforming of methane. Int. J. Hydrog. Energy 2014, 39, 778–787. [Google Scholar] [CrossRef]
- Fakeeha, A.H.; Khan, W.U.; Al-Fatesh, A.S.; Abasaeed, A.E.; Naeem, M.A. Production of hydrogen and carbon nanofibers from methane over Ni-Co-Al catalysts. Int. J. Hydrog. Energy 2015, 40, 1774–1781. [Google Scholar] [CrossRef]
- Al-Hassani, A.A.; Abbas, H.F.; Wan Daud, W.M.A. Production of COx-free hydrogen by the thermal decomposition of methane over activated carbon: Catalyst deactivation. Int. J. Hydrog. Energy 2014, 39, 14783–14791. [Google Scholar] [CrossRef]
- Anjaneyulu, C.; Kumar, S.N.; Kumar, V.V.; Naresh, G.; Bhargava, S.K.; Chary, K.V.R.; Venugopal, A. Influence of La on reduction behavior and Ni metal surface area of Ni-Al2O3 catalysts for COx free H2 by catalytic decomposition of methane. Int. J. Hydrog. Energy 2015, 40, 3633–3641. [Google Scholar] [CrossRef]
- Al-Hassani, A.A.; Abbas, H.F.; Wan Daud, W.M.A. Hydrogen production via decomposition of methane over activated carbons as catalysts: Full factorial design. Int. J. Hydrog. Energy 2014, 39, 7004–7014. [Google Scholar] [CrossRef]
- Pereira, M.F.R.; Figueiredo, J.L.; Órfão, J.J.M.; Serp, P.; Kalck, P.; Kihn, Y. Catalytic activity of carbon nanotubes in the oxidative dehydrogenation of ethylbenzene. Carbon 2004, 42, 2807–2813. [Google Scholar] [CrossRef]
- Serp, P.; Corrias, M.; Kalck, P. Carbon nanotubes and nanofibers in catalysis. Appl. Catal. A Gen. 2003, 253, 337–358. [Google Scholar] [CrossRef]
- Iijima, S. Helical microtubes of graphitic carbon. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Lee, M.S.; Lee, S.Y.; Park, S.J. Preparation and characterization of multi-walled carbon nanotubes impregnated with polyethyleneimine for carbon dioxide capture. Int. J. Hydrog. Energy 2015, 40, 3415–3421. [Google Scholar] [CrossRef]
- Takenaka, S.; Ishida, M.; Serizawa, M.; Tanabe, E.; Otsuka, K. Formation of carbon nanofibers and carbon nanotubes through methane decomposition over supported cobalt catalysts. J. Phys. Chem. B 2004, 108, 11464–11472. [Google Scholar] [CrossRef]
- Li, D.; Chen, J.; Li, Y. Evidence of composition deviation of metal particles of a NieCu/Al2O3 catalyst during methane decomposition to COx-free hydrogen. Int. J. Hydrog. Energy 2009, 34, 299–307. [Google Scholar] [CrossRef]
- Jana, P.; de la Pena, V.A.; Shea, O.; Coronado, J.M.; Serrano, D.P. Cobalt based catalysts prepared by pechini method for CO2-free hydrogen production by methane decomposition. Int. J. Hydrog. Energy 2010, 35, 10285–10294. [Google Scholar] [CrossRef]
- Pinilla, J.L.; Utrilla, R.; Lazaro, M.J.; Moliner, R.; Suelves, I.; García, A.B. Ni- and Fe-based catalysts for hydrogen and carbon nanofilament production by catalytic decomposition of methane in a rotary bed reactor. Fuel Process. Technol. 2011, 92, 1480–1488. [Google Scholar] [CrossRef]
