2. Structural Discussion of Platinum Group Metal Phosphides
Metals and phosphorus are known to form a large number of crystalline compounds, which display a high variety of metal to phosphorus ratios and a variety of crystalline structures [
1]. The ICSD counts 760 entries for binary and 2300 entries for ternary phosphides. As expected, this variety in stoichiometry and structure can also be observed within the binary phosphides of the platinum group metals (PGM), where 23 binary compounds with 16 different structures are reported (
Table 1). The phosphides of the PGM, similar to the transition metal phosphides, can be divided into three groups according to the metal to phosphorus ratio [
1]:
polyphosphides (with nP > nM),
monophosphides (with nP = nM),
metal-rich phosphides (with nP < nM).
The following review will start with a short overview of some compounds from the three groups, focusing on the phosphides of rhodium since catalytic materials containing rhodium phosphides have the largest amount of reported investigations as materials for catalytic applications in hydroprocessing and syngas-related catalysis. Further overviews of the structure of other binary and ternary metal phosphides can be found in Pöttgen [
1], Kuz’ma [
2], and Aronsson [
3].
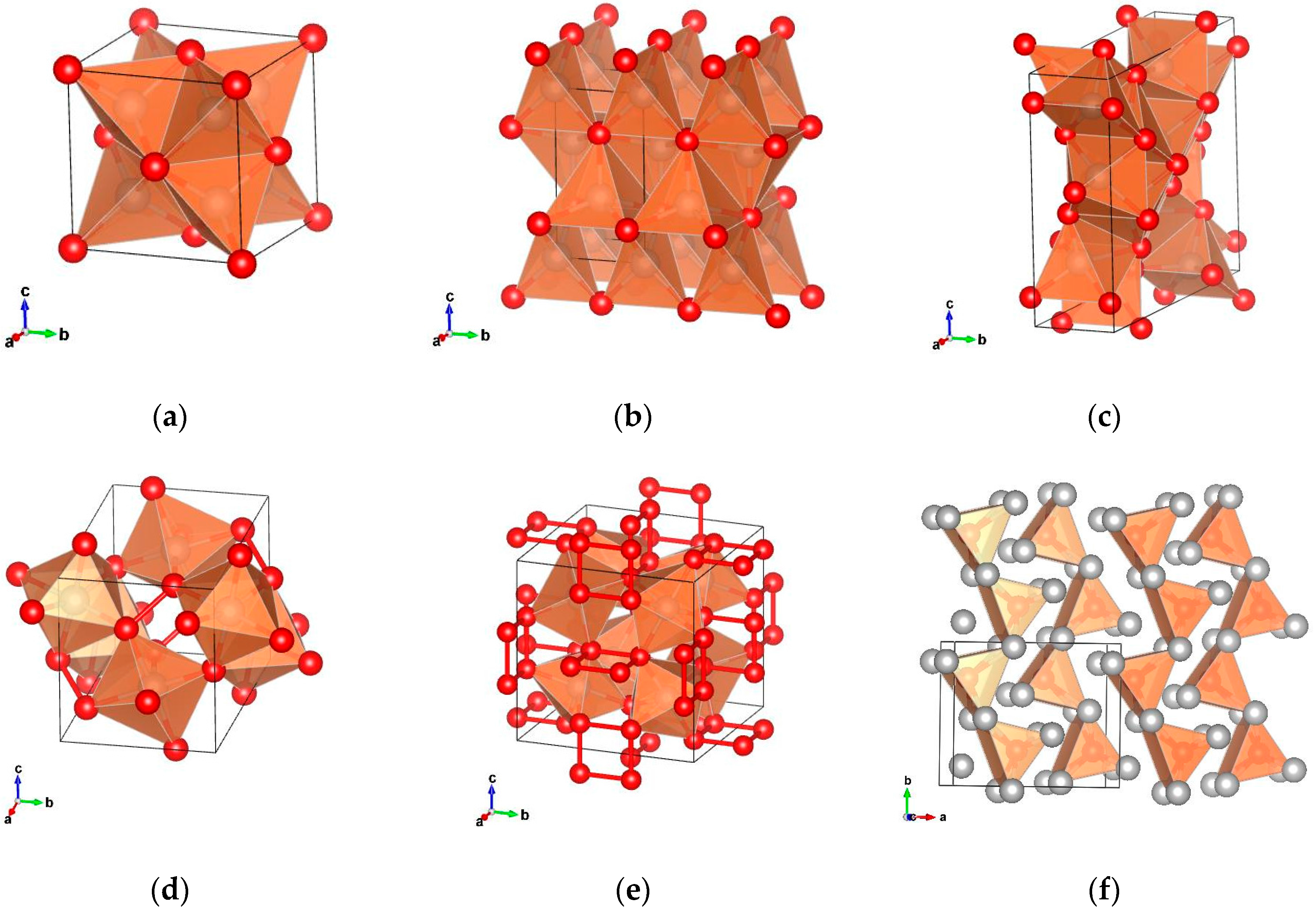
The polyphosphides of the PGMs are, as other polyphosphides, characterized by the formation of P–P bonds. The compounds RhP
3, PdP
3, and IrP
3 crystallize in the CoAs
3 structure [
13] where the phosphorus atoms are coordinated to octahedrons. Further, the formation of P
4−4 rings can be observed (
Figure 1e). Other binary polyphosphides crystallize in the pyrite structure and in other related structures. RuP
2 crystallizes in the FeS
2 structure (cubic) [
8] and PtP
2 and PdP
2 crystallize in the NiP
2 structure [
10,
22]. RhP
2 (
Figure 1d) and IrP
2 both crystallize in the CoSb
2 structure (monoclinic) [
4]. In all three structures the metal coordinates the phosphorus to octahedrons, which are connected to each other by the formation P
2 dimers (
Figure 1d).
The only reported monophosphide of the platinum metal group is RuP, which crystallizes in the MnP structure [
5], just as the other transition metal phosphides (NiP, MnP). This structure can be distinguished by the formation of phosphorus zigzag chains along the c-axis.
Metal-rich phosphides are characterized by the formation of metal to metal bonds. Certain metal-rich PGM phosphides crystallize in a number of unique structure types compared to other transition metal phosphides that cannot be found in any other phosphide compound (Pd
15P
2, Pd
6P, Pt
5P
2, Pd
7P
3, and Rh
4P
3). Ru
2P crystallizes in the Co
2P structure, which is related to the anti-PbCl structure [
16]. Characteristic of this structure is the formation of a distorted tetragonal prism ordered in chains along the b-axis (
Figure 1f). Rh
2P and Ir
2P crystallize in the anti-CaF
2 structure [
1,
18]; in this structure, the metal coordinates the phosphorus atoms to tetrahedrons (
Figure 1a).
The phosphides of rhodium are among the most investigated compounds of all the PGM phosphides in the field of heterogeneous catalysis. There are five different phosphides of rhodium (Rh
2P, Rh
3P
2, Rh
4P
3, RhP
2, and RhP
3) [
23]. The compounds Rh
3P
2 and Rh
4P
3 both crystallize in specific structure types. The Rh
3P
2 structure is characterized by the coordination of the phosphorus to tetrahedrons and square pyramids; planes of tetrahedrons and pyramids are stapled over each other (
Figure 1b). In the Rh
4P
3 structure, the phosphorus is coordinated to square pyramids (
Figure 1c).
The structures of some rhodium phosphides are isomorphic with some other metal phosphides. The phosphides RhP
3, IrP
3, CoP
3, and NiP
3 all crystallize in the CoAs
3 structure. The phosphides RhP
2, IrP
2, and CoP
2 all crystallize in the CoSb
2 structure (
Figure 1f). Finally, the metal-rich phosphides Rh
2P and Ir
2P both crystallize in the anti-CaF
2 structure; up to now, no ternary compounds have been reported. Common to all of the compounds is that with increasing metal to phosphorus ratio, the metallic properties of the phosphides increase. This can be observed especially regarding the distance between the rhodium atoms of the metal-rich rhodium phosphides. The distance between the rhodium atoms in Rh
4P
3 lies between 0.280 and 0.294 nm, in Rh
3P
2 between 0.282 and 0.285 nm, and in Rh
2P only 0.276 nm [
1,
3]. This last distance is just slightly larger than the distance between two rhodium atoms in metallic rhodium (0.27 nm).
3. Synthetic Pathways to Supported Platinum Group Metal Phosphides
As platinum metals are scarce and per unit mass expensive, the application of bulk materials for any application of commercial relevance can only have model character. A number of groups have embarked on developing ways to support nanoparticles of PGM phosphides. A variety of pathways for the preparation of transition and platinum group metal phosphides, either as a bulk material or as a support material for catalysis applications, have been reported. An overview of the different preparation pathways has been given by Aronsson et al. [
3], Kuz’ma et al. [
2,
4], Pöttgen et al. [
1], Prins et al. [
5,
24] and Oyama et al. [
25,
26]. Aronsson, Kuz’ma, and Pöttgen described the preparation of bulk binary and ternary phosphides and their structure while Prins and Oyama focused mainly on the preparation of supported transition metal phosphides for catalysis applications.
