Direct Catalytic Conversion of CO2 to Cyclic Organic Carbonates under Mild Reaction Conditions by Metal—Organic Frameworks


Abstract
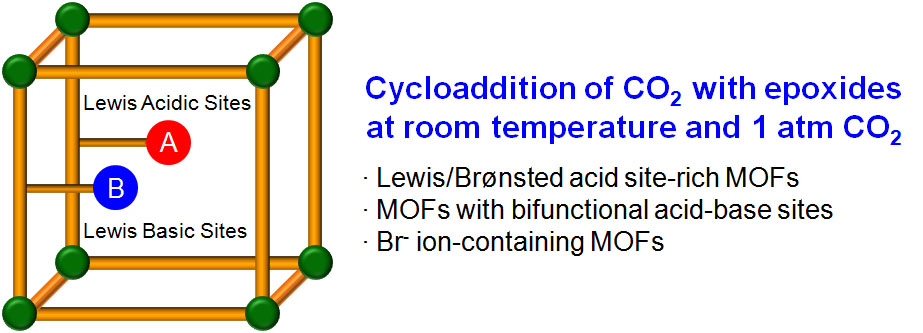
: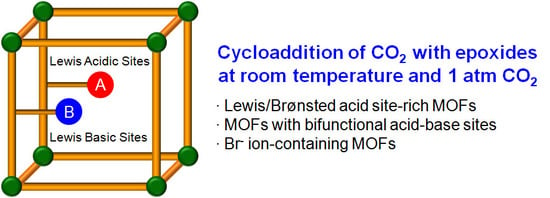
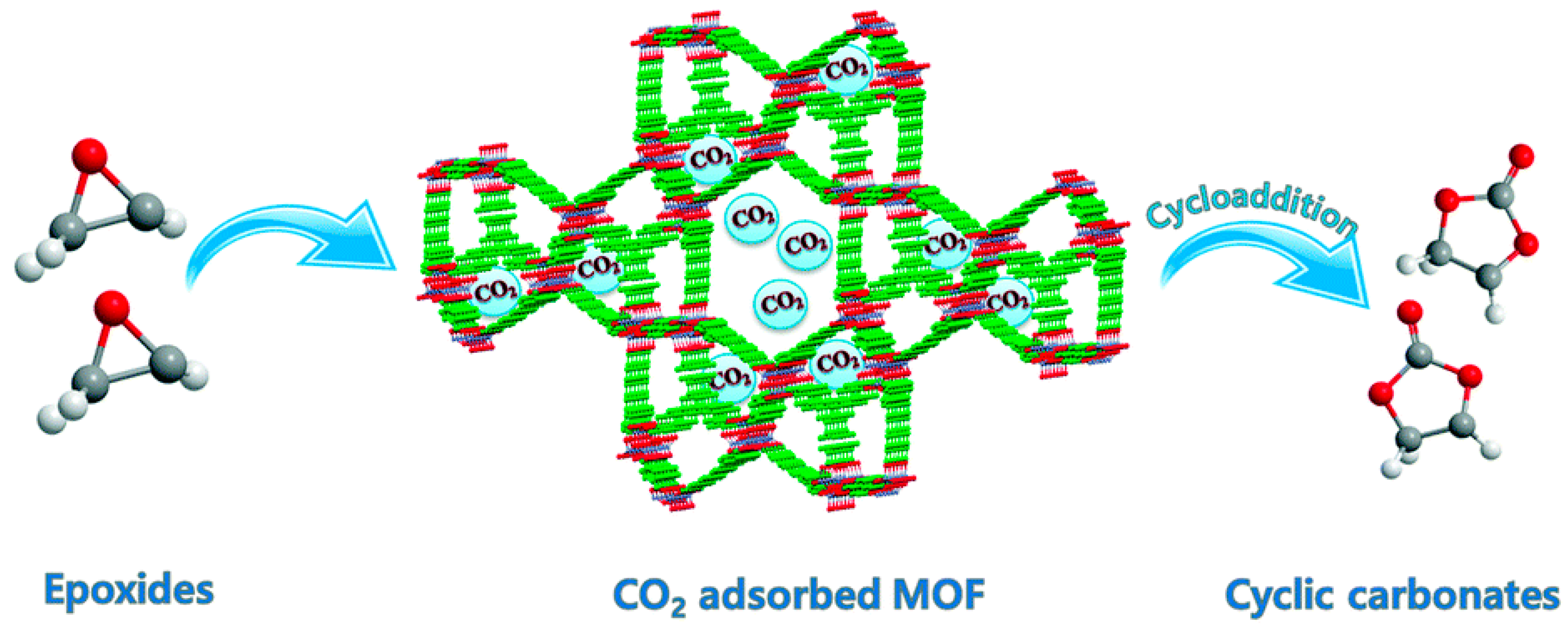
1. Introduction
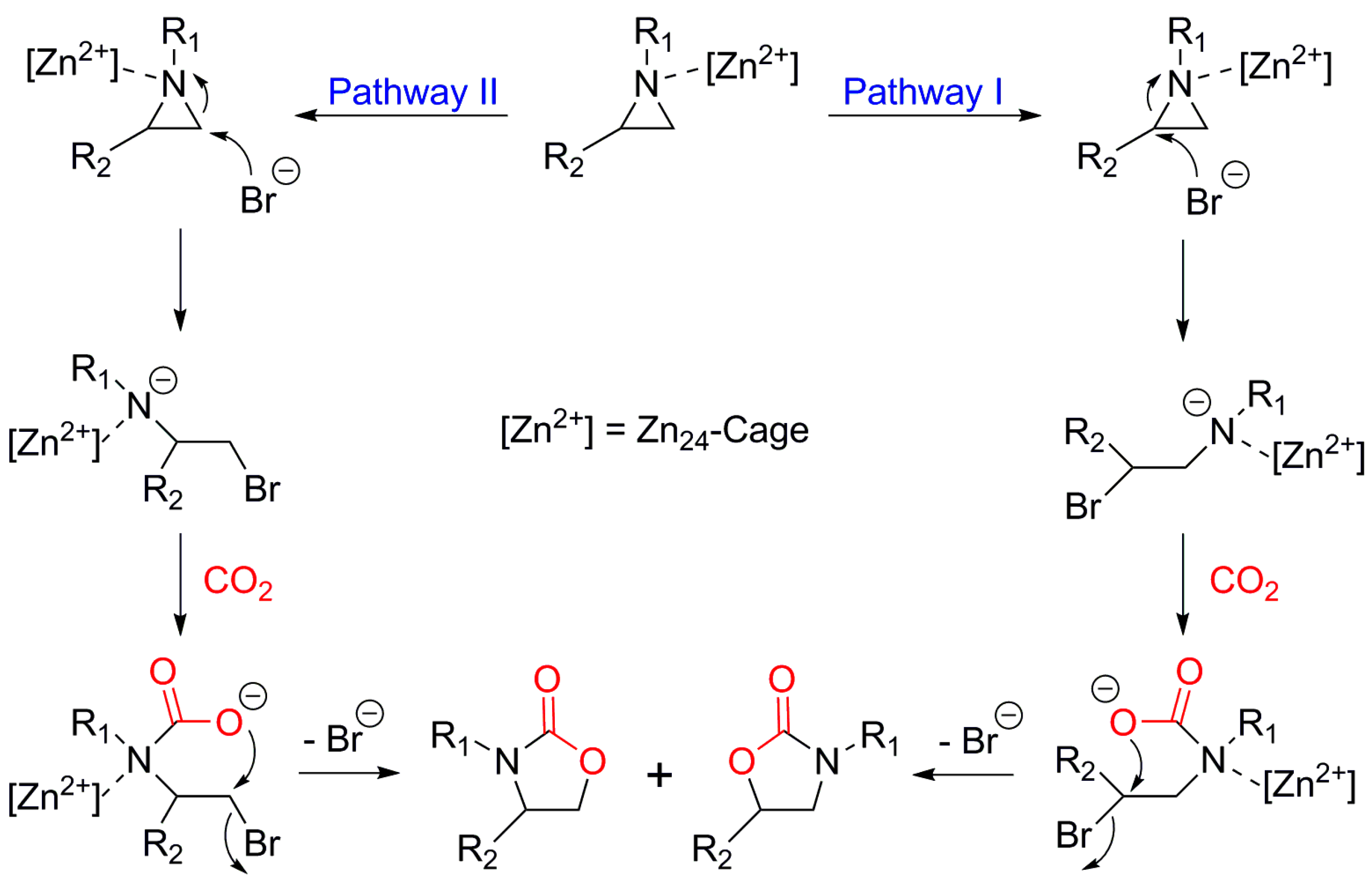
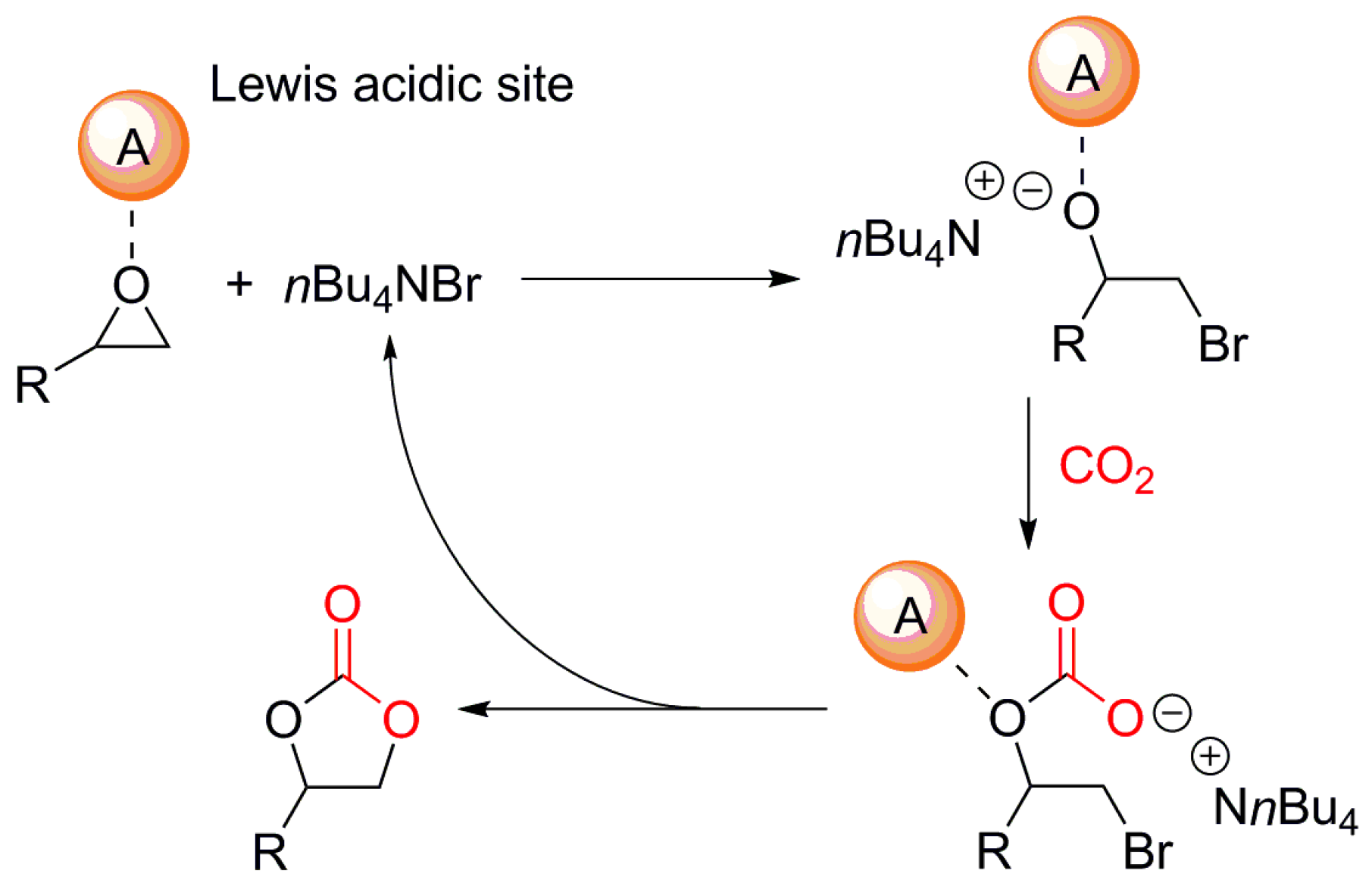
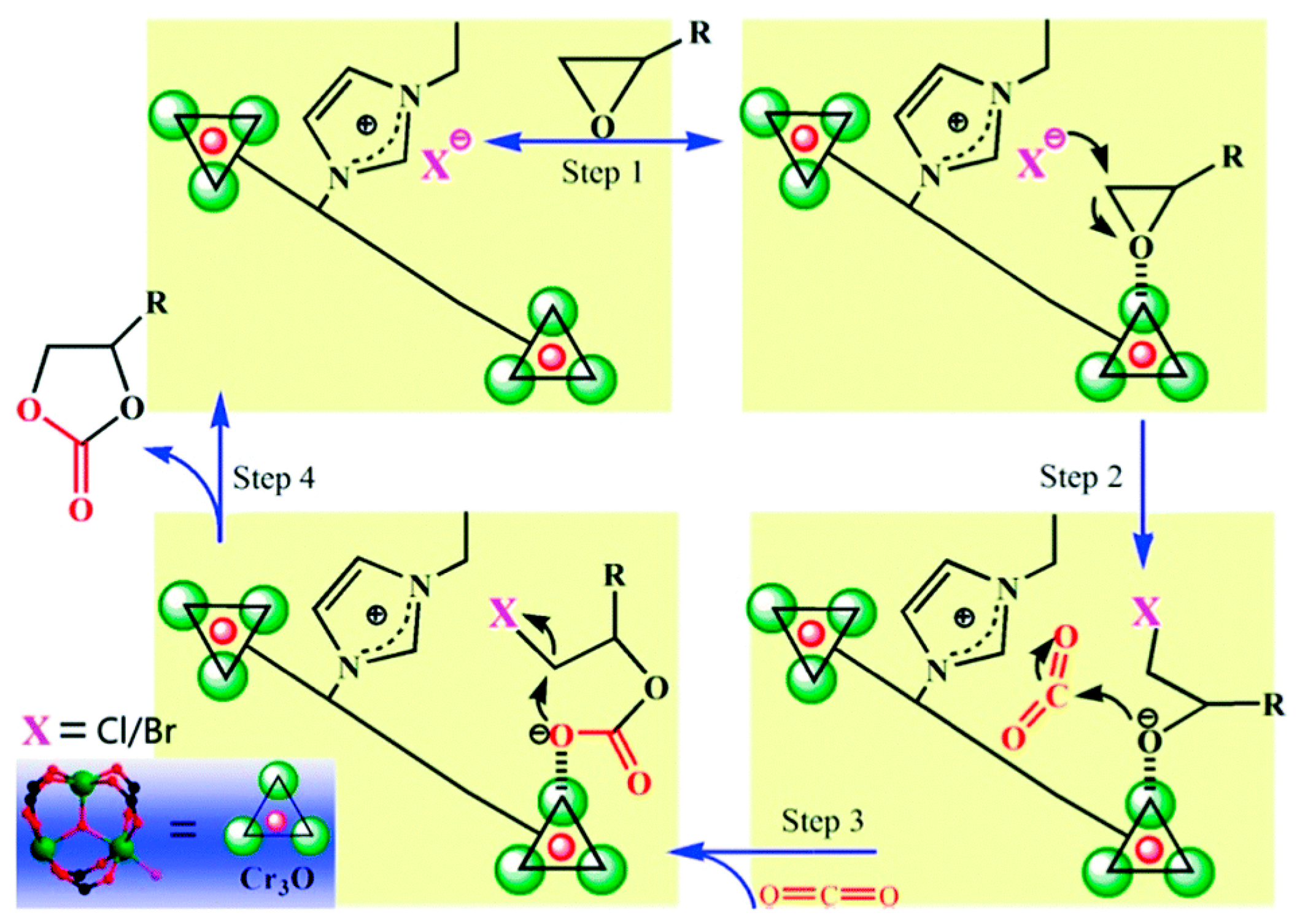
2. Mechanism of the Cycloaddition of CO2 with Epoxides
- (i)
- Lewis acid catalyst activates the epoxide.
- (ii)
- Nucleophilic attack by a cocatalyst (usually Br− ion from TBABr) to lead to an alkoxide.
- (iii)
- The intermediate alkoxide reacts with CO2 to provide the required cyclic carbonate.
3. MOF-Based Catalytic Systems
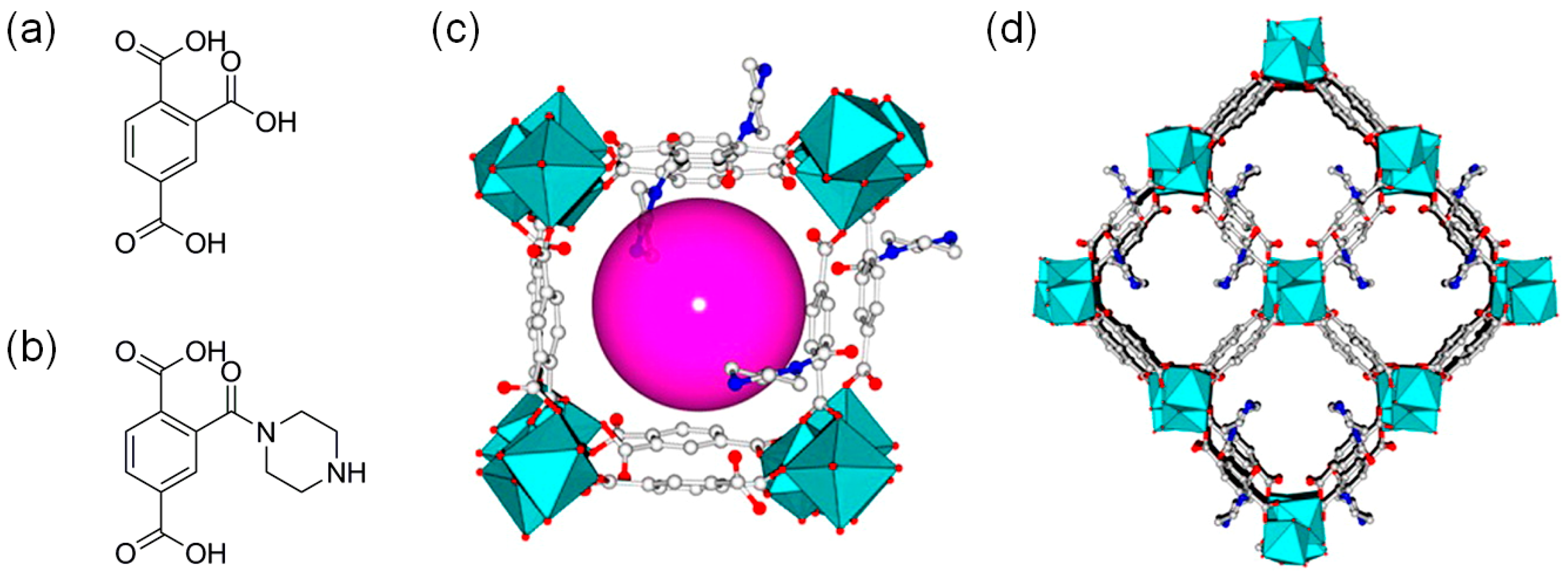
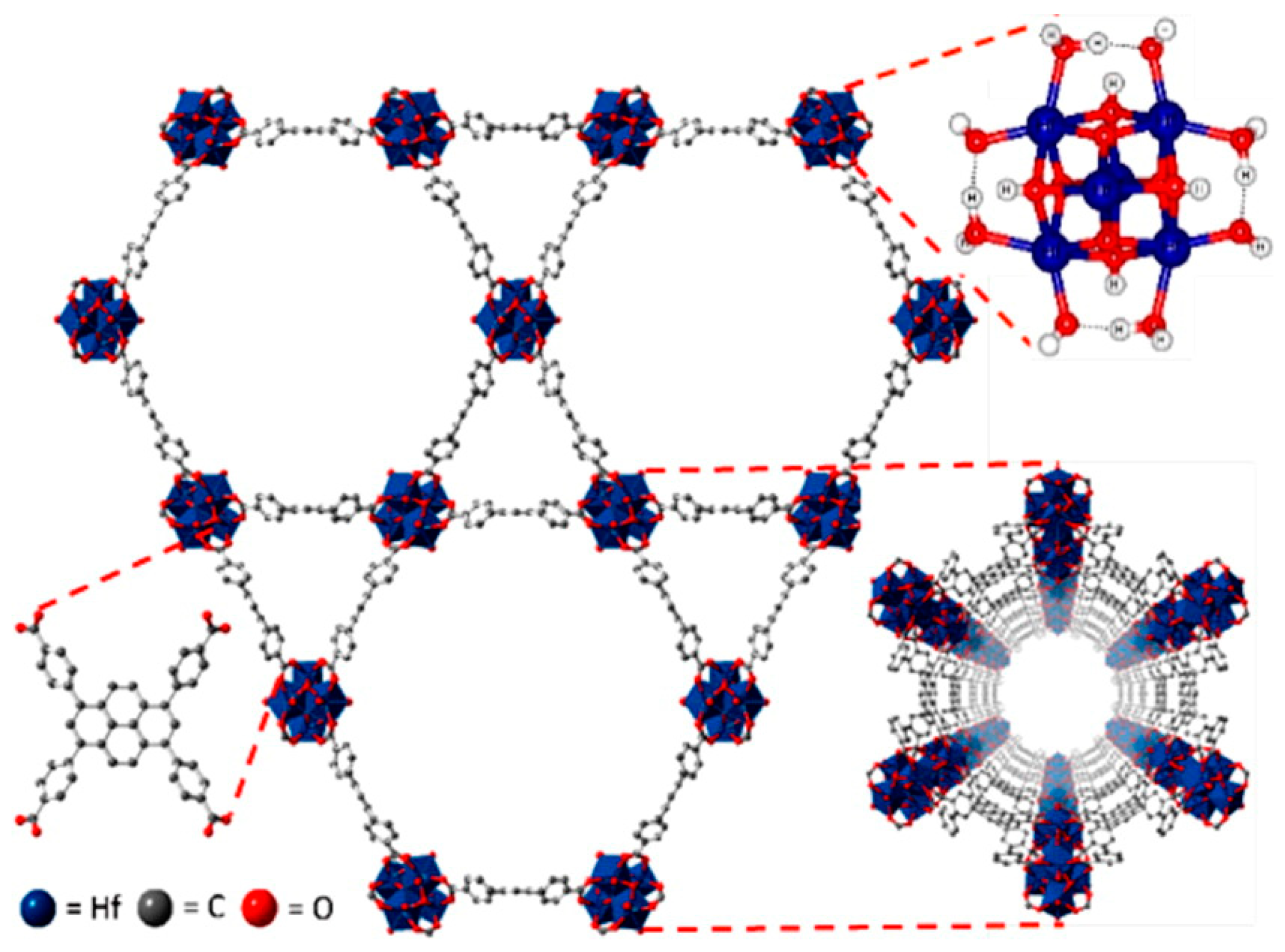
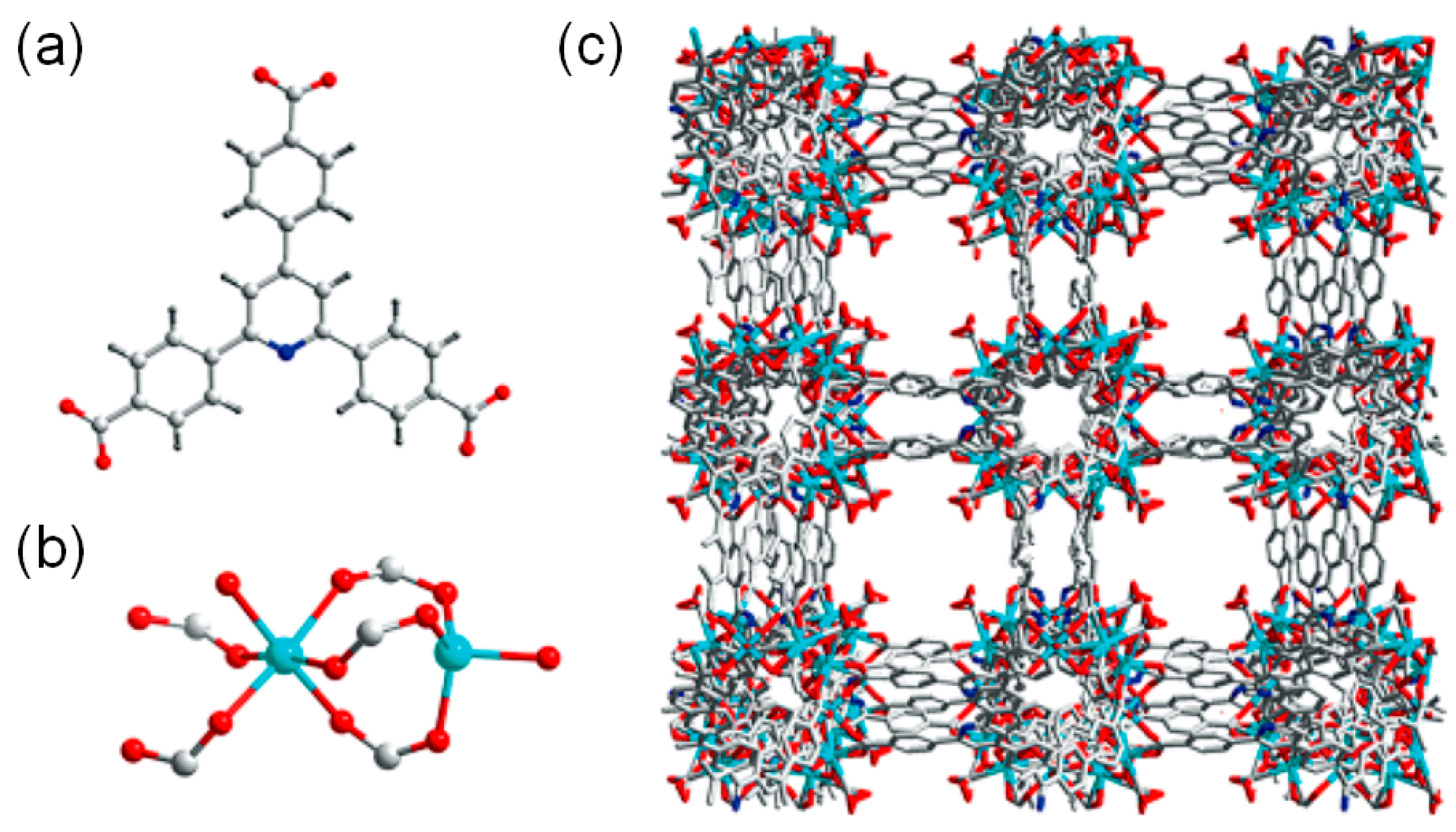
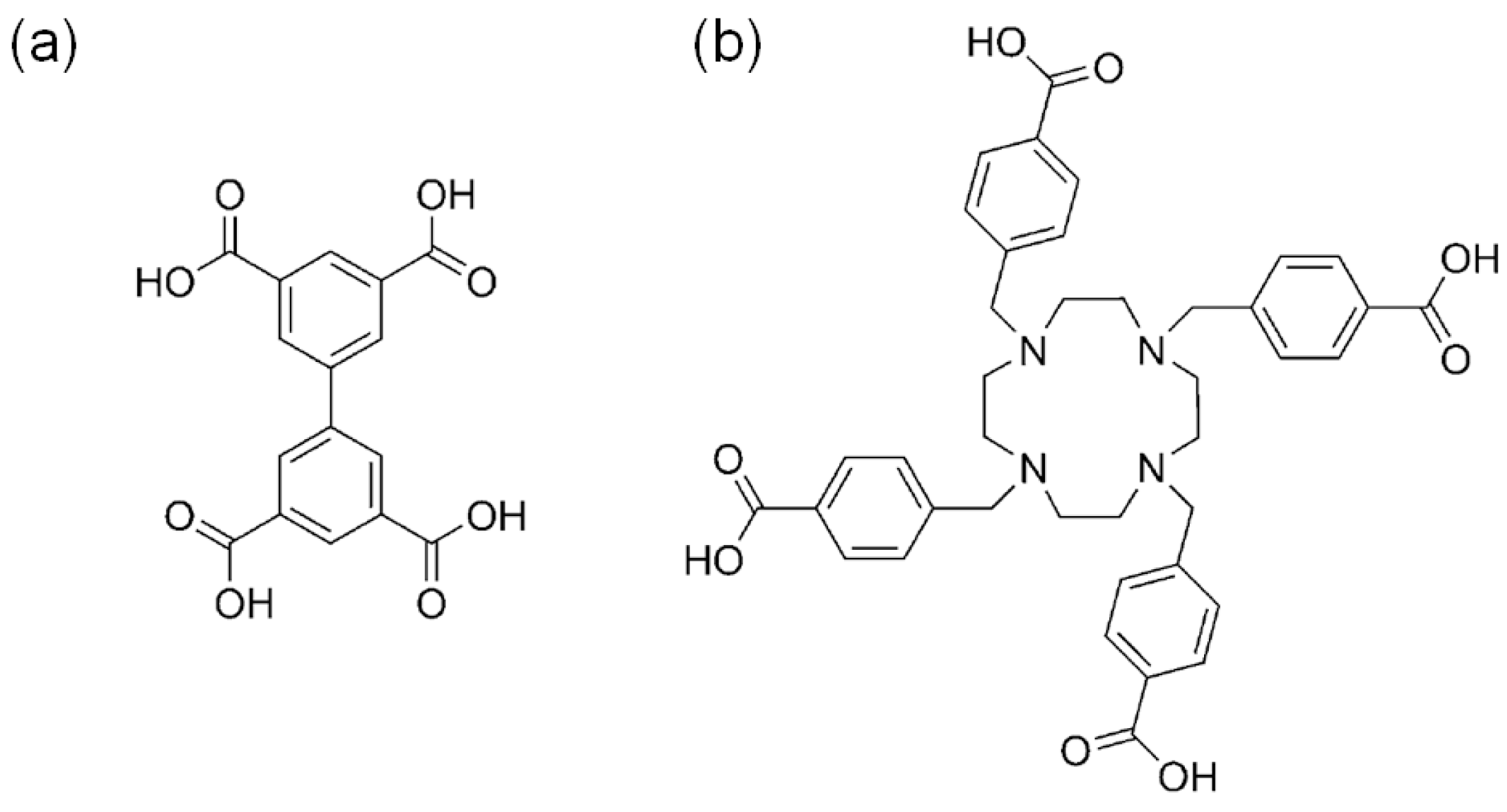
3.1. Acid Site-Rich MOF Catalytic Systems
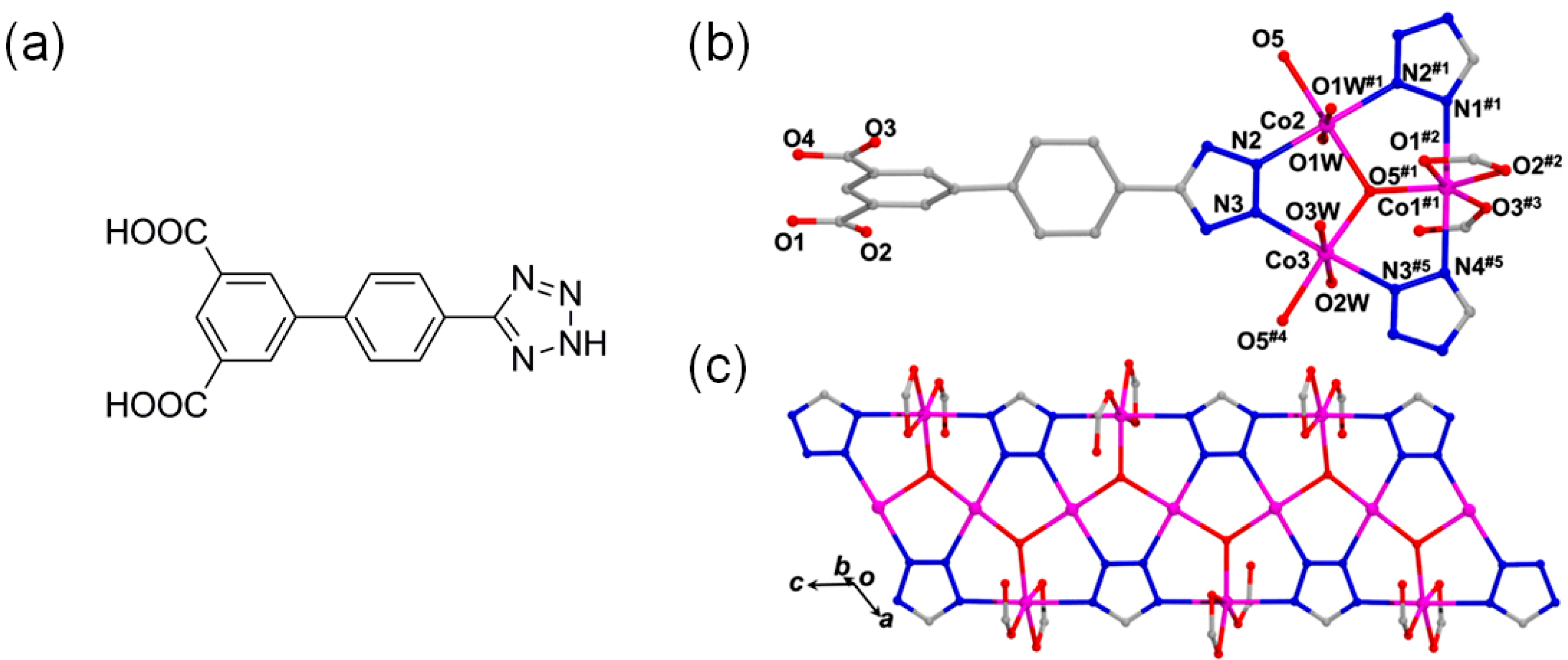
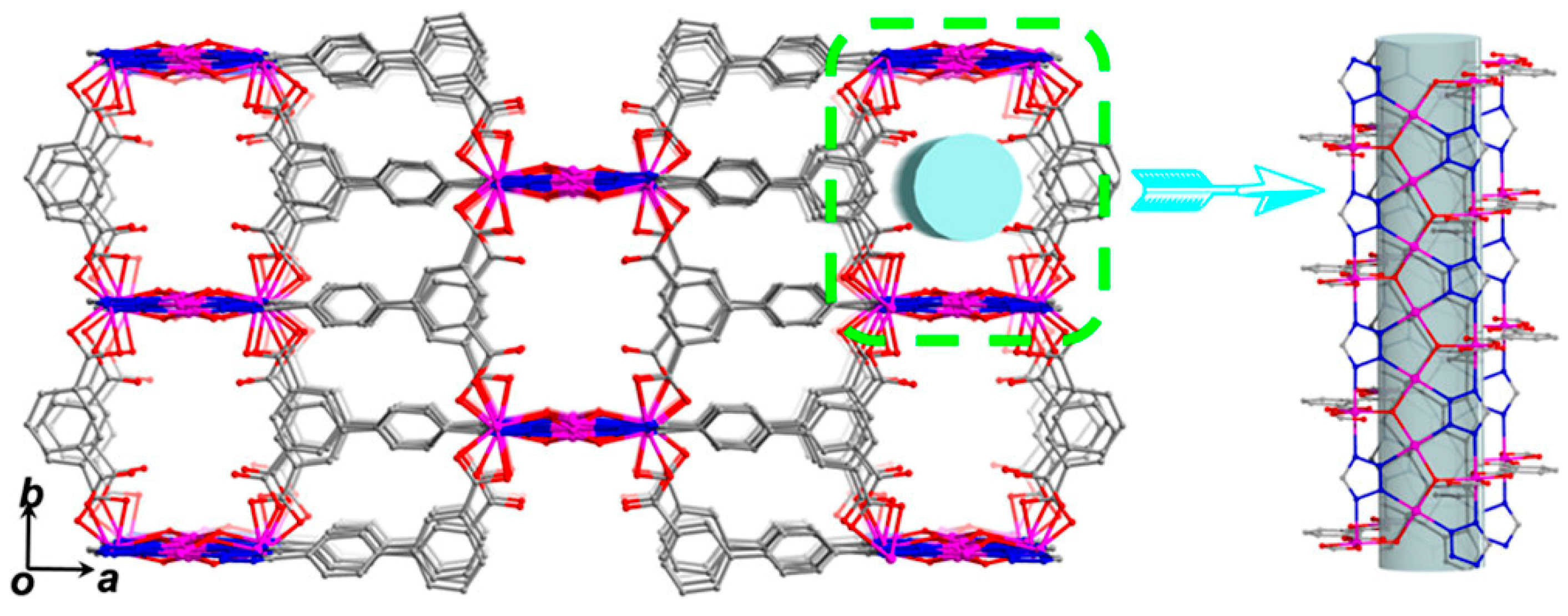
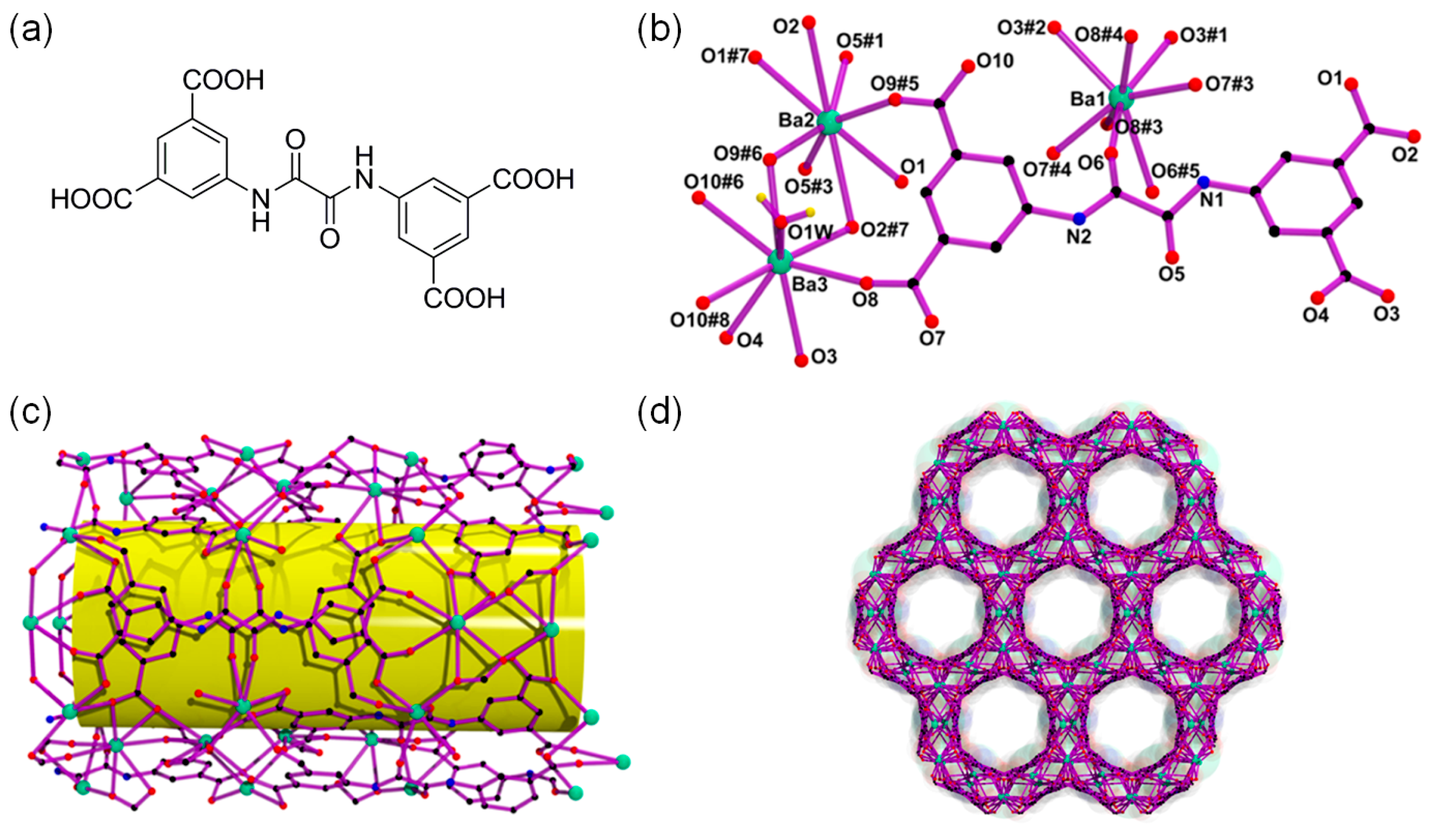
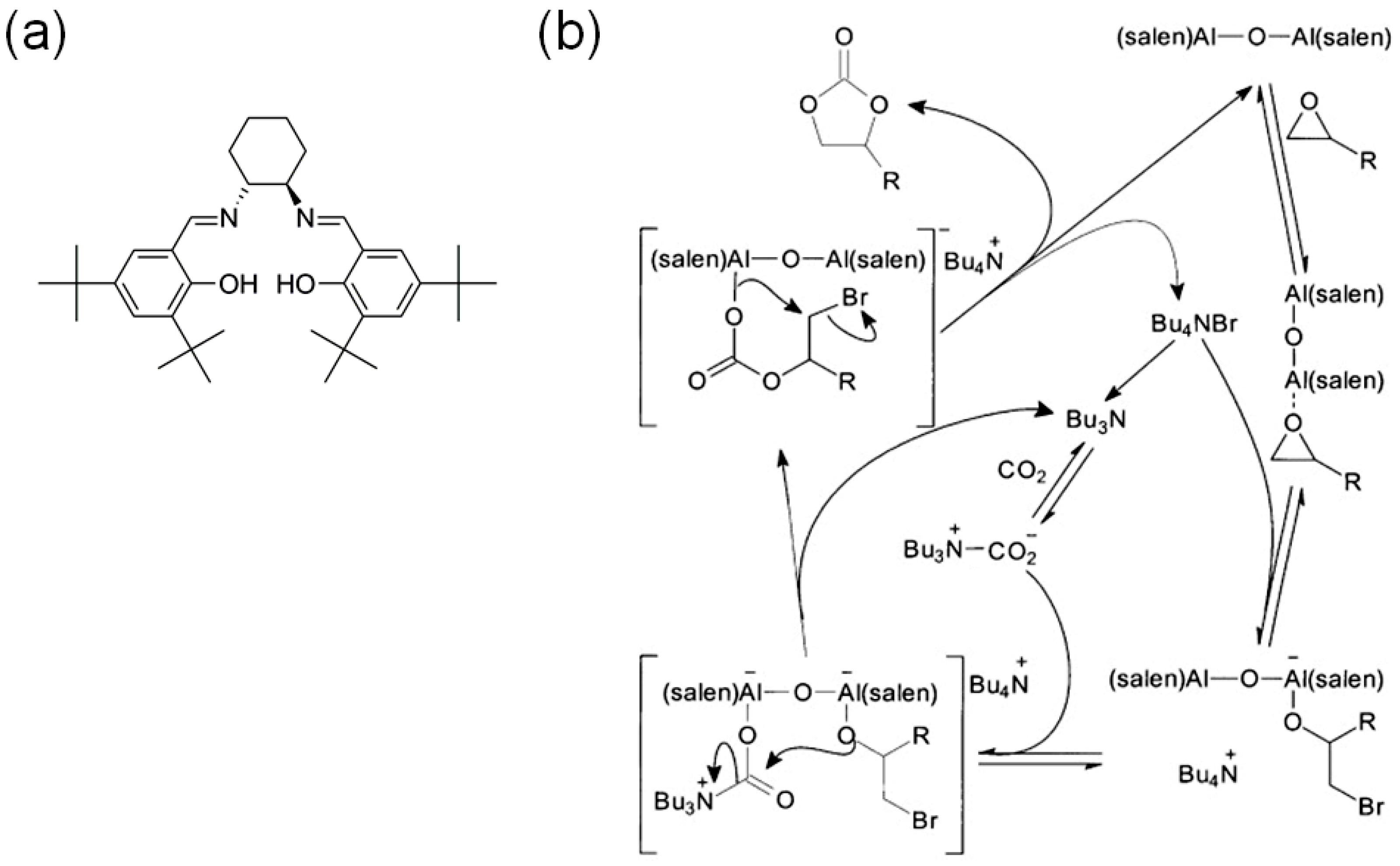
3.2. Cooperative Bifunctional Acid-Base Catalytic Systems
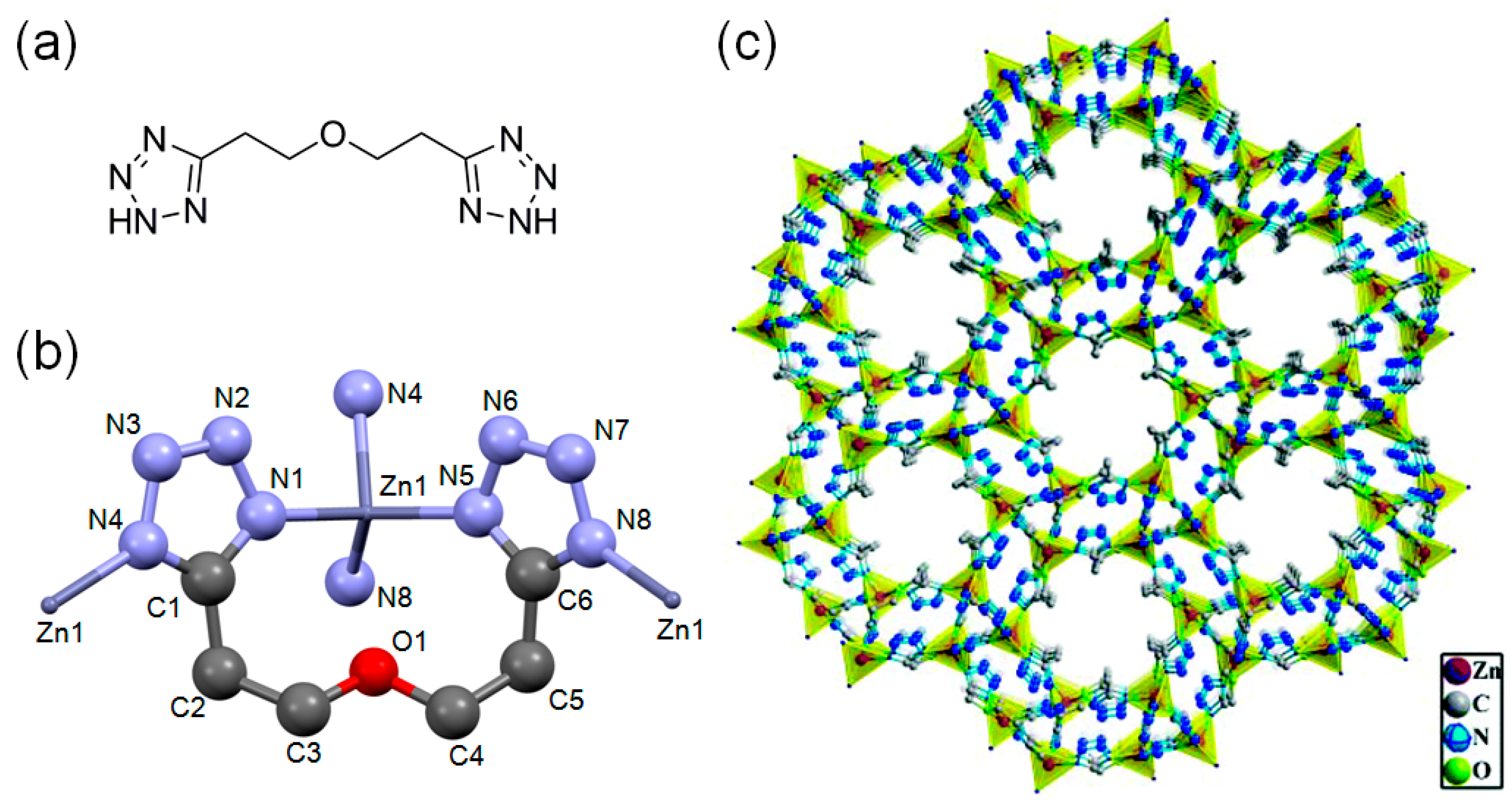
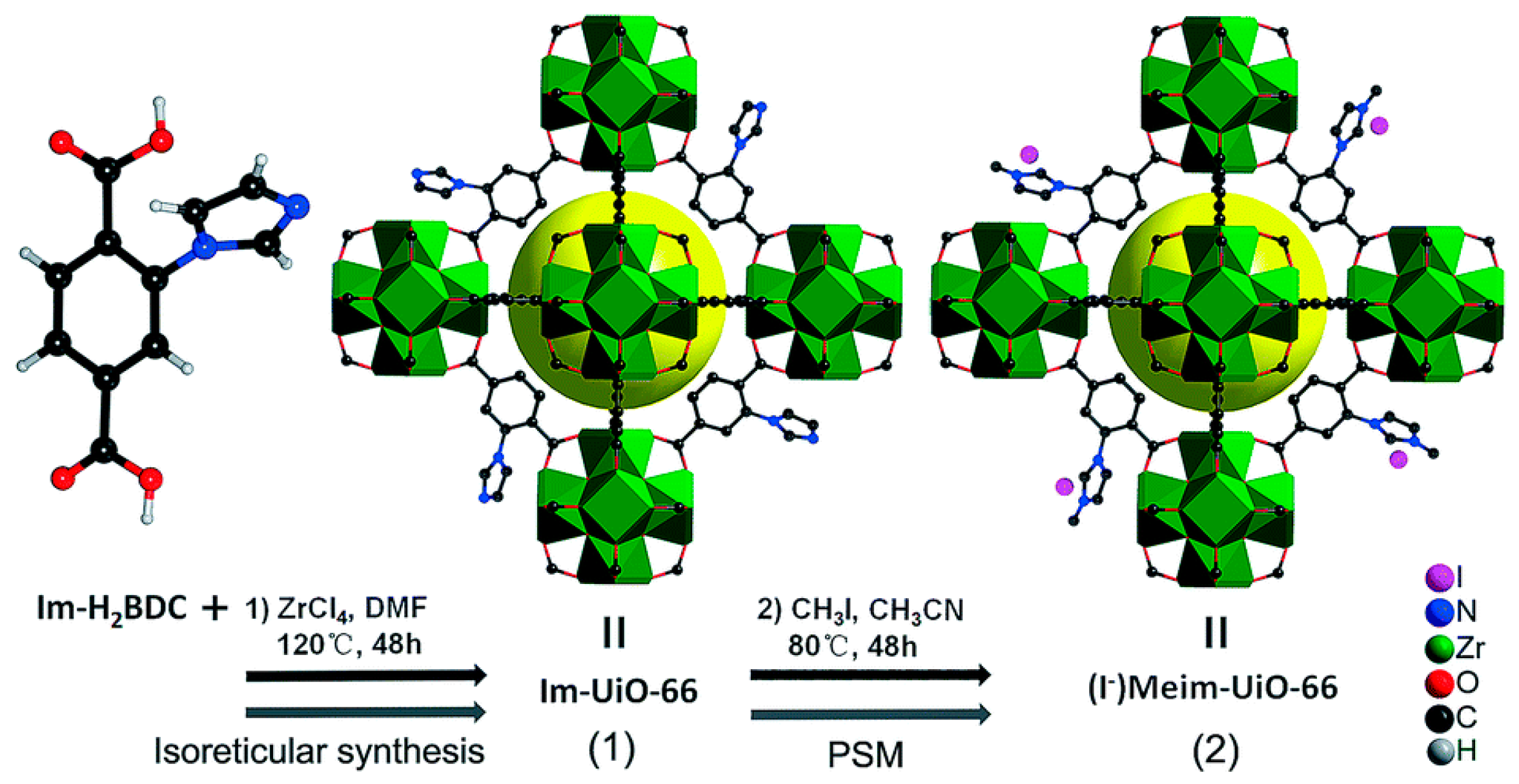
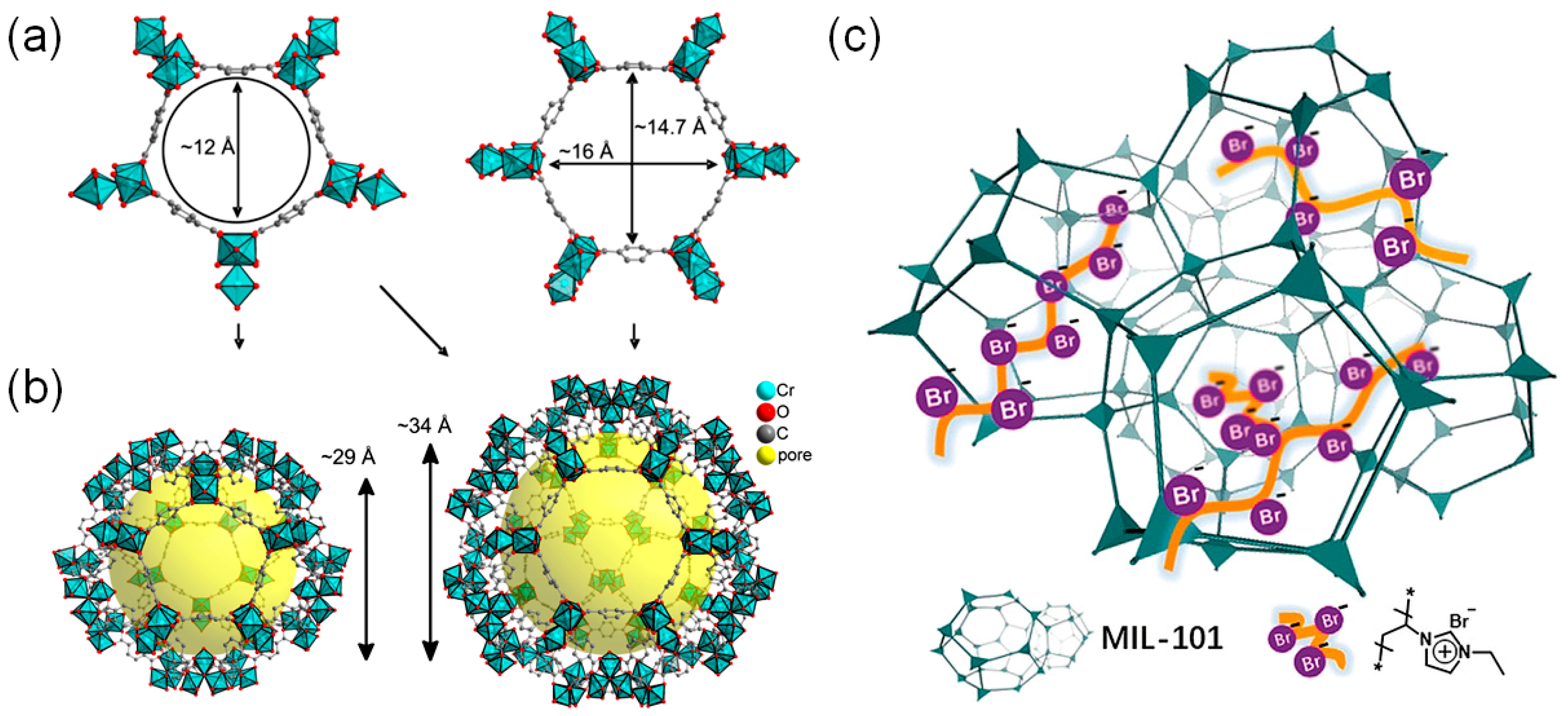
3.3. Direct Incorporation of Br- Nucleophile into MOF Channels
4. Conclusions and Prospects
- (i)
- MOFs with rich Lewis/Brønsted acid sites or well-defined Lewis acidic reaction centers,
- (ii)
- MOFs with cooperative bifunctional acid-base catalytic sites, and
- (iii)
- MOFs with directly incorporated Br− nucleophiles inside channels.
Acknowledgments
Conflicts of Interest
References
