Chiral Dirhodium(II) Carboxylates: New Insights into the Effect of Ligand Stereo-Purity on Catalyst Structure and Enantioselectivity




Abstract
:1. Introduction
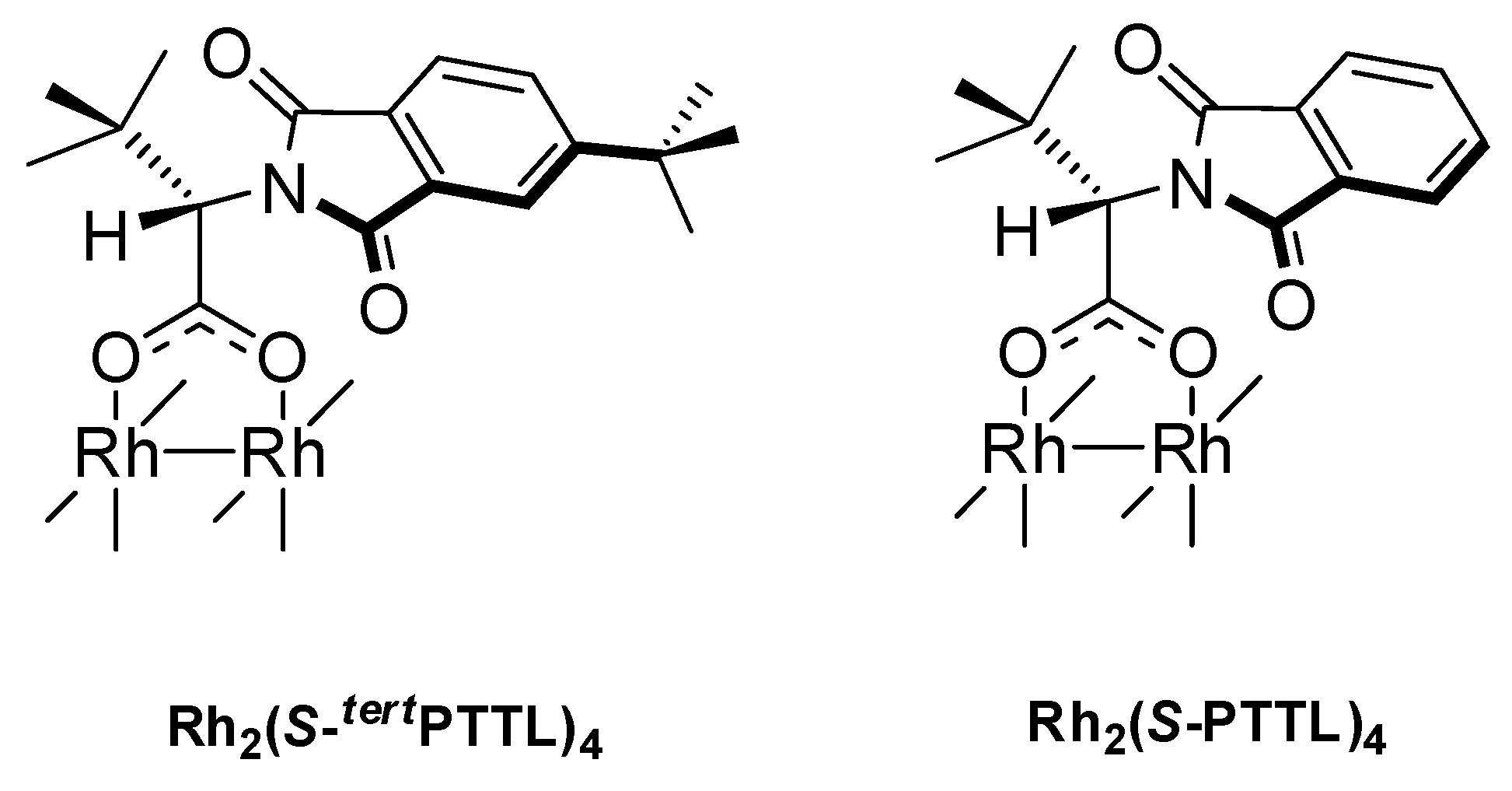
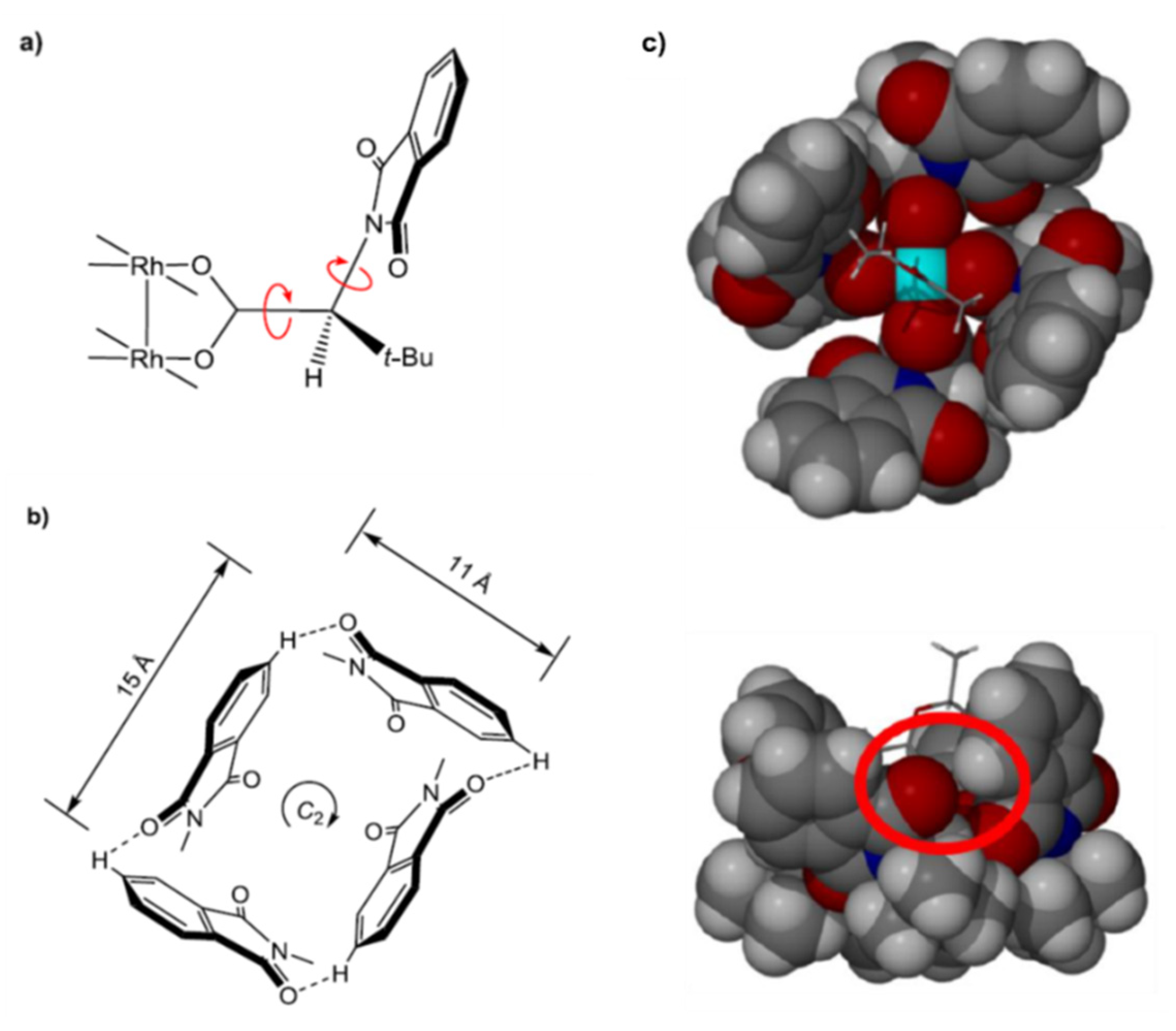
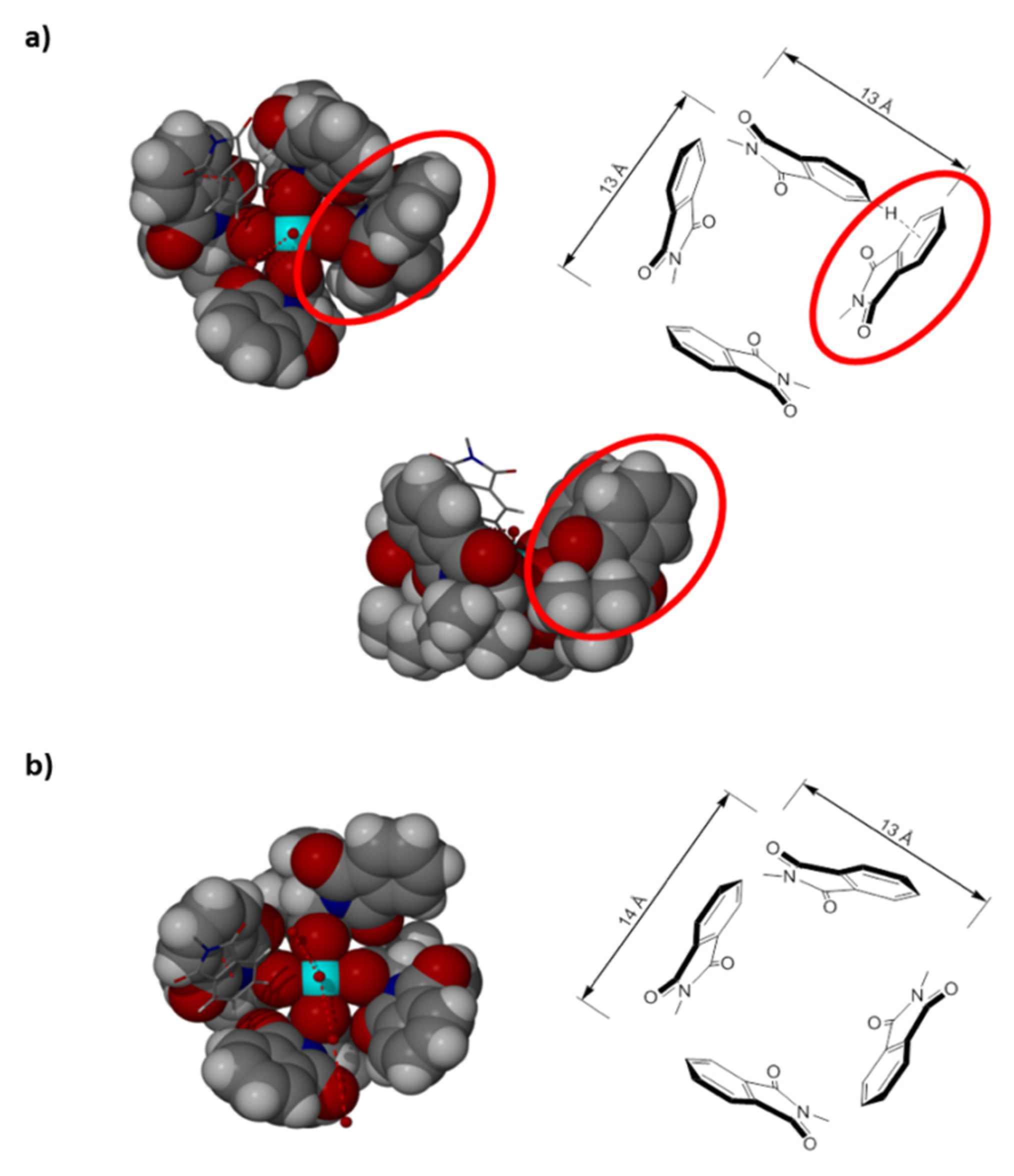
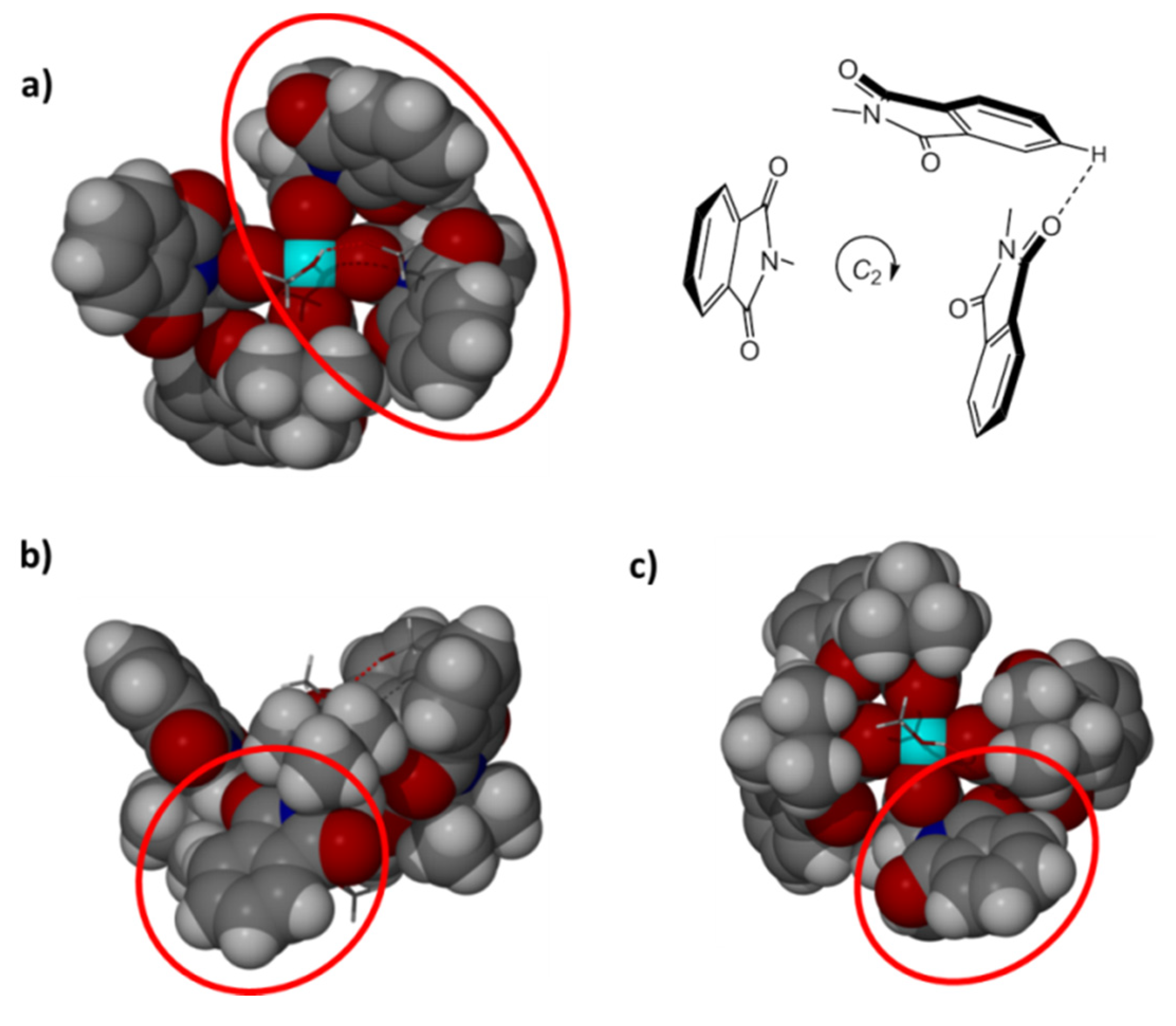
