Rational Design of Chiral Selenium-π-Acid Catalysts


Abstract
:1. Introduction
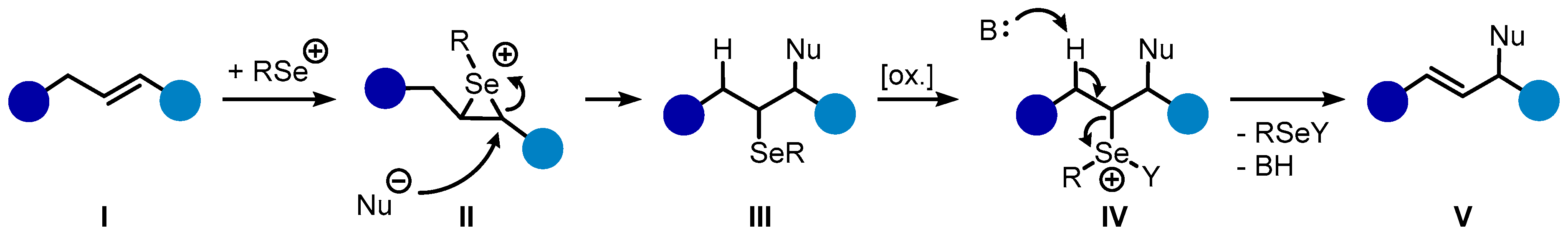
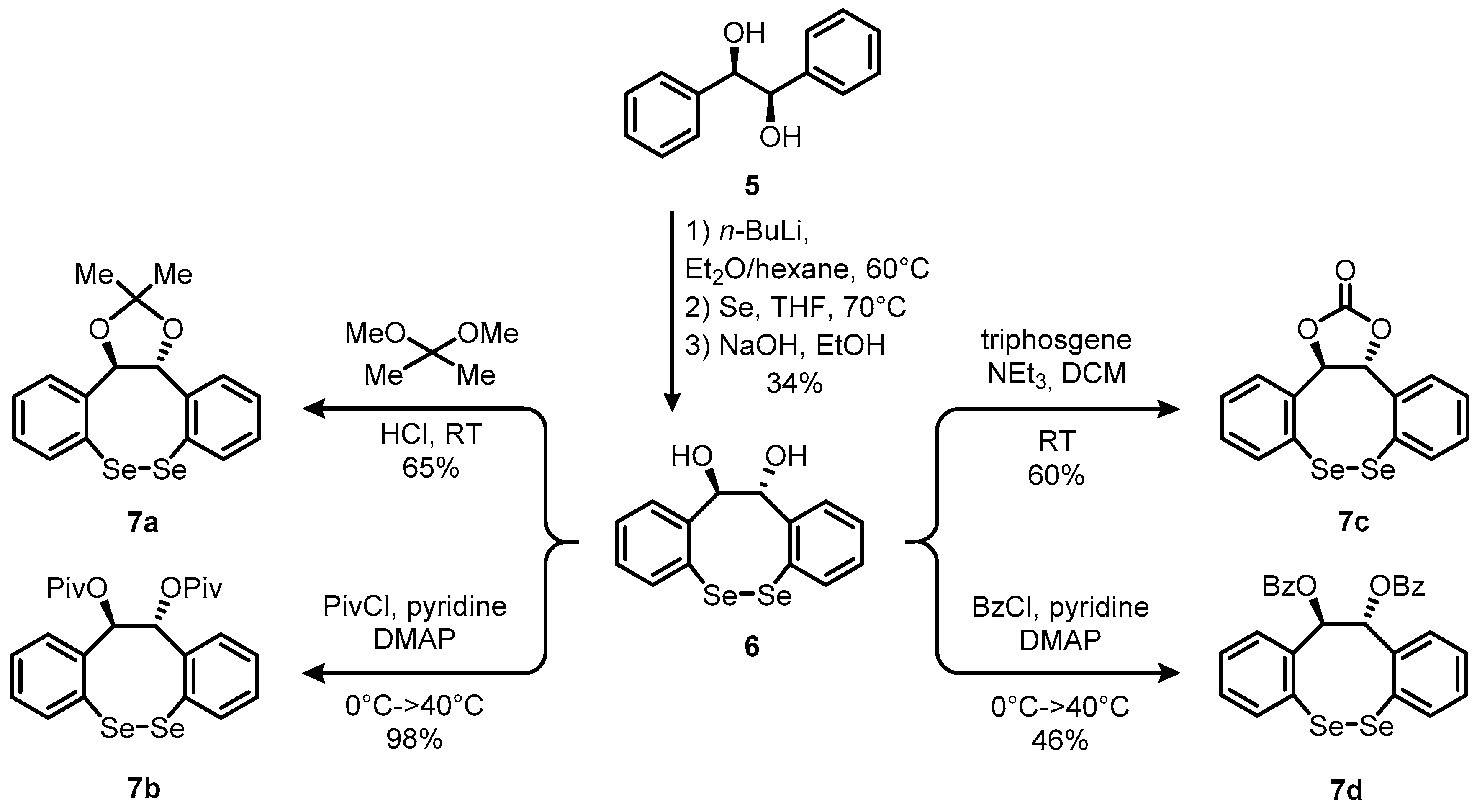
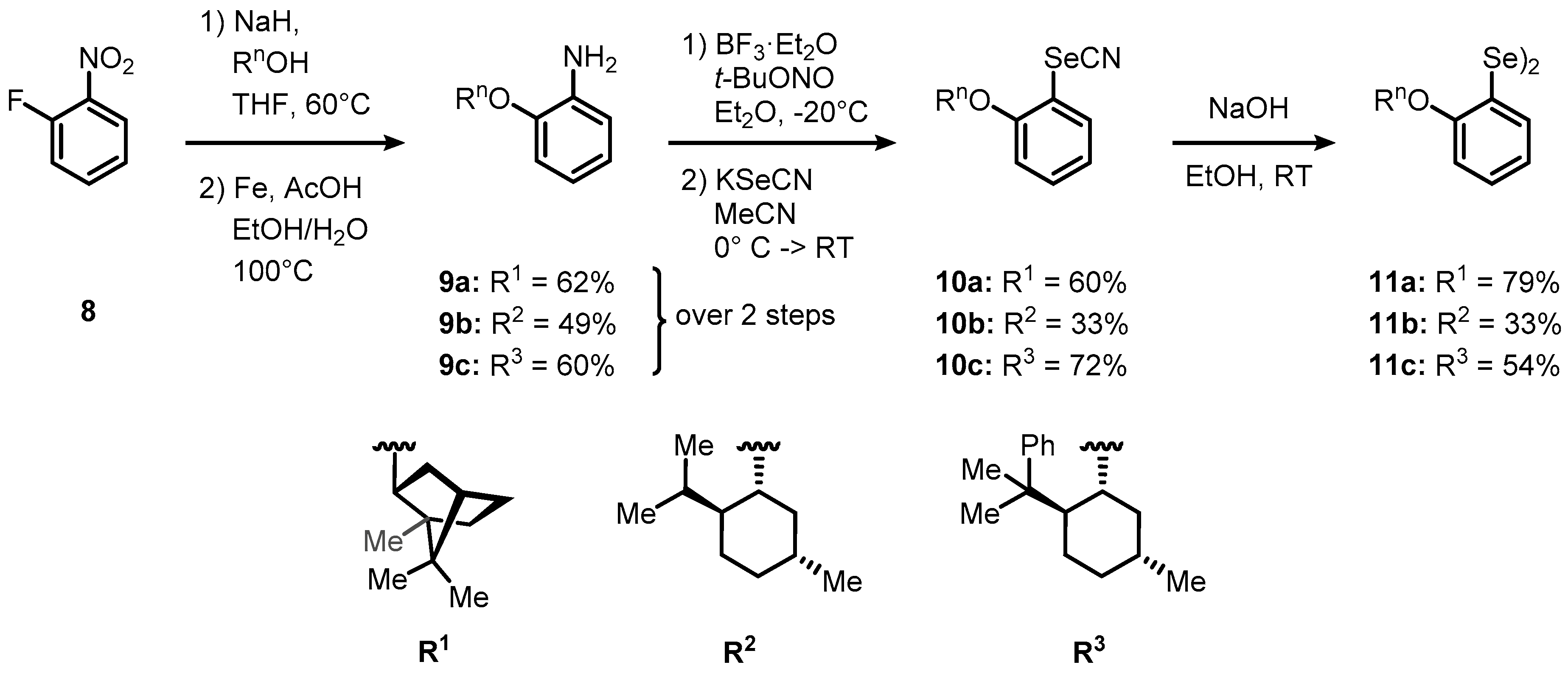
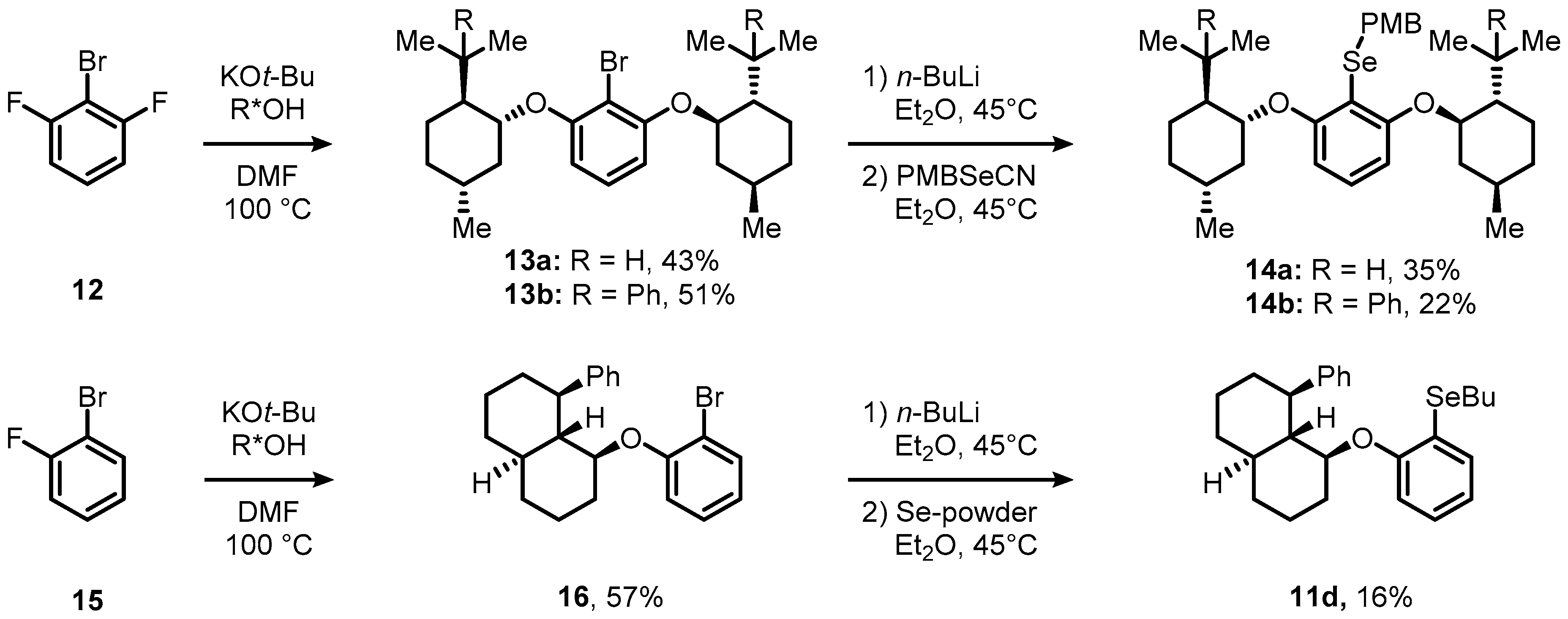
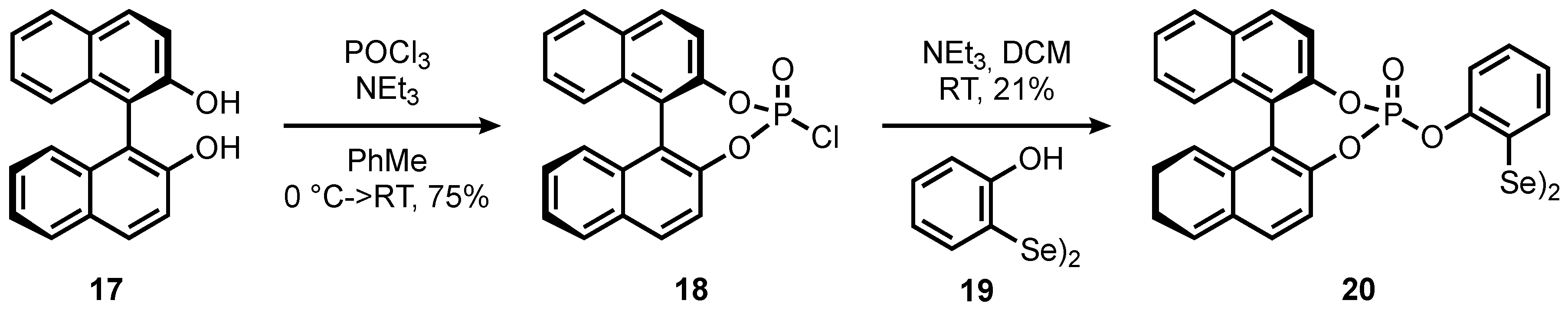
2. Results and Discussion

3. Materials and Methods
4. Conclusion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Comprehensive Asymmetric Catalysis; Jacobsen, E.N.; Pfaltz, A.; Yamamoto, H. (Eds.) Springer: Berlin, Germany, 1999. [Google Scholar]
- Torres, R.R. Stereoselective Organocatalysis—Bond Formation Methodologies and Activation Modes; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Dalko, P.I. Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications; WileyalystVerlag GmbH & Co. KGaA: Weinheim, Germany, 2013. [Google Scholar]
- Denmark, S.E.; Kuester, W.E.; Burk, M.T. Catalytic, Asymmetric Halofunctionalization of Alkenes—A Critical Perspective. Angew. Chem. Int. Ed. 2012, 51, 10938–10953. [Google Scholar] [CrossRef]
- Tan, C.K.; Yeung, Y.-Y. Recent Advances in Stereoselective Bromofunctionalization of Alkenes using N-Bromoamide Reagents. Chem. Commun. 2013, 49, 7985–7996. [Google Scholar] [CrossRef] [PubMed]
- Breder, A.; Ortgies, S. Recent Developments in Sulfur- and Selenium-Catalyzed Oxidative and Isohypsic Functionalization Reactions of Alkenes. Tetrahedron Lett. 2015, 56, 2843–2852. [Google Scholar] [CrossRef]
- Ortgies, S.; Breder, A. Oxidative Alkene Functionalizations via Selenium-π-Acid Catalysis. ACS Catal. 2017, 7, 5828–5840. [Google Scholar] [CrossRef]
