The Mechanism of Adsorption, Diffusion, and Photocatalytic Reaction of Organic Molecules on TiO2 Revealed by Means of On-Site Scanning Tunneling Microscopy Observations


Abstract
:1. Introduction
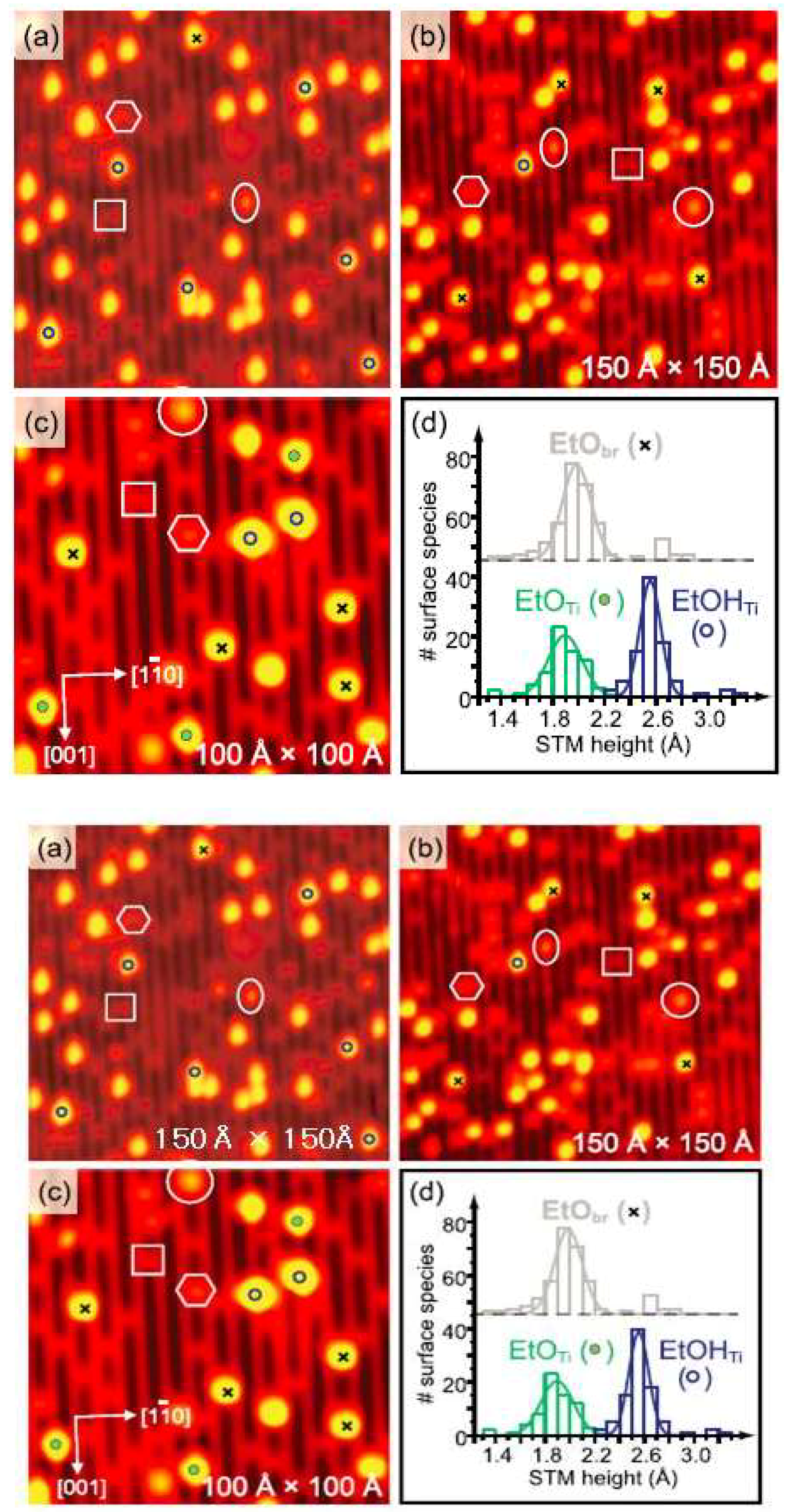
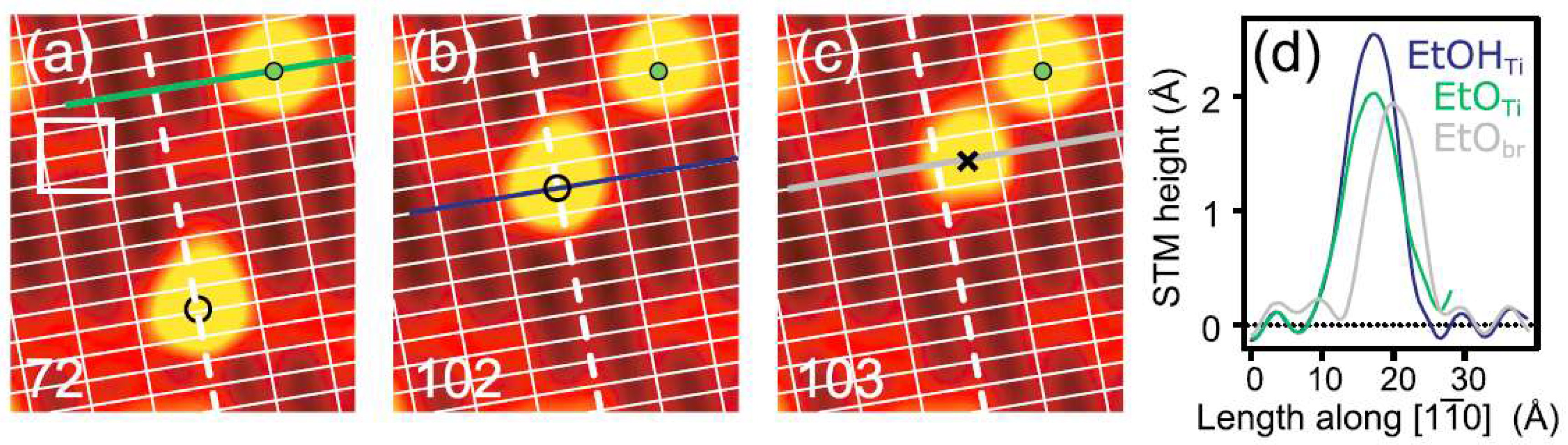
2. Adsorption Structure
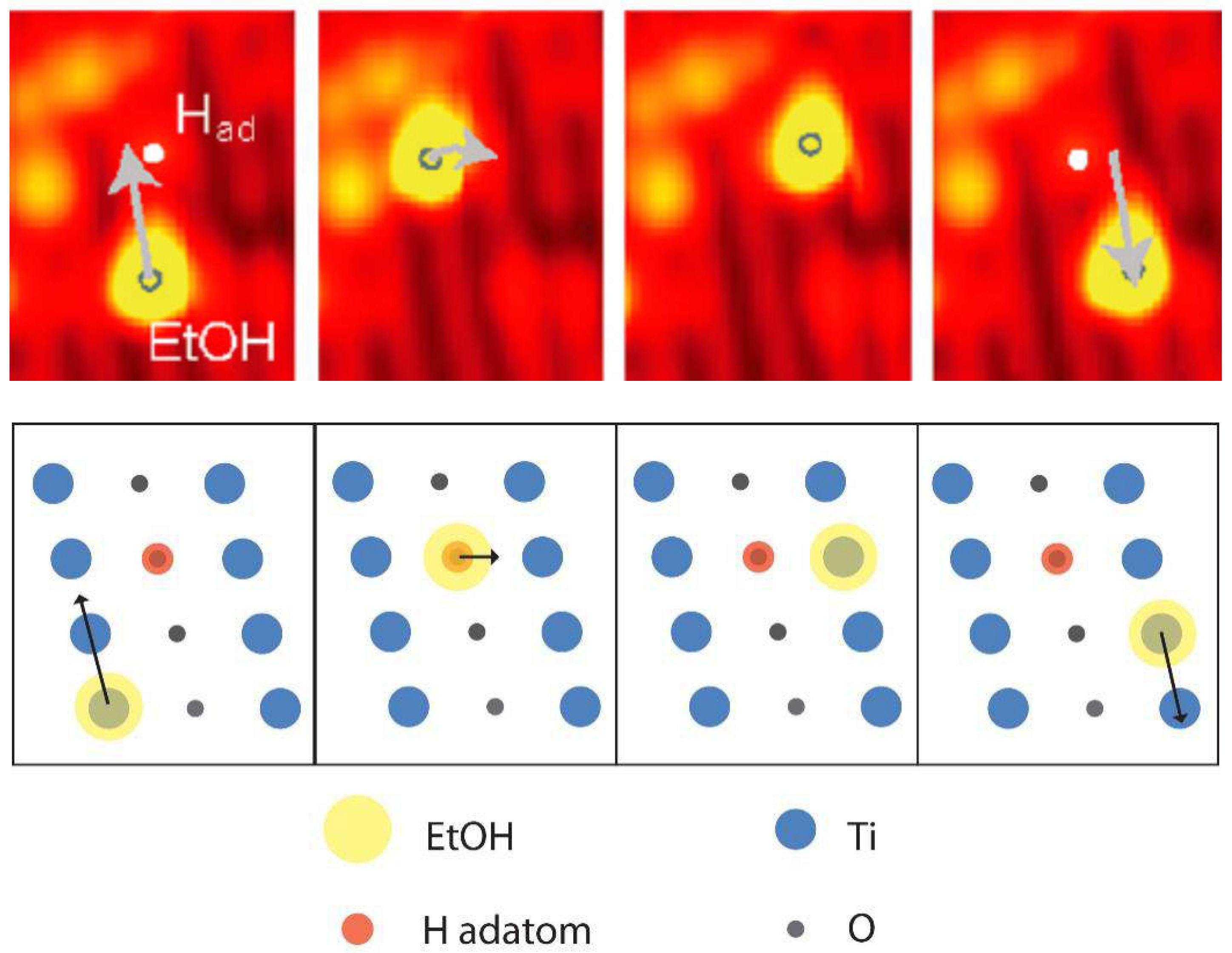
3. Diffusion
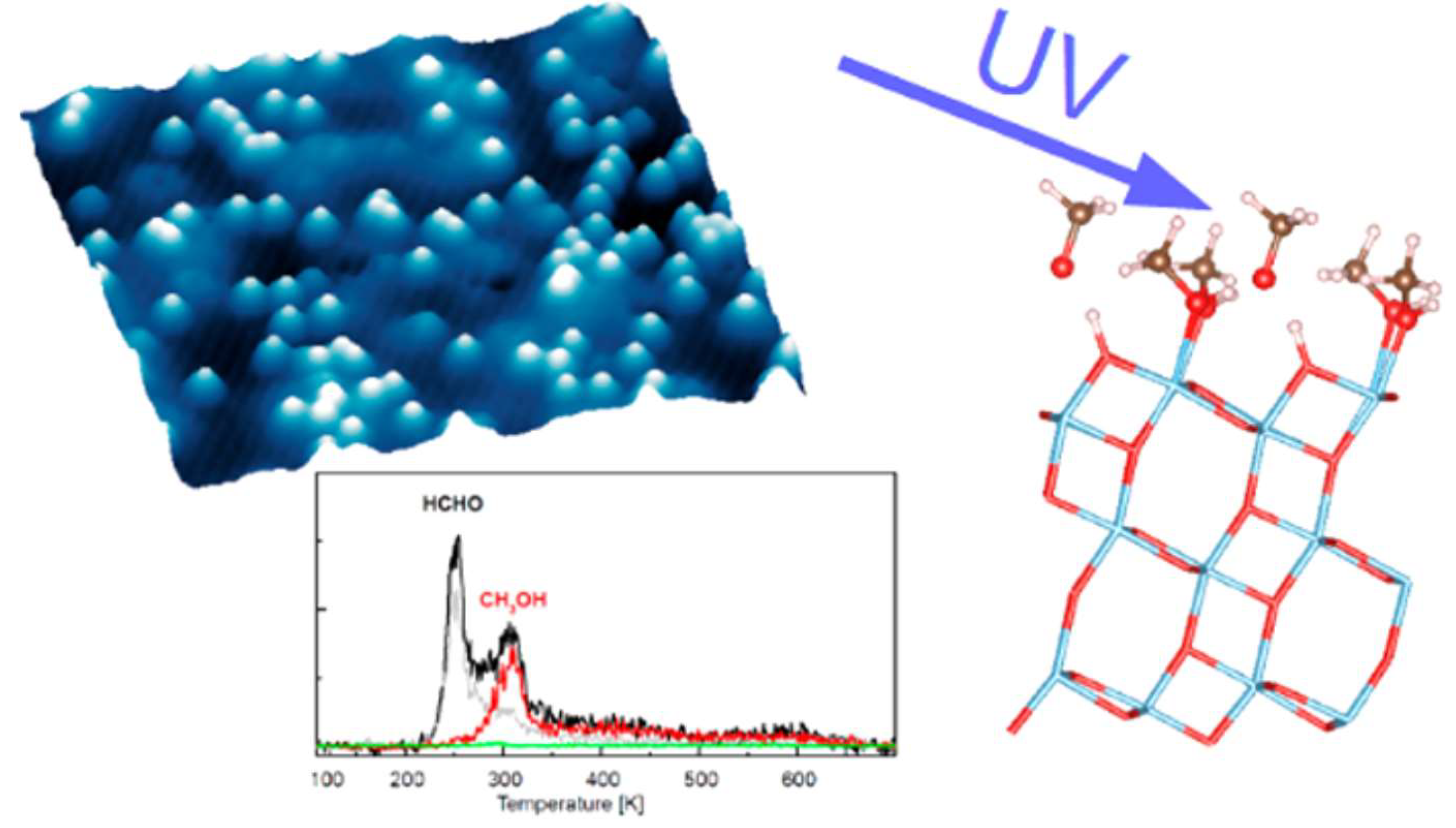
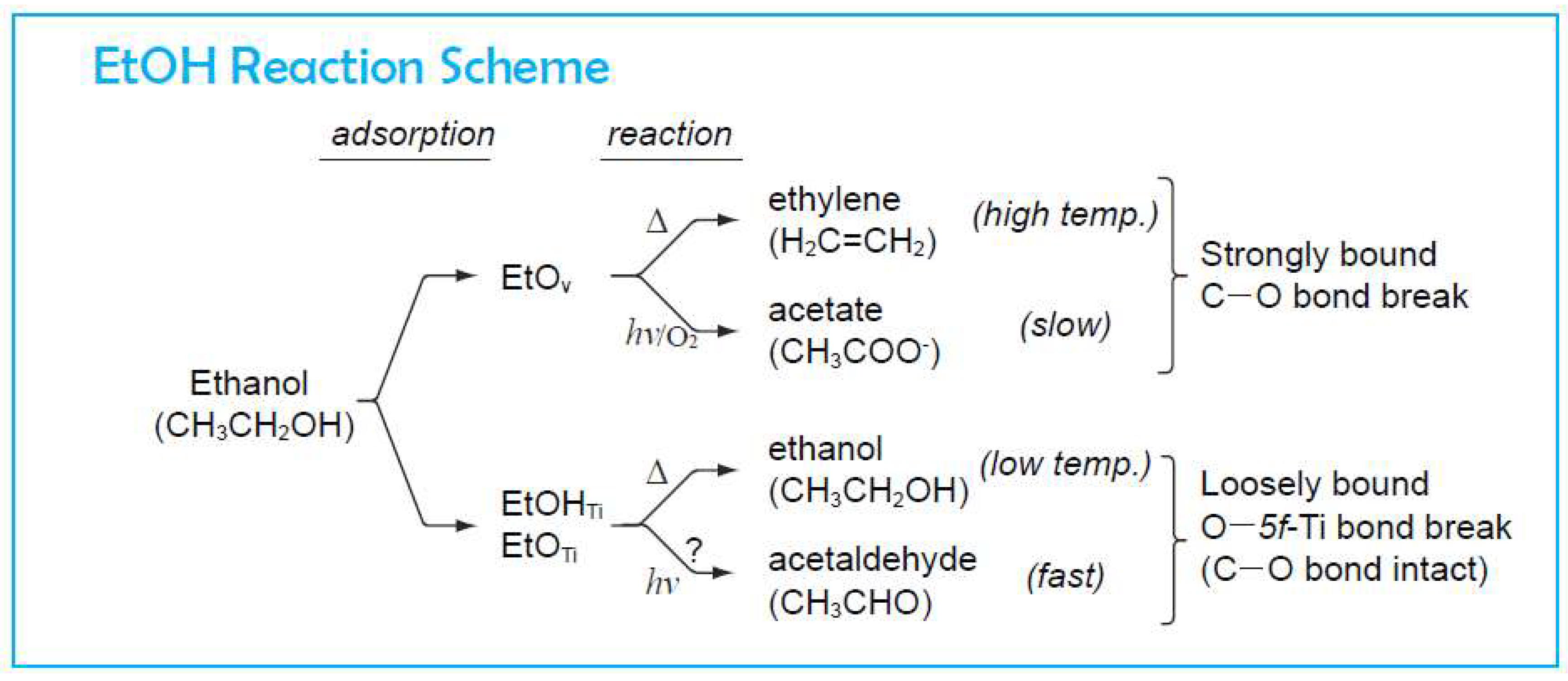
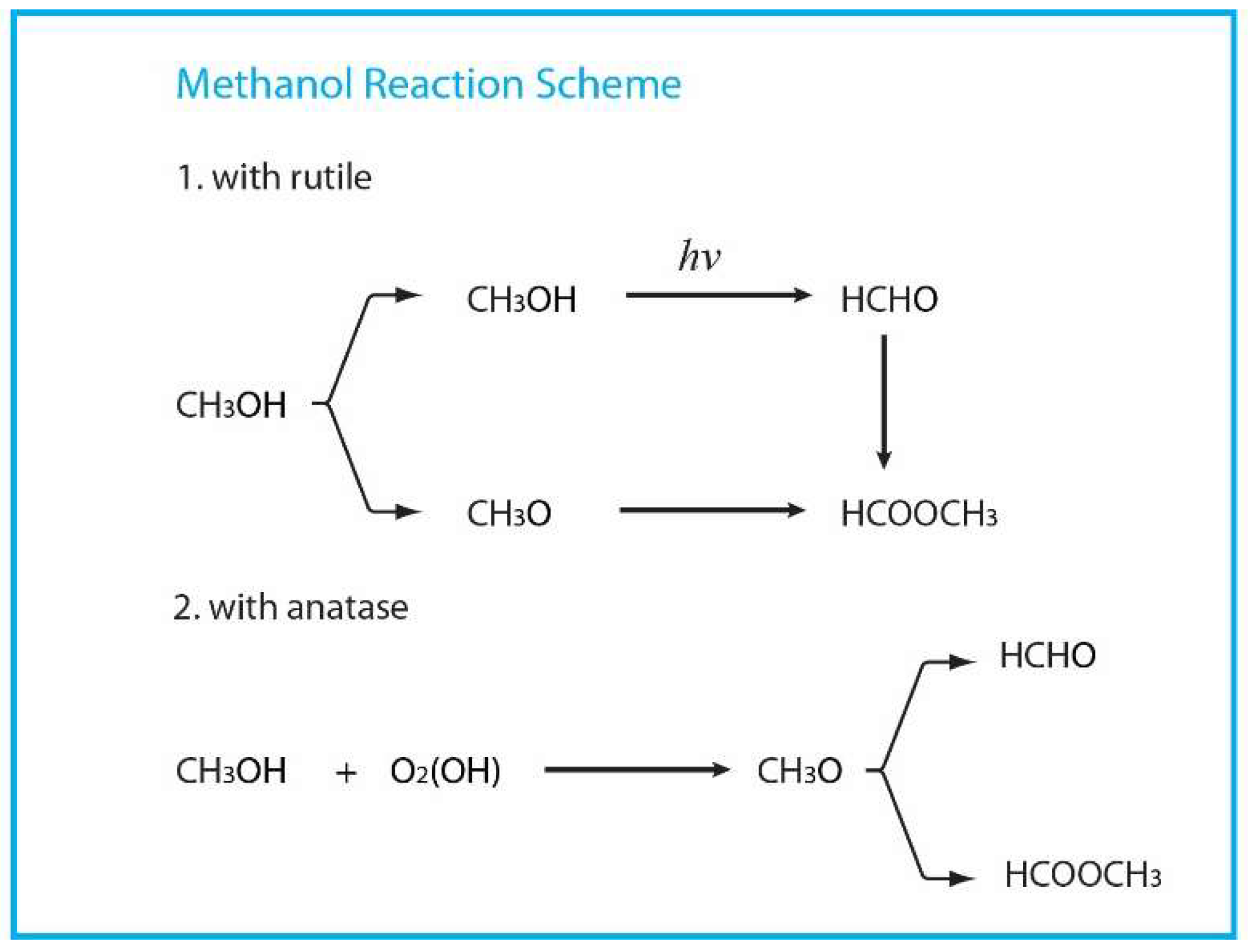
4. Photo-Catalytic Reaction
5. Challenges and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Irie, H.; Fujishima, A. TiO2 photocatalysis: A historical overview and future prospects. Jpn. J. Appl. Phys. 2005, 44, 8269–8285. [Google Scholar] [CrossRef]
- Baba, R.; Konda, R.; Fujishima, A.; Honda, K. Photoelectrochemical deposition of metals on TiO2 powders in the presence of alcohols. Chem. Lett. 1986, 15, 1307–1310. [Google Scholar] [CrossRef]
- Wang, R.; Hashimoto, K.; Fujishima, A.; Chikuni, M.; Kojima, E.; Kitamura, A.; Shimohigoshi, M.; Watanabe, T. Light-induced amphiphilic surfaces. Nature 1997, 388, 431–432. [Google Scholar] [CrossRef]
- Nakabayashi, S.; Fujishima, A.; Honda, K. Single charge accumulation dynamics on photocatalytic TiO2 particles in ethanol slurries by time domain reflectometry. J. Am. Chem. Soc. 1985, 107, 250–251. [Google Scholar] [CrossRef]
- O’Regan, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Bahnemann, W.; Muneer, M.; Haque, M.M. Titanium dioxide-mediated photocatalysed degradation of few selected organic pollutants in aqueous suspensions. Catal. Today 2007, 124, 133–148. [Google Scholar] [CrossRef]
- Sunada, K.; Watanabe, T.; Hashimoto, K. Studies on photokilling of bacteria on TiO2 thin film. J. Photochem. Photobiol. A Chem. 2003, 156, 227–233. [Google Scholar] [CrossRef]
- Li, F.B.; Li, X.Z.; Hou, M.F. Photocatalytic degradation of 2-mercaptobenzothiazole in aqueous La3+-TiO2 suspension for odor control. Appl. Catal. B Environ. 2004, 48, 185–194. [Google Scholar] [CrossRef]
- Imagawa, H.; Tanaka, T.; Takahashim, N.; Matsunaga, S.; Suda, A.; Shinjoh, H. Synthesis and characterization of Al2Ow and ZrO2-TiO2 nano-composite as a support for NOx storage-reduction catalyst. J. Catal. 2007, 251, 315–320. [Google Scholar] [CrossRef]