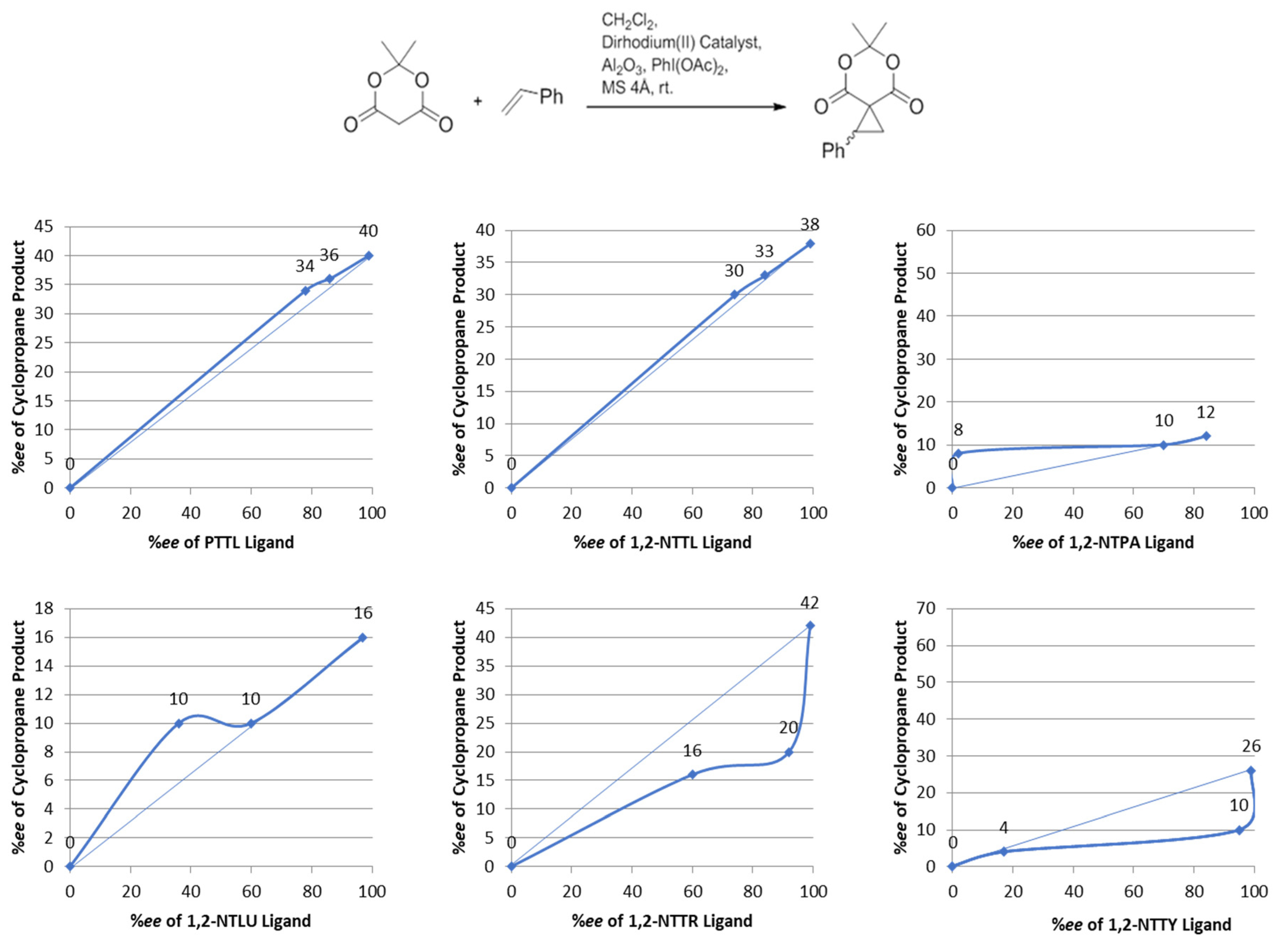
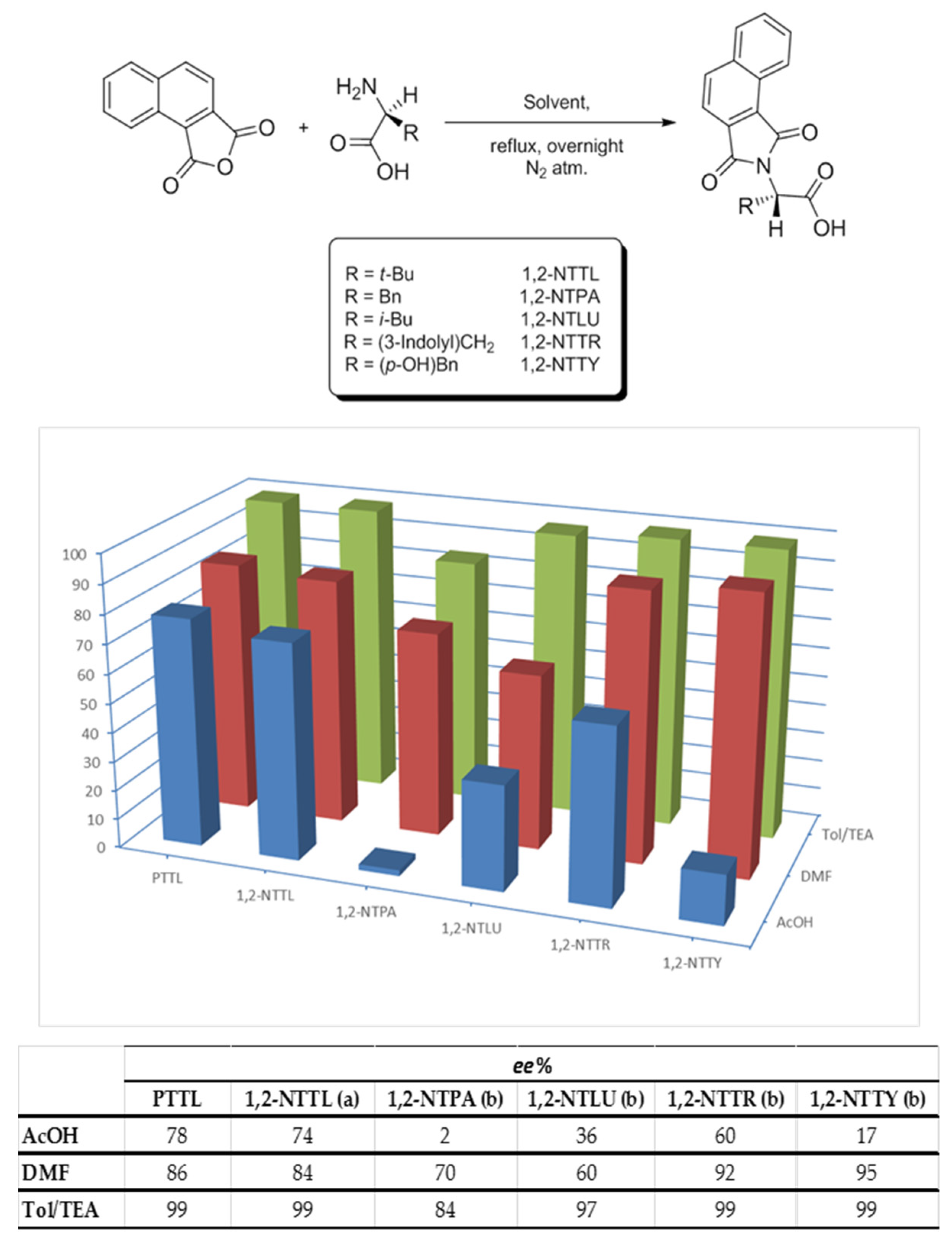
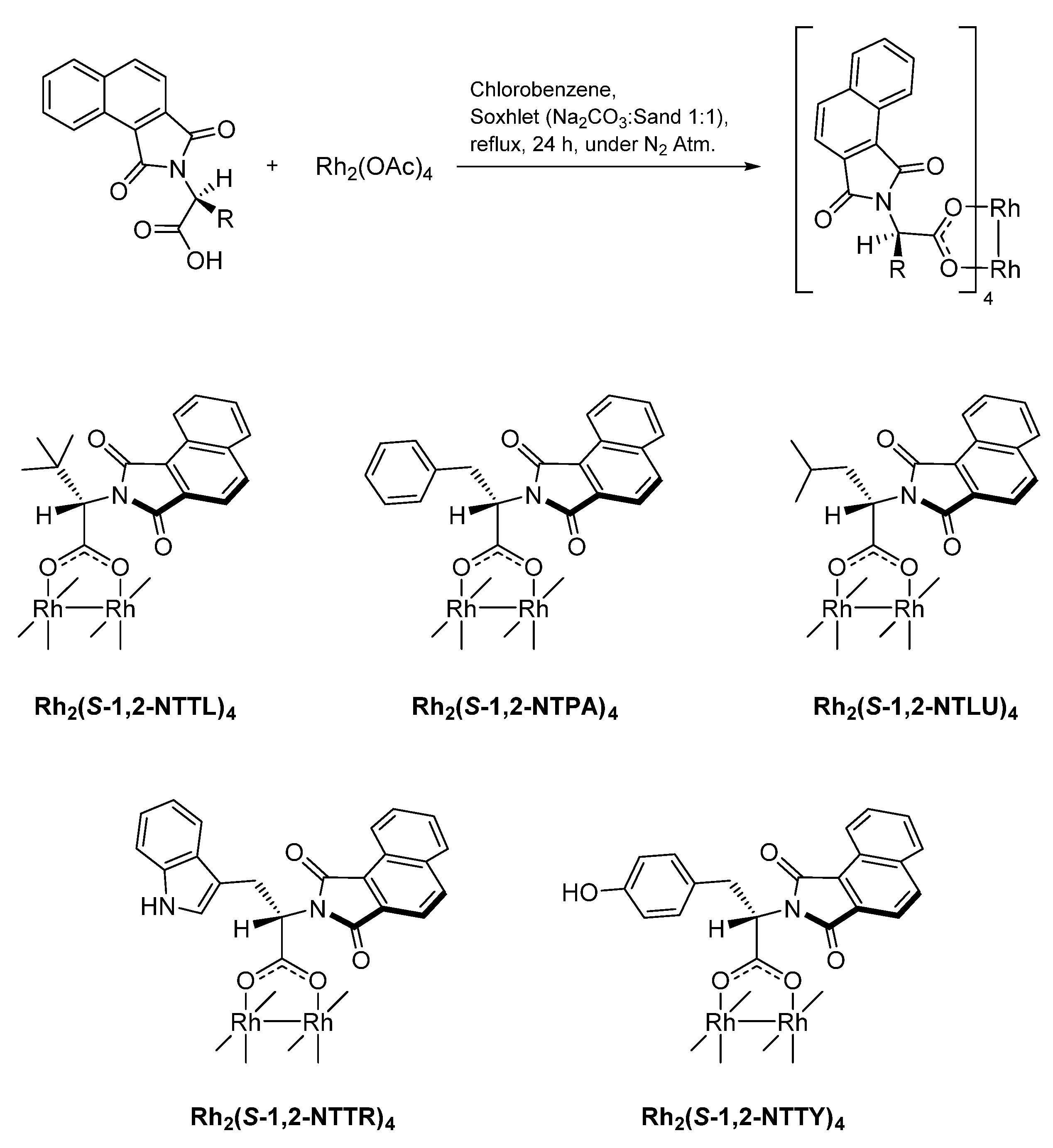
2. Results and Discussion
3. Materials and Methods
3.1. Chemicals
3.2. Instruments
3.3. HPLC Analysis
3.4. X-ray Crystallography for Dirhodium(II) Complexes
3.5. General Procedure for Ligand Preparation
3.6. Reaction Workup Procedures
- (a)
- When using acetic acid as solventThe reaction solvent was evaporated in vacuo and the residue was directly purified on silica gel column chromatography using ethyl acetate: n-hexane as an eluent to afford the desired product.
- (b)
- When using DMF as solventThe reaction mixture was diluted in water and extracted with ethyl acetate twice. The organic layer was washed with water three times, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was then purified on silica gel column chromatography using ethyl acetate: n-hexane as an eluent to afford the desired product.
- (c)
- When using Toluene/TEA as solventThe mixture was diluted with ethyl acetate, washed twice with 0.1 M hydrochloric acid solution, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was then purified on silica gel column chromatography using ethyl acetate: n-hexane as an eluent to afford the desired product.