- Romero, R.M.; Wöste, T.H.; Muñiz, K. Vicinal Difunctionalization of Alkenes with Iodine(III) Reagents and Catalysts. Chem. Asian J. 2014, 9, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Haubenreisser, S.; Wöste, T.H.; Martínez, C.; Ishihara, K.; Muñiz, K. Structurally Defined Molecular Hypervalent Iodine Catalysts for Intermolecular Enantioselective Reactions. Angew. Chem. Int. Ed. 2016, 55, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, Y.; Hashimoto, T.; Maruoka, K. A Chiral Electrophilic Selenium Catalyst for Highly Enantioselective Oxidative Cyclization. J. Am. Chem. Soc. 2016, 138, 5206–5209. [Google Scholar] [CrossRef]
- Wirth, T.; Häuptli, S.; Leuenberger, M. Catalytic Asymmetric Oxyselenenylation–Elimination Reactions Using Chiral Selenium Compounds. Tetrahedron 1998, 9, 547–550. [Google Scholar] [CrossRef]
- Fujita, K.; Iwaoka, M.; Tomoda, S. Synthesis of Diaryl Diselenides Having Chiral Pyrrolidine Rings with C2 Symmetry. Their Application to the Asymmetric Methoxyselenenylation of trans-β-Methylstyrenes. Chem. Lett. 1994, 23, 923–926. [Google Scholar] [CrossRef]
- Tiecco, M.; Testaferri, L.; Santi, C.; Tomassini, C.; Marini, F.; Bagnoli, L.; Temperini, A. Asymmetric Azidoselenenylation of Alkenes: A Key Step for the Synthesis of Enantiomerically Enriched Nitrogen-Containing Compounds. Angew. Chem. Int. Ed. 2003, 42, 3131–3133. [Google Scholar] [CrossRef]
- Zhao, L.; Li, Z.; Wirth, T. Asymmetric Methoxyselenenylations with Chiral Selenium Electrophiles. Eur. J. Org. Chem. 2011, 2011, 7080–7082. [Google Scholar] [CrossRef]
- Browne, D.; Wirth, T. New Developments with Chiral Electrophilic Selenium Reagents. Curr. Org. Chem. 2006, 10, 1893–1903. [Google Scholar] [CrossRef]