- Sumida, K.; Rogow, D.L.; Mason, J.A.; McDonald, T.M.; Bloch, E.D.; Herm, Z.R.; Bae, T.-H.; Long, J.R. Carbon Dioxide Capture in Metal-Organic Frameworks. Chem. Rev. 2012, 112, 724–781. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, D.M.; Smit, B.; Long, J.R. Carbon Dioxide Capture: Prospects for New Materials. Angew. Chem. Int. Ed. 2010, 49, 6058–6082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-R.; Ma, Y.; McCarthy, M.C.; Sculley, J.; Yu, J.; Jeong, H.-K.; Balbuena, P.B.; Zhou, H.-C. Carbon dioxide capture-related gas adsorption and separation in metal-organic frameworks. Coord. Chem. Rev. 2011, 255, 1791–1823. [Google Scholar] [CrossRef]
- Lu, A.-H.; Hao, G.-P. Porous materials for carbon dioxide capture. Annu. Rep. Prog. Chem. Sect. A Inorg. Chem. 2013, 109, 484–503. [Google Scholar] [CrossRef]
- Liu, J.; Thallapally, P.K.; McGrail, B.P.; Brown, D.R.; Liu, J. Progress in adsorption-based CO2 capture by metal-organic frameworks. Chem. Soc. Rev. 2012, 41, 2308–2322. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; D’Alessandro, D.M. Tuning the functional sites in metal-organic frameworks to modulate CO2 heats of adsorption. CrystEngComm 2015, 17, 706–718. [Google Scholar] [CrossRef]
- Kim, Y.; Huh, S. Pore engineering of metal-organic frameworks: Introduction of chemically accessible Lewis basic sites inside MOF channels. CrystEngComm 2016, 18, 3524–3550. [Google Scholar] [CrossRef]
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The Chemistry and Applications of Metal-Organic Frameworks. Science 2013, 341, 1230444. [Google Scholar] [CrossRef]
- Datta, S.J.; Khumnoon, C.; Lee, Z.H.; Moon, W.K.; Docao, S.; Nguyen, T.H.; Hwang, I.C.; Moon, D.; Oleynikov, P.; Terasaki, O.; et al. CO2 capture from humid flue gases and humid atmosphere using a microporous coppersilicate. Science 2015, 350, 302–306. [Google Scholar] [CrossRef]
- Würfel, P. Problems of the Energy Economy. In Physics of Solar Cells; Wiley-VCH: Weinheim, Germany, 2009; pp. 1–10. [Google Scholar]
