1. Introduction
Notwithstanding a number of investigations to prevent sintering, supported-metal catalyst deactivation owing to metal sintering is inevitable [
1], and redispersion of sintered metal clusters is the key in catalyst rejuvenation. Based on scientific and patent literature surveys, halogen acid gas, oxygen, chlorine, and nitric oxide can be used as agents to redisperse sintered Pt catalysts [
1,
2,
3,
4,
5,
6,
7,
8,
9]. Among them, oxychlorination, which uses oxygen- and chlorine-containing compounds as the dispersion agent, is the most common method employed in the industry to redisperse noble metals on non-reducible metal oxides, such as silica, alumina, zeolite, and cordierite [
1,
4,
10,
11].
In petroleum refineries, the oxychlorination technique has been applied to redisperse Pt/Al
2O
3, Pt–Re/Al
2O
3, and Pt–Ir/Al
2O
3 reforming catalysts for decades [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
12]. However, the fundamental understanding of detailed surface chemistry could further the process of evolution and development, and might spark new inventions as well; for instance, through the application of in situ XAS (X-ray Absorption Spectroscopy) and TEM (Transmission Electron Microscopy), a rapid and effective redispersion of Pt particles supported on ceria-based oxide catalysts three-way catalysts (TWCs) has been developed by Nagai et. al [
13,
14].
With the Pt/alumina catalyst as an example, the redispersion process typically involves four steps: (1) formation of redispersed Pt complexes by the reaction of Pt atoms with dispersion agents; (2) dissociation and liberation of the complexes from sintered Pt clusters; (3) migration and deposition of these species on the support surface; and (4) the reduction of the complexes to Pt clusters [
2,
3,
4,
5]. The structure of redispersed complexes will vary with the operation parameters, such as the Air/EDC (ethylene dichloride) mole ratio, residence time, duration of operation, and redispersion temperature. Among them, temperature is the most critical operation parameter because it directly influences the structure, the stability of the redispersed complexes, and thus the redispersion efficacy [
10,
15,
16,
17,
18].
Lee and Kim have reported that the oxychlorination redispersion capacity for Pt/γ-Al
2O
3 increases with operation temperature from 400 °C to about 550 °C, and then decreases abruptly [
3]. Consistent with Lee and Kim’s results, oxychlorination redispersion of commercial reforming catalysts is typically performed at 510–530 °C for about 4 h [
19]. Recently, Decoodt et al., in a patent disclosure, suggested that the inlet temperature of oxychlorination zone should be 510 °C [
20].
The goals of this research are to understand the effects of redispersion temperature on the structure of redispersed complexes and the corresponding redispersion efficacy. Commercial catalysts are inherently nonuniform in structure. Redispersed complexes derived from these catalysts are difficult to be characterized. Model Pt/γ-Al2O3 was thus prepared with the goal of preparing nearly fully redispersed (uniform) complexes to facilitate structure identification.
γ-Al2O3 was chosen as a catalyst support, because: (1) this material has been widely used as a catalyst support in refinery, petro-chemical, and vehicle emission control; (2) the surface area is high (>150 m2/g); (3) stronger interactions with precious metals, as opposed to silica and zeolite, allowing sintered catalysts to be fully redispersed; and (4) its structure is well-known and nearly uniform.
Instead of H2PtCl6, which is commonly used in preparing supported Pt catalysts, Pt(NH3)4(NO3)2 was used, because residual chlorine contained in the H2PtCl6 prepared catalysts complicates the oxychlorination process and the structure characterization of redispersed complexes. Instead of chlorine and HCl, which are commonly used as redispersion agents, ethylenedichloride (EDC) was employed, because: (1) it has long been used as a chlorine source in redispersing sintered Pt catalysts for the catalytic reformer, as well as for converters fitted to gasoline and diesel cars; and (2) its toxicity is less than chlorine, and its corrosiveness is less than HCl.
EXAFS (Extended X-ray Absorption Fine Structure) can provide quantitative structure characterization of surface species formed in the redispersion process [
21], whereas an accurate structure characterization strongly relies on the identification of back-scatter atoms (atoms bonded to absorb-atoms, Pt) and the determination of the coordination number (
N) for each shell. To identify the back-scatter atom, phase-corrected Fourier transform was performed on the EXAFS function with different phase functions. The ligands bonded to Pt were then characterized by a rather symmetric peak with a bond distance (
R) close to that of crystallographic data [
21]. However, the identification may be interfered by other back-scatter atoms, resulting in a distortion of the symmetry of the peak [
22,
23]. For the elimination of the interference, a difference file technique was used in data analysis [
24]. To determine number of ligands bonded to Pt, the oxidation state of Pt based on XANES (X-ray absorption near edge structure) analysis results was used as an initial guess in structure parameter estimation. Moreover, in the EXAFS function, N and Δσ
2 (the Debye-Waller factor between the sample and reference compound) are highly correlated—hence, data-fitting was performed both in different weightings, [k
1χ(
k)] and in [k
3χ(
k)], so as to decouple the correlation of these two parameters [
24]. Alternatively, this correlation could be minimized by calculating Debye-Waller factors ab initio [
25,
26,
27,
28,
29,
30].
