Preparation of Sterically Demanding 2,2-Disubstituted-2-Hydroxy Acids by Enzymatic Hydrolysis


 , ,
, ,
Abstract
:
1. Introduction
2. Results
2.1. Screening of Biocatalysts and Substrates
2.2. Optimization
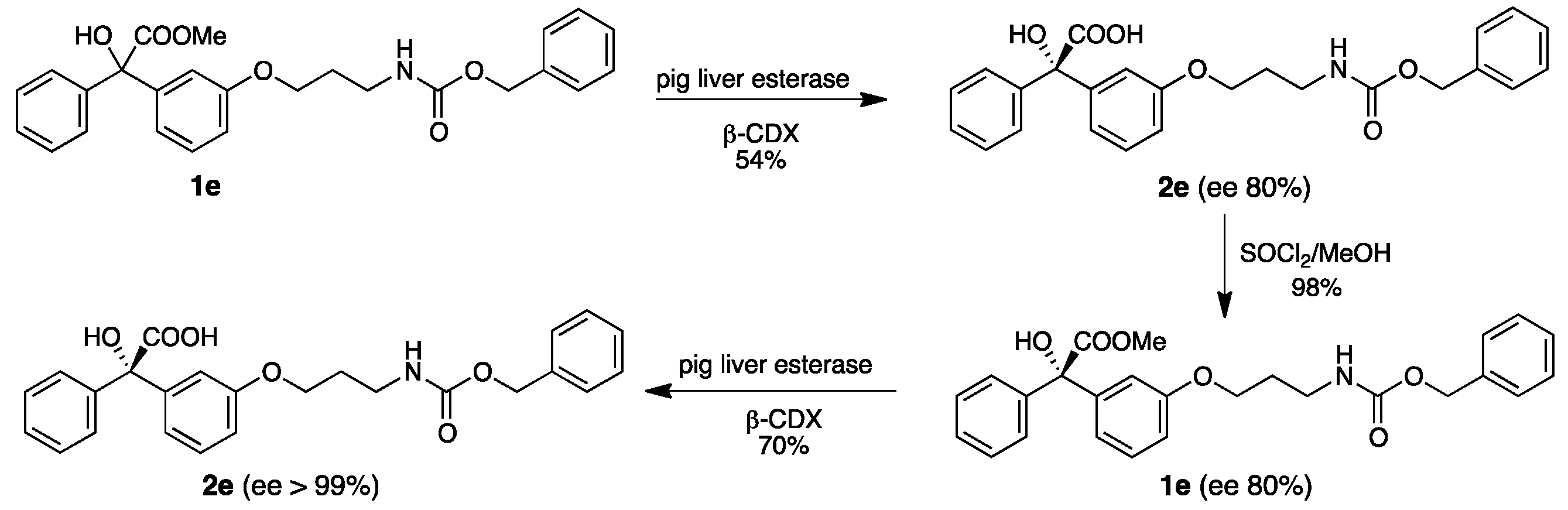
2.3. Preparative Biotransformation
3. Discussion
4. Materials and Methods
4.1. Analyticals
4.2. Procedure for the Synthesis of methyl 2-(4-(benzyloxy)phenyl)-2-hydroxy-2-phenylacetate (1a)
4.3. Procedure for the Synthesis of methyl 2-hydroxy-2-(3-hydroxyphenyl)-2-phenylacetate (1b)
4.4. Procedure for the Synthesis of 2-(dimethylamino)ethyl 2-(3-(benzyloxy)phenyl)-2-hydroxy-2-phenylacetate (1c)
4.5. Procedure for the Synthesis of (1-benzylpiperidin-4-yl)methyl 2-(3-(benzyloxy)phenyl)-2-hydroxy-2-phenylacetate (1d)
4.6. Procedure for the Synthesis of methyl 2-(3-(3-(((benzyloxy)carbonyl)amino)propoxy)phenyl)-2-hydroxy-2-phenylacetate (1e)
4.7. Enantiomeric Excess Determination
4.8. General Procedure for Biotransformations
4.8.1. (S)-2-(3-(Benzyloxy)phenyl)-2-hydroxy-2-phenylacetic acid (2a)
4.8.2. (S)-2-Hydroxy-2-(3-hydroxyphenyl)-2-phenylacetic acid (2b)
4.8.3. (S)-2-(3-(3-(((Benzyloxy)carbonyl)amino)propoxy)phenyl)-2-hydroxy-2-phenylacetic acid (2e)
4.9. Procedure for the Synthesis of methyl (S)-2-(3-(3-(((benzyloxy)carbonyl)amino)propoxy)phenyl)-2-hydroxy-2-phenylacetate (optically enriched 1e)
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bornscheuer, U.T.; Kazlauskas, R.T. Hydrolases in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 1999. [Google Scholar]
- Gotor-Fernandez, V.; Brieva, R.; Gotor, V. Lipases: Useful biocatalysts for the preparation of pharmaceuticals. J. Mol. Catal. B. 2006, 40, 111–120. [Google Scholar] [CrossRef]
- De Miranda, A.S.; Miranda, L.S.M.; de Souza, R.O.M.A. Lipases: Valuable catalysts for dynamic kinetic resolutions. Biotechnol. Adv. 2015, 33, 372–393. [Google Scholar] [CrossRef] [PubMed]
- Romano, D.; Bonomi, F.; de Mattos, M.C.; de Sousa Fonseca, T.; de Oliveira, M.C.F.; Molinari, F. Esterases as stereoselective biocatalysts. Biotechnol. Adv. 2015, 33, 547–565. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.C.; de Sousa Fonseca, T.; de Mattos, M.C.; de Oliveira, M.C.F.; de Lemos, T.L.; Molinari, F.; Romano, D.; Serra, I. Recent advances in lipase-mediated preparation of pharmaceuticals and their intermediates. Int. J. Mol. Sci. 2015, 16, 29682–29716. [Google Scholar] [CrossRef] [PubMed]
- Bornscheuer, U.T. Methods to increase enantioselectivity of lipases and esterases. Curr. Opin. Biotechnol. 2002, 13, 543–547. [Google Scholar] [CrossRef]