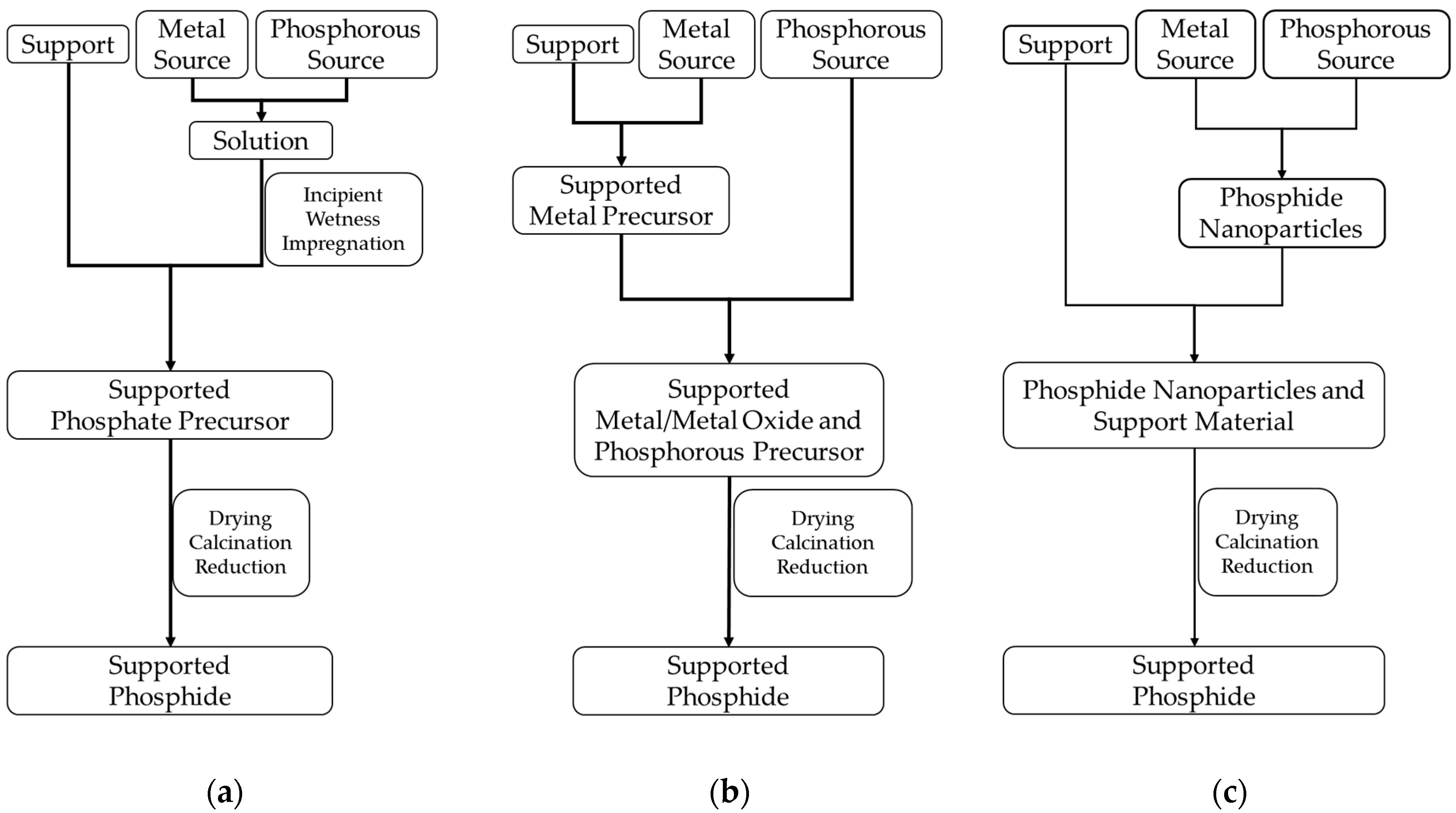
The three routes to be described next are the most frequently reported pathways in the literature for the preparation of supported platinum group metal phosphides:
the incipient wetness impregnation method combined with reductive treatment,
the conversion of supported metals or metal oxides with a phosphorus-containing compound combined with reductive treatment,
the deposition of metal phosphide nanoparticles prepared via colloidal preparation routes on a support material.
Figure 2 gives a schematic overview of the three pathways and
Table 2 lists different supported PGM phosphides, the pathways by which they are prepared, and the relevant references from the literature.
The incipient wetness impregnation method (
Figure 2a) consists in the preparation of a supported metal phosphate precursor that is then heated under a reductive atmosphere (H
2 or minor amounts of H
2 in N
2) to form the metal phosphide. First, the support material is impregnated with a solution containing the metal and phosphorus precursor. Nitrates and chlorides of the metals and (NH
4)
2HPO
4 are the most commonly used precursors. The samples are dried after the impregnation and further fired under air to decompose the nitrate or chloride compounds and to form the oxide and phosphate precursors. Last, the sample is reduced under hydrogen-containing atmosphere to form the corresponding phosphides.
The incipient wetness impregnation method combined with reductive treatment can be applied to prepare a broad range of metal phosphides, metal to phosphorus ratios (mainly metal-rich phosphides), and support materials for the preparation of dispersed PGM phosphides.
Hayes et al. [
27] applied the incipient wetness impregnation for the preparation of Rh
2P on SiO
2 for the hydrodesulfurization (HDS) of thiophen with a rhodium (Rh) loading of 5 wt %. First, a solution of RhCl
3xH
2O and NH
4H
2PO
4 with an Rh:P = 1 in ultra-pure water is prepared and impregnated on the support material SiO
2, followed by a drying step at 318 K, calcination at 773 K, and reduction at 923 K under pure H
2 flow. The characterization via XRD shows the formation of Rh
2P without metallic Rh impurities. TEM investigations show well dispersed Rh
2P particles with an average size of approximately 5 nm.
Habas et al. [
28] applied the method of the incipient wetness impregnation, among others, for the preparation of Rh
2P and Pd
3P on SiO
2 in order to compare the performance of the samples prepared by the different pathways as catalysts for the hydrodeoxygenation (HDO) of acetic acid as a model compound. Kanda et al. [
6,
29] applied the incipient wetness impregnation method for the preparation of Ru
2P, PtP
2, Rh
2P, and Pd
4.8P on SiO
2 to test the metal phosphides as catalysts in the HDS of thiophen. On later publications, Kanda et al. [
30] focused on the preparation of Rh
2P on different support materials (SiO
2, TiO
2, Al
2O
3, MgO, and ZrO
2) and the effect of the reduction temperature and the effect of phosphorus loading [
31] on the formation of Rh
2P. The compound Rh
2P can be successfully prepared on all the support materials investigated. Further, Kanda observed the formation of Rh
2P on SiO
2 at lower reduction temperatures (350 °C) with a high excess of phosphorus (P:Rh = 2), while higher temperatures are required (550 °C) to form phase-pure Rh
2P on SiO
2 samples with a lower excess of phosphorus (P:Rh = 1). This shows that the addition of P enhances the formation of Rh
2P at lower temperatures, but an excess of P leads to the formation of RhP
2 at higher reduction temperatures. Further, the TEM investigation shows that the average particle diameter of the Rh
2P particles on the catalysts increases with increasing reduction temperature and P content. A sample reduced at 550 °C with 1.5 wt % P loading has an average particle diameter of 6.4 nm, while a sample with 3.0 wt % has an average particle diameter of 8.8 nm. Kanda et al. [
32] also compared the use of acetylacetonato (acac) precursors and triphenylphosphine (TPP) with the use of chloride and phosphate precursors for the preparation of Rh
2P and Pd
4.8P on SiO
2. When the acac precursor and TPP are employed, slightly lower reduction temperatures are needed for the formation of the phosphides. In the PXRD patterns, the group observed phase-pure Rh
2P on SiO
2 at a reduction temperature of 450 °C when the acac and TPP precursors are used, while a temperature of 650 °C is needed when chloride and phosphate are used as precursors.
Sawada et al. [
33,
34] prepared Rh
2P catalysts supported on SiO
2, Al
2O
3, and different zeolites (Na-beta, Na-MFI, Na-MOR) for the HDS of thiophen via the incipient wetness impregnation method. Sawanda et al. [
33] investigated the effect of Na doping on the formation of Rh
2P. The addition of Na to samples lowers the required reduction temperature of the samples supported on zeolites or Al
2O
3 by weakening the interactions between the Al and the phosphate precursor [
33].
Alvarado et al. [
35] applied the incipient wetness method for the preparation of Rh
2P on SiO
2 and investigated the effect of the reduction temperature on the performance of the samples in the hydroformylation of ethene and propene. XRD patterns show the formation of phase-pure Rh
2P at temperatures of 500 °C that remain stable at temperatures of 900 °C. TEM images reveal a slight increase in the particle size by increasing the reduction temperature from 250 to 900 °C. Further, HRTEM analysis of a single particle from the sample reduced at 900 °C reveals high crystallinity of Rh
2P with cubic structure.
Guan et al. [
36] and Wang et al. [
37] also applied the method of the incipient wetness for the preparation of supported RuP and Ru
2P on MCM-41 and on SiO
2, respectively. Guan used RuCl
3·3H
2O and NaH
2PO
2·H
2O as precursors for the incipient wetness impregnation and a static Ar environment for the thermal treatment of the samples. From the results, Guan proposed a mechanism for the formation of the phosphides: first, PH
3 is formed by the decomposition of NaH
2PO
4, which reduces RuCl
3 to Ru. Further formation of PH
3 allows Ru to form Ru
2P or RuP, depending on the amount of PH
3 present.