2. Results and Discussion
2.1. Ligand Identification
In order to separate the contributions from different shells and to identify ligands of the redispersed complexes (back-scatter atoms), phase- and amplitude-corrected Fourier transform was performed to the EXAFS function, x(
k). The radial distribution function was formulated as:
where
k is the electron wave number,
F(
k) is the backscattering amplitude characteristic of a particular type of neighboring atom, and
ϕj(
k) is a phase shift function, approximated by linear dependence on
k, i.e.,
ϕ(
k) =
ϕ0 +
σ × k.
The phase shift function causes peaks in the radical distribution function to shift to a lower R (bond length) value, while the
k dependent amplitude function can result in a peak broadening and/or a sidelobe [
22,
31]; especially in the case of high-Z backscatter atoms, more than one peak will be present for a single shell for the transformation without amplitude correction [
32].
For an absorber-backscattering pair X–Y, if the right phase and amplitude function is applied, an imaginary part of the Fourier transform (FT) of the EXAFS function will peak positively and center at the maximum of the magnitude. The correct coordination distance determined by the peak location [
22,
24,
31] allows us to identify ligand atoms.
In this study, only the phase correction was applied in identifying ligand atoms, because: (1) the amplitude for low-Z is relatively smooth over the EXAFS range, and the estimated distance between absorber and backscatter will not deviate from real distance significantly; and (2) the signal-to-noise ratio at the end spectrum is relatively small, and dividing χ(
k) by
F(
k) will yield a high level of background, leading to a deviation in estimating bond distance [
24].
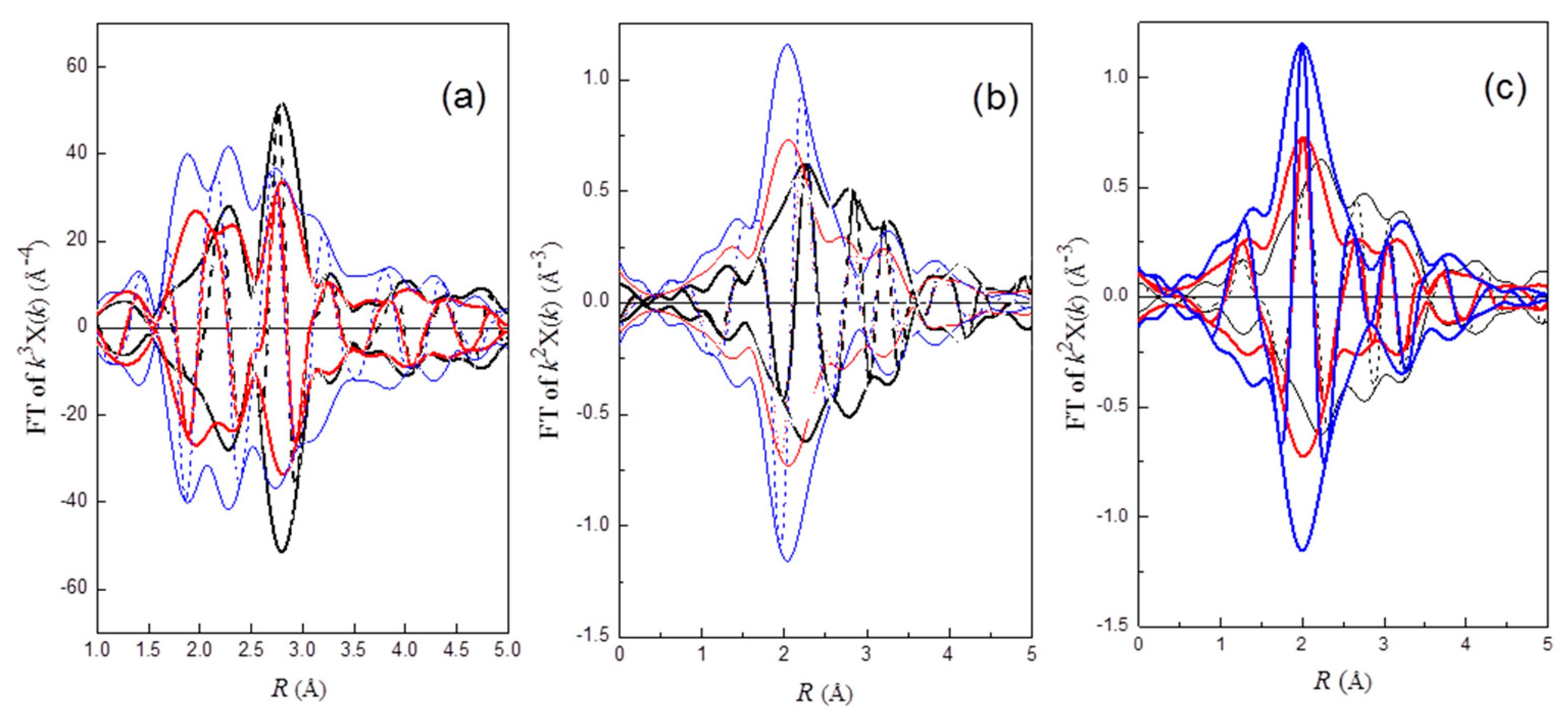
In order to examine the efficacy of redispersion qualitatively, a
k3-weighted Pt–Pt phase and amplitude-corrected Fourier-transform in the range of 4.0 < k < 14.0 Å
−1 was performed to the EXAFS functions of Pt
rd46, Pt
rd52, and Pt
rd56. As shown in
Figure 1a, a positive peak at about 2.75 Å appears in the imaginary part of FT for Pt
rd46, Pt
rd56, while a rather broad peak with a shoulder at about 3.2 Å was observed for Pt
rd52. The positive peak at 2.75 Å for Pt
rd46, Pt
rd56 is consistent with the first shell Pt–Pt bond distance of Pt crystal structure data [
33]. Among these three samples, Pt
rd56 with the highest peak amplitude indicated that the redispersion efficacy of this sample was lowest. In contrast, the lowest amplitude with peak broadening for Pt
rd52 suggested that this sample was almost fully re-dispersed. The peak broadening was caused by the coupling of the characteristic peak of Pt–Pt with that of ligands.
