Functional Analysis of Methylomonas sp. DH-1 Genome as a Promising Biocatalyst for Bioconversion of Methane to Valuable Chemicals


Abstract
:1. Introduction
2. Results
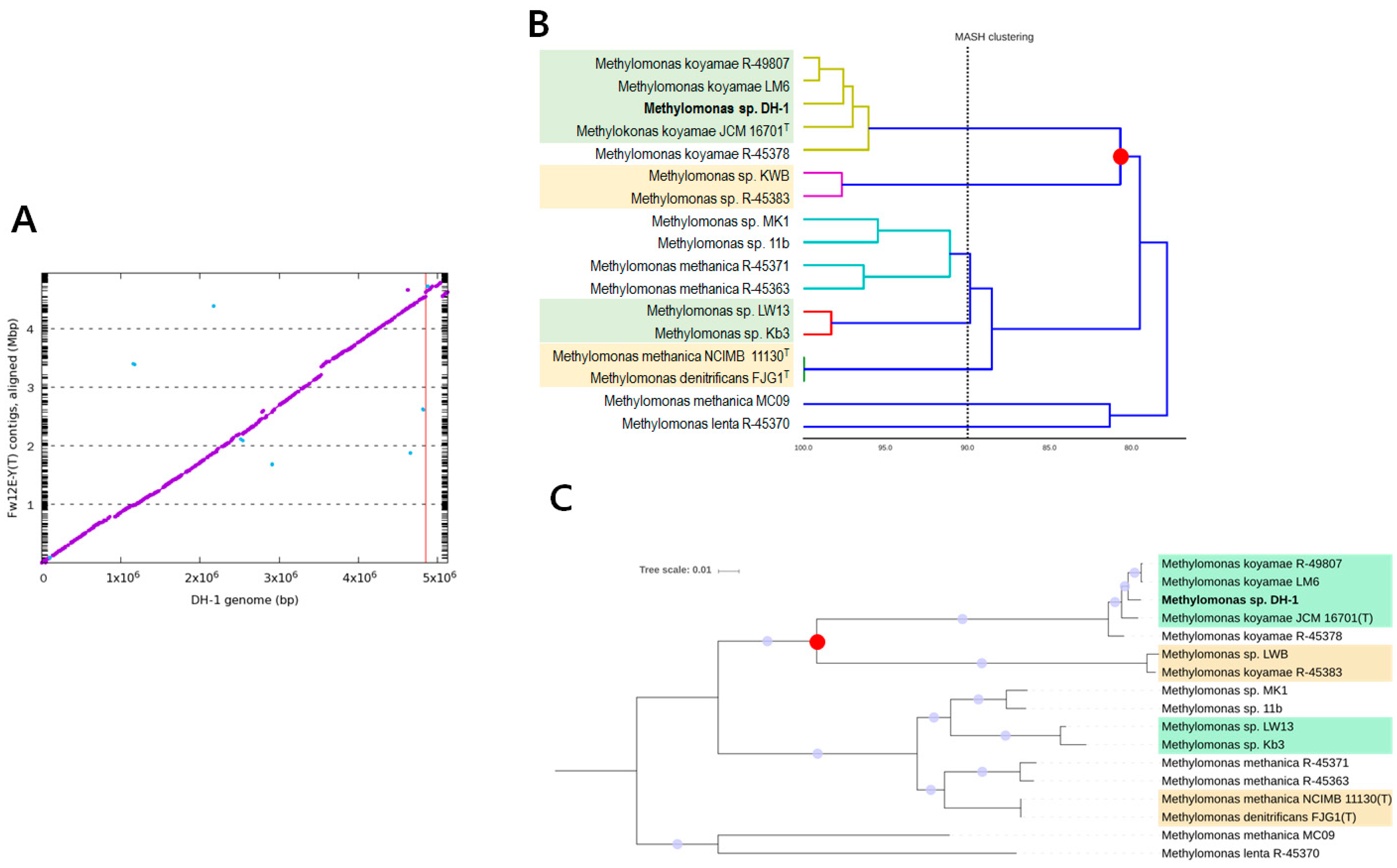
2.1. Genome Statistics and General Features
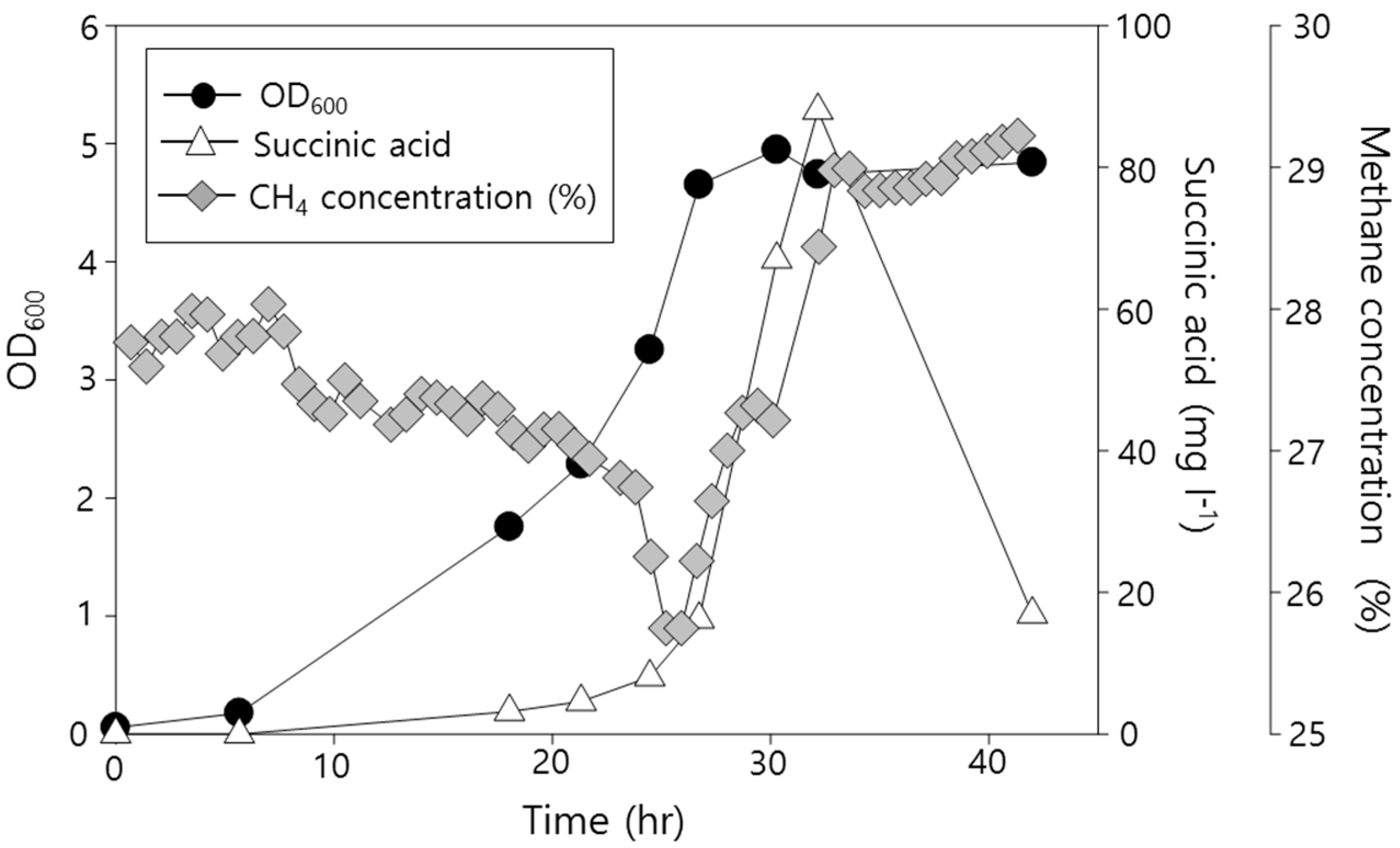
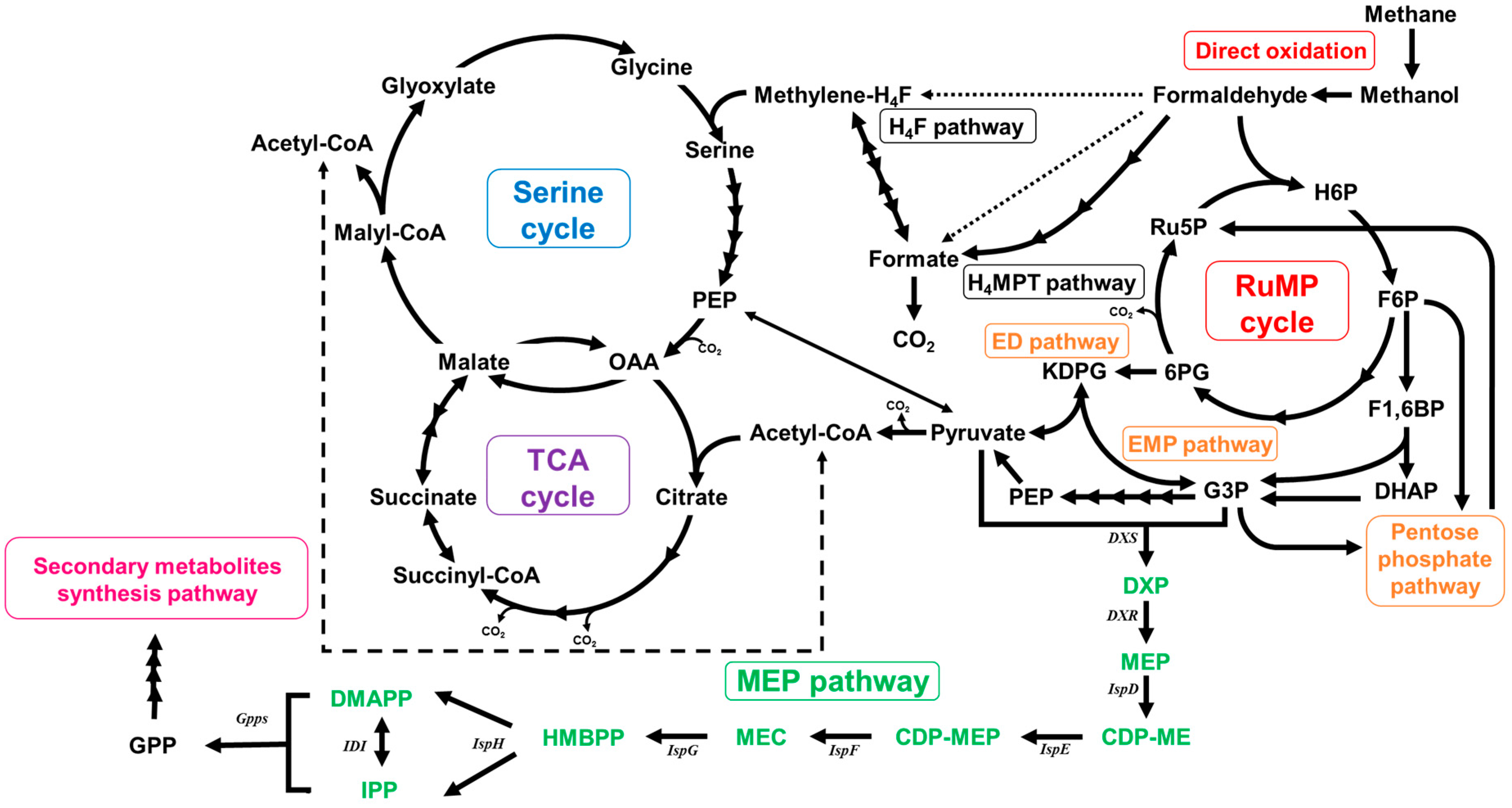
2.2. Functional Analysis of the Complete Genome Sequence of Methylomonas sp. DH-1 and the Production of Succinate from Methane
2.3. Nucleotide Sequence Accession Number
3. Conclusions
4. Materials and Methods
4.1. Bacterial Growth, DNA Isolation, Genome Assembly and Annotation
4.2. Analytical Methods
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hwang, I.Y.; Hur, D.H.; Lee, J.H.; Park, C.H.; Chang, I.S.; Lee, J.W.; Lee, E.Y. Batch conversion of methane to methanol using Methylosinus trichosporium OB3b as biocatalyst. J. Microbiol. Biotechnol. 2015, 25, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.A.; Gonzalez, R. Rethinking biological activation of methane and conversion to liquid fuels. Nat. Chem. Biol. 2014, 10, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.K.; Hur, D.H.; Nguyen, D.T.N.; Lee, E.Y. Metabolic engineering of methanotrophs and its application to production of chemicals and biofuels from methane. Biofuels Bioprod. Bioref. 2016, 10, 848–863. [Google Scholar] [CrossRef]
- Hur, D.H.; Na, J.G.; Lee, E.Y. Highly efficient bioconversion of methane to methanol using a novel type I Methylomonas sp. DH-1 newly isolated from brewery waste sludge. J. Chem. Technol. Biotechnol. 2017, 92, 311–318. [Google Scholar] [CrossRef]