3.7. N-(Phthaloyl)-Tert-Leucine (PTTL)
3.8. N-(1,2-Naphthaloyl)-Tert-Leucine (1,2-NTTL)
3.9. N-(1,2-Naphthaloyl)-Phenylalanine (1,2-NTPA)
3.10. N-(1,2-Naphthaloyl)-Leucine (1,2-NTLU)
3.11. N-(1,2-Naphthaloyl)-Tryptophan (1,2-NTTR)
3.12. N-(1,2-Naphthaloyl)-Tyrosine (1,2-NTTY)
3.13. General Procedure for Ligand Exchange
3.14. General Procedure for Cyclopropanation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Adly, F.G.; Ghanem, A. Enantiomerically pure compounds by enantioselective synthetic chiral metal complexes. In Asymmetric Synthesis of Drugs and Natural Products; Nag, A., Ed.; CRC Press: Raton, FL, USA, 2018. [Google Scholar]
- Adly, F.G.; Ghanem, A. Chiral dirhodium(II) carboxylates and carboxamidates as effective chemzymes in asymmetric synthesis of three-membered carbocycles. Chirality 2014, 26, 692–711. [Google Scholar] [CrossRef] [PubMed]
- El-Deftar, M.; Adly, F.G.; Gardiner, M.G.; Ghanem, A. Chiral dirhodium catalysts: A new era for asymmetric catalysis. Curr. Org. Chem. 2012, 16, 1808–1836. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Manning, J.R. Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Qiu, H.; D Srinivas, H.; P Doyle, M. Chiral dirhodium(II) catalysts for selective metal carbene reactions. Curr. Org. Chem. 2016, 20, 61–81. [Google Scholar] [CrossRef]
- Trindade, A.F.; Coelho, J.A.S.; Afonso, C.A.M.; Veiros, L.F.; Gois, P.M.P. Fine tuning of dirhodium(II): Expolring the axial modification. ACS Catal. 2012, 2, 370–383. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Morton, D. Guiding principles for site selective and stereoselective intermolecular C-H functionalization by donor/acceptor rhodium carbenes. Chem. Soc. Rev. 2011, 40, 1857–1869. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Bois, J.D.; Yu, J.-Q. C-H functionalization in organic synthesis. Chem. Soc. Rev. 2011, 40, 1855–1856. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; Duffy, R.; Ratnikov, M.; Zhou, L. Catalytic carbene insertion into C–H bonds. Chem. Rev. 2010, 110, 704–724. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Hedley, S.J. Intermolecular reactions of electron-rich heterocycles with copper and rhodium carbenoids. Chem. Soc. Rev. 2007, 36, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Merlic, C.A.; Zechman, A.L. Selectivity in rhodium(II) catalyzed reactions of diazo compounds: Effects of catalyst electophilicity, diazo substitution, and substrate substitution. From chemoselectivity to enantioselectivity. Synthesis 2003, 34, 1137–1156. [Google Scholar] [CrossRef]
- Colacot, T.J. An overview on the application of “doyle catalysts” in asymmetric cyclopropanation, cyclopropenation and C-H insertion reactions. Proc. Indian Acad. Sci. (J. Chem. Sci.) 2000, 11, 197–207. [Google Scholar] [CrossRef]
- Liao, K.; Negretti, S.; Musaev, D.G.; Bacsa, J.; Davies, H.M.L. Site-selective and stereoselective functionalization of unactivated C–H bonds. Nature 2016, 533, 230. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.; Yang, H.; Ellis-Guardiola, K.; Lewis, J.C. Engineering a dirhodium artificial metalloenzyme for selective olefin cyclopropanation. Nat. Commun. 2015, 6, 7789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-J.; Yan, M.; Huang, D. Catalyzed addition of diazoacetoacetates to imines: Synthesis of highly functionalized aziridines. Org. Biomol. Chem. 2009, 7, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Yamawaki, M.; Tanaka, M.; Abe, T.; Anada, M.; Hashimoto, S. Catalytic enantioselective aziridination of alkenes using chiral dirhodium(II) carboxylates. Heterocycles 2007, 72, 709–721. [Google Scholar] [CrossRef]
- Catino, A.J.; Nichols, J.M.; Forslund, R.E.; Doyle, M.P. Efficient aziridination of olefins catalyzed by mixed-valent dirhodium(II,III) caprolactamate. Org. Lett. 2005, 7, 2787–2790. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Denton, J.R. Application of donor/acceptor-carbenoids to the synthesis of natural products. Chem. Soc. Rev. 2009, 38, 3061–3071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubiak, R.W.; Mighion, J.D.; Wilkerson-Hill, S.M.; Alford, J.S.; Yoshidomi, T.; Davies, H.M. Enantioselective intermolecular C-H functionalization of allylic and benzylic sp3 C–H bonds using n-sulfonyl-1, 2, 3-triazoles. Org. Lett. 2016, 18, 3118–3121. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P. Perspective on dirhodium carboxamidates as catalysts. J. Org. Chem. 2006, 71, 9253–9260. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; McKervey, M.A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; Wiley: New York, NY, USA, 1998. [Google Scholar]
- Hodgson, D.M.; Stupple, P.A.; Pierard, F.Y.T.M.; Labande, A.H.; Johnstone, C. Development of dirhodium(II)-catalyzed generation and enantioselective 1,3-dipolar cycloaddition of carbonyl ylides. Chem. Eur. J. 2001, 7, 4465–4476. [Google Scholar] [CrossRef]
- Doyle, M.P.; Forbes, D.C.; Vasbinder, M.M.; Peterson, C.S. Enantiocontrol in the generation and diastereoselective reactions of catalytically generated oxonium and iodonium ylides. Metal-stabilized ylides as reaction intermediates. J. Am. Chem. Soc. 1998, 120, 7653–7654. [Google Scholar] [CrossRef]