- Zornoza, B.; Martinez-Joaristi, A.; Serra-Crespo, P.; Tellez, C.; Coronas, J.; Gascon, J.; Kapteijn, F. Functionalized flexible MOFs as fillers in mixed matrix membranes for highly selective separation of CO2 from CH4 at elevated pressures. Chem. Commun. 2011, 47, 9522–9524. [Google Scholar] [CrossRef]
- Hu, Y.; Dong, X.; Nan, J.; Jin, W.; Ren, X.; Xu, N.; Lee, Y.M. Metal-organic framework membranes fabricated via reactive seeding. Chem. Commun. 2011, 47, 737–739. [Google Scholar] [CrossRef] [PubMed]
- Rodenas, T.; Luz, I.; Prieto, G.; Seoane, B.; Miro, H.; Corma, A.; Kapteijn, F.; Llabrés i Xamena, F.X.; Gascon, J. Metal-organic framework nanosheets in polymer composite materials for gas separation. Nat. Mater. 2015, 14, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A. Utilisation of CO2 as a chemical feedstock: Opportunities and challenges. Dalton Trans. 2007, 2975–2992. [Google Scholar] [CrossRef] [PubMed]
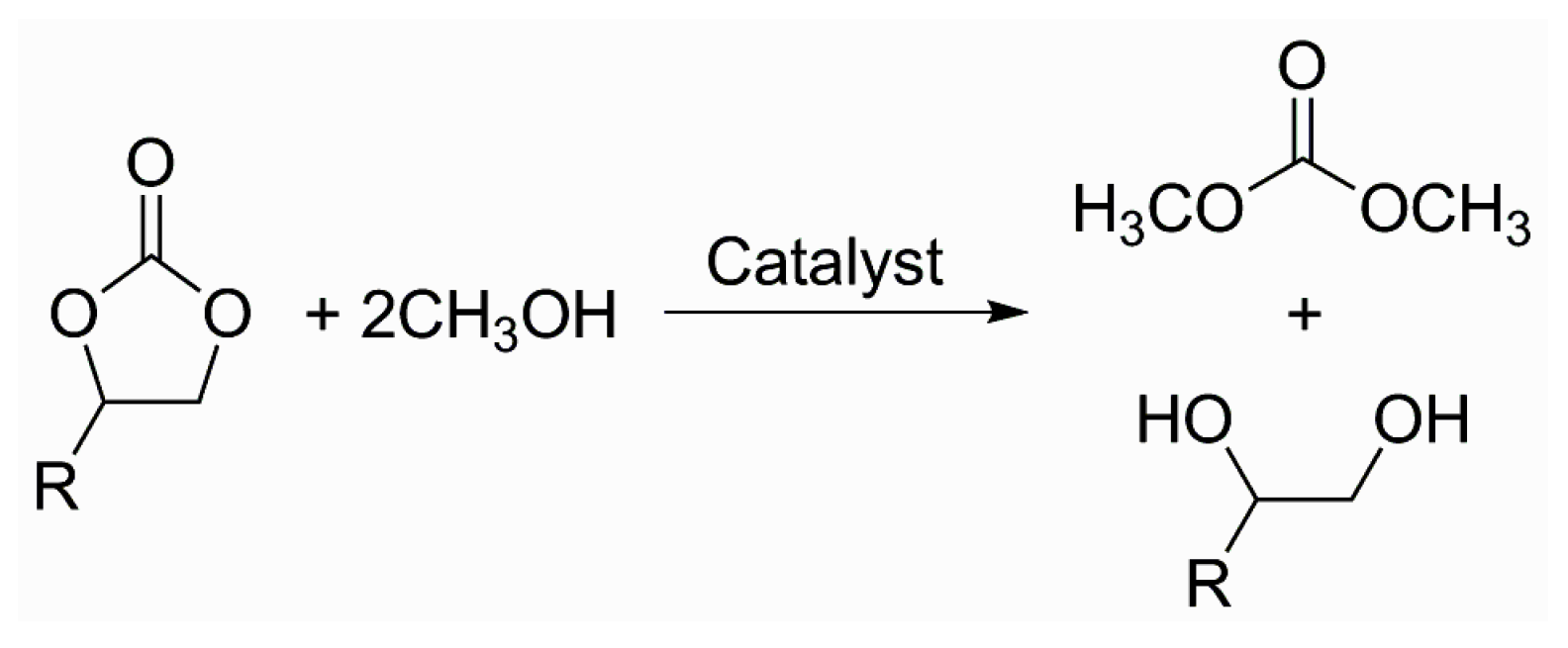
- Sakakura, T.; Kohno, K. The synthesis of organic carbonates from carbon dioxide. Chem. Commun. 2009, 1312–1330. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Qi, C.; Wu, W.; Jiang, H. Recent advances in organic synthesis with CO2 as C1 synthon. Curr. Opin. Green Sustain. Chem. 2017, 3, 22–27. [Google Scholar] [CrossRef]
- Yang, Z.-Z.; He, L.-N.; Dou, X.-Y.; Chanfreau, S. Dimethyl carbonate synthesis catalyzed by DABCO-derived basic ionic liquids via transesterification of ethylene carbonate with methanol. Tetrahedron Lett. 2010, 51, 2931–2934. [Google Scholar] [CrossRef]
- Beyzavi, M.H.; Stephenson, C.J.; Liu, Y.; Karagiaridi, O.; Hupp, J.T.; Farha, O.K. Metal-organic framework-based catalysts: Chemical fixation of CO2 with epoxides leading to cyclic organic carbonates. Front. Energy Res. 2015, 2, 63. [Google Scholar] [CrossRef]