- Hur, D.H.; Nguyen, T.T.; Kim, D.; Lee, E.Y. Selective bio-oxidation of propane to acetone using methane-oxidizing Methylomonas sp. DH-1. J. Ind. Microbiol. Biotechnol. 2017, 44, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Kalyuzhnaya, M.G.; Lamb, A.E.; McTaggart, T.L.; Oshkin, I.Y.; Shapiro, N.; Woyke, T.; Chistoserdova, L. Draft genome sequences of gammaproteobacterial methanotrophs isolated from lake Washington sediment. Genome Announc. 2015, 3, e00103-15. [Google Scholar] [CrossRef] [PubMed]
- Heylen, K.; De Vos, P.; Vekeman, B. Draft genome sequences of eight obligate methane oxidizers occupying distinct niches based on their nitrogen metabolism. Genome Announc. 2016, 4, e00421-16. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Reva, O.; Tümmler, B. Think big—Giant genes in bacteria. Environ. Microbiol. 2008, 10, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Tavormina, P.L.; Orphan, V.J.; Kalyuzhnaya, M.G.; Jetten, M.S.M.; Klotz, M.G. A novel family of functional operons encoding methane/ammonia monooxygenase-related proteins in gammaproteobacterial methanotrophs. Environ. Microbiol. Rep. 2011, 3, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, R.; Kits, K.D.; Ramonovskaya, V.A.; Rozova, O.N.; Yurimoto, H.; Iguchi, H.; Khmelenina, V.N.; Sakai, Y.; Dunfield, P.F.; Klotz, M.G.; et al. Draft genomes of gammaproteobacterial methanotrophs isolated from terrestrial ecosystems. Genome Announc. 