The possibility of the Cl ligand being bound to Pt can be examined by a
k2-weighted Pt–Cl phase-corrected Fourier transform. As shown in
Figure 1b, a rather symmetric positive peak at about 2.3 Å appears in the imaginary part of FT for Pt
rd56, while the peak for Pt
rd46 and Pt
rd52 is rather asymmetric. These results suggested the existence of the Cl ligand on the redispersed Pt complex of Pt
rd56.
Since oxygen gas is present in the oxychlorination process, oxygen-containing species could be a ligand of the redispersed complexes. After the Pt–O phase-corrected Fourier-transform (
k2-weighted, 2.8 < k < 11.0 Å
−1) was performed on the EXAFS functions, the appearance of a symmetric positive peak at about 2.05 Å for Pt
rd52 and Pt
rd46 (
Figure 1c) suggested that oxygen was bonded to Pt. In addition, the appearance of a negative peak in the imaginary part at about 3.2 Å suggested that Cl was bonded to the O atom of Pt–O (
Figure 1c); FT of Pt–Cl* (Cl* is the Cl atom of O–Cl ligand) corrected by the Pt–O phase function yielding a negative peak was due to an approximate π radian difference between the Pt–Cl and Pt–O phase function (
ϕPt–Cl −
ϕPt–O), as was calculated by FEFF8 [