- Juhl, P.B.; Doderer, K.; Hollmann, F.; Thum, O.; Pleiss, J. Engineering of Candida antarctica lipase B for hydrolysis of bulky carboxylic acid esters. J. Biotechnol. 2010, 150, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Pogorevc, M.; Faber, K. Biocatalytic resolution of sterically hindered alcohols, carboxylic acids and esters containing fully substituted chiral centers by hydrolytic enzymes. J. Mol. Catal. B. 2000, 10, 357–376. [Google Scholar] [CrossRef]
- Kourist, R.; Bornscheuer, U.T. Biocatalytic synthesis of optically active tertiary alcohols. Appl. Microbiol. Biotechnol. 2011, 91, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rodríguez, J.A.; Gotor, V.; Brieva, R. Lipase catalyzed resolution of the quaternary stereogenic center in ketone-derived benzo-fused cyclic cyanohydrins. Tetrahedron: Asymmetry 2011, 22, 1218–1224. [Google Scholar] [CrossRef]
- Lalonde, J.J.; Bergbreiter, D.E.; Wong, C.-H. Enzymatic kinetic resolution of α-nitro α-methyl carboxylic acids. J. Org. Chem. 1988, 53, 2323–2327. [Google Scholar] [CrossRef]
- Kometani, T.; Isobe, T.; Goto, M.; Takeuchi, Y.; Haufe, G. Enzymatic resolution of 2-fluoro-2-arylacetic acid derivatives. J. Mol. Catal. B 1988, 5, 171–174. [Google Scholar] [CrossRef]
- Miyazawa, T.; Shimaoka, M.; Yamada, T. Resolution of 2-cyano-2-methylalkanoic acids via porcine pancreatic lipase-catalyzed enantioselective ester hydrolysis: effect of the alcohol moiety of the substrate ester on enantioselectivity. Biotechnol. Lett. 1999, 21, 309–312. [Google Scholar] [CrossRef]
- Domínguez de María, P.; García-Burgos, C.A.; Bargeman, G.; van Gemert, R.W. Pig Liver Esterase (PLE) as biocatalyst in organic synthesis: from nature to cloning and to practical applications. Synthesis 2007, 10, 1439–1452. [Google Scholar] [CrossRef]
- Namiki, Y.; Fujii, T.; Nakada, M. Preparation of chiral building blocks for the enantioselective total synthesis of ent-kauranoids by the pig liver esterase-catalyzed asymmetric hydrolysis of a dialkyl malonate-type prochiral diester. Tetrahedron: Asymmetry 2014, 25, 718–724. [Google Scholar] [CrossRef]
- Rancati, F.; Rizzi, A.; Carzaniga, L.; Linney, I.; Knight, C.; Schmidt, W. Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity. Patent WO 2016/128456 Al, August 2016. [Google Scholar]
- Rancati, F.; Rizzi, A.; Carzaniga, L.; Linney, I.; Knight, C.; Schmidt, W. Compounds having muscarinic receptor antagonist and beta2 adrenergic receptor agonist activity. Patent WO 2018/011090 A1, January 2018. [Google Scholar]