- Mills, A.; Davies, R.H.; Worsley, D. Water purification by semiconductor photocatalysis. Chem. Soc. Rev. 1993, 22, 417–425. [Google Scholar] [CrossRef]
- Hoffmann, M.R.; Martin, S.T.; Choi, W.; Bahnemann, D.W. Environmental applications of semiconductor photocatalysis. Chem. Rev. 1995, 95, 69–96. [Google Scholar] [CrossRef]
- Kamat, P.V. Photophysical, photochemical and photocatalytic aspects of metal nanoparticles. J. Phys. Chem. C 2002, 106, 7729–7744. [Google Scholar] [CrossRef]
- Carp, O.; Huisman, C.L.; Reller, A. Photoinduced reactivity of titanium dioxide. Prog. Solid State Chem. 2004, 32, 33–177. [Google Scholar] [CrossRef]
- Fujishima, A.; Zhang, X.; Tryk, D.A. TiO2 photocatalysis and related surface phenomena. Surf. Sci. Rep. 2008, 63, 515–582. [Google Scholar] [CrossRef]
- Karunagaran, B.; Uthirakumar, P.; Chung, S.J.; Velumani, S.; Suh, E.K. TiO2, thin film gas sensor for monitoring ammonia. Mater. Charact. 2007, 58, 680–684. [Google Scholar] [CrossRef]
- Hu, L.H.; Dai, S.Y.; Weng, J.; Xiao, S.F.; Sui, Y.F.; Huang, Y.; Chen, S.H.; Kong, F.T.; Pan, X.; Liang, L.Y.; Wang, K.J. Microstructure design of nanoporous TiO2 photoelectrodes for dye-sensitized solar cell modules. J. Phys. Chem. B 2007, 111, 358–362. [Google Scholar] [CrossRef]
- Hong, S.P.; Park, J.; Bhat, S.S.M.; Lee, T.H.; Lee, S.A.; Hong, K.; Choi, M.J.; Shokouhimehr, M.; Jang, H.W. Comprehensive study on the morphology control of TiO2 nanorods on foreigh substrates by the hydrothermal method. Cryst. Growth Des. 2018, 18, 6504–6512. [Google Scholar] [CrossRef]
- Schubert, G.; Bánsági, T.; Solymosi, F. Photocatalytic decomposition of methyl formate over-supported Pt metals. J. Phys. Chem. C 2013, 117, 22797–22804. [Google Scholar] [CrossRef] [Green Version]
- Neatu, S.; Maciá-Agulló, J.A.; Concepción, P.; Garcia, H. Gold-copper nanoalloys supported on TiO2 as photocatalysts for CO2 reduction by water. J. Am. Chem. Soc. 2014, 136, 15969–15976. [Google Scholar] [CrossRef]
- Cong, Y.; Zhang, J.L.; Chen, F.; Anpo, M.; He, D.N. Preparation, photocatalytic activity, and mechanism of nano-TiO2 co-doped with nitrogen and iron (III). J. Phys. Chem. C 2007, 111, 10618–10623. [Google Scholar] [CrossRef]