- Politano, A.; Chiarello, G. Unveiling the Oxidation Processes of Pt3Ni(111) by Real-Time Surface Core-Level Spectroscopy. ChemCatChem 2016, 8, 713–718. [Google Scholar] [CrossRef]
- Ren, H.; Humbert, M.P.; Carl, A.; Menning, C.A.; Chen, J.G.; Shu, Y.; Singh, U.G.; Cheng, W.C. Inhibition of coking and CO poisoning of Pt catalysts by the formation of Au/Pt bimetallic surfaces. Appl. Catal. A 2010, 375, 303–309. [Google Scholar] [CrossRef]
- Baraldi, A.; Bianchettin, L.; Gironcoli, S.D.; Vesselli, E.; Lizzit, S.; Petaccia, L.; Comelli, G.; Rosei, R. Enhanced chemical reactivity of under-coordinated atoms at Pt-Rh bimetallic surfaces: A spectroscopic characterization. J. Phys. Chem. C 2011, 115, 3378–3384. [Google Scholar] [CrossRef]
- Nilekar, A.U.; Xu, Y.; Zhang, J.; Vukmirovic, M.B.; Sasaki, K.; Adzic, R.; Mavrikakis, M. Bimetallic and Ternary Alloys for Improved Oxygen Reduction Catalysis. Top. Catal. 2007, 46, 276–284. [Google Scholar] [CrossRef]
- Reshetenko, T.V.; Avdeeva, L.B.; Ushakov, V.A.; Moroz, E.M.; Shmakov, A.N.; Kriventsov, V.V.; Kochubey, D.I.; Pavlyukhin, Y.T.; Chuvilin, A.L.; Ismagilov, Z.R. Coprecipitated iron-containing catalysts (Fe-Al2O3, Fe-Co-Al2O3, Fe-Ni-Al2O3) for methane decomposition at moderate temperatures. Part II: Evolution of the catalysts in reaction. Appl. Catal. A 2004, 270, 87–99. [Google Scholar] [CrossRef]
- Shen, W.; Huggins, F.E.; Shah, N.; Jacobs, G.; Wang, Y.; Shi, X.; Huffman, G.P. Novel Fe–Ni nanoparticle catalyst for the production of CO- and CO2-free H2 and carbon nanotubes by dehydrogenation of methane. Appl. Catal. A 2008, 351, 102–110. [Google Scholar] [CrossRef]
- Avdeeva, L.B.; Reshetenko, T.V.; Ismagilov, Z.R.; Likholobov, V.A. Iron-containing catalysts of methane decomposition: Accumulation of filamentous carbon. Appl. Catal. A 2002, 228, 53–63. [Google Scholar] [CrossRef]
- Latorre, N.; Cazana, F.; Martínez-Hansen, V.M.; Royo, C.; Romeo, E.; Monzón, A. Ni-Co-Mg-Al catalysts for hydrogen and carbonaceous nanomaterials production by CCVD of methane. Catal. Today 2011, 172, 143–151. [Google Scholar] [CrossRef]
- Shah, N.; Panjala, D.; Huffman, G.P. Hydrogen production by catalytic decomposition of methane. Energy Fuels 2001, 15, 1528–1534. [Google Scholar] [CrossRef]
- Pudukudy, M.; Yaakob, Z.; Zubair, Z.; Akmal, S. Direct decomposition of methane over Pd promoted Ni/SBA-15 catalysts. Appl. Surf. Sci. 2015, 353, 127–136. [Google Scholar] [CrossRef]
- Awadallah, A.E.; Aboul-Enein, A.A.; El-Desouki, D.S.; Aboul-Gheit, A.K. Catalytic thermal decomposition of methane to COx-free hydrogen and carbon nanotubes over MgO supported bimetallic group VIII catalysts. Appl. Surf. Sci. 2014, 296, 100–107. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Carbon deposition in steam reforming and methanation. Catal. Rev. Sci. Eng. 1982, 24, 67–112. [Google Scholar] [CrossRef]