- Maina, J.W.; Pozo-Gonzalo, C.; Kong, L.; Schütz, J.; Hill, M.; Dumée, L.F. Metal organic framework based catalysts for CO2 conversion. Mater. Horiz. 2017, 4, 345–361. [Google Scholar] [CrossRef]
- Zou, B.; Hu, C. Halogen-free processes for organic carbonate synthesis from CO2. Curr. Opin. Green Sustain. Chem. 2017, 3, 11–16. [Google Scholar] [CrossRef]
- He, H.; Perman, J.A.; Zhu, G.; Ma, S. Metal-Organic Frameworks for CO2 Chemical Transformations. Small 2016, 12, 6309–6324. [Google Scholar] [CrossRef]
- North, M.; Pasquale, R. Mechanism of Cyclic Carbonate Synthesis from Epoxides and CO2. Angew. Chem. Int. Ed. 2009, 48, 2946–2948. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, J.; North, M.; Pasquale, R. Synthesis of Cyclic Carbonates from Atmospheric Pressure Carbon Dioxide Using Exceptionally Active Aluminium(salen) Complexes as Catalysts. Eur. J. Inorg. Chem. 2007, 3323–3326. [Google Scholar] [CrossRef]
- Chui, S.S.-Y.; Lo, S.M.-F.; Charmant, J.P.H.; Orpen, A.G.; Williams, I.D. A Chemically Functionalizable Nanoporous Material [Cu3(TMA)2(H2O)3]n. Science 1999, 283, 1148–1150. [Google Scholar] [CrossRef]
- Ji, X.-H.; Zhu, N.-N.; Ma, J.-G.; Cheng, P. Conversion of CO2 into cyclic carbonates by a Co(II) metal-organic framework and the improvement of catalytic activity via nanocrystallization. Dalton Trans. 2018, 47, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.-Y.; Chen, Y.; Niu, Y.; Williams, K.; Cash, L.; Perez, P.J.; Wojtas, L.; Cai, J.; Chen, Y.-S.; Ma, S. Crystal Engineering of an nbo Topology Metal-Organic Framework for Chemical Fixation of CO2 under Ambient Conditions. Angew. Chem. Int. Ed. 2014, 53, 2615–2619. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, S.-M.; Han, Z.-B.; Ding, M.; Yuan, D.-Q.; Jiang, H.-L. Exceptionally Robust In-Based Metal-Organic Framework for Highly Efficient Carbon Dioxide Capture and Conversion. Inorg. Chem. 2016, 55, 3558–3565. [Google Scholar] [CrossRef] [PubMed]