Wang et al. [
37] compared two similar preparation methods: the conventional method, which implies the use of RuCl
3 and NH
4H
2PO
4 as precursors for the incipient wetness impregnation; and a new method applying RuCl
3 and triphenylphosphine (TPP) dissolved in ethanol for a slurry impregnation. Both compounds, RuP and Ru
2P, can be prepared with the two methods by modifying the metal to phosphorus ratios; however, lower reduction temperatures are needed for the formation of the phosphide when TPP is used.
Later publications investigated the doping of Ni
2P with noble metals such as Ir, Rh, Ru, and Pt on SiO
2 [
7,
38] or Zeolites [
39]. The groups applied the method of the incipient wetness, preparing first a solution with the two metals and the phosphorus precursor and impregnating it on the support material, followed by a drying, calcination, and reduction step. XRD patterns show the formation of Ni
2P with no impurities.
A further synthetic pathway for the preparation of supported metal phosphides consists of the conversion of supported metals or metal oxides with a phosphorus precursor to the corresponding phosphides (
Figure 2b).
First, the preparation of a supported metal or metal oxide takes place. This can be carried out, for example, via incipient wetness of the support material with a solution of the metal precursor, followed by a calcination step and subsequent reduction to obtain the supported metal. For the preparation of the phosphides, a phosphorus precursor is added to the supported metal or metal oxide followed by a thermal treatment.
Muetterties et al. [
40] described this method in 1974 for the preparation of the phosphides of Ru, Rh, Pd, Pt, and other transition metals (Ni, W, Co, Fe) on Al
2O
3. First, the metal or metal oxides are prepared on the support material. The supported metals and metal oxides are then treated under a mixture of He and PH
3 in a temperature range of 250–300 °C to form the phosphides. Unfortunately, the materials were not characterized by XRD to confirm the formation of the phases.
Bowker et al. [
41] prepared first RuO
4 and PdO on SiO
2, which is then impregnated with NH
4H
2PO
2 and heated to 700 °C under a H
2 atmosphere for the formation of the phosphides. By changing the phosphorus to metal ratio during the preparation, the phosphides RuP or Ru
2P and Pd
5P
2 or Pd
3P can be prepared.
Kucernak et al. [
42] used commercial Pd on active carbon, which is then heated in trioctylphosphine (TOP) to 300 °C for the formation of Pd
5P
2 and PdP
2. A similar method was applied by Kanda et al. [
43] for the preparation of Rh
2P on Al
2O
3. First, Rh on Al
2O
3 is prepared via incipient wetness impregnation, calcination, and reduction. The Rh on Al
2O
3 is impregnated with a TPP/hexane solution, dried under inert conditions, and reduced under a 5% H
2 in N
2 flow at different temperatures. Kanda also prepared a further catalyst using the (NH
4)
2HPO
4 to compare the reduction temperature of the two samples. By using TPP as a phosphorus precursor instead of using a phosphate precursor, the reduction temperature for the formation of the phosphide can be decreased from 800 to 650 °C.
The last method discussed here consists in the deposition of phosphide nanoparticles, prepared via colloidal methods, on the support material (
Figure 2c). First, the platinum group metal phosphide nanoparticles are prepared and, on a second step, deposited on the support material. The preparation of the nanoparticles beforehand seems to allow better control of the size and size distribution of the PGM phosphides. The preparation of phosphide nanoparticles can be carried out by preparing first the metal nanoparticles and then reacting them with PH
3 [
44]
. Another highly reactive possible phosphorus precursor is P(SiMe
3)
3, which allows a one-pot preparation of the phosphide nanoparticles and has been successfully applied for the preparation of Ni
2P nanoparticles. A further, less reactive but versatile and more and more common phosphorus precursor for the preparation of phosphide nanoparticles is TOP. Carenco et al. [
44] reviewed the preparation of nano-scaled metal phosphides where they reported the development of the synthetic pathways from highly active precursors such as PH
3 and metal carbonyls to more stable ones such as TOP and metal acetylacetonates. Henkes et al. [
45] showed the versatility of TOP as a phosphorus precursor using it for the preparation of different metal phosphides (Cu, Fe, Ni, Co, Ag, In, Rh, Pd, Zn) as nanoparticles, powder, foils, wires, and thin films. Henkes et al. [
46] also prepared Rh nanoparticles with different shapes and morphologies and converted them to Rh
2P using TOP. The phosphides keep the same shape as the metal templates, as shown on TEM images. A further method for the preparation of phosphide nanoparticles is the use of single-source molecular precursors such as Ni(PPh
3)
2(CO)
2, Rh(PPh
3)
2(CO)Cl, or Pd(PPh
3)
4, mixed in oleylamine and 1-octadecene, heated to 300 °C for 1 h and cooled to room temperature. The nanoparticles are then washed with ethanol and centrifuged. Single-source precursors were used by Habas et al. [
28] and by Griffin et al. [
47] for the preparation of Ni
2P, Rh
2P, and Pd
3P on SiO
2. The Rh
2P nanoparticles are formed using Rh(PPh
3)
2(CO)Cl mixed in an oleylamine and 1-octadecen solution, which is heated up to 300 °C. The nanoparticles are recovered, washed with ethanol, re-dispersed in CHCl
3, and added to a suspension of SiO
2 in CHCl
3, sonicated and dried to form the finished catalyst.
4. Rh2P as a Prototypic Example for PGM Phosphides in Hydrotreating Reactions
Of all the rhodium phosphides, only Rh
2P has been reported to be a suitable catalyst material for different reactions, mainly the HDS and HDO of hydrocarbons. Most of the investigations regarding the application of metal phosphides in hydrotreating reactions have been driven by the motivation to find improved hydrotreating catalysts due to the increasing environmental regulations limiting the content of sulfur in transportation fuels and due to the diminishing quality of oil feedstocks [
25,
26,
49]. The most investigated metal phosphide is Ni
2P [
24,
25,
26,
49], which shows high activity in the HDS of dibenzothiophene (DBT) where it hydrogenates preferably the sulfur compound than the aromatic ring [
8,
50]. The activity of the Ni
2P catalysts supported on SiO
2 is comparable to the activity of a commercially available Ni-Mo-S/Al
2O
3 catalyst. Further investigations show the higher activity of CoP for the HDN of chinolin [
50], of MoP and Ni
2P for the HDO of palmitic acid [
51], or Ni
2P for the HDO of benzofuran [
50]. Within the frame of these investigations, different PGM phosphides have been tested as catalyst materials. Here, the Rh
2P/SiO
2 material shows a significantly higher activity in the HDS of DBT than Rh/SiO
2 and higher than the Ni
2P/SiO
2 material [
27]. Further investigations have shown that Rh
2P has a higher activity in the HDS of thiophene than all the other PGM phosphides [
29]. Due to the higher intrinsic activity of the Rh
2P catalyst, different publications have focused on the application of Rh
2P as a catalyst material for hydrotreating reactions [
27,
28,
29,
30,
31,
32,
33,
34,
43,
47,
52], for the hydrogen evolution reaction [
53], and for the hydroformylation [
35]. Reviews focusing on the metal phosphides in general and their applications as catalyst materials can be found elsewhere [
24,
25,
26,
49,
54,
55].
The next section of this review deals with the application of Rh2P as a catalyst material for the reactions of HDS and HDO. Investigations regarding the application of Rh2P as a catalyst for the HDN have not been published yet to our knowledge.