34]. The results suggested the existence of a OCl
− ligand on Pt
rd46 and Pt
rd52.
2.2. XANES (X-Ray Absorption Near Edge Structure) Analysis
The intensity of the threshold resonance of the Pt
LIII-edge XANES is related to the transition probability of exciting 2p
3/2 mainly into 5d
5/2 and 5d
3/2 orbitals. Nonetheless, XANES has been widely used to study the coordination chemistry and oxidation state of Pt complexes. Due to the lack of a simple equation for XANES, the majority of studies based on the analysis of XANES data still rely on reference materials [
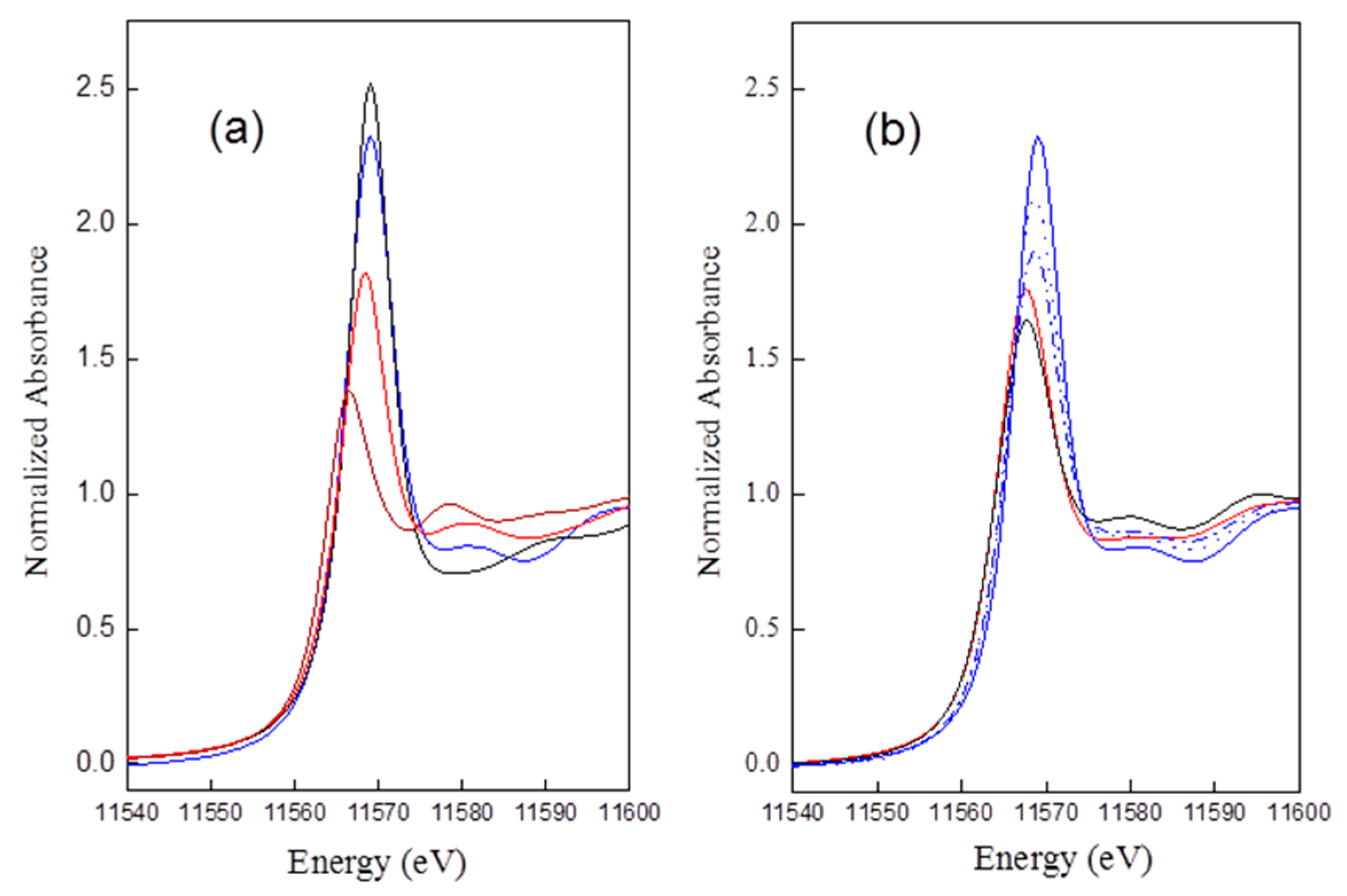
35]. A comparison of Pt
LIII edge data for references PtCl
2, PtCl
4, H
2Pt(OH)
6, and Pt
rd52 is shown in
Figure 2a, and that for Pt
rd46, Pt
rd56, Pt
rd52,
Air/ED=20, Pt
rd52,
WHSV=0.187, and Pt
rd52 is shown in
Figure 2b.
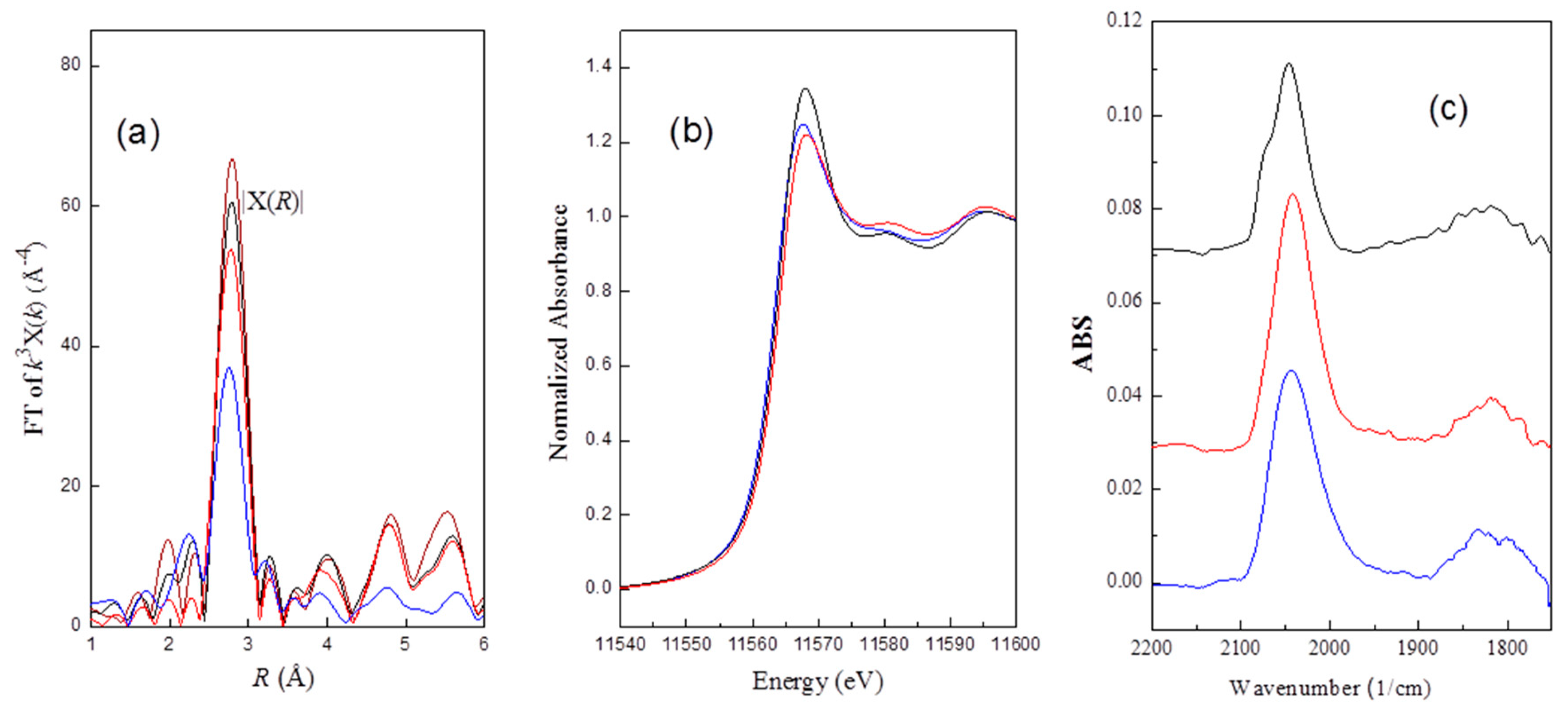
The intensity of the normalized threshold resonance of the absorption edge is in the order of: H2Pt(OH)6 ≈ (slightly higher than) Ptrd52 > PtCl4 ≈ Ptrd46 ≈ Ptrd56 > PtCl2. The results suggested a redispersed complex for Ptrd52 bearing about six ligands, in contrast to about four for Ptrd46 and Ptrd56.