- Höltzle, G.; Jenny, W. Zur Kenntnis der Sulfen und Selenensäuren und ihrer Derivate: Additions- und Substitutionsreaktionen organischer Selenverbindungen mit unpolaren und polaren Äthylenen. Helv. Chim. Acta 1958, 73, 593–603. [Google Scholar] [CrossRef]
- Deziel, R.; Goulet, S.; Grenier, L.; Bordeleau, J.; Bernier, J. Asymmetric Selenomethoxylation of Olefins Involving a Chiral C2 Symmetrical Electrophilic Organoselenium Reagent. J. Org. Chem. 1993, 58, 3619–3621. [Google Scholar] [CrossRef]
- Denmark, S.E.; Collins, W.R. Lewis Base Activation of Lewis Acids: Development of a Lewis Base Catalyzed Selenolactonization. Org. Lett. 2007, 9, 3801–3804. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Edwards, M.G. On the Mechanism of the Selenolactonization Reaction with Selenenyl Halides. J. Org. Chem. 2006, 71, 7293–7306. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T. Organoselenium Chemistry in Stereoselective Reactions. Angew. Chem. Int. Ed. 2000, 39, 3740–3749. [Google Scholar] [CrossRef]
- Trenner, J.; Depken, C.; Weber, T.; Breder, A. Direct Oxidative Allylic and Vinylic Amination of Alkenes through Selenium Catalysis. Angew. Chem. Int. Ed. 2013, 52, 8952–8956. [Google Scholar] [CrossRef] [PubMed]
- Braga, A.L.; Silva, S.J.N.; Lüdtke, D.S.; Drekener, R.L.; Silveira, C.C.; Rocha, J.B.T.; Wessjohann, L.A. Chiral Diselenide Ligands for the Asymmetric Copper-Catalyzed Conjugate Cddition of Grignard Reagents to Enones. Tetrahedron Lett. 2002, 43, 7329–7331. [Google Scholar] [CrossRef]
- Mukherjee, A.J.; Zade, S.S.; Singh, H.B.; Sunoj, R.B. Organoselenium Chemistry: Role of Intramolecular Interactions. Chem. Rev. 2010, 110, 4357–4416. [Google Scholar] [CrossRef]