4.1. Rh2P as a Catalyst Material for HDS
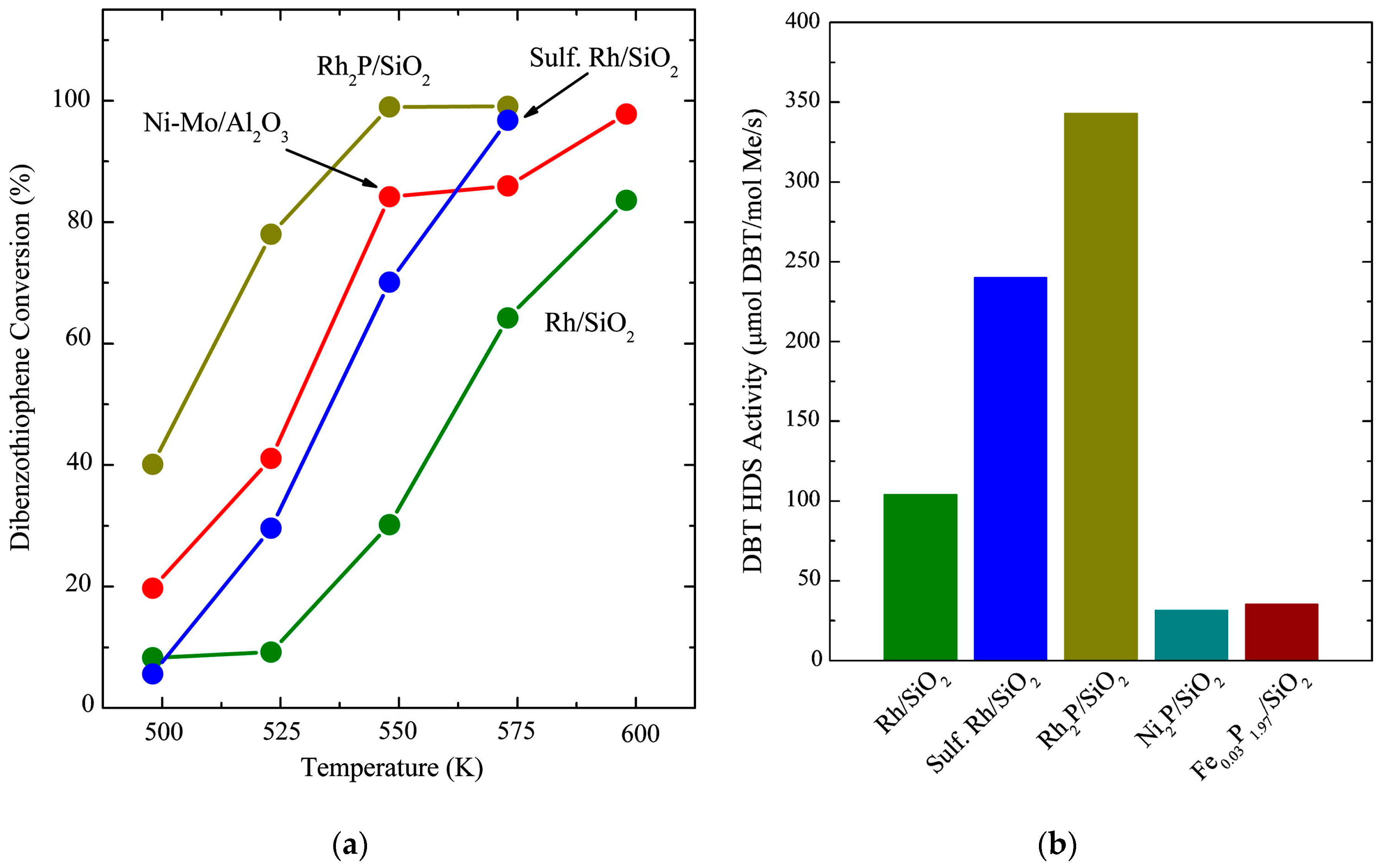
Among the first investigations on the application of Rh
2P as a catalyst material in HDS are the articles published by Hayes et al. [
27] who prepared a series of supported Rh-based catalysts (Rh
2P, Rh, sulfonated Rh on SiO
2, Rh loading of 5 wt %) and compared their performance in the HDS of DBT. The findings of their investigation show that the Rh
2P/SiO
2 catalyst material is more active in the HDS of DBT (fixed-bed reactor, temperature range of 225–300 °C, 3.0 MPa, Feed: decalin solution with 3000 ppm DBT) than the sulfonated Rh/SiO
2 and the Rh/SiO
2 catalysts; and is also more active than the commercially available Ni-Mo/Al
2O
3 catalyst used as a benchmark, as shown on
Figure 3a.
Table 3 shows the results of the investigation at 275 °C where Rh
2P/SiO
2 shows significantly higher conversion values (99.0%) than the benchmark catalyst Ni-Mo/Al
2O
3 (84.2%). The product distribution shows that the Rh
2P/SiO
2 catalyst favors the hydrogenation of the first aromatic ring and then the removal of S, and thus the high selectivity values for cyclohexane–benzene.
Further findings of the investigation are the higher tolerance of the Rh
2P catalyst toward H
2S than the Rh/SiO
2 catalyst. Both materials were tested before and after co-feeding of 50 kPa H
2S. The conversion of DBT at 275 °C decreases from 80% to 22% for the Rh
2P catalyst and from 65% to 12% for the Rh/SiO
2 catalyst. The normalization of the HDS activity per mol of metal allowed the authors to make a comparison with other catalyst systems such as a 25 wt % Ni
2P/SiO
2 catalyst. Hayes et al. showed that the per mol metal HDS activity of the Rh
2P/SiO
2 catalyst at 275 °C is approx. eleven times higher than the activity of the Ni
2P/SiO
2 catalyst, as
Figure 3b and the values in
Table 4 show, and so increasing the interest in Rh
2P as a suitable catalyst material for the HDS reaction.
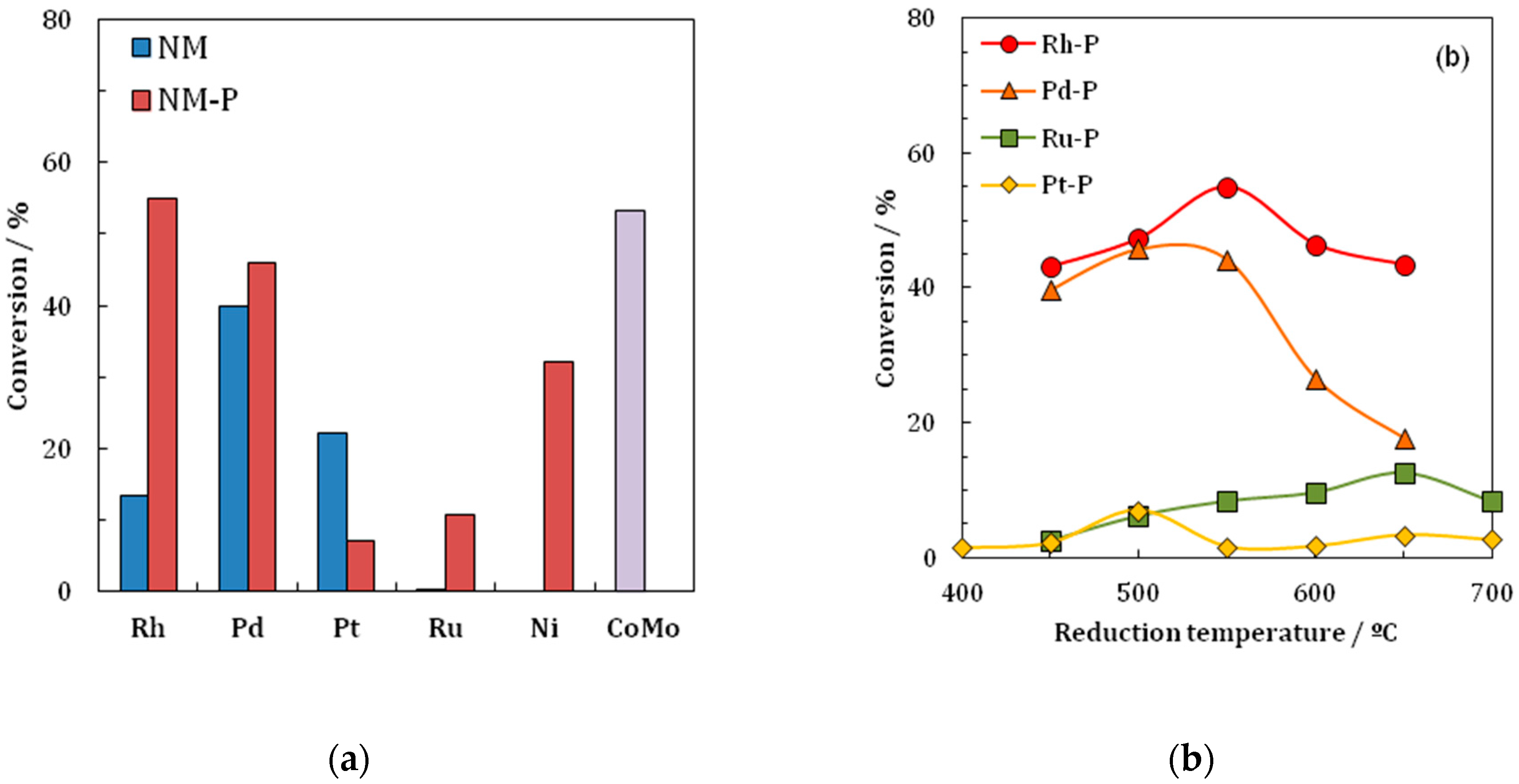
Most of the work performed on Rh
2P as a hydrogenolysis active catalyst material for the HDS reaction has been reported by the group of Yasuharu Kanda. In a first publication [
29], the performance of PGM metals and PGM phosphides supported on SiO
2 (Rh
2P, Pd
4.6P, Ru
2P, and PtP
2) were compared in the HDS reaction of thiophene. The findings of the investigation are that the Rh
2P/SiO
2 catalyst is the most active PMG phosphide in the HDS of thiophene, as shown by the results in
Table 5 and
Figure 4. The Rh
2P/SiO
2 catalyst shows the highest values for the conversion of thiophene in the HDS reaction. Further findings are that Rh
2P/SiO
2 is more active than the Ni
2P/SiO
2 catalyst; that Rh
2P/SiO
2 has a comparable activity to the commercial CoMoP/Al
2O
3 catalyst they use as a benchmark; that the PGM phosphides are superior to the PGMs themselves and, therefore, phosphide formation is essential for the activity of the material, as shown in the results in
Table 5. The Rh/SiO
2 catalyst achieves a conversion value of 13.32% while Rh
2P/SiO
2 achieves a value of 54.95%; the only phosphide displaying a lower activity than the metal is PtP
2.