- Jia, Y.N.; Zhan, S.H.; Ma, S.L.; Zhou, Q.X. Fabrication of TiO2-Bi2WO6 binanosheet for enhanced solar photocatalytic disinfection of E. coli: Insights on the mechanism. ACS Appl. Mater. Interfaces 2016, 8, 6841–6851. [Google Scholar] [CrossRef] [PubMed]
- Boschloo, G.K.; Goossens, A. Electron trapping in porphyrin-sensitized porous nanocrystalline TiO2 electrodes. J. Phys. Chem. 1996, 100, 19489–19494. [Google Scholar] [CrossRef]
- Wang, X.L.; Feng, Z.C.; Shi, J.Y.; Jia, G.Q.; Shen, S.; Zhou, J.; Li, C. Trap states and carrier dynamics of TiO2 studied by photoluminescence spectroscopy under weak excitation condition. Phys. Chem. Chem. Phys. 2012, 12, 7083–7090. [Google Scholar] [CrossRef]
- Leytner, S.; Hupp, J.T. Evaluation of the energetics of electron trap states at the nanocrystalline titanium dioxide/aqueous solution interface via time-resolved photoacoustic spectroscopy. Chem. Phys. Lett. 2000, 330, 231–236. [Google Scholar] [CrossRef]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.L.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 photocatalysis: Mechanisms and materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef] [PubMed]
- Wendt, S.; Sprunger, P.T.; Lira, E.; Madsen, G.K.H.; Li, Z.; Hansen, J.Ø.; Mattuesen, J.; Blekinge-Rasmussen, A.; Lægsgaard, E.; Hammer, B.; et al. The role of interstitial sites in the Ti3d defect state in the band gap of titania. Science 2008, 320, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- Sasahara, A.; Pang, C.L.; Onishi, H. STM observation of a ruthenium dye adsorbed on a TiO2(110) surface. J. Phys. Chem. B 2006, 110, 4751–4755. [Google Scholar] [CrossRef]
- Li, S.C.; Diebold, U. Reactivity of TiO2 rutile and anatase toward nitroaromatics. J. Am. Chem. Soc. 2009, 132, 64–66. [Google Scholar] [CrossRef]
- Potapenko, D.V.; Li, Z.; Osgood, R.M. Dissociation of single 2-chloroanthracene molecules by STM-tip electron injection. J. Phys. Chem. C 2012, 116, 4679–4685. [Google Scholar] [CrossRef]
- Hansen, J.Ø.; Mattiesen, J.; Lira, E.; Lammich, L.; Wendt, S. A new recipe for preparing oxidized TiO2(110) surfaces: An STM study. Surf. Sci. 2017, 666, 113–122. [Google Scholar] [CrossRef]