- Beyzavi, M.H.; Klet, R.C.; Tussupbayev, S.; Borycz, J.; Vermeulen, N.A.; Cramer, C.J.; Stoddart, J.F.; Hupp, J.T.; Farha, O.K. A Hafnium-Based Metal-Organic Framework as an Efficient and Multifunctional Catalyst for Facile CO2 Fixation and Regioselective and Enantioretentive Epoxide Activation. J. Am. Chem. Soc. 2014, 136, 15861–15864. [Google Scholar] [CrossRef]
- Wang, H.-H.; Hou, L.; Li, Y.-Z.; Jiang, C.-Y.; Wang, Y.-Y.; Zhu, Z. Porous MOF with Highly Efficient Selectivity and Chemical Conversion for CO2. ACS Appl. Mater. Interfaces 2017, 9, 17969–17976. [Google Scholar] [CrossRef]
- Li, X.-Y.; Ma, L.-N.; Liu, Y.; Hou, L.; Wang, Y.-Y.; Zhu, Z. Honeycomb Metal-Organic Framework with Lewis Acidic and Basic Bifunctional Sites: Selective Adsorption and CO2 Catalytic Fixation. ACS Appl. Mater. Interfaces 2018, 10, 10965–10973. [Google Scholar] [CrossRef]
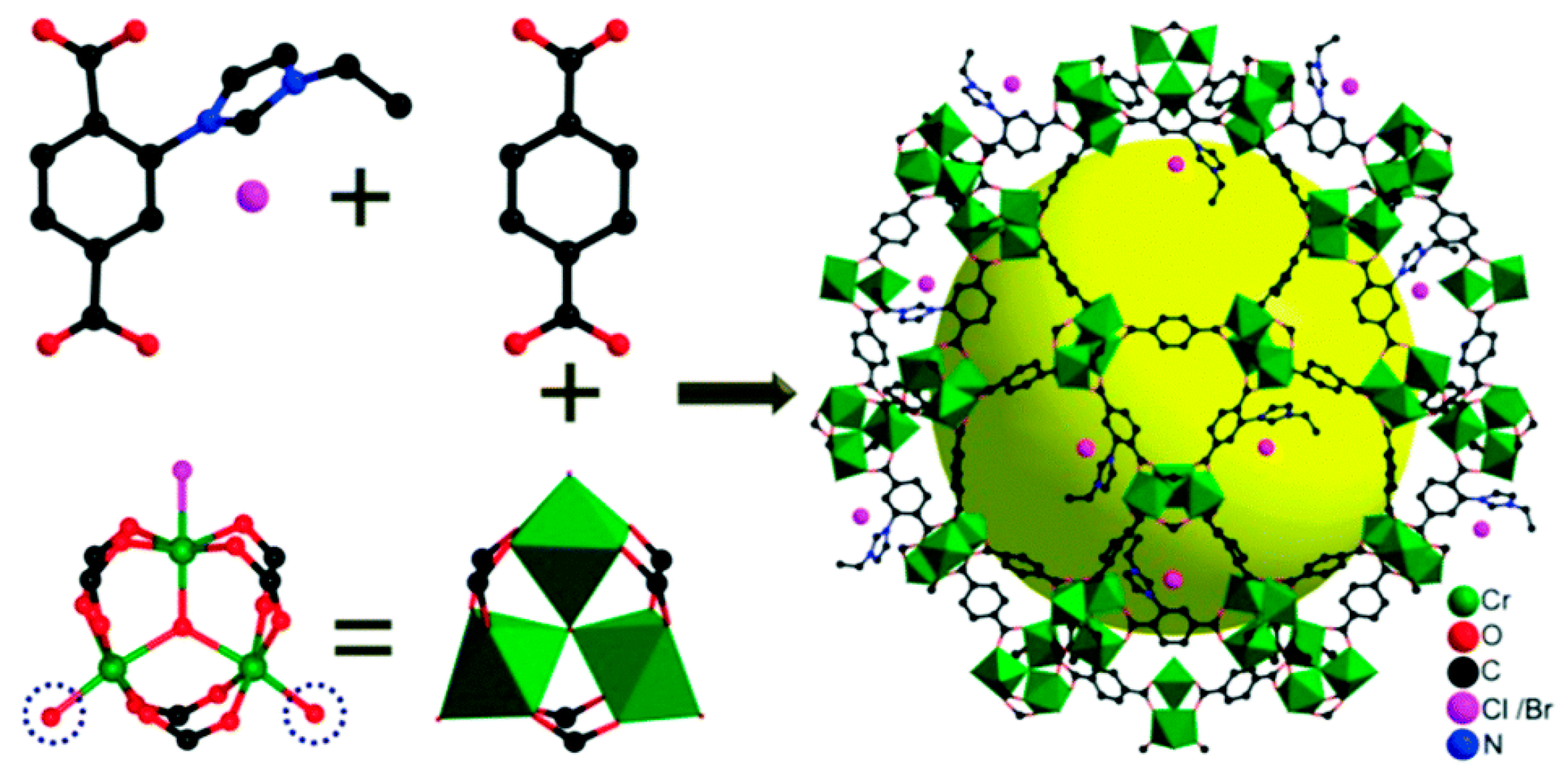
- Cao, C.-S.; Shi, Y.; Xu, H.; Zhao, B. A multifunctional MOF as a recyclable catalyst for the fixation of CO2 with aziridines or epoxides and as a luminescent probe of Cr(VI). Dalton Trans. 2018, 47, 4545–4553. [Google Scholar] [CrossRef]
- Kumar, S.; Verma, G.; Gao, W.-Y.; Niu, Z.; Wojtas, L.; Ma, S. Anionic Metal-Organic Framework for Selective Dye Removal and CO2 Fixation. Eur. J. Inorg. Chem. 2016, 4373–4377. [Google Scholar] [CrossRef]
- Babu, R.; Kathalikkattil, A.C.; Roshan, R.; Tharun, J.; Kim, D.-W.; Park, D.-W. Dual-porous metal organic framework for room temperature CO2 fixation via cyclic carbonate synthesis. Green Chem. 2016, 18, 232–242. [Google Scholar] [CrossRef]