Besides H
2Pt(OH)
6, a small peak appearing at postedge, 11,580 eV in the spectra of Cl contained reference standards and the redispersed samples. This peak was assigned as a hybridization peak by the authors in [
36], which could be arisen by the hybridization of the Pt d photoelectron state with the unoccupied atomic Cl 3d states, mediated by multiple scattering. The results suggested the presence of Cl neighboring with Pt clusters after oxychlorination.
2.3. Detail EXAFS Analysis
A k2-weighted Fourier transformation was performed on the EXAFS function over the range of 3.0 < k < 14.5 Å−1 for all samples. The major contributions were isolated by inverse Fourier transformation of the data in the ranges of 1.15 < r < 3.30 Å. For Ptrd56, the structural parameters characteristic of the Pt–Pt contribution were roughly determined by fitting the k3-weighted Fourier isolated EXAFS function in the range 6.0 < k < 13.0 Å−1 to de-emphasize the low-Z contribution. An EXAFS function calculated from these parameters was then subtracted from the raw data (Fourier isolated EXAFS function). The residual spectra were expected to represent the ligands of the redispersed complexes; for example, Pt–Cl. The EXAFS parameters of the Pt–Cl contribution was then estimated with the coordination distance that gave the best agreement with the Pt–Cl phase-corrected FT of the residual spectra.
To check other contributions, a difference file was calculated by subtracting Pt–Pt and Pt–Cl contributions from the experimental data [
37]. It is evident from the difference file that two other scatters were present, as indicted by the peaks at about 2.0 and 3.2 Å. These two peaks were assigned as characteristic peaks (Pt–O and Pt–Cl*) for Pt–OCl. The structural parameters characteristic of Pt–O and Pt–Cl* were then fitted with the residual data. The calculated EXAFS functions for Pt–O and Pt–Cl* were then subtracted from the raw data, and better parameters for Pt–Pt and Pt–Cl contributions were estimated. The refinement through this iteration was continued until a good overall agreement was obtained.
EXAFS analysis steps for Pt
rd46 and Pt
rd52 are similar to those for Pt
rd56, except that Pt–O was fitted first, because it is the main contribution of the EXAFS. The final fitting results are summarized in
Table 1. Based on the results, the Pt(O
s)
3–4(O–Cl)
2–3 (O
s represents support oxygen or hydroxyl oxygen) complex was suggested to be formed on γ-Al
2O
3 of Pt
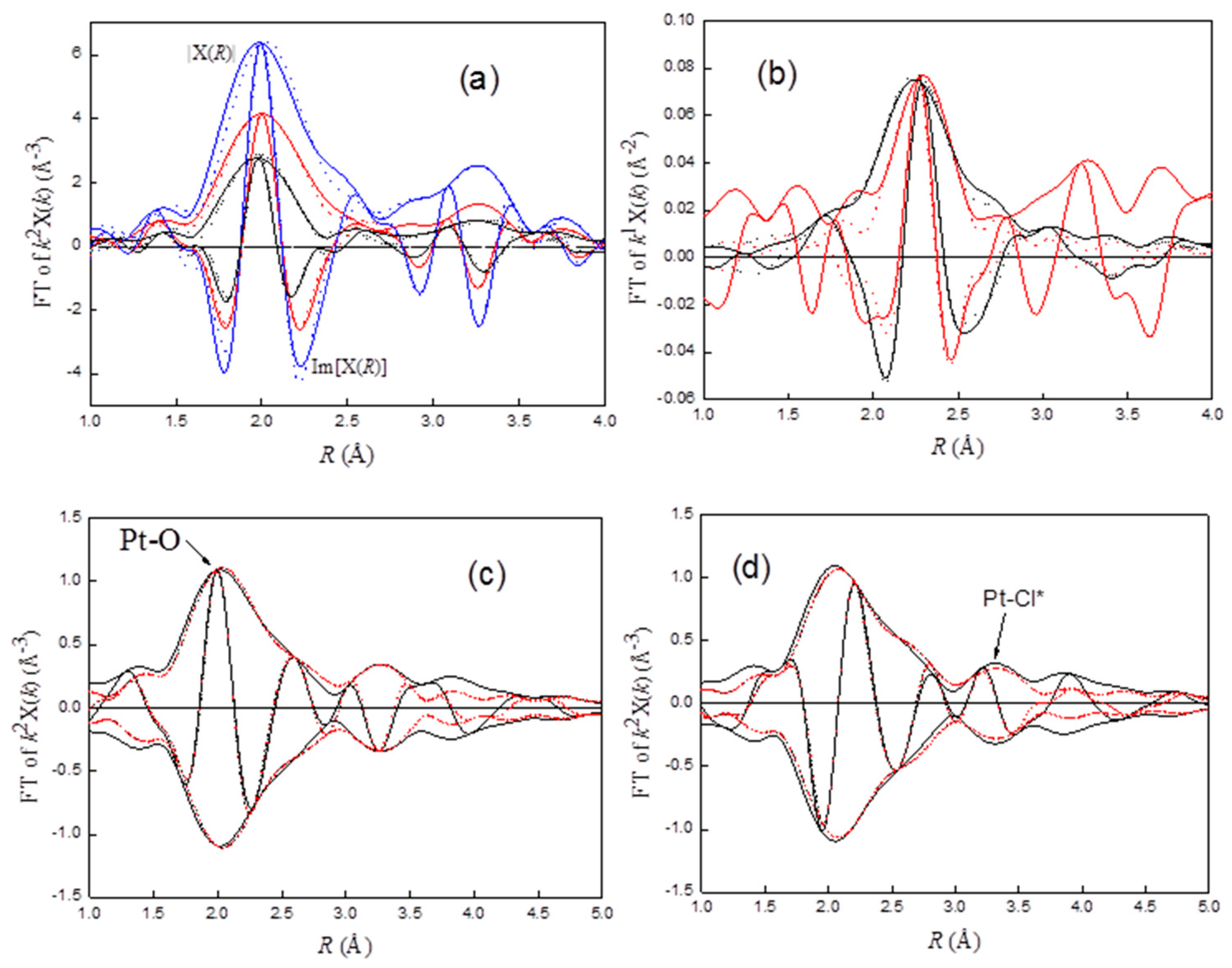
rd52. The comparisons of the experimental EXAFS characterizing Pt–O–Cl and Pt–Cl with the fits are shown in
Figure 3a,b, respectively, while the characteristic peak for Pt–O and Pt–Cl* are shown in
Figure 3c,d, respectively.
2.4. Oxychlorination Redispersion Mechanism Proposed Based on XAS
The redispersion of sintered catalysts involves the direct reaction between the Pt clusters and reactive gaseous components, such as Cl