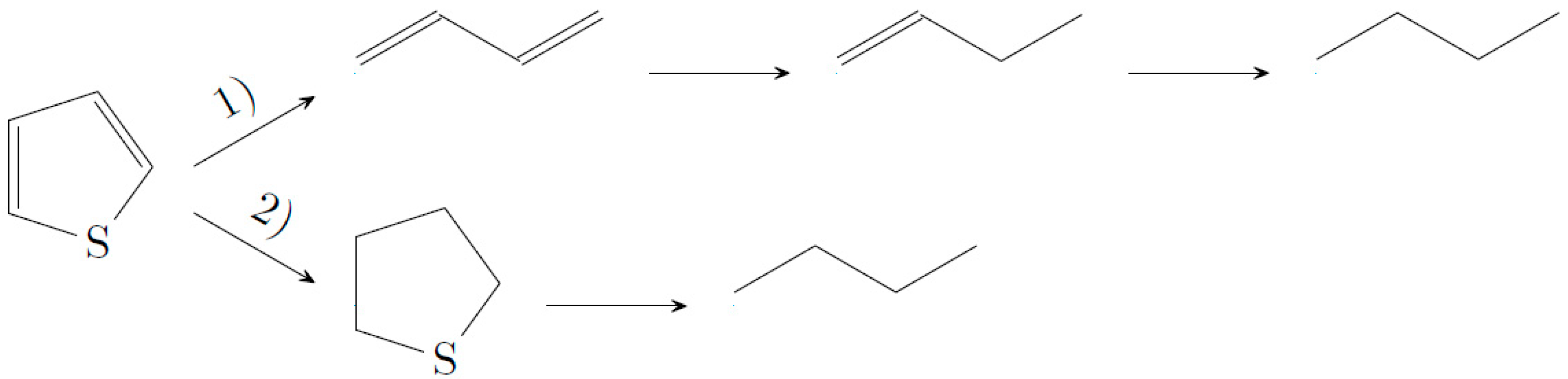
In investigations concerning the reaction mechanism, it was proven that direct desulfurization is favored over dehydrogenation of the double bonds of thiophene (reaction route 1 on
Figure 5). The product with the highest selectivity is 1-butene, followed by butane and small amounts of tetrahydrothiophene, as seen in
Table 5.
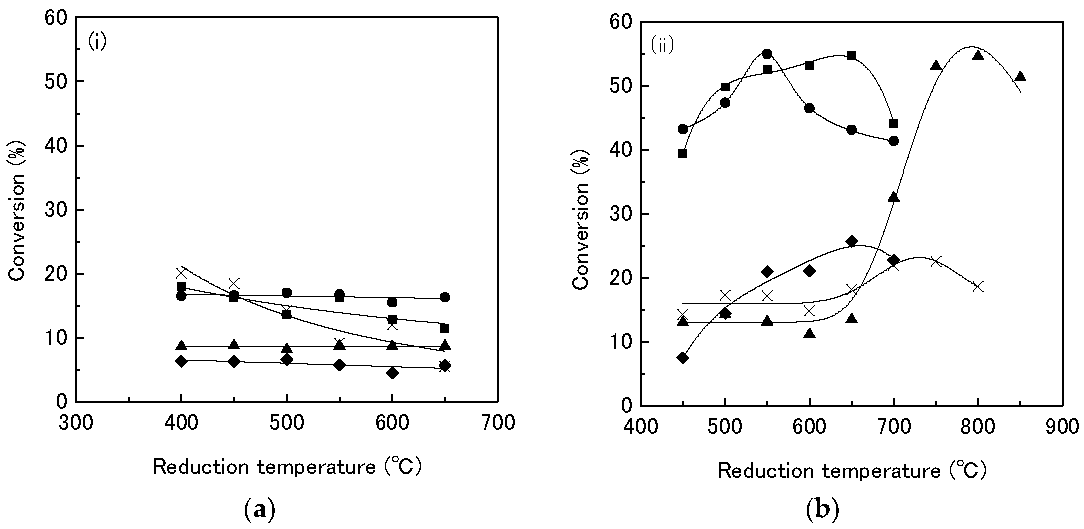
In a further publication [
30], Kanda investigated the effect of the support material and reduction temperature on the performance of the Rh
2P catalysts on the HDS of thiophene. The activity of the sample changes with the support material. The catalysts on SiO
2, Al
2O
3, and TiO
2 are the most active followed by MgO and last ZrO
2, as shown in
Table 6. Further, as mentioned above, the results show that the formation of the phosphides enhances the activity of the materials, as
Table 6 shows. The highest conversion value for a supported Rh catalyst material is 20.03% (Rh/ZrO
2), while the Rh
2P material supported on SiO
2, Al
2O
3, or TiO
2 reaches conversion values of approx. 54%. Further, TPR experiments show that the reduction behavior of the phosphate precursors to the phosphides changes with the support material, due probably to metal-support interactions. A reduction temperature as high as 800 °C is needed to convert the phosphate precursors to Rh
2P on Al
2O
3, while the material on SiO
2 is converted to Rh
2P at a temperature of 550 °C. Last, the group investigated the effect of the reduction temperature. The materials are reduced at different temperatures in the range of 450–850 °C and their performance in the HDS of thiophene was compared. All materials show an increase in turnover frequency (TOF) with an increasing reduction temperature. The most significant increase is observed with the materials supported on Al
2O
3 (
Table 7,
Figure 6). The sample reduced at 450 °C displays a TOF of only 22.9 h
−1 while the sample reduced at 850 °C shows a TOF of 251.4 h
−1. The increase in the TOF is due to the formation of Rh
2P at higher reduction temperatures, which is more active than Rh or the phosphate species.
A further investigation by Kanda et al. [
31] focused on the preparation of Rh
2P on SiO
2 and on the effect of the P loading and the reduction temperature on the HDS of thiophene. The highest HDS activity is observed on the catalyst with a loading of 5 wt % Rh and 1.5 wt % P on SiO
2, reduced at a temperature of 550 °C, which achieves conversion values of up to 55.0 % (
Table 8) and arate constant of approximately 21 mmol h
−1 g
−1. The higher activity of the 1.5 wt % P loaded catalyst is attributed to the formation of small Rh
2P particles at a relatively low reduction temperature (550 °C), as shown by the results from the TEM and PXRD investigations. The authors assumed that catalysts with lower P loadings (0.8 wt %) need higher temperatures for the formation of Rh
2P with Rh impurities and that high reduction temperatures lead to sintering of the active phase. Catalysts with higher P loadings (2.2–3.0 wt %) show the formation of highly dispersed Rh
2P already at lower temperatures (350 °C), but the excess P covers the active phase. A moderate P loading of 1.5 wt % leads to the formation of well-dispersed Rh
2P at moderate temperatures without the drawback of excess P covering or sintering due to the high reduction temperature.