- Hartenstein, H.U.; Hoffman, T. Method of Regeneration of SCR Catalyst. U.S. Patent 7723251B2, 25 May 2010. [Google Scholar]
- Hazzim, F.; Abbas, W.M.A.; Wan, D. Thermocatalytic decomposition of methane for hydrogen production using activated carbon catalyst: Regeneration and characterization studies. Int. J. Hydrog. Energy 2009, 34, 8034–8045. [Google Scholar]
- Al-Fatesh, A.S.; Fakeeha, A.H.; Khan, W.U.; Ibrahim, A.A.; He, S.; Seshan, K. Production of hydrogen by catalytic methane decomposition over alumina supported mono-, bi- and tri-metallic catalysts. Int. J. Hydrog. Energy 2016, 41, 22932–22940. [Google Scholar] [CrossRef]
- Osman, A.I.; Abu-Dahrieh, J.K.; McLaren, M.; Laffir, F.; Rooney, D.W. Characterization of Robust Combustion Catalyst from Aluminum Foil Waste. ChemistrySelect 2018, 3, 1545–1550. [Google Scholar] [CrossRef]
- Zárate, J.; Rosas, G.; Pérez, R. Structural Transformations of the Pseudo boehmite to a-Alumina. Adv. Technol. Mater. Mater. Process. J. 2005, 1. [Google Scholar] [CrossRef]
- Jorio, A.; Saito, R.; Hafner, J.H.; Lieber, C.M.; Hunter, M.; McClure, T.; Dresselhaus, G.; Dresselhaus, M.S. Structural (n, m) determination of isolated single-wall carbon nanotubes by resonant Raman scattering. Phys. Rev. Lett. 2001, 86, 1118–1121. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.X.; Xu, S.T.; Ying, Z.; Yang, Q.H.; Liu, C.; Cheng, H.M. Hydrogen adsorption/desorption behavior of multi-walled carbon nanotubes with different diameters. Carbon 2003, 41, 2471–2476. [Google Scholar] [CrossRef]
- Miners, S.A.; Rance, G.A.; Khlobystov, A.N. Chemical reactions confined within carbon nanotubes. Chem. Soc. Rev. 2016, 45, 4727–4746. [Google Scholar] [CrossRef] [PubMed]
- Cullity, B.D. Elements of X-ray Diffractions; Addison-Wesley: New York, NY, USA, 1956. [Google Scholar]
- Al-Fatesh., A.S.; Barama, S.; Ibrahim, A.A.; Barama, A.; Khan, W.U.; Fakeeha, A. Study of Methane Decomposition on Fe/MgO-Based Catalyst Modified by Ni, Co, and Mn Additives. Chem. Eng. Commun. 2017, 204, 739–749. [Google Scholar] [CrossRef]
- Al-Fatesh, A.S.; Amin, A.; Ibrahim, A.A.; Khan, W.U.; Soliman, M.A.; AL-Otaibi, R.L.; Fakeeha, A.H. Effect of Ce and Co Addition to Fe/Al2O3 for Catalytic Methane Decomposition. Catalysts 2016, 6, 40. [Google Scholar] [CrossRef]
- Ferrari, A.C. Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and non-adiabatic effects. Solid State Commun. 2007, 143, 47–57. [Google Scholar] [CrossRef]
- Shi, B.; Zhang, Z.; Zha, B.; Liu, D. Structure evolution of spinel Fe-MII (M=Mn, Fe, Co, Ni) ferrite in CO hydrogeneration. Mol. Catal. 2018, 456, 31–37. [Google Scholar] [CrossRef]