2, HCl, O
2, and H
2O [
38,
39]. The EXAFS results showed that the formation of PtOCl species was accompanied by the fragmentation of Pt clusters (
Figure 1a,
Figure 3a, and
Table 1). The details of the chemistry—whereby the formation of PtOCl caused the dispersion of Pt clusters—are yet to be elucidated. Our experimental results suggest the role of an interaction between the redispersed complexes and supports.
In the reaction system, chlorine was formed by the reaction of HCl with oxygen (Equation (4) the reaction is a well-known Deacon reaction for the production of chlorine from hydrogen chloride. The chlorine molecules, Cl2, may be adsorbed on the active sites, S, of Pt/γ-Al2O3 and then dissociated to Cl⋅ (Equation (6)). The very reactive Cl⋅ then attacks PtO2 to form Pt–O–Cl species (Equation (7)). The species may peel off from Pt clusters and get trapped on the surface of the support (Equation (8)), thereby redispersing the sintered Pt clusters.
The interactions between Pt–O–Cl species and γ-Al
2O
3 can be characterized by the loss of multiple scattering effects of the Pt–Cl* peak. According to Teo [
40], multiple scattering in EXAFS is significant when atoms (e.g., Pt, O, Cl) are arranged in an approximately linear array. The photoelectron emitted from Pt is strongly forward-scattered by the intervening O atom, resulting in a significant amplitude enhancement. Another indication of multiple scattering effects is the phase shift of approximately 180° of the Pt–Cl* peak, which results in a negative peak located at the maximum of the magnitude of the Pt–Cl phase-corrected FT. As shown in
Figure 3d, FT corrected by Pt–Cl, a positive peak without amplitude enhancement, was observed at about 3.25 Å. These results suggest the interactions of Pt–O–Cl with support (Equation (8)); the peak appearing at about 3.8 Å for Pt
rd52 (solid black line,
Figure 3c) could be the characteristic peak of the interactions.
In summary, the possible reactions involved in the oxychlorination redispersion reaction were proposed by the following equations:
The reaction chemistry involved in the oxychlorination reaction is very complicated. The mechanism proposed is simplified, as there may be a variety of mechanisms, rather than just one. Specifically, the mechanism of the formation of reactive Cl⋅ (Equation (6)) and the roles of the support in redispersed complex formation remain unclear (Equation (7)).
Reactive Cl⋅ was suggested to be formed from the dissociation of Cl
2 (Equation (6)), whereas—inferred from the paper reported by Moser et al. [
41]—reactive Cl⋅ may also be formed by the dissociation of O
2 on the active sites, S (Equation (9)), followed by stripping proton from HCl (Equation (10)). The stripped proton was then associated with intermediate O⋅⋅S to form HO⋅⋅S (Equation (10)).