2015, 3, e00515-15. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.E.; Smirnova, A.V.; Kalyuzhnaya, M.G.; Bringel, F.; Hirayama, H.; Jetten, M.S.; Khmelenina, V.N.; Klotz, M.G.; Knief, C.; Kyrpides, N.; et al. Draft genome sequence of the moderately halophilic methanotroph Methylohalobius crimeensis strain 10Ki. Genome Announc. 2015, 3, e00644-15. [Google Scholar] [CrossRef] [PubMed]
- Trotsenko, Y.A.; Murrell, J.C. Metabolic aspects of aerobic obligate methanotrophy. Adv. Appl. Microbiol. 2008, 63, 183–229. [Google Scholar] [PubMed]
- Fu, Y.; Li, Y.; Lidstrom, M. The oxidative TCA cycle operates during methanotrophic growth of the Type I methanotroph Methylomicrobium buryatense 5GB1. Metab. Eng. 2017, 42, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Müller, R.; Wohlleben, W.; et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef] [PubMed]
- Schöner, T.A.; Gassel, S.; Osawa, A.; Tobias, N.J.; Okuno, Y.; Sakakibara, Y.; Shindo, K.; Sandmann, G.; Bode, H.B. Aryl polyenes, a highly abundant class of bacterial natural products, are functionally related to antioxidative carotenoids. ChemBioChem 2016, 17, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Jospin, G.; Lowe, E.; Matsen, F.A.; Bik, H.M.; Eisen, J.A., IV. PhyloSift: Phylogenetic analysis of genomes and metagenomes. PeerJ 2014, 2, e243. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. 2010. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Chromosome | Plasmid 1 |