- Rode, K.; Palomba, M.; Ortgies, S.; Rieger, R.; Breder, A. Aerobic Allylation of Alcohols with Non-Activated Alkenes Enabled by Light-Driven Selenium-π-Acid Catalysis. Synthesis 2018, 50, 3875–3885. [Google Scholar] [CrossRef]
- Ortgies, S.; Depken, C.; Breder, A. Oxidative Allylic Esterification of Alkenes by Cooperative Selenium-Catalysis Using Air as the Sole Oxidant. Org. Lett. 2016, 18, 2856–2859. [Google Scholar] [CrossRef] [PubMed]
- Depken, C.; Krätzschmar, F.; Rieger, R.; Rode, K.; Breder, A. Photocatalytic Aerobic Phosphatation of Alkenes. Angew. Chem. Int. Ed. 2018, 57, 2459–2463. [Google Scholar] [CrossRef] [PubMed]
- Ortgies, S.; Rieger, R.; Rode, K.; Koszinowski, K.; Kind, J.; Thiele, C.M.; Rehbein, J.; Breder, A. Mechanistic and Synthetic Investigations on the Dual Selenium-π-Acid/Photoredox Catalysis in the Context of the Aerobic Dehydrogenative Lactonization of Alkenoic Acids. ACS Catal. 2017, 7, 7578–7586. [Google Scholar] [CrossRef]
- Leisering, S.; Riaño, I.; Depken, C.; Gross, L.J.; Weber, M.; Lentz, D.; Zimmer, R.; Stark, C.B.W.; Breder, A.; Christmann, M. Synthesis of (+)-Greek Tobacco Lactone via a Diastereoablative Epoxidation and a Selenium-Catalyzed Oxidative Cyclization. Org. Lett. 2017, 19, 1478–1481. [Google Scholar] [CrossRef] [PubMed]
- Hori, T.; Sharpless, K.B. Conversion of Allylic Phenylselenides to the Rearranged Allylic Chlorides by N-Chlorosuccinimide. Mechanism of Selenium-Catalyzed Allylic Chlorination of beta-Pinene. J. Org. Chem. 1979, 44, 4208–4210. [Google Scholar] [CrossRef]
- Tunge, J.A.; Mellegaard, S.R. Selective Selenocatalytic Allylic Chlorination. Org. Lett. 2004, 6, 1205–1207. [Google Scholar] [CrossRef] [PubMed]