- Lira, E.; Huo, P.P.; Hansen, J.Ø.; Rieboldt, F.; Bechstein, R.; Wei, Y.Y.; Streber, R.; Porsgaard, S.; Li, Z.S.; Lægsgaard, E.; et al. Effects of the crystal reduction state on the interaction of oxygen with rutile TiO2(110). Catal. Today 2012, 182, 25–38. [Google Scholar] [CrossRef]
- Henderson, M.A.; White, J.M.; Uetsuka, H.; Onishi, H. Selectivity changes during organic photooxidation on TiO2 role of O2 pressure and organic coverage. J. Catal. 2006, 238, 153–164. [Google Scholar] [CrossRef]
- Yang, F.; Chen, M.S.; Goodman, D.W. Sintering of Au particles supported on TiO2(110) during CO oxidation. J. Phys. Chem. C 2009, 113, 254–260. [Google Scholar] [CrossRef]
- Hansen, J.Ø.; Huo, P.P.; Martinez, U.; Lira, E.; Wei, Y.Y.; Streber, R.; Lægsgaard, E.; Hammer, B.; Wendt, S.; Besenbacher, F. Direct evidence for ethanol dissociation on rutile TiO2(110). Phys. Rev. Lett. 2011, 107, 136102. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.; Lu, D.; Senanayake, S.D.; Starr, D.E. Monoethanolamine adsorption on TiO2(110): Bonding, structure, and implications for use as a model solid-supported CO2 capture material. J. Phys. Chem. C 2014, 118, 1576–1586. [Google Scholar] [CrossRef]
- Zhang, Z.; Bondarchuk, O.; White, J.M.; Kay, B.D.; Dohnálek, Z. Imaging adsorbate O–H bond cleavage: Methanol on TiO2(110). J. Am. Chem. Soc. 2006, 128, 4198–4199. [Google Scholar] [CrossRef]
- Grätzel, M. Photoelectrochemical cells. Nature 2001, 414, 338–344. [Google Scholar] [CrossRef]
- Lyubinetsky, I.; Yu, Z.Q.; Henderson, M.A. Direct observation of adsorption evolution and bonding configuration of TMAA on TiO2(110). J. Phys. Chem. C 2007, 111, 4342–4346. [Google Scholar] [CrossRef]
- Lyubinetsky, I.; Deskins, N.A.; Du, Y.; Vestergaard, E.K.; Kim, D.J.; Dupuis, M. Adsorption state and mobility of trimethylacetic acid molecules on reduced TiO2(110) surface. Phys. Chem. Chem. Phys. 2010, 12, 5986–5992. [Google Scholar] [CrossRef]
- Grinter, D.C.; Woolcot, T.; Pang, C.L.; Thornton, G. Ordered carboxylates on TiO2(110) formed at aqueous interfaces. J. Phys. Chem. Lett. 2014, 5, 4265–4269. [Google Scholar] [CrossRef] [PubMed]