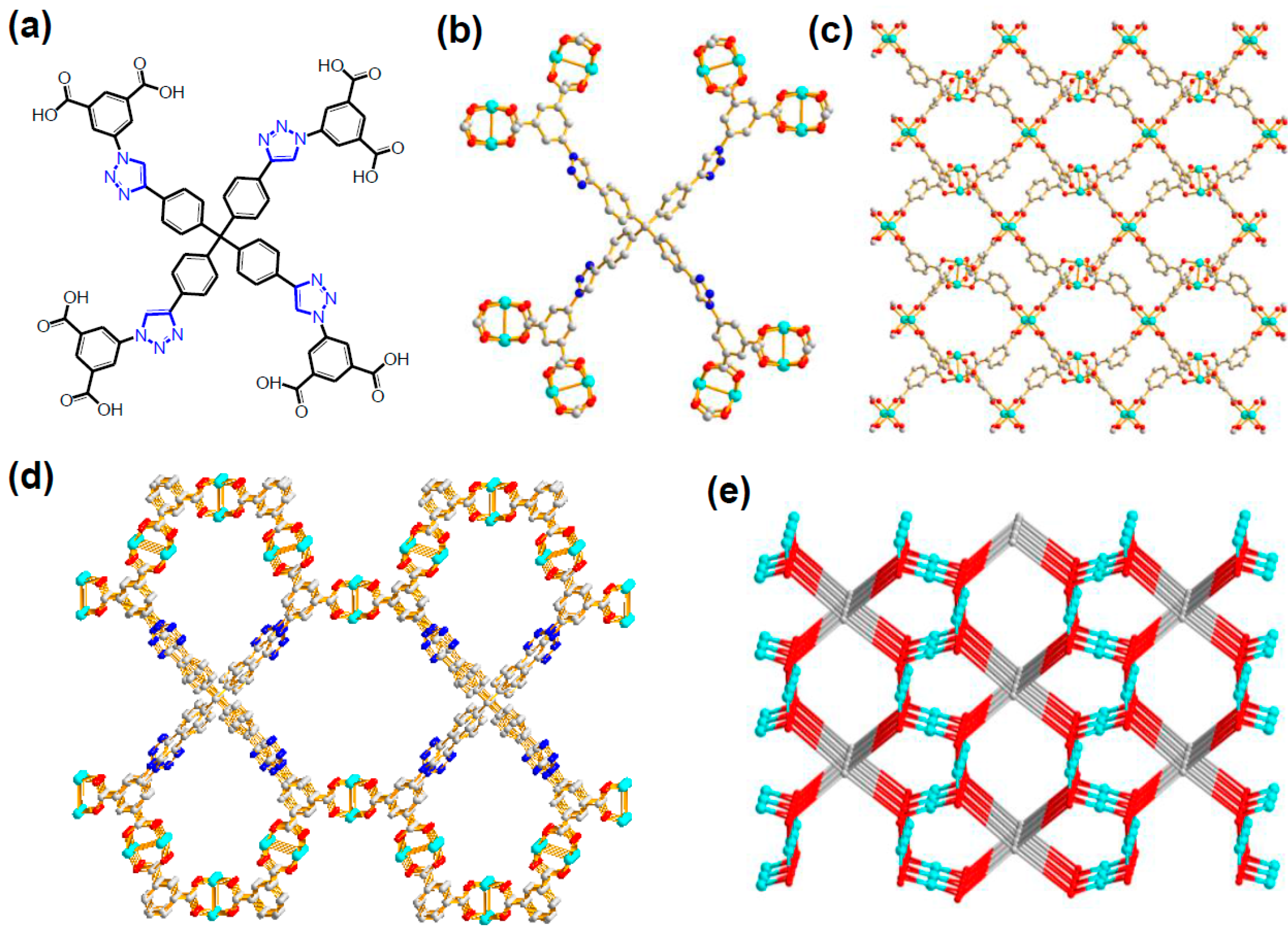
- Li, P.-Z.; Wang, X.-J.; Liu, J.; Lim, J.S.; Zou, R.; Zhao, Y. A Triazole-Containing Metal-Organic Framework as a Highly Effective and Substrate Size-Dependent Catalyst for CO2 Conversion. J. Am. Chem. Soc. 2016, 138, 2142–2145. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Xie, Y.-Q.; Wang, X.-S.; Wang, Q.; Liu, T.-T.; Huang, Y.-B.; Cao, R. An imidazolium-functionalized mesoporous cationic metal-organic framework for cooperative CO2 fixation into cyclic carbonate. Chem. Commun. 2018, 54, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Chen, R.-P.; Wang, X.-Y.; Liu, T.-T.; Wang, X.-S.; Huang, Y.-B.; Cao, R. Postsynthetic ionization of an imidazole-containing metal-organic framework for the cycloaddition of carbon dioxide and epoxides. Chem. Sci. 2017, 8, 1570–1575. [Google Scholar] [CrossRef] [PubMed]
- Aguila, B.; Sun, Q.; Wang, X.; O’Rourke, E.; Al-Enizi, A.M.; Nafady, A.; Ma, S. Lower Activation Energy for Catalytic Reactions through Host-Guest Cooperation within Metal-Organic Frameworks. Angew. Chem. Int. Ed. 2018, 57, 10107–10111. [Google Scholar] [CrossRef] [PubMed]
- Jeazet, H.B.T.; Koschine, T.; Staudt, C.; Raetzke, K.; Janiak, C. Correlation of Gas Permeability in a Metal-Organic Framework MIL-101(Cr)–Polysulfone Mixed-Matrix Membrane with Free Volume Measurements by Positron Annihilation Lifetime Spectroscopy (PALS). Membranes 2013, 3, 331–353. [Google Scholar] [CrossRef] [Green Version]
- Dibenedetto, A.; Angelini, A.; Stufano, P. Use of carbon dioxide as feedstock for chemicals and fuels: Homogeneous and heterogeneous catalysis. J. Chem. Technol. Biotechnol. 2014, 89, 334–353. [Google Scholar] [CrossRef]
- Lu, X.-B.; Darensbourg, D.J. Cobalt catalysts for the coupling of CO2 and epoxides to provide polycarbonates and cyclic carbonates. Chem. Soc. Rev. 2012, 41, 1462–1484. [Google Scholar] [CrossRef]