- Khan, A.; Smirniotis, P.G. Relationship between temperature-programmed reduction profile and activity of modified ferrite-based catalysts for WGS reaction. J. Mol. Catal. A Chem. 2008, 280, 43–51. [Google Scholar] [CrossRef]
- Jo, S.B.; Chae, H.J.; Kim, T.Y.; Lee, C.H.; Oh, J.U.; Kang, S.-H.; Kim, J.W.; Jeong, M.; Lee, S.C.; Kim, J.C. Selective CO hydrogenation over bimetallic Co-Fe catalysts for the production of light paraffin hydrocarbons (C2-C4): Effect of H2/CO ratio and reaction temperature. Catal. Commun. 2018, 117, 74–78. [Google Scholar] [CrossRef]
- Zhou, L.; Enakonda, L.R.; Harb, M.; Saih, Y.; A-Tapiaa, A.; Ould-Chikh, S.; Hazemann, J.L.; Li, J.; Wei, N.; Gary, D.; et al. Fe catalysts for methane decomposition to produce hydrogen and carbon nano materials. Appl. Catal. B Environ. 2017, 208, 44–59. [Google Scholar] [CrossRef]
- Muradov, N.; Chen, Z.; Smith, F. Fossil hydrogen with reduced CO2 emission: Modeling thermocatalytic decomposition of methane in a fluidized bed of carbon particles. Int. J. Hydrog. Energy 2005, 30, 1149–1158. [Google Scholar] [CrossRef]
- Moliner, R.; Suelves, I.; Lázaro, M.J.; Moreno, O. Thermo-catalytic decomposition of methane over activated carbons: Influence of textural properties and surface chemistry. Int. J. Hydrog. Energy 2005, 30, 293–300. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
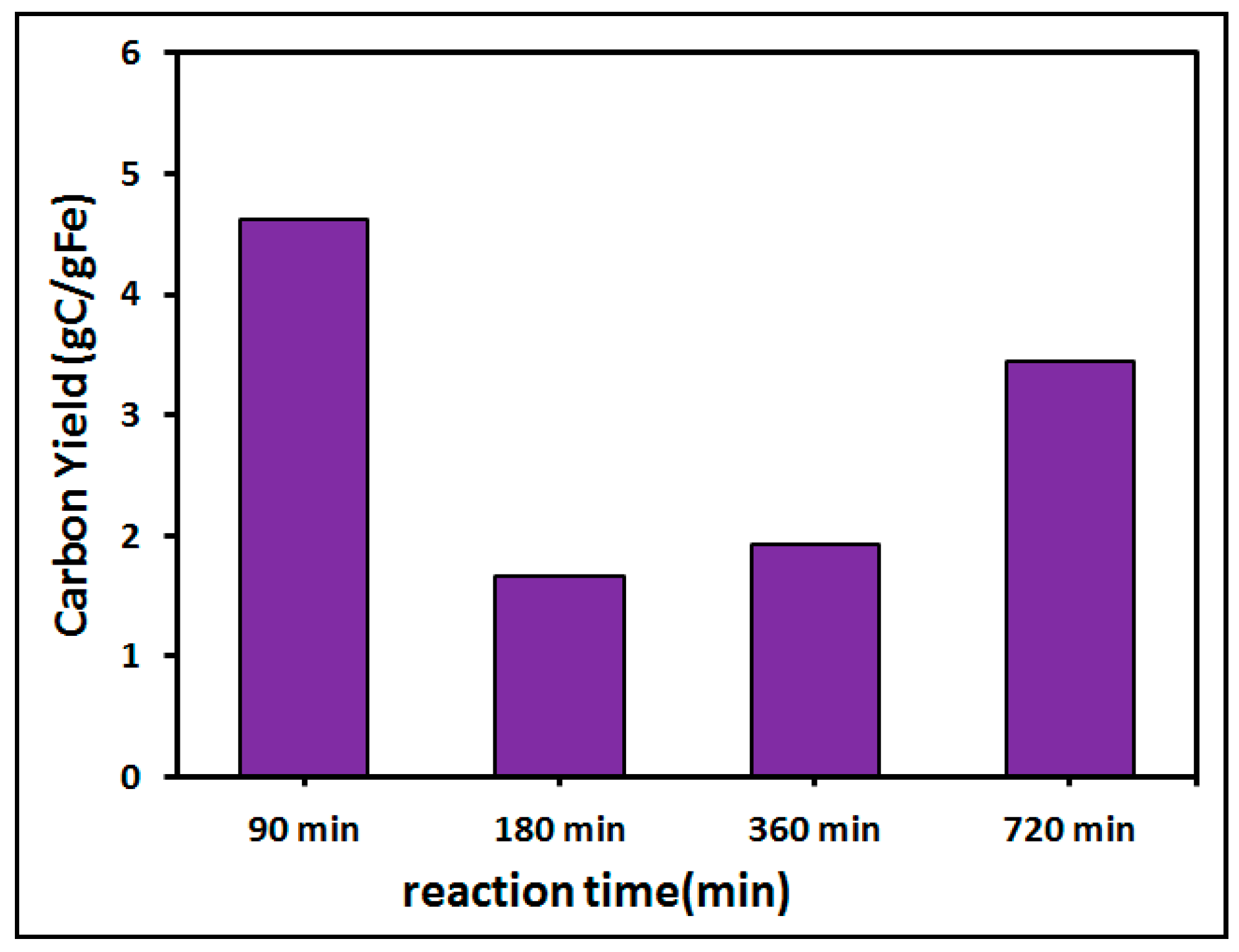{kind=link}
| Samples (SP) | Crystallite Size (*) of Carbon Graphite (Å) | Average Size (**) of Carbon Particles (nm) |
|---|---|---|
| 90 min | 49.7 | 52.3 |
| 180 min | 42.6 | 49.3 |
| 360 min | 59.6 | 53.2 |
| 720 min | 39.7 | 27.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fakeeha, A.H.; Barama, S.; Ibrahim, A.A.; Al-Otaibi, R.-L.; Barama, A.; Abasaeed, A.E.; Al-Fatesh, A.S. In Situ Regeneration of Alumina-Supported Cobalt–Iron Catalysts for Hydrogen Production by Catalytic Methane Decomposition. Catalysts 2018, 8, 567. https://doi.org/10.3390/catal8110567
Fakeeha AH, Barama S, Ibrahim AA, Al-Otaibi R-L, Barama A, Abasaeed AE, Al-Fatesh AS. In Situ Regeneration of Alumina-Supported Cobalt–Iron Catalysts for Hydrogen Production by Catalytic Methane Decomposition. Catalysts. 2018; 8(11):567. https://doi.org/10.3390/catal8110567
Chicago/Turabian StyleFakeeha, Anis H., Siham Barama, Ahmed A. Ibrahim, Raja-Lafi Al-Otaibi, Akila Barama, Ahmed E. Abasaeed, and Ahmed S. Al-Fatesh. 2018. "In Situ Regeneration of Alumina-Supported Cobalt–Iron Catalysts for Hydrogen Production by Catalytic Methane Decomposition" Catalysts 8, no. 11: 567. https://doi.org/10.3390/catal8110567
APA StyleFakeeha, A. H., Barama, S., Ibrahim, A. A., Al-Otaibi, R. -L., Barama, A., Abasaeed, A. E., & Al-Fatesh, A. S. (2018). In Situ Regeneration of Alumina-Supported Cobalt–Iron Catalysts for Hydrogen Production by Catalytic Methane Decomposition. Catalysts, 8(11), 567. https://doi.org/10.3390/catal8110567