|---|---|---|
| Size (bp) | 4,849,532 | 277,875 |
| G + C content (%) | 56.47 | 51.66 |
| Protein coding genes 2 | 4441 | 228 |
| Pseudogenes | 85 | 13 |
| tRNAs | 47 | 0 |
| rRNAs | 3, 3, 3 (16S, 23S, 5S) | 0 |
| ncRNAs | 4 | 0 |
| CRISPR arrays | 4 | 0 |
| GenBank accession | CP014360 | CP014361 |
| Category | Functional Classification | Chromosome | Plasmid |
|---|---|---|---|
| A | RNA processing and modification | 1 | 1 |
| B | Chromatin structure and dynamics | 2 | 0 |
| C | Energy production and conversion | 200 | 1 |
| D | Cell cycle control, cell division, chromosome partitioning | 53 | 4 |
| E | Amino acid transport and metabolism | 191 | 3 |
| F | Nucleotide transport and metabolism | 58 | 0 |
| G | Carbohydrate transport and metabolism | 111 | 0 |
| H | Coenzyme transport and metabolism | 157 | 1 |
| I | Lipid transport and metabolism | 73 | 0 |
| J | Translation, ribosomal structure and biogenesis | 172 | 0 |
| K | Transcription | 183 | 12 |
| L | Replication, recombination and repair | 242 | 32 |
| M | Cell wall/membrane/envelope biogenesis | 248 | 13 |
| N | Cell motility | 123 | 0 |
| O | Posttranslational modification, protein turnover, chaperones | 162 | 5 |
| P | Inorganic ion transport and metabolism | 223 | 9 |
| Q | Secondary metabolites biosynthesis, transport and catabolism | 63 | 2 |
| General function prediction only | 366 | 15 | |
| S | Function unknown | 329 | 10 |
| T | Signal transduction mechanisms | 340 | 9 |