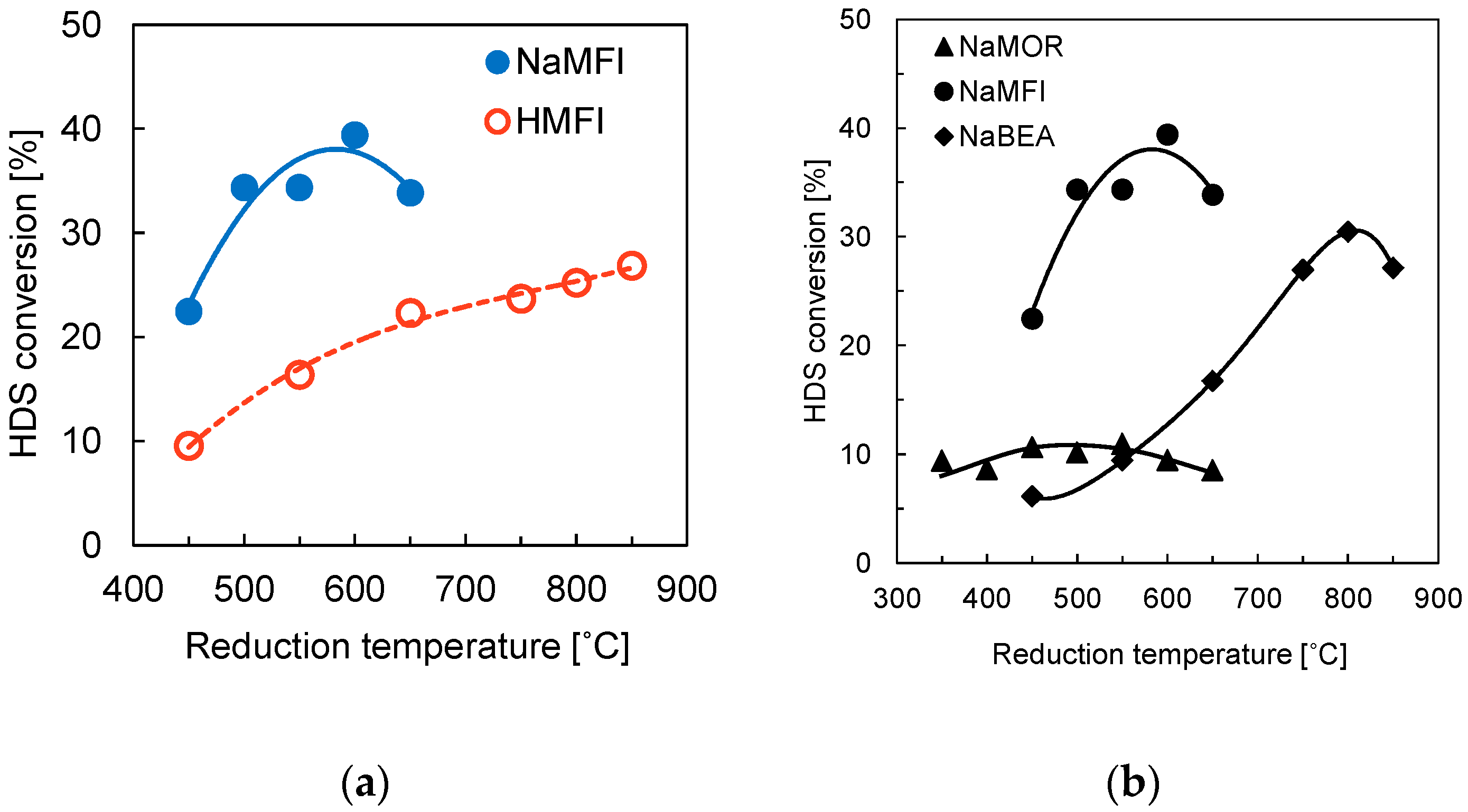
Sawanda et al. [
33,
34] investigated the HDS of thiophene with Rh
2P supported on zeolites. In the first investigation [
10,
33] the effect of adding Na to the Rh
2P catalysts supported on MFI zeolite, SiO
2, and Al
2O
3 on the HDS of thiophene was studied. The addition of Na to the zeolites increases the activity of the catalysts. The Rh
2P catalyst supported on NaMFI is higher than the catalysts supported on HMFI. The highest conversion values for the NaMFI catalyst is ca. 40% (
Figure 7), while the HMFI reaches a maximum conversion value of ca 25% under identical conditions (fixed-bed reactor, 0.1 g catalyst, 350 °C, 0.1 MPa, H
2/thiophene = 30, W/F = 37.9 g h mol
−1). Sawanda suggested that the addition of Na decreases the interaction between the Al and the phosphate and thus enhances the reducibility of the phosphate species to Rh
2P and increases the activity of the catalysts. In a subsequent investigation [
34], different Na-form zeolites (Na-beta, NaMFI, NaMOR) were used as supports for the Rh
2P catalyst and tested in the HDS of thiophen. The order of the thiophene conversion values is NaMFI (~35%) > Na-beta (~17%) » NaMOR (~8%) under identical reaction conditions (fixed-bed reactor, 0.1 g catalyst, 350 °C, 0.1 MPa, H
2/thiophene = 30, W/F = 37.9 g h mol
−1). Sawanda et al. attributed the low activity of NaMOR supported Rh
2P catalyst to the one-dimensional channel structure of NaMOR, so that the Rh and phosphate species do not easily diffuse into the micropores of the support material, leading to low CO uptake values and low catalyst activity. The catalyst supported on NaMOR displays CO uptake values between 2 and 7 μmol/g while the catalysts supported on NaMFI and Na-beta achieve CO uptake values as high as 40 μmol/g. Further, a significantly higher reduction temperature is needed for the formation of phase-pure Rh
2P supported on the Na-beta zeolite. Due to the high reduction temperature (850 °C), the structure of the support partially collapses, leading to lower activity of the Rh
2P/Na-beta catalyst.
Finally, Kanda compared the performance of two Rh2P/Al2O3 catalysts prepared over two different pathways, the reduction of a supported phosphate precursor, and the conversion of supported Rh particles with TPP. The use of TPP reduces the reduction temperature from 800 to 450 °C for the formation of Rh2P on Al2O3. The highest conversion values in the HDS of thiophene (fixed-bed reactor, 0.1 g catalyst, 350 °C, 0.1 MPa, H2/thiophene = 30, W/F = 37.9 g h mol−1) are achieved with catalysts prepared with TPP and reduced at 650 °C (the catalyst achieves a reaction rate of 38.5 mmol g−1 h−1, while the catalyst prepared over the phosphate reduction pathway achieves a reaction rate of 20.9 mmol g−1 h−1). At a reduction temperature of 450 °C, an excess of P covers the Rh2P surface, which decreases the HDS activity of the catalysts. A reduction temperature of 650 °C eliminates the P excess and enhances the crystallinity of the Rh2P nanoparticles and so increases the activity of the HDS. The higher activity of the catalysts prepared with TPP is attributed to the significantly smaller Rh2P nanoparticles that have an average particle diameter of approx. 1.2 nm, while the particles prepared over the reduction of the phosphates have an average particle diameter of ca. 8.5 nm, according to the TEM studies performed in the investigation.
The investigations performed by Kanda and Sawanda and other research groups on the performance of supported Rh
2P catalysts and other supported noble metal phosphides as hydrotreating catalysts were reviewed in 2015 by Kanda and Uemichi [
52].
4.2. Rh2P as a Catalyst Material for HDO
In the last decades, transition metal phosphides have gained a lot of attention in hydroprocessing with the main focus on HDS [
25,
56]. PGM phosphides for HDO have only recently come into focus [
24,
25,
57], also due to the emerging perspective of renewable resources for the production of fuels and chemicals. The most investigated phosphides in this field are non-noble metal phosphides such as Ni
2P, MoP, and Co
2P [
56], and only recently Rh
2P and RuP have been considered as viable candidates.
Hydrodeoxygenation is a reaction that can be applied to different feeds and the specific process will determine the choice of the appropriate catalyst. Typically, HDO occurs parallel to HDS and HDN during hydrotreatment of conventional petroleum fractions. The oxygen content of most petroleum feedstocks is usually lower than 2% and sulfides of CoMo/γ-Al
2O
3 or NiMo/γ-Al
2O
3 [
58] are the conventional and validated catalysts employed in this case (reaction condition range 3–10 MPa H
2 pressure, 300–500 °C). Bio-oils derived from biomass have a completely different composition, being remarkable for their high oxygen content (usually up to 40%) and water content (usually up to 30%) [
59]. In the case of bio-oils, efficient HDO is relevant to improve their stability, increase the heating value, and decrease the viscosity [
58]. However, conventional hydrotreating catalysts are usually not stable in the presence of feeds containing high oxygen content without an additional supply of a S source [
58]. Up to now, the identification of optimal catalyst systems for HDO has been a challenging task and several solutions have been proposed and investigated. The main challenge for identifying an appropriate HDO catalyst is that a large versatility to convert different functional groups with different reactivity is demanded and stability under the corrosive reaction conditions is vital. Indeed, typical bio-oils are complex mixtures constituted of oxygenated components, such as phenolics, furans, ethers, carboxylic acids, aldehydes, ketones, and alcohols (an example of the reactivity scale for HDO is provided by Elliott [
60]). Phenols [
61] and dibenzofurans [
62] are generally chosen as model compounds to discriminate and estimate the HDO reactivity of a given catalyst system since they are the least reactive compounds to HDO.
Transition metal phosphides are potential candidates in HDO. Especially, metal-rich phosphides that have excellent heat and electrical conductivities and high thermal and chemical stabilities can match the properties desired for hydrotreatment [
25,
54,
63]. This can be attributed to their good hydrogen transfer properties that are enhanced by the presence of phosphorus [
64,
65].