- Mellegaard, S.R.; Tunge, J.A. Selenium-Catalyzed Halolactonization: Nucleophilic Activation of Electrophilic Halogenating Reagents. J. Org. Chem. 2004, 69, 8979–8981. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Guo, R.; Zhao, X. Organoselenium-Catalyzed Regioselective C-H Pyridination of 1,3-Dienes and Alkenes. Angew. Chem. Int. Ed. 2017, 56, 3201–3205. [Google Scholar] [CrossRef]
- Iwaoka, M.; Tomoda, S. Catalytic Conversion of Alkenes into Allylic Ethers and Esters using Diselenides having Internal Tertiary Amines. J. Chem. Soc. Chem. Commun. 1992, 1165–1167. [Google Scholar] [CrossRef]
- Cresswell, A.J.; Eey, S.T.-C.; Denmark, S.E. Catalytic, Stereospecific syn-Dichlorination of Alkenes. Nat. Chem. 2015, 7, 146–152. [Google Scholar] [CrossRef]
- Niyomura, O.; Cox, M.; Wirth, T. Electrochemical Generation and Catalytic Use of Selenium Electrophiles. Synlett 2006, 2, 251–254. [Google Scholar] [CrossRef]
- Altermann, S.M.; Richardson, R.D.; Page, T.K.; Schmidt, R.K.; Holland, E.; Mohammed, U.; Paradine, S.M.; French, A.N.; Richter, C.; Bahar, A.M.; et al. Catalytic Enantioselective α-Oxysulfonylation of Ketones Mediated by Iodoarenes. Eur. J. Org. Chem. 2008, 2008, 5315–5328. [Google Scholar] [CrossRef]
- Kattuboina, A.; Li, G. Chiral N-Phosphonyl Imine Chemistry: New Reagents and their Applications for Asymmetric Reactions. Tetrahedron Lett. 2008, 49, 1573–1577. [Google Scholar] [CrossRef]
- Martiny, M.; Steckhan, E.; Esch, T. Cycloaddition Reactions Initiated by Photochemically Excited Pyrylium Salts. Chem. Ber. 1993, 126, 1671–1682. [Google Scholar] [CrossRef]