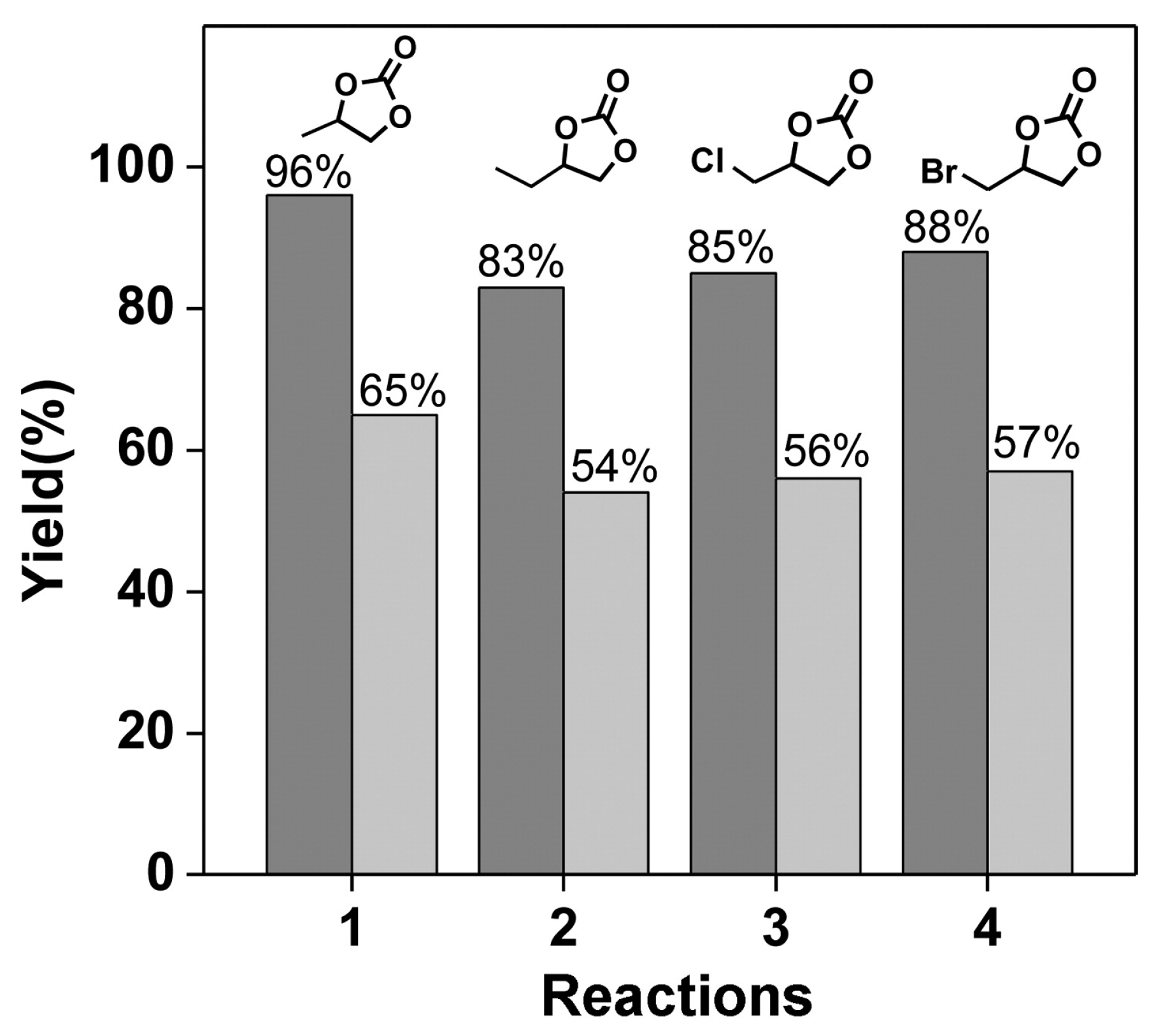
- Noh, J.; Kim, Y.; Park, H.; Lee, J.; Yoon, M.; Park, M.H.; Kim, Y.; Kim, M. Functional group effects on a metal-organic framework catalyst for CO2 cycloaddition. J. Ind. Eng. Chem. 2018, 64, 478–483. [Google Scholar] [CrossRef]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Epoxides | Products | Yield (%) b | TOF (h−1) |
|---|---|---|---|---|
| 1 |  |  | 77.9 | 7.1 |
| 2 |  |  | 60.1 | 5.4 |
| 3 |  |  | 44.2 | 4.0 |
| 4 |  |  | 31.6 | 2.9 |
| Entry | Catalyst | Epoxides | Products | Temp. (°C) | Press. (atm) | Time (h) | Yield c (%) |
|---|---|---|---|---|---|---|---|
| 1a a | Hf-NU-1000 |  |  | r.t. | 1 | 56 | 100 |
| 1b a | NU-1000 | r.t. | 1 | 56 | 46 | ||
| 1c b | - | r.t. | 1 | 56 | 0 | ||
| 1d a | Hf-NU-1000 | 55 | 1 | 13 | 100 | ||
| 2 a | Hf-NU-1000 |  |  | 55 | 1 | 19 | 100 |
| 3 a | Hf-NU-1000 |  |  | r.t. | 1 | 26 | 100 |
| Entry | Epoxides | Products | Conversion (%) | TON b |
|---|---|---|---|---|
| 1 |  |  | 98.1 | 196.2 |
| 2 |  |  | 95.7 | 191.4 |
| 3 |  |  | 94.5 | 189.0 |
| 4 |  |  | 90.2 | 180.4 |
| 5 |  |  | 19.8 | 39.6 |
| 6 |  |  | 52.3 | 104.6 |
| Entry | Epoxides | Products | Yield (%) |
|---|---|---|---|
| 1 |  |  | 92.1 |
| 2 |  |  | 88.6 |
| 3 |  |  | 78.4 |
| 4 |  |  | 69.7 |
| 5 |  |  | 39.2 |
| Entry | Epoxides | Products | Time (h) | Yields (%) |
|---|---|---|---|---|
| 1 |  |  | 48 | 99 |
| 2 |  |  | 48 | 95 |
| 3 |  |  | 96 | 82 |
| 4 |  |  | 96 | 84 |
| 5 |  |  | 72 | 33 |
| Types of MOFs | MOF Catalysts | Epoxides | Temp. (°C) | CO2 (atm) | Time (h) | Conversions (%) | Ref. |
|---|---|---|---|---|---|---|---|
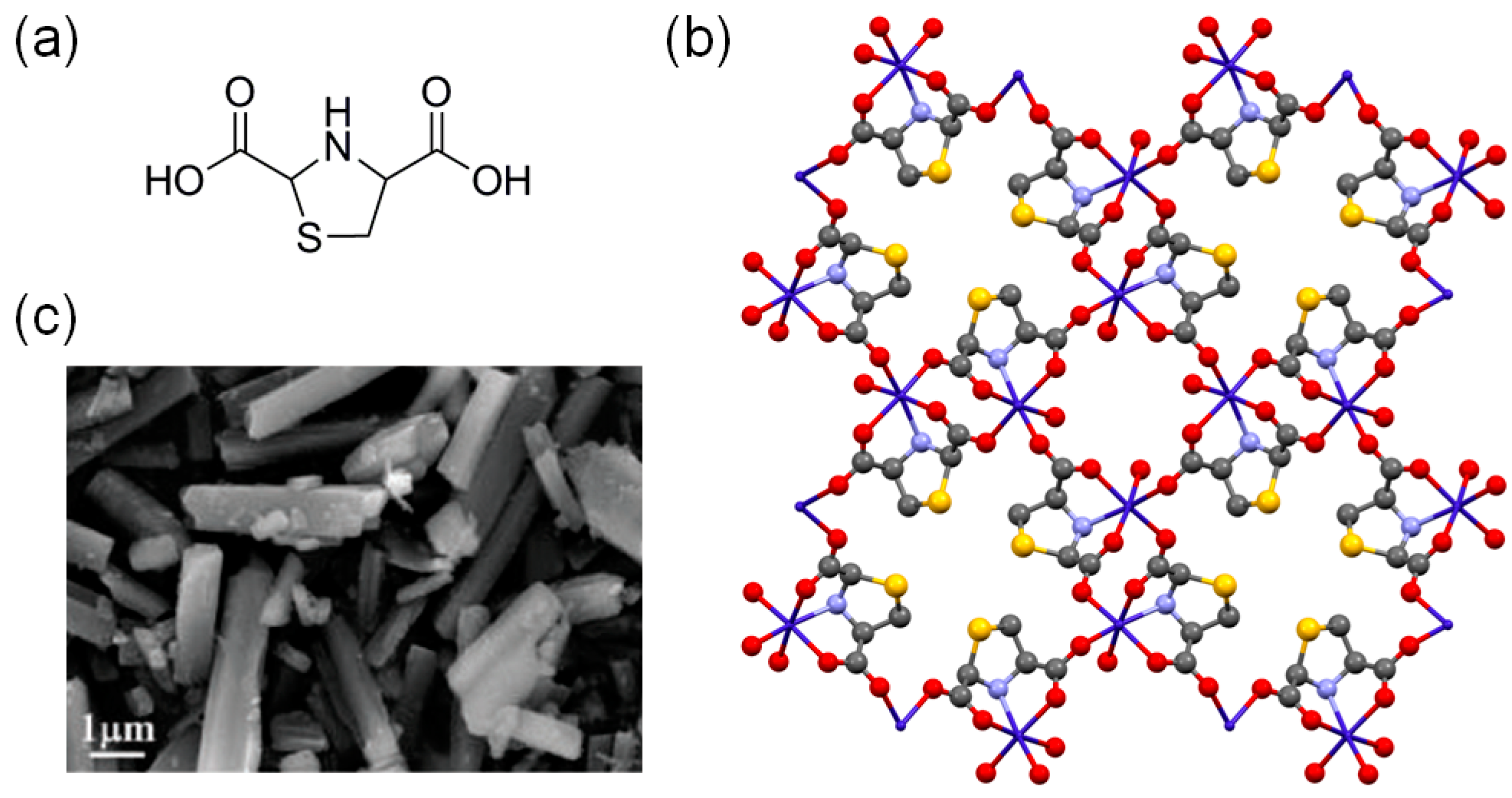
| Acid site-rich (with TBABr) | {Co(μ3-L)(H2O)]·0.5H2O}n a |  | 25 | 1 | 48 | 19.3 | 25 |