- Montuschi, Z.; Macagno, F.; Valente, S.; Fuso, L. Inhaled muscarinic acetylcholine receptor antagonists for treatment of COPD. Curr Med Chem. 2013, 20, 1464–1476. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, R.; Marinelli, F.; Lazzarini, A.; Molinari, F. Cell-bound and extracellular carboxylesterases from Streptomyces: hydrolytic and synthetic activities. J. Appl Microbiol 2000, 89, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Converti, A.; Del Borghi, A.; Gandolfi, R.; Lodi, A.; Molinari, F.; Palazzi, E. Simplified kinetics and thermodynamics of geraniol acetylation by lyophilized cells of Aspergillus oryzae. Biotechnol. Bioeng. 2002, 77, 232–237. [Google Scholar] [CrossRef]
- Molinari, F.; Romano, D.; Gandolfi, R.; Kroppenstedt, R.M.; Marinelli, F. Newly isolated Streptomyces spp. as enantioselective biocatalysts: hydrolysis of 1,2-O-isopropylidene glycerol racemic esters. J. Appl. Microbiol. 2005, 99, 960–967. [Google Scholar] [CrossRef]
- Molinari, F.; Cavenago, K.S.; Romano, A.; Romano, D.; Gandolfi, R. Enantioselective hydrolysis of (RS)-isopropylideneglycerol acetate with Kluyveromyces marxianus. Tetrahedron: Asymmetry 2004, 15, 1945–1947. [Google Scholar] [CrossRef]
- Monti, D.; Ferrandi, E.E.; Righi, M.; Romano, D.; Molinari, F. Purification and characterization of the enantioselective esterase from Kluyveromyces marxianus CBS 1553. J. Biotechnol. 2008, 133, 65–72. [Google Scholar] [CrossRef]
- De Vitis, V.; Nakhnoukh, C.; Pinto, A.; Contente, M.L.; Barbiroli, A.; Milani, M.; Bolognesi, M.; Molinari, F.; Gourlay, L.; Romano, D. A stereospecific carboxyl esterase from Bacillus coagulans hosting nonlipase activity within a lipase-like fold. FEBS J. 2018, 285, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Hummel, A.; Brüsehaber, E.; Böttcher, D.; Trauthwein, H.; Doderer, K.; Bornscheuer, U.T. Isoenzymes of Pig-Liver Esterase reveal striking differences in enantioselectivities. Angew. Chem. Int. Ed. Engl. 2007, 46, 8492–8494. [Google Scholar] [CrossRef] [PubMed]
- Romano, D.; Gandolfi, R.; Guglielmetti, S.; Molinari, F. Enzymatic hydrolysis of capsaicins for the production of vanillylamine using ECB deacylase from Actinoplanes utahensis. Food Chem. 2011, 124, 1096–1098. [Google Scholar] [CrossRef]
- Bjorkling, F.; Boutelje, J.; Gatenbeck, S.; Hult, K.; Norin, T. Enzyme catalysed hydrolysis of dialkylated propanedioic acid diesters, chain length dependent reversal of enantioselectivity. Appl. Microbiol. Biotechnol. 1985, 21, 16–19. [Google Scholar] [CrossRef]
- Del Valle, E.M.M. Cyclodextrins and their uses: a review. Process Biochem. 2004, 39, 1033–1046. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
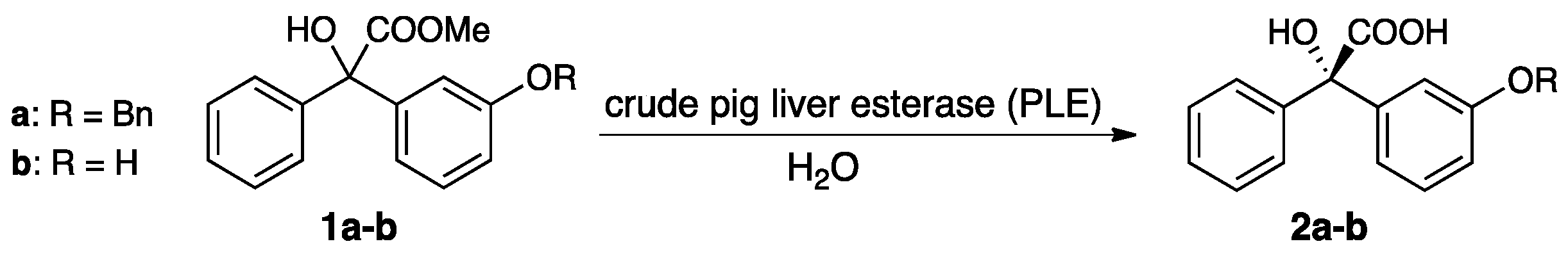{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Conv. (%) | ee (R)-ester (%) | ee (S)-acid (%) | E | Time (h) |
|---|---|---|---|---|---|---|