Pt clusters could be the active sites (S) responsible for the formation of reactive Cl⋅ However, reactive Cl⋅ could also be formed from the cleavage of chlorine molecular followed by the penetration of Cl⋅ into the lattice of PtO
2—hence, concomitant with Cl· formation, the cleaved Cl atom (Cl·) may replace the lattice oxygen atom of PtO
2 to form a Pt⋅⋅Cl bond (Equation (11)). The formation of a Pt–Cl bond was evidenced by the appearance of a PtCl characteristic peak of 2.3 Å for Pt
rd56 (
Figure 3b).
Moreover, based on the paper reported by Fung [
38,
39], chlorine could be dissociated on γ-Al
2O
3 to form HOCl or H
2O associated with the formation of a Al–Cl bond (Equations (12) and (13)). The interactions of PtO
2 with the surface chlorides may promote the formation Pt–OCl and/or Pt–Cl concomitant with the dissociation these Cl-containing species from Pt clusters. Some of the Al-bound Cl may be located at the interface between Pt clusters and support, resulting in an appearance of a Pt–Cl characteristic peak (
Figure 3b).
2.5. Effects of Operation Conditions on Redispersion Efficacy
The deacon reaction is mildly exothermic (Δ
H0 = −113.6 kJ mol
−1) and equilibrium-limited. Lower reaction temperatures favor the formation of Cl
2 [
42]. For Pt
sin redispersed at 520 °C, equilibrium chlorine concentration in the reaction system is lower than that which is redispersed at 460 °C, whereas more PtOCl species were formed (
Figure 3a,
Table 1) and a higher redispersion efficacy were observed (
Table 1). The results suggest that factors influencing the redispersion efficacy depend not only on the chlorine concentration, but also the dissociation of the chlorine molecule and the interactions between redispersed complexes and the support.
Even though increasing the temperature to 560 °C may prompt Cl
2 dissociation to enhance the redispersion efficacy, it also decreases the complex-support interactions and the available chlorine concentration, leading to a lower redispersion. Hence, as shown in
Table 1, CN
Pt–Pt of 4.3 for Pt
rd56 is the biggest among all three samples. Further increasing redispersion temperature, the mobile redispersed complexes may collide to form bigger particles, leading to Pt sintering, rather than redispersion.
DFT (density function theory) calculations indicated that an increased O coverage promotes stripping protons from HCl (Equation (10)) [
43,
44]. Hence, maintaining a higher O
2 content in the redispersion process could result in increased redispersion efficacy. Our experimental results show that sintered Pt clusters (Pt
sin) were almost fully redispersed under the redispersion conditions of temp = 520 °C, WHSV = 0.087, and Air/EDC = 30 to 40. As air/EDC was decreased to 20 (Pt
rd52,
Air/EDC=20), concomitant with the decrease of white line intensity (
Figure 2a), CN
Pt–Cl* decreases from 2.6 to 2.2, while CN
Pt–Pt increases from 1.7 to 1.9 (
Table 1).
PtO2 on the surface may react with Cl (Equation (7)) and/or HCl (Equation (5)). Increasing WHSV drives chlorine away from its equilibrium concentration, leading to an increase in the HCl/Cl2 ratio, which favors the formation of PtCl in the competitive reactions (Equations (5) and (7)).
When increasing WHSV from 0.07 to 0.187 (Pt
rd52,
WHSV=0.187), with the appearance of a PtCl characteristic peak at about 2.3 Å (
Figure 4b), CN
Pt–Cl* was decreased from 2.6 to 1.5 (
Table 1,
Figure 4a). Actually, some PtCl may also have been formed in Pt
rd46, Pt
rd52,
Air/EDC=20, and Pt
rd52. However, due to strong coupling between the characteristic peak of Pt–O (d
Pt–O ≈ 2.0 Å) and Pt–Cl (d
Pt–O ≈ 2.3 Å), the rather small Pt–Cl contribution for these samples could not be decoupled by the Fourier filter and a difference file-fitting technique. Hence, the estimation of structure parameters for the Pt–Cl contribution of these samples was not attempted.