| | 50 | 1 | 36 | 98.2 | 25 | ||
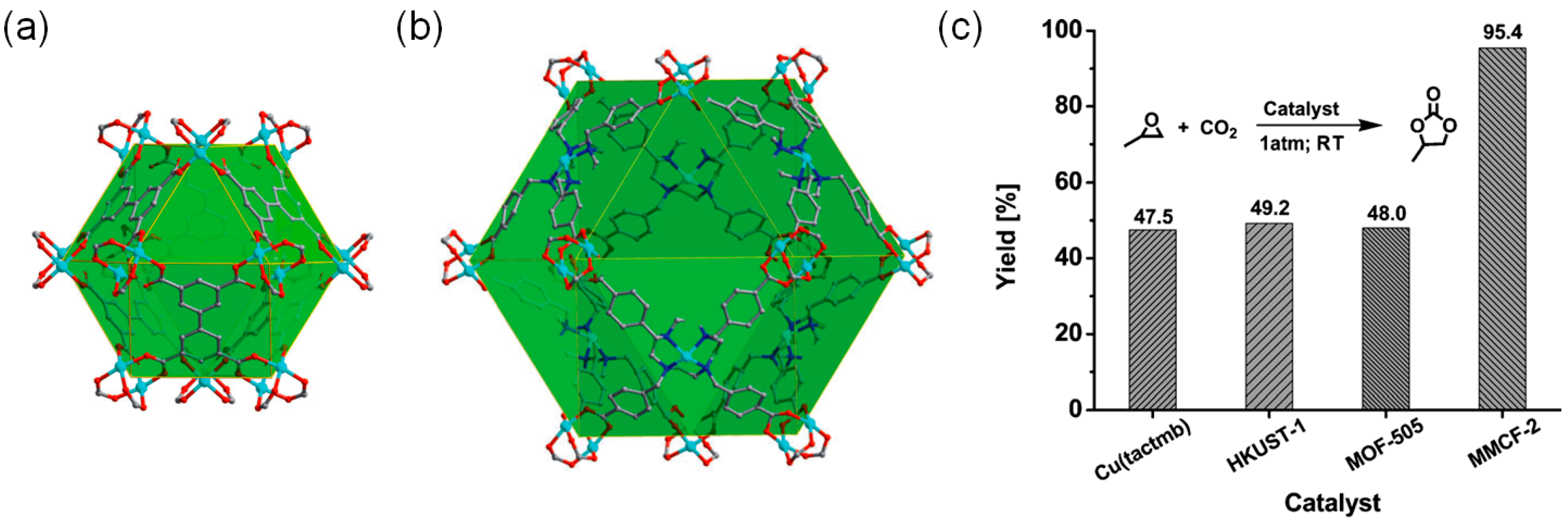
| [Cu2(Cu-tactmb)(H2O)3 (NO3)2] (MMCF-2) b | | r.t. | 1 | 48 | 95.4 | 26 | |
| In2(OH)(btc)(Hbtc)0.4(L)0.6· 3H2O c | | r.t. | 1 | 48 | 77.9 | 27 | |
| Hf-NU-1000 | | r.t. | 1 | 26 | 100 | 28 | |
| {[Co2(tzpa)(OH)(H2O)2]· DMF}n d | | r.t. | 1 | 48 | 93.8 | 29 | |
| Bifunctional acid–base | [Ba2(BDPO)(H2O)] e | | r.t. | 1 | 48 | 98.1 | 30 |
| (with TBABr) | {[Zn(btz)]·DMF·0.5H2O}n f |  | 30 | 1 | 12 | 39 | 31 |
| | 70 | 1 | 12 | 99 | 31 | ||
| Zn-PTB g | | r.t. | 1 | 48 | 92.1 | 32 | |
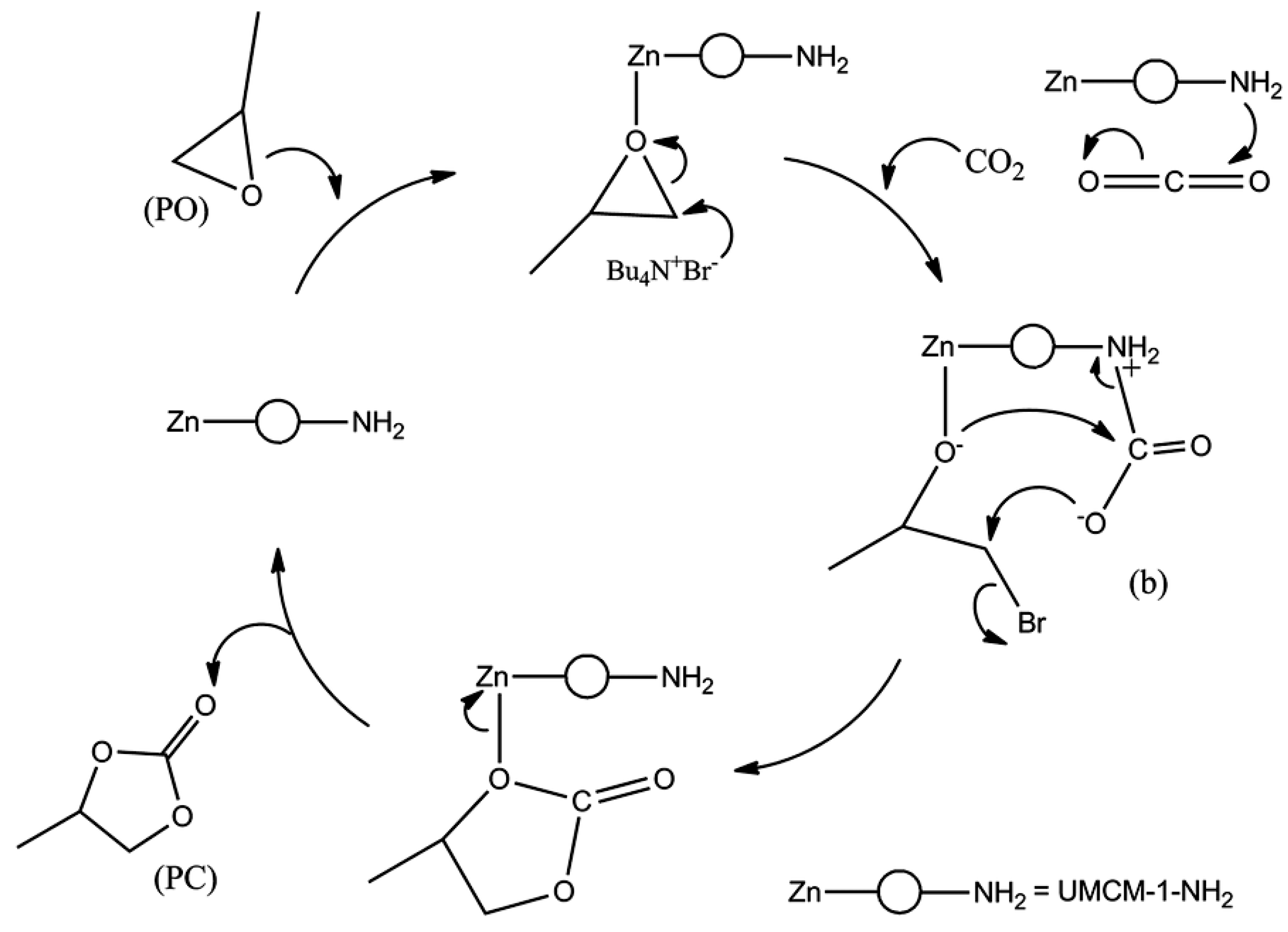
| UMCM-1-NH2 | | r.t. | 3.95 | 4 | 78 | 33 | |
| Cu-L1 h | | r.t. | 1 | 48 | 96 | 34 | |
| Incorporated Br− nucleophile | FJI-C10 |  | r.t. | 1 | 24 | 27.3 | 35 |
| (no TBABr) | | 60 | 1 | 24 | 99.7 | 35 | |
| (I−)Meim-UiO-66 | | 100 | 1 | 24 | 88.5 | 36 | |
| MIL-101-IP | | r.t. | 1 | 48 | 99 | 37 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huh, S. Direct Catalytic Conversion of CO2 to Cyclic Organic Carbonates under Mild Reaction Conditions by Metal—Organic Frameworks. Catalysts 2019, 9, 34. https://doi.org/10.3390/catal9010034
Huh S. Direct Catalytic Conversion of CO2 to Cyclic Organic Carbonates under Mild Reaction Conditions by Metal—Organic Frameworks. Catalysts. 2019; 9(1):34. https://doi.org/10.3390/catal9010034
Chicago/Turabian StyleHuh, Seong. 2019. "Direct Catalytic Conversion of CO2 to Cyclic Organic Carbonates under Mild Reaction Conditions by Metal—Organic Frameworks" Catalysts 9, no. 1: 34. https://doi.org/10.3390/catal9010034
APA StyleHuh, S. (2019). Direct Catalytic Conversion of CO2 to Cyclic Organic Carbonates under Mild Reaction Conditions by Metal—Organic Frameworks. Catalysts, 9(1), 34. https://doi.org/10.3390/catal9010034