- Tanner, R.E.; Sasahara, A.; Liang, Y.; Altman, E.I.; Onishi, H. Formic acid adsorption on anatase TiO2(001)-(1×4) thin films studied by NC-AFM and STM. J. Phys. Chem. B 2002, 106, 8211–8222. [Google Scholar] [CrossRef]
- Grinter, D.C.; Nicotra, M.; Thornton, G. Acetic acid adsorption on anatase TiO2(101). J. Phys. Chem. C 2012, 116, 11643–11651. [Google Scholar] [CrossRef]
- Pang, C.L.; Watkins, M.; Cabailh, G.; Ferrero, S.; Ngo, L.T.; Chen, Q.; Humphrey, D.S.; Shluger, A.L.; Thornton, G. Bonding of methyl phosphonate to TiO2(110). J. Phys. Chem. C 2010, 114, 16983–16988. [Google Scholar] [CrossRef]
- Guo, Q.; Cocks, I.; Williams, E.M. The adsorption of benzoic acid on a TiO2 (110) surface studied using STM, ESDIAD and LEED. Surf. Sci. 1997, 393, 1–11. [Google Scholar] [CrossRef]
- Prauzner-Bechcicki, J.S.; Godlewski, S.; Tekiel, A.; Cyganik, P.; Budzioch, J.; Szymonski, M. High-resolution STM studies of terephthalic acid molecules on rutile TiO2(110)-(1 × 1) surfaces. J. Phys. Chem. C 2009, 113, 9309–9315. [Google Scholar] [CrossRef]
- Tekiel, A.; Prauzner-Bechcicki, J.S.; Godlewski, S.; Budzioch, J.; Szymonski, M. Self-assembly of terephthalic acid on rutile TiO2(110): Toward chemically functionalized metal oxide surfaces. J. Phys. Chem. C 2008, 112, 12606–12609. [Google Scholar] [CrossRef]
- Suzuki, S.; Yamaguchi, Y.; Onishi, H.; Sasaki, T.; Fukui, K.I.; Iwasawa, Y. Study of pyridine and its derivatives adsorbed on a TiO2(110)-(1×1) surface by means of STM, TDS, XPS and MD calculation in relation to surface acid-base interaction. J. Chem. Soc. Faraday Trans. 1998, 94, 161–166. [Google Scholar] [CrossRef]
- Otero-Irurueta, G.; Martínez, J.I.; Lovat, G.; Lanzilotto, V.; Méndez, J.; López, M.F.; Floreano, L.; Martín-Gago, J.A. Densely packed perylene layers on the rutile TiO2(110)-(1 × 1) surface. J. Phys. Chem. C 2015, 119, 7809–7816. [Google Scholar] [CrossRef]
- Lanzilotto, V.; Lovat, G.; Otero, G.; Sanchez, L.; López, M.F.; Méndez, J.; Martín-Gago, J.A.; Bavdek, G.; Floreano, L. Commensurate growth of densely packed PTCDI islands on the rutile TiO2(110) surface. J. Phys. Chem. C 2013, 117, 12639–12647. [Google Scholar] [CrossRef]
- Potapenko, D.V.; Choi, N.J.; Osgood, R.M. Adsorption geometry of anthracene and 4-bromobiphenyl on TiO2(110) surfaces. J. Phys. Chem. C 2010, 114, 19419–19424. [Google Scholar] [CrossRef]