As described in the case of Ni
2P, the presence of phosphorus has an important effect on the HDO activity [
65,
66]. Phosphorus has a ligand effect on the metal site of the phosphide, modifying the electron density on the metal cation (M
δ+) and resulting in an easier hydrogen dissociation on this surface, and the catalytic center can act as a Lewis acid [
55,
66]. The group P–OH, which is co-located on the metal phosphide phase as result of an incomplete reduction of phosphorus on the surface, constitutes a Brønsted acid of moderate acidity [
63,
66]. The synergy between the two groups (metallic and acidic) promotes catalyst activity comparable to that of a noble metal on an acidic support material and can be described as a cooperative mechanism over more than one neighboring catalytic center. The metal centers are active for hydrogenation, hydrogenolysis, and demethylation reactions [
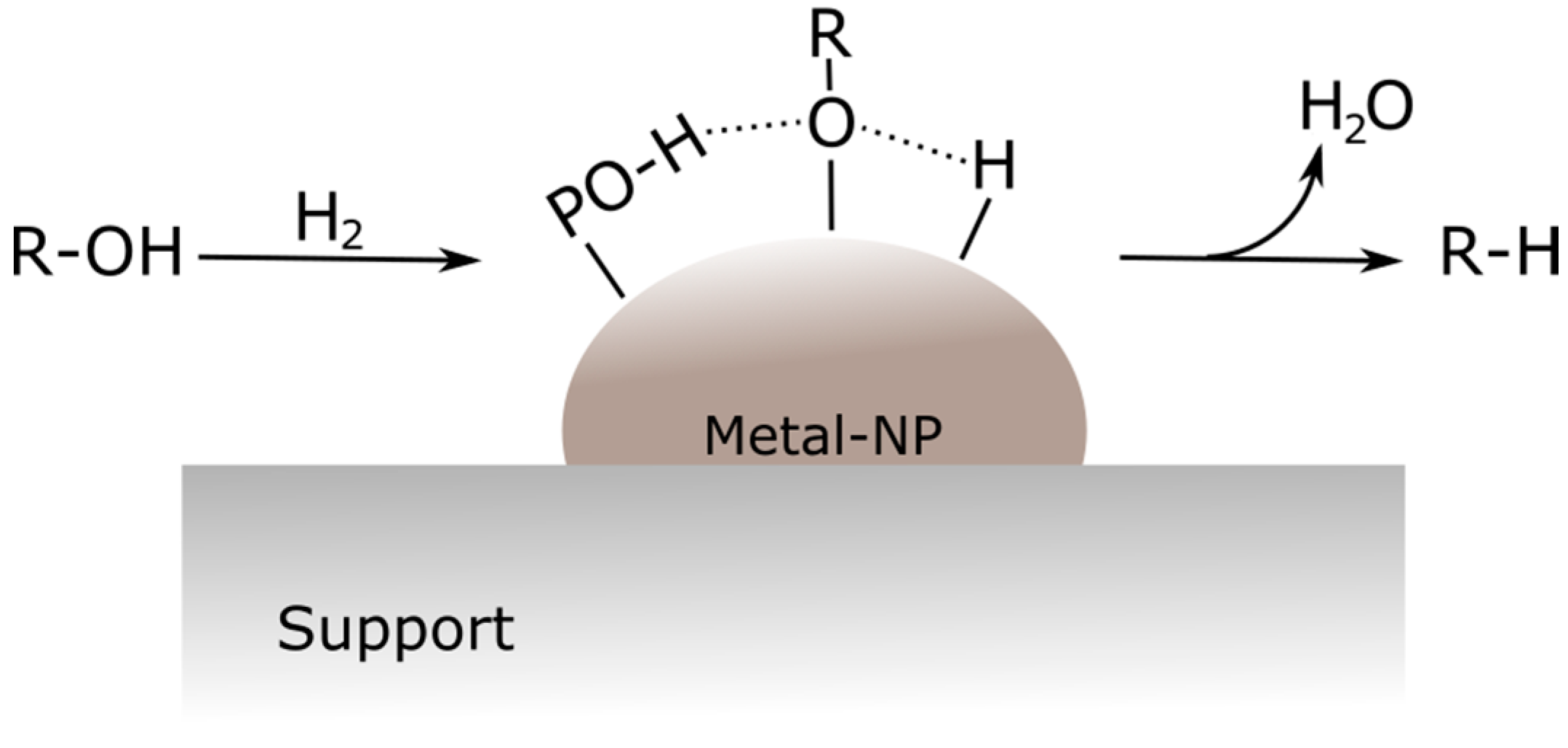
66], while PO-H groups can donate active hydrogen species as well, but they are much less active than the metal site. A higher d electron density of the metal is described as beneficial for an improved activity. A proposed HDO mechanism is reported in
Figure 8. The oxygenated compounds and adsorbed H
2 are activated on the metal center and hydrogen atoms from the metal center and P–OH site react with the oxygenated species adsorbed on the surface; finally, water and the deoxygenated product species are formed [
55].
Ni
2P is one of the most studied metal phosphides for hydrotreatment showing good HDO activity as well for model compounds and biomass-derived feeds [
57,
61,
67]. Zhao et al. [
61] demonstrated that Ni
2P shows superior activity compared to other base metals. The following sequence in activity was reported for several phosphides in the HDO of guaiacol in a fixed-bed reactor at 300 °C and atmospheric pressure:
All the catalysts are less active than Pd/Al
2O
3, and the least stable system proved to be Co-MoS
2/Al
2O
3, representing an advantage of the phosphide materials. Another study related to phenol HDO underlines that phosphides can be active in hydrogenation as well as in isomerization [
68]. For the phosphide materials, this results in higher selectivity for methylcyclopentane than cyclohexane during conversion of phenol. Koike et al. [
57] explored Ni
2P/SiO
2 in the HDO of bio-oil produced from cedar chips in a two-stage fluidized bed reactor (0.1 MPa, 300 °C and 350 °C, residence time 0.64–1.0 s). Phenolic compounds were unreactive under the reaction condition used and a lower deoxygenation degree was recorded compared to the test with 2-methyltetrahydrofuran as the model compound.
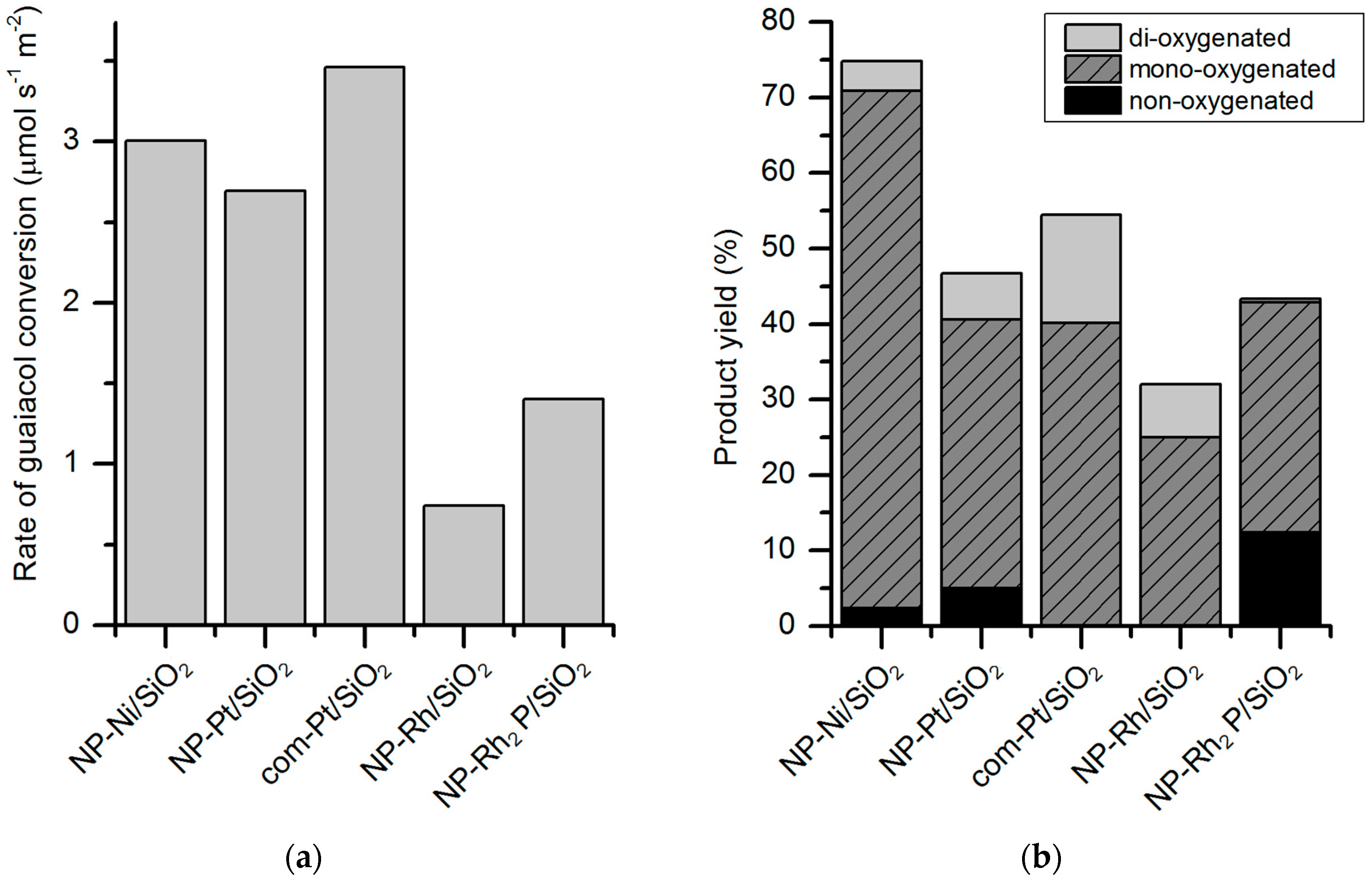
Rh
2P has received less attention than Ni
2P but recent studies have demonstrated a high activity in HDO, superior also to that of supported noble metals in metallic form [
25,
47]. Griffin et al. [
47] employed Rh
2P nanoparticles, obtained via solution-phase synthesis, supported on silica for guaiacol HDO. The catalytic tests were carried out in a reactor system allowing continuous flow at 350 °C, 0.5 MPa with a ratio 12:1 H
2 to guaiacol, and WHSV 5 h
−1. The synthesis method of the catalyst has an influence on the catalytic performance; a better selectivity towards deoxygenated species has been found for nanoparticles synthesized in solution-phase compared to incipient wetness (IW) impregnation (
Figure 9). The activity of Rh
2P/SiO
2 in guaiacol conversion is not the highest among the tested catalysts but it has the highest selectivity for deoxygenated products such as cyclohexane and benzene (26% combined). Compared to the other catalysts (such as Rh/SiO
2, Ni/SiO
2, Ni
2P/SiO
2), Rh
2P/SiO
2 is more selective for C–O cleavage of the aryl–OH bond, giving benzene and anisole as the products.