| U | Intracellular trafficking, secretion, and vesicular transport | 127 | 9 |
| V | Defense mechanisms | 83 | 5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, A.D.; Hwang, I.Y.; Lee, O.K.; Hur, D.H.; Jeon, Y.C.; Hadiyati, S.; Kim, M.-S.; Yoon, S.H.; Jeong, H.; Lee, E.Y. Functional Analysis of Methylomonas sp. DH-1 Genome as a Promising Biocatalyst for Bioconversion of Methane to Valuable Chemicals. Catalysts 2018, 8, 117. https://doi.org/10.3390/catal8030117
Nguyen AD, Hwang IY, Lee OK, Hur DH, Jeon YC, Hadiyati S, Kim M-S, Yoon SH, Jeong H, Lee EY. Functional Analysis of Methylomonas sp. DH-1 Genome as a Promising Biocatalyst for Bioconversion of Methane to Valuable Chemicals. Catalysts. 2018; 8(3):117. https://doi.org/10.3390/catal8030117
Chicago/Turabian StyleNguyen, Anh Duc, In Yeub Hwang, Ok Kyung Lee, Dong Hoon Hur, Young Chan Jeon, Susila Hadiyati, Min-Sik Kim, Sung Ho Yoon, Haeyoung Jeong, and Eun Yeol Lee. 2018. "Functional Analysis of Methylomonas sp. DH-1 Genome as a Promising Biocatalyst for Bioconversion of Methane to Valuable Chemicals" Catalysts 8, no. 3: 117. https://doi.org/10.3390/catal8030117
APA StyleNguyen, A. D., Hwang, I. Y., Lee, O. K., Hur, D. H., Jeon, Y. C., Hadiyati, S., Kim, M. -S., Yoon, S. H., Jeong, H., & Lee, E. Y. (2018). Functional Analysis of Methylomonas sp. DH-1 Genome as a Promising Biocatalyst for Bioconversion of Methane to Valuable Chemicals. Catalysts, 8(3), 117. https://doi.org/10.3390/catal8030117