- Wang, Y.; Ye, Y.; Wu, K. Adsorption and assembly of copper phthalocyanine on cross-linked TiO2(110)-(1×2) and TiO2(210). J. Phys. Chem. B 2006, 110, 17960–17965. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Liu, L.; Dong, S.; Cui, X.; Zhao, J.; Wang, B. Dynamic processes of formaldehyde at terminal Ti sites on the rutile TiO2(110) surface. J. Phys. Chem. C 2016, 120, 24287–24293. [Google Scholar] [CrossRef]
- Huo, P.P.; Hansen, J.Ø.; Martinez, U.; Lira, E.; Streber, R.; Wei, Y.; Lægsgaard, E.; Hammer, B.; Wendt, S.; Besenbacher, F. Ethanol diffusion on rutile TiO2(110) mediated by H adatoms. J. Phys. Chem. Lett. 2012, 3, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Xia, Y.; Tang, M.; Wang, Z.T.; Lyubinetsky, I.; Ge, Q.; Dohnálek, Z.; Park, K.T.; Zhang, Z. Low-temperature reductive coupling of formaldehyde on rutile TiO2(110). J. Phys. Chem. C 2015, 119, 18452–18457. [Google Scholar] [CrossRef]
- Zhu, K.; Xia, Y.; Zhang, Z.; Park, K. Probing structure of cross-linked (1 × 2) rutile TiO2(110): Adsorption of trimethyl acetic acid. J. Phys. Chem. C 2016, 120, 15257–15264. [Google Scholar] [CrossRef]
- Jensen, S.C.; Shank, A.; Madix, R.J.; Friend, C.M. Butyrophenone on O-TiO2(110): One-dimensional motion in a weakly confined potential well. ACS Nano 2012, 6, 2925–2930. [Google Scholar] [CrossRef]
- Hansen, J.Ø.; Bebensee, R.; Martinez, U.; Porsgaard, S.; Lira, E.; Wei, Y.Y.; Lammich, L.; Li, Z.S.; Idriss, H.; Besenbacher, F.; et al. Unravelling site-specific photo-reactions of ethanol on rutile TiO2(110). Sci. Rep. 2006, 6, 21990. [Google Scholar] [CrossRef]
- Katsiev, K.; Harrison, G.; Alghamdi, H.; Alsalik, Y.; Wilson, A.; Thornton, G.; Idriss, H. Mechanism of ethanol photooxidation on single-crystal anatase TiO2(101). J. Phys. Chem. C 2017, 121, 2940–2950. [Google Scholar] [CrossRef]
- Zhou, C.; Ren, Z.; Tan, S.; Ma, Z.; Mao, X.; Dai, D.; Fan, H.; Yang, X.; LaRue, J.; Cooper, R.; et al. Site-specific photocatalytic splitting of methanol on TiO2(110). Chem. Sci. 2010, 1, 575–580. [Google Scholar] [CrossRef]