- Doyle, M.P.; Hu, W. Macrocycle formation from catalytic metal carbene transformations. Synlett 2001, 2001, 1364–1370. [Google Scholar] [CrossRef]
- Doyle, M.P.; Phillips, I.M.; Hu, W. A new class of chiral lewis acid catalysts for highly enantioselective hetero-diels-alder reactions: Exceptionally high turnover numbers from dirhodium(II) carboxamidates. J. Am. Chem. Soc. 2001, 123, 5366–5367. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; Valenzuela, M.; Huang, P. Asymmetric hetero-diels-alder reaction catalyzed by dirhodium(II) carboxamidates. Proc. Natl. Acad. Sci. USA 2004, 101, 5391–5395. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wolf, J.; Zavalij, P.; Doyle, M.P. Cationic chiral dirhodium carboxamidates are activated for lewis acid catalysis. Angew. Chem. Int. Ed. 2008, 47, 1439–1442. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Shimada, N.; Anada, M.; Hashimoto, S. Enantio- and diastereoselective hetero-diels-alder reactions between 4-methyl-substituted rawal’s diene and aldhydes catalyzed by chiral dirhodium(II) carboxamidates: Catalytic asymmetric synthesis of (-)-cis-aerrangis lactone. Tetrahedron: Asymmetry 2014, 25, 63–73. [Google Scholar] [CrossRef]
- Anada, M.; Washio, T.; Shimada, N.; Kitagaki, S.; Nakajima, M.; Shiro, M.; Hashimoto, S. A new dirhodium(II) carboxamidate complex as a chiral lewis acid catalyst for enantioselective heteo-diels-alder reactions. Angew. Chem. Int. Ed. 2004, 43, 2665–2668. [Google Scholar] [CrossRef]
- Hansen, J.H.; Parr, B.T.; Pelphrey, P.; Jin, Q.; Autschbach, J.; Davies, H.M.L. Rhodium(II)-catalyzed cross-coupling of diazo compounds. Angew. Chem. Int. Ed. 2011, 50, 2544–2548. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; Protopopova, M.N. New aspecs of catalytic asymmetric cyclopropanation. Tetrahedron 1998, 54, 7919–7946. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Antoulinakis, E.G. Intermolecular metal-catalyzed carbenoid cyclopropanations. Org. React. 2001, 57, 1–326. [Google Scholar]
- Lebel, H.; Marcoux, J.-F.; Molinaro, C.; Charette, A.B. Stereoselective cyclopropanation reactions. Chem. Rev. 2003, 103, 977–1050. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, J. Modern Rhodium-Catalyzed Organic Reactions; Wiley: Weinheim, Germany, 2005. [Google Scholar]
- Adly, F.G.; Gardiner, M.G.; Ghanem, A. Design and synthesis of novel chiral dirhodium(II) carboxylate complexes for asymmetric cyclopropanation reactions. Chem. Eur. J. 2016, 22, 3447–3461. [Google Scholar] [CrossRef] [PubMed]
- Adly, F.G.; Maddalena, J.; Ghanem, A. Rh2(S-1,2-NTTL)4: A novel Rh2(S-PTTL)4 analog with lower ligand symmetry for asymmetric synthesis of chiral cyclopropylphosphonates. Chirality 2014, 26, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.P.; Lee, G.H.; Davies, H.M.L. Dirhodium tetracarboxylate derived from adamantylglycine as chiral catalyst for carbenoid reactions. Org. Lett. 2006, 8, 3437–3440. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Bruzinski, P.R.; Fall, M.J. Effect of diazoalkane structure on the stereoselectivity of rhodium(II) (S)-N-(arylsulfonyl)prolinate catalyzed cyclopropanations. Tetrahedron Lett. 1996, 37, 4133–4136. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Panaro, S.A. Novel dirhodium tetraprolinate catalysts containing bridging prolinate ligands for asymmetric carbenoid reactions. Tetrahedron Lett. 1999, 40, 5287–5290. [Google Scholar] [CrossRef]
- Boruta, D.T.; Dmitrenko, O.; Yap, G.P.A.; Fox, J.M. Rh2(s-PTTL)3TPA—A mixed-ligand dirhodium(II) catalyst for enantioselective reactions of α-alkyl-α-diazoesters. Chem. Sci. 2012, 3, 1589–1593. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, V.N.G.; Charette, A.B. Design and synthesis of chiral heteroleptic rhodium(II) carboxylate catalysts: Experimental investigations of halogen bond rigidification effects in asymmetric cyclopropanation. ACS Catal. 2012, 2, 1221–1225. [Google Scholar] [CrossRef]
- Yamawaki, M.; Tsutsui, H.; Kitagaki, S.; Anada, M.; Hashimoto, S. Dirhodium(II) tetrakis[N-tetrachlorophthaloyl-(S)-tert-leucinate]: A new chiral Rh(II) catalyst for enantioselective amidation of C–H bonds. Tetrahedron Lett. 2002, 43, 9561–9564. [Google Scholar] [CrossRef]
- Hashimoto, S.; Watanabe, N.; Ikegami, S. Enantioselective intramolecular C-H insertion of α-diazo β-keto esters catalyzed by homochiral rhodium(II) carboxylates. Tetrahedron Lett. 1990, 31, 5173–5174. [Google Scholar] [CrossRef]
- Qin, C.; Boyarskikh, V.; Hansen, J.H.; Hardcastle, K.I.; Musaev, D.G.; Davies, H.M.L. D2-symmetric dirhodium catalyst derived from a 1,2,2-triarylcyclopropanecarboxylate ligand: Design, synthesis and application. J. Am. Chem. Soc. 2011, 133, 19198–19204. [Google Scholar] [CrossRef] [PubMed]
- Adly, F.G.; Ghanem, A. Polymer monolith-supported dirhodium(II)-catalyzed continuous flow cyclopropanation in capillary format. Tetrahedron Lett. 2016, 57, 852–857. [Google Scholar] [CrossRef]