Further studies on Rh
2P on different support materials were also carried out by Griffin et al. [
69]. Similar reaction conditions compared to the previous study [
47] were used with Rh
2P on TiO
2, Al
2O
3, and MgO as support materials in order to assess the influence of the support in HDO of guaiacol as the model compound. Rh
2P/TiO
2 and Ni/TiO
2 exhibit higher guaiacol conversion showing products with lower O:C ratio for C
5+ fraction compared to the catalysts supported on active carbon. This is attributed to the properties of TiO
2, which displays reducible oxygen vacancies capable of facilitating C–O bond scission without aromatic ring hydrogenation. Ni/Al
2O
3 and Rh
2P/Al
2O
3 have also been found to be highly active catalysts, but they are less selective toward deoxygenation, and they also promote alkylation. MgO as support material produces the worst performance among the catalyst set investigated; it is speculated that the basic nature of the support is the main reason for the observation.
Habas et al. [
28] investigated the activity of Ni
2P, Rh
2P, and Pd
3P on the HDO of acetic acid in a temperature range from 200 to 500 °C. The metal phosphide nanoparticles were synthesized by commercially available, air-stable metal−phosphine complexes in a one-pot reaction and supported on silica. Analogous catalysts were prepared by incipient wetness as a comparison. Both Rh
2P and Ni
2P show hydrogen-assisted decarbonylation reactions with CO, CH
4, and H
2O as products. Pd
3P does not show remarkable activity.
Cecilia et al. [
38] were able to enhance the catalyst activity of Ni
2P by adding Rh, Ru, Ir, and Pt (molar ratio metal/Ni = 10%). The catalyst activity for HDO of dibenzofuran (P = 3.0 MPa, 175–300 °C, fixed-bed reactor) follows the sequence:
The structural interaction between the phosphide Ni2P and the noble metal was not clarified but it was observed that Rh, Ru, and Ir could effectively enhance the catalyst activity and stability, while Pt provokes the opposite effect. The presence of a noble metal seems to improve the adsorption and activation of hydrogen in the metal support interface and it increases the presence of P–OH.
Bowker et al. [
70] described the synthesis and HDO activity of silica-supported ruthenium phosphide (Ru
2P, RuP) catalysts. At 400 °C in a continuous reactor, Ru
2P and RuP are more active in HDO of furan than the conventional CoMo/Al
2O
3 catalyst, as reported in the sequence:
Ru2P/SiO2 is the most active and stable catalyst of the set (TOS 48 h). Higher selectivity for C4 hydrocarbons is attributed to the presence of phosphorus, compared to Ru metal that produces more C3 compounds.
One of the biggest dilemmas concerning transition metal phosphides is their stability in contact with complex mixtures such as petroleum fractions or real bio-oils in the process reaction conditions. Only a few studies have reported the stability of these catalysts and offer insight into the eventual deactivation processes of these catalysts. Typically, water created in the reaction or present initially in the feed can deactivate or degrade the catalyst through the formation of phosphates or the respective metal oxides from the corresponding phosphides [
66]. Griffin et al. [
69] reported that metal phosphides show a tolerance to water under the acidic conditions present in pyrolysis vapors. Cecilia et al. [
62] asserted that Ni
2P shows high resistance to deactivation towards coke, although the water produced as a byproduct could be responsible for the slight decrease in activity. Koike et al. [
57] identified coke formation as the main source of deactivation for Ni
2P/SiO
2. Griffin et al. [
69] reported also for Rh
2P an increase in the coke deposited on the catalyst surface during guaiacol HDO.
In conclusion, metal phosphides appear promising for HDO although their activity may be lower compared to their intrinsic activity in HDS or HDN. The advantage of these materials is that their properties can be tuned depending on the metal chosen or by varying the metal/phosphorus ratio or the particle size of the phosphides [
54]. It will be interesting to see the advances in the field in order to design an optimal phosphide-based catalyst for HDO.
4.3. Other Succesful Examples of Catalysis with Rh2P
The homogeneously catalyzed hydroformylation of olefins is one of the most important large-scale industrial processes. Cobalt carbonyl complex-based catalyst systems are still used today for the hydroformylation of mid- and long-chain olefins, while the inherently more active rhodium carbonyl complexes stabilized with phosphine or phosphite ligands are applied for the hydroformylation of short olefins. The application of expensive rhodium catalyst systems demands an effective separation of the catalyst from the reaction products to minimize the risk of rhodium loss and catalyst deactivation. This explains why rhodium is only applied for the hydroformylation of short olefins where the separation of the products by distillation is feasible or alternatively the biphasic process where the catalyst complex remains in the aqueous phase while the products are removed over the organic phase.
Technical solutions involving heterogeneous catalysts in the hydroformylation with the catalyst in the solid phase and the products in the gas or liquid phase could offer alternatives that may also lead to new solutions regarding the process design. Research activities in academia and in industry are focusing on the synthesis and development of heterogeneous or heterogenized molecular hydroformylation catalysts that have proven to be inferior to the current state of the art in homogeneous catalysis [
71,
72,
73,
74,
75].
As shown in the previous section, PGM phosphides, and especially rhodium phosphide, have proven to be useful heterogeneous catalysts for hydrotreating reactions. Although the phosphides show high catalytic activity in different reactions, they have not been widely considered as candidates in reactions involving the activation of carbon monoxide such as the hydroformylation of lower olefins.
We have carried out tests with Rh
2P on SiO
2 as a catalyst for the gas phase hydroformylation of ethylene and propylene [
35]. The preparation of these catalysts was carried out via the incipient wetness impregnation of phosphate precursors followed by a reductive thermal treatment at different temperatures. The samples show the formation of phase-pure Rh
2P particles on SiO
2 at a reduction temperature of 500 °C, as PXRD investigation illustrated; highly crystalline Rh
2P nano-particles could be retained even at a reduction temperature of 900 °C, as proven by HRTEM investigations.
The samples were tested in a fixed-bed reactor system for the gas phase hydroformylation of ethylene, where the best results are achieved at low H2/CO ratios (50 bar; GHSV = 2773 h−1; 170–240 °C; 10% C2H4; 10% H2; 70% CO; 10% Ar). A further increase in the selectivity of the catalysts towards the aldehyde is observed when water is added to the feed. It is reasoned that by adding water to the system the formation of the hydrogenation product ethane and the aldol condensation of the aldehyde are suppressed so that the selectivity of the desired aldehyde increases.
A comparison between the performance of the catalysts reduced at different temperatures has proven that the most active and selective catalysts are the catalyst types reduced at the highest temperatures. Such catalysts are highly stable: the spent catalyst of the sample reduced at 900 °C displays phase-pure Rh
2P on SiO
2. Further evidence to rationalize the higher activity of the catalyst reduced at 900 °C can be taken from the IR absorption spectra after CO adsorption. The IR spectra of the catalyst reduced at 500 °C shows different signals in the region around 2000 cm
−1 that correspond to both the linearly and bridged bonded CO on Rh atoms. The sample reduced at 900 °C shows only one signal at 2700 cm
−1, which corresponds to the CO linearly bonded to the Rh atom, and which formation favors the hydroformylation of the olefin as reported by Chuang and Pien [
73,
74,
75]. Considering these facts, the authors suggest that this finding can be taken as evidence that Rh
2P in the form of highly crystalline-supported nanoparticles features surface sites that allow catalytic functionality similar to that of a phosphine- or phosphite-modified rhodium complex [
76].
Based on the results presented and the fact that single-site catalysis is the proposed mode of action for Rh
2P, the question may be raised as to what the nature of the catalytically active site is. From the evidence we have gathered [
35,
76] and the structural considerations, we can draw the following conclusions: Rh
2P crystallizes in the cubic CaF
2 phase prototype. The nano-particle catalysts are highly crystalline and one can take as first approximation the bulk structure as the model for potential surface terminations. The CO-adsorption experiments prove that the surface must be terminated with exposed metal species. Cuts through the cubic CaF
2 phase prototype, even along high index surfaces, suggest that for all geometries separation of rhodium sites by phosphorus is obtained. In light of the fact that the spectroscopic data indicate that the presence of single rhodium sites available on highly crystalline nano-particles is a prerequisite for good performance, we suggest that single-site catalysis is the mode of action relevant for the materials discussed. The rhodium sites are separated and coordinated by phosphorus and the prevailing motive of these solids can be summarized as “frustrated” single catalytic sites that do not cooperate, which is in contrast to the examples mentioned above in Rh
2P as hydroprocessing catalysis [
76]. Although Rh
2P is present as an extended solid, the specific structure of the solid and the fact that a metal and a non-metal are the constituents do create an environment suitable for single site catalysis: the motif of “frustration” can be taken as the new descriptor for the development of alternative catalytic materials for single site catalysis.
Further, we also report the application of the catalysts in the hydroformylation of larger olefins, such as propylene, with a selectivity of 90% towards butyraldehyde with an n/iso ratio of 2.4 in the gaseous phase. Here again, a positive effect can be observed when water is added to the feed stream. Last, the catalysts are tested in the methoxycarbonylation of ethylene by exchanging hydrogen in the feed gas mixtures by methanol. Under the conditions applied, no ester formation could be observed. By adding small amounts of hydrogen, noticeable amounts of the product methyl propionate, together with major amounts of propionaldehyde and ethane, could be observed. From this observation, a reaction mechanism in which the ester is not formed via alkoxycarbonylation but via dehydrogenation of an intermediate hemiacetal is suggested.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}