- Wei, D.; Jin, X.; Huang, C.; Dai, D.; Ma, Z.; Li, W.X.; Yang, X. Direct imaging single methanol molecule photocatalysis on titania. J. Phys. Chem. C 2015, 119, 17748–17754. [Google Scholar] [CrossRef]
- Phillips, K.R.; Jensen, S.C.; Baron, M.; Li, S.C.; Friend, C.M. Sequential photo-oxidation of methanol to methyl formate on TiO2(110). J. Am. Chem. Soc. 2013, 135, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Setvin, M.; Shi, X.; Hulva, J.; Simschitz, T.; Parkinson, G.S.; Schmid, M.; Valentin, C.D.; Selloni, A.; Diebold, U. Methanol on anatase TiO2(101): Mechanistic insights into photocatalysis. ACS Catal. 2017, 7, 7081–7091. [Google Scholar] [CrossRef] [PubMed]
- Ariga, H.; Taniike, T.; Morikawa, H.; Tada, M.; Min, B.K.; Watanabe, K.; Watanabe, Y.; Ikeda, S.; Saiki, K.; Iwasawa, Y. Surface-mediated visible-light photo-oxidation on pure TiO2(001). J. Am. Chem. Soc. 2009, 2, 14670–14672. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.A.; Epling, W.S.; Peden, C.H.F.; Perkins, C.L. Insights into photoexcited electron scavenging processes on TiO2 obtained from studies of the reaction of O2 with OH groups adsorbed at electronic defects on TiO2(110). J. Phys. Chem. B 2003, 107, 534–545. [Google Scholar] [CrossRef]
- White, J.M.; Henderson, M.A. Trimethyl acetate on TiO2(110): Preparation and anaerobic photolysis. J. Phys. Chem. B 2005, 109, 12417–12430. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.T.; Deskins, N.A.; Henderson, M.A.; Lyubinetsky, I. Inhibitive influence of oxygen vacancies for photoactivity on TiO2(110). Phys. Rev. Lett. 2012, 109, 266103. [Google Scholar] [CrossRef]
- Wang, Z.T.; Henderson, M.A.; Lyubinetsky, I. Origin of coverage dependence in photoreactivity of carboxylate on TiO2(110): Hindering by charged coadsorbed hydroxyls. ACS Catal. 2015, 5, 6463–6467. [Google Scholar] [CrossRef]
- Denis, V. Potapenko, photoreactions on a single isolated TiO2 nanocrystal on Au(111): Photodecomposition of TMAA. J. Phys. Chem. C 2015, 119, 28946–28953. [Google Scholar] [CrossRef]
- Landis, E.C.; Jensen, S.C.; Phillips, K.R.; Friend, C.M. Photostability and thermal decomposition of benzoic acid on TiO2. J. Phys. Chem. C 2012, 116, 21508–21513. [Google Scholar] [CrossRef]