- Joshi-Pangu, A.; Lévesque, F.; Roth, H.G.; Oliver, S.F.; Campeau, L.-C.; Nicewicz, D.; DiRocco, D.A. Acridinium-Based Photocatalysts: A Sustainable Option in Photoredox Catalysis. J. Org. Chem. 2016, 81, 7244–7249. [Google Scholar] [CrossRef]
- Wilken, M.; Ortgies, S.; Breder, A.; Siewert, I. Mechanistic Studies on the Anodic Functionalization of Alkenes Catalyzed by Diselenides. ACS Catal. 2018, 8, 10901–10912. [Google Scholar] [CrossRef]
- Cardoso do Vale, M.L.; Rodríguez-Borges, H.E.; Caamaño, O.; Fernández, F.; García-Mera, X. The Use of (−)-8-Phenylisoneomenthol and (−)-8-Phenylmenthol in the Enantioselective synthesis of 3-functionalized 2-azabicyclo[2.2.1]heptane derivatives via aza-Diels–Alder reaction. Tetrahedron 2006, 62, 9475–9482. [Google Scholar] [CrossRef]
- Whitesell, J.K.; Lawrence, R.M.; Chen, H.H. Auxiliary Structure and Asymmetric Induction in the Ene Reactions of Chiral Glyoxylates. J. Org. Chem. 1986, 51, 4779–4784. [Google Scholar] [CrossRef]
- Corey, E.J.; Ensley, H.E. Preparation of an Optically Active Prostaglandin Intermediate via Asymmetric Induction. J. Am. Chem. Soc. 1975, 97, 6908–6909. [Google Scholar] [CrossRef]
- Letzel, M.C.; Schäfer, H.J.; Fröhlich, R. Diastereoselective Anodic Hetero- and Homo-Coupling of Menthol-, 8-Methylmenthol- and 8-Phenylmenthol-2-Alkylmalonates. Beilstein J. Org. Chem. 2017, 13, 33–42. [Google Scholar] [CrossRef]
- Maruoka et al. reported an e.r. of 97.5:2.5 for the same substrate when NFSI was used as the terminal oxidant at room temperature in toluene (cf. Ref. [10]).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Se-Catalyst | Solvent | er | Yield [%] |
|---|---|---|---|---|
| 1 | 7a | CyH | - | 0% |