- Adly, F.G. On the structure of chiral dirhodium(II) carboxylate catalysts: Stereoselectivity relevance and insights. Catalysts 2017, 7, 347. [Google Scholar] [CrossRef]
- DeAngelis, A.; Boruta, D.T.; Lubin, J.-B.; Plampin, J.N.; Yap, G.P.A.; Fox, J.M. The chiral crown conformation in paddlewheel complexes. Chem. Commun. 2010, 46, 4541–4543. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, A.; Dmitrenko, O.; Yap, G.P.A.; Fox, J.M. Chiral crown conformation of Rh2(PTTL)4: Enantioselective cyclopropanation with α-alkyl-α-diazoesters. J. Am. Chem. Soc. 2009, 131, 7230–7231. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.; Gardiner, M.G.; Williamson, R.M.; Müller, P. First X-ray structure of a N-naphthaloyl-tethered chiral dirhodium(II) complex: Structural basis for tether substitution improving asymmetric control in olefin cyclopropanation. Chem. Eur. J. 2010, 16, 3291–3295. [Google Scholar] [CrossRef] [PubMed]
- Mattiza, J.T.; Fohrer, J.G.G.; Duddeck, H.; Gardiner, M.G.; Ghanem, A. Optimizing dirhodium(II) tetrakiscarboxylates as chiral nmr auxiliaries. Org. Biomol. Chem. 2011, 9, 6542–6550. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, V.N.G.; Lin, W.; Charette, A.B. Experimental evidence for the all-up reactive conformation of chiral rhodium(II) carboxylate catalysts: Enantioselective synthesis of cis-cyclopropane α-amino acids. J. Am. Chem. Soc. 2009, 131, 16383–16385. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, T.; Abraham, S.; Kagan, H.B. Nonlinear effects in asymmetric catalysis. Angew. Chem. Int. Ed. 2009, 48, 456–494. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Abe, T.; Nakamura, S.; Anada, M.; Hashimoto, S. Practical synthesis of dirhodium(II) tetrakis[N-phthaloyl-(S)-tert-leucinate]. Chem. Pharm. Bull. 2005, 53, 1366–1368. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.; Aboul-Enein, H.Y.; Müller, P. One-pot synthesis and chiral analysis of cyclopropane derivatives. Chirality 2005, 17, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Kitagaki, S.; Anada, M.; Kataoka, O.; Matsuno, K.; Umeda, C.; Watanabe, N.; Hashimoto, S. Enantiocontrol in tandem carbonyl ylide formation and intermolecular 1,3-dipolar cycloaddition of α-diazo ketones mediated by chiral dirhodium(II) carboxylate catalyst. J. Am. Chem. Soc. 1999, 121, 1417–1418. [Google Scholar] [CrossRef]
- Yao, G.; Dai, W.; Ye, M.; Huang, R.; Pan, Y.-M.; Liao, Z.-X.; Wang, H.-S. Synthesis and antitumor properties of novel alizarin analogs. Med. Chem. Res. 2014, 23, 5031–5042. [Google Scholar] [CrossRef]
- Ghanem, A.; Lacrampe, F.; Aboul-Enein, H.Y.; Schurig, V. Diazo compounds and phenyliodonium ylides in inter- and intramolecular cyclopropanations catalyzed by dirhodium(II). Synthesis and chiral resolution by GC versus HPLC. Monatsh. Chem. 2005, 136, 1205–1219. [Google Scholar] [CrossRef]
- Adly, F.G.; Gardiner, M.G.; Ghanem, A. Unpublished X-ray Crystal Structure of Rh2(NTTY)4. The obtained X-ray crystal structure of Rh2(NTTY)4 revealed a polymeric structure where some of the p-OH groups of the –CH2C6H4–OH substituents of one complex molecule acts as the axial ligands on each Rh centre of adjacent complex molecules, 2014.
- McPhillips, T.M.; McPhillips, S.E.; Chiu, H.J.; Cohen, A.E.; Deacon, A.M.; Ellis, P.J.; Garman, E.; Gonzalez, A.; Sauter, N.K.; Phizackerley, R.P.; et al. Blu-Ice and the Distributed Control System: Software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 2002, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Shelx-97, Programs for Crystal Structure Analysis; Universität of Göttingen: Göttingen, Germany, 1998. [Google Scholar]
- Barbour, L.J. X-seed—A software tool for supramolecular crystallography. J. Supramol. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst Used | Product Yield (%) 1 |
|---|---|---|
| 1 | Rh2(S-PTTL)4 | 45 |
| 2 | Rh2(S-1,2-NTTL)4 | 60 |
| 3 | Rh2(S-1,2-NTPA)4 | 47 |
| 4 | Rh2(S-1,2-NTLU)4 | 54 |
| 5 | Rh2(S-1,2-NTTR)4 | 18 |
| 6 | Rh2(S-1,2-NTTY)4 | 14 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adly, F.G.; Bollard, H.; Gardiner, M.G.; Ghanem, A. Chiral Dirhodium(II) Carboxylates: New Insights into the Effect of Ligand Stereo-Purity on Catalyst Structure and Enantioselectivity. Catalysts 2018, 8, 268. https://doi.org/10.3390/catal8070268
Adly FG, Bollard H, Gardiner MG, Ghanem A. Chiral Dirhodium(II) Carboxylates: New Insights into the Effect of Ligand Stereo-Purity on Catalyst Structure and Enantioselectivity. Catalysts. 2018; 8(7):268. https://doi.org/10.3390/catal8070268
Chicago/Turabian StyleAdly, Frady G., Hannah Bollard, Michael G. Gardiner, and Ashraf Ghanem. 2018. "Chiral Dirhodium(II) Carboxylates: New Insights into the Effect of Ligand Stereo-Purity on Catalyst Structure and Enantioselectivity" Catalysts 8, no. 7: 268. https://doi.org/10.3390/catal8070268
APA StyleAdly, F. G., Bollard, H., Gardiner, M. G., & Ghanem, A. (2018). Chiral Dirhodium(II) Carboxylates: New Insights into the Effect of Ligand Stereo-Purity on Catalyst Structure and Enantioselectivity. Catalysts, 8(7), 268. https://doi.org/10.3390/catal8070268