- Jensen, S.C.; Phillips, K.R.; Baron, M.; Landis, E.C.; Friend, C.M. Norrish type I surface photochemistry for butyrophenone on TiO2(110). Phys. Chem. Chem. Phys. 2013, 15, 5193–5201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, P.; Yuan, B.; Ji, W.; Cheng, Z.; Qiu, X. Real-space identification of intermolecular bonding with atomic force microscopy. Science 2013, 342, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Guo, J.; Hapala, P.; Cao, D.; Ma, R.; Cheng, B.; Xu, L.; Ondráček, M.; Jelínek, P.; Wang, E.; et al. Weakly perturbative imaging of interfacial water with submolecular resolution by atomic force microscopy. Nat. Commun. 2018, 9, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Meng, X.; Chen, J.; Peng, J.; Sheng, J.; Li, X.Z.; Xu, L.; Shi, J.R.; Wang, E.; Jiang, Y. Real-space imaging of interfacial water with submolecular resolution. Nat. Mater. 2014, 5, 184–189. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organic Molecule | Adsorption | Diffusion | Photocatalytic Reaction |
|---|---|---|---|
| Alcohols |
|
|
|
| Carboxylic acids |
|
|
|
| Aromatic compounds |
|
|
|
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huo, P.; Kumar, P.; Liu, B. The Mechanism of Adsorption, Diffusion, and Photocatalytic Reaction of Organic Molecules on TiO2 Revealed by Means of On-Site Scanning Tunneling Microscopy Observations. Catalysts 2018, 8, 616. https://doi.org/10.3390/catal8120616
Huo P, Kumar P, Liu B. The Mechanism of Adsorption, Diffusion, and Photocatalytic Reaction of Organic Molecules on TiO2 Revealed by Means of On-Site Scanning Tunneling Microscopy Observations. Catalysts. 2018; 8(12):616. https://doi.org/10.3390/catal8120616
Chicago/Turabian StyleHuo, Peipei, Parveen Kumar, and Bo Liu. 2018. "The Mechanism of Adsorption, Diffusion, and Photocatalytic Reaction of Organic Molecules on TiO2 Revealed by Means of On-Site Scanning Tunneling Microscopy Observations" Catalysts 8, no. 12: 616. https://doi.org/10.3390/catal8120616
APA StyleHuo, P., Kumar, P., & Liu, B. (2018). The Mechanism of Adsorption, Diffusion, and Photocatalytic Reaction of Organic Molecules on TiO2 Revealed by Means of On-Site Scanning Tunneling Microscopy Observations. Catalysts, 8(12), 616. https://doi.org/10.3390/catal8120616