| 2 | 7a | PhMe | 57:43 | 16% |
| 3 | 7a | Et2O | 57:43 | 27% |
| 4 | 7a | 1,4-dioxane | 57.5:42.5 | 17% |
| 5 | 7a | MTBE | 58:42 | 28% |
| 6 | 7a | THF | 59.5:40.5 | 29% |
| 7 | 7a | DCM | 59:41 | 18% |
| 8 | 7a | MeNO2 | 54:46 | 16% |
| 9 | 7a | THF/MeCN | 53.5:46.5 | 37% |
| 10 | 7a | MeCN | 51.5:48.5 | 50% |
| 11 | 7b | THF | 54:46 | 49% |
| 12 | 7d | THF | 58:42 | 52% |
| 13 | 7c | THF | 75:25 | 81% |

| Entry | Photocatalyst | Solvent | T | t | Yield | er4 |
|---|---|---|---|---|---|---|
| 1 | TAPT | MeCN | 35 °C | 20 h | 24% 1 | 74:26 |
| 2 | TAPT | MeCN | 50 °C | 96 h | 99% 2 | 62:38 |
| 3 | TAPTP | MeCN | 35 °C | 17 h | 13% 2 | 56:44 |
| 4 | TAPTP 3 | MeCN | 35 °C | 18 h | 10% 2 | 56:44 |
| 5 | DMAT | PhMe | 35 °C | 16 h | 33% 1 | 54:46 |
| 6 | rhodamine G | MeCN | 35 °C | 16 h | 0% | nd |
| 7 | Ru(bpz)3(PF6)2 | MeCN | 45 °C | 16 h | 19% 2 | 52:48 |

 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krätzschmar, F.; Ortgies, S.; Willing, R.Y.N.; Breder, A. Rational Design of Chiral Selenium-π-Acid Catalysts. Catalysts 2019, 9, 153. https://doi.org/10.3390/catal9020153
Krätzschmar F, Ortgies S, Willing RYN, Breder A. Rational Design of Chiral Selenium-π-Acid Catalysts. Catalysts. 2019; 9(2):153. https://doi.org/10.3390/catal9020153
Chicago/Turabian StyleKrätzschmar, Felix, Stefan Ortgies, Robert Y. N. Willing, and Alexander Breder. 2019. "Rational Design of Chiral Selenium-π-Acid Catalysts" Catalysts 9, no. 2: 153. https://doi.org/10.3390/catal9020153
APA StyleKrätzschmar, F., Ortgies, S., Willing, R. Y. N., & Breder, A. (2019). Rational Design of Chiral Selenium-π-Acid Catalysts. Catalysts, 9(2), 153. https://doi.org/10.3390/catal9020153