| 1 | 1a | 52 | 67 | 63 | 8 | 5 |
| 2 | 1a | >97 | <5 | <5 | - | 24 |
| 3 | 1b | 50 | 67 | 67 | 10 | 5 |
| 4 | 1b | > 97 | <5 | <5 | - | 24 |
| Entry | Substrate | Conv. (%) | ee (R)-ester (%) | ee (S)-acid (%) | E | Time (h) |
|---|---|---|---|---|---|---|
| 1 | Crude PLEs | 30 | 37 | 87 | 21 | 5 |
| 2 | ECS-PLE01 | <5 | - | - | - | 24 |
| 3 | ECS-PLE02 | <5 | - | - | - | 24 |
| 4 | ECS-PLE03 | 19 | 8 | 30 | <5 | 24 |
| 5 | ECS-PLE04 | 8 | - | n.d. | - | 24 |
| 6 | ECS-PLE05 | <5 | - | - | - | 24 |
| 7 | ECS-PLE06 | 37 | 51 | 87 | 24 | 24 |
| Entry | Co-solvent (% v/v) | Conv. (%) | ee (R)-ester (%) | ee (S)-acid (%) | E |
|---|---|---|---|---|---|
| 1 | none | 30 | 23 | 90 | 23 |
| 2 | EtOH (8) | <5 | - | - | - |
| 3 | iPrOH (8) | <5 | - | - | - |
| 4 | DMSO (8) | 30 | 39 | 90 | 28 |
| 5 | THF (8) | 10 | 9 | 79 | 10 |
| 6 | acetone (8) | <5 | - | - | - |
| 7 | Et2O (30) | <5 | - | - | - |
| 8 | toluene (30) | <5 | - | - | - |
| 9 | n-heptane (30) | 22 | 25 | 90 | 21 |
| 10 | isooctane (30) | 25 | 30 | 88 | 21 |
| Entry | β-Cyclodextrin | Conv. (%) | eeester (%) | eeacid (%) | E |
|---|---|---|---|---|---|
| 1 | underivatized | 45 | 70 | 86 | 28 |
| 2 | triacetyl | 40 | 59 | 88 | 28 |
| 3 | methyl | 33 | 44 | 88 | 24 |
| 4 | trimethylammonium | 25 | 31 | 91 | 28 |
| Entry | Ratio [β-CDX]/[S] | Conv. (%) | eeester (%) | eeacid (%) | E |
|---|---|---|---|---|---|
| 1 | 1 | 41 | 60 | 86 | 24 |
| 2 | 1.25 | 45 | 70 | 86 | 28 |
| 3 | 1.5 | 45 | 70 | 86 | 28 |
| 4 | 2 | 48 | 78 | 84 | 28 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, A.; Serra, I.; Romano, D.; Contente, M.L.; Molinari, F.; Rancati, F.; Mazzucato, R.; Carzaniga, L. Preparation of Sterically Demanding 2,2-Disubstituted-2-Hydroxy Acids by Enzymatic Hydrolysis. Catalysts 2019, 9, 113. https://doi.org/10.3390/catal9020113
Pinto A, Serra I, Romano D, Contente ML, Molinari F, Rancati F, Mazzucato R, Carzaniga L. Preparation of Sterically Demanding 2,2-Disubstituted-2-Hydroxy Acids by Enzymatic Hydrolysis. Catalysts. 2019; 9(2):113. https://doi.org/10.3390/catal9020113
Chicago/Turabian StylePinto, Andrea, Immacolata Serra, Diego Romano, Martina Letizia Contente, Francesco Molinari, Fabio Rancati, Roberta Mazzucato, and Laura Carzaniga. 2019. "Preparation of Sterically Demanding 2,2-Disubstituted-2-Hydroxy Acids by Enzymatic Hydrolysis" Catalysts 9, no. 2: 113. https://doi.org/10.3390/catal9020113
APA StylePinto, A., Serra, I., Romano, D., Contente, M. L., Molinari, F., Rancati, F., Mazzucato, R., & Carzaniga, L. (2019). Preparation of Sterically Demanding 2,2-Disubstituted-2-Hydroxy Acids by Enzymatic Hydrolysis. Catalysts, 9(2), 113. https://doi.org/10.3390/catal9020113