2.6. Metal-Support Interface of the Re-Dispersed Pt/γ-Al2O3
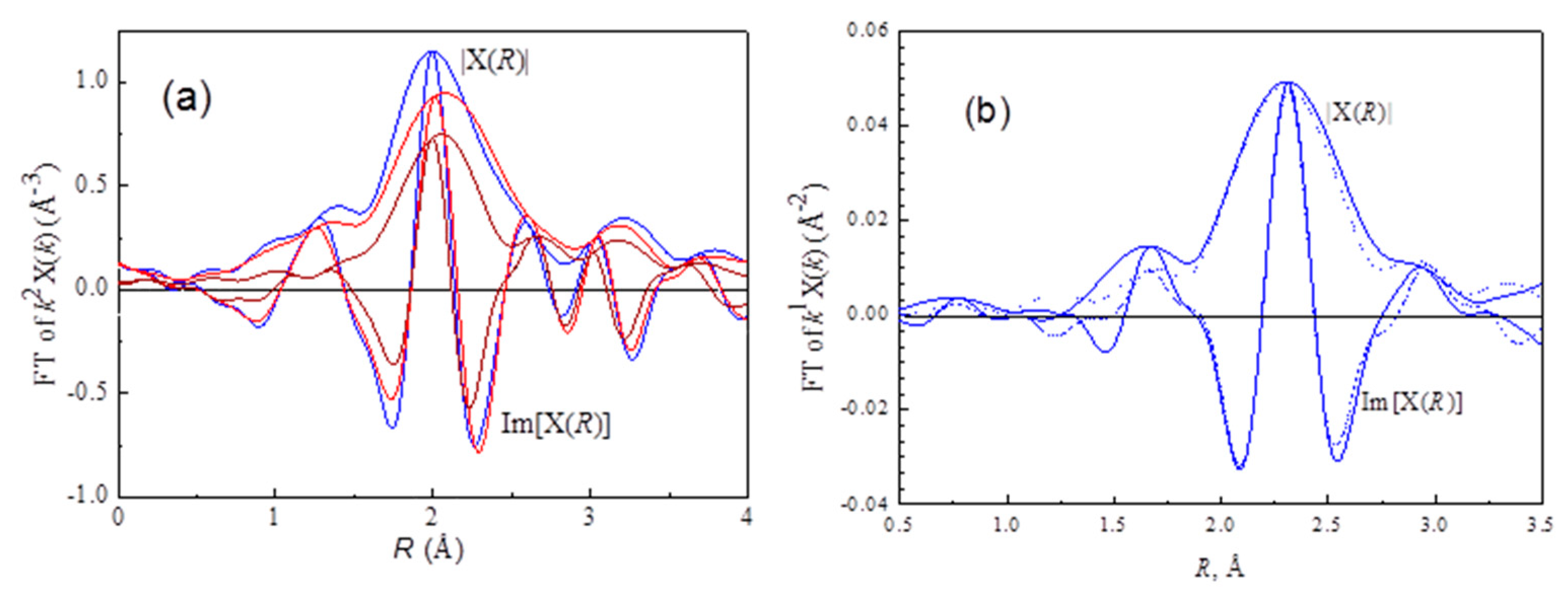
The Pt–Pt
k3 weighted phase- and amplitude-corrected Fourier transform characterizing Pt
sin Ptr
rd46, Ptr
rd52, and Ptr
rd56 samples are shown in
Figure 5a. The peaks at about 2.7, 3.9, 4.7, 5.6 Å are consistent with the first, second, third, and fourth shell for bulk FCC (faced centered cubic) platinum [
27]. These samples are characterized by a first-shell Pt–Pt contribution with a coordination number of 5.8, 7.2, 8.1, and 8.4, corresponding 0.9, 1.3, 2.2, and 3.1 nm for Ptr
rd52, Ptr
rd46, Ptr
rd56, and Pt
sin, respectively. The particle size for the redispersed samples (
Table 2) is inversely proportional to the Pt–O–Cl contribution (CN
Pt–Cl*,
Table 1), suggesting that the formation of Pt–O–Cl plays a key role in oxychlorination redispersion.
In investigating the metal-support interface by X-ray absorption spectroscopy, an EXAFS function calculated from structure parameters of the first shell Pt–Pt contribution was subtracted from the Fourier-isolated EXAFS function. The residual spectra were expected to represent platinum-support interactions. For Ptr
rd52, the residual spectra were contributed from Pt–O
l of the bond distance at 2.58 Å and Pt–O
s at 2.16 Å (
Table 2). In contrast, for Ptr
rd56, the residual spectra were contributed from Pt–Cl at 2.26 Å (
Table 2). These results suggested that Pt clusters on Ptr
rd52 were bonded to the oxygen of γ-Al
2O
3 support
, whereas those on Ptr
rd56 were bonded to Cl on support.
A metal-oxygen bond length of about the sum of the covalent radii of the metal and oxygen (about 2.1 Å) is normally observed when the catalyst is reduced at a high temperature (>450 °C). However, when the supported-metal catalysts are treated at low temperature in H
2, a longer Pt–O (2.5–2.7 Å) is observed. The longer distance could be due to the interactions between platinum and support oxide with a hydride presented at the interface between the metal and oxygen atoms [
46,
47].
Bazin et al. reported a strong correlation existing between the electronic density and the metal cluster size of a nanometer-scale through ab initio calculation [
48]. Based on the results, we expected that XANES intensity decreased with increasing Pt particle size. Except for the Ptr
rd46 sample, the experimental results were as expected. As shown in
Figure 5b, the intensity of the normalized threshold resonance edge decreases in the order: Ptr
rd56 > Ptr
rd52 > Ptr
rd46. The unexpected higher XANES intensity for Ptr
rd56 may be due to Pt–Cl interactions. Electrons withdrawn from Pt clusters to Cl atom result in an increase in XANES intensity.
2.7. FT-IR (Fourier-Transform Infrared)Spectroscopy Characterizing the CO Adsorbed on the Pt of the Re-Dispersed Pt/γ-Al2O3
The FT-IR (Fourier-transform infrared) spectra of the CO adsorbed on the re-dispersed catalyst samples were consistent with spectra reported for CO adsorbed on supported Pt catalysts [
49,
50]. The band peaking at about 2045 cm
−1 for all three samples was assigned as the terminal CO adsorbed on Pt clusters, and the broad absorption band located at about 1840 cm
−1 could be the characteristic peak for bridging CO adsorption (
Figure 5c).
In contrast to Ptr
rd46, Ptr
rd52 where it can be seen that the terminal peak is a rather symmetrical peak, an additional shoulder at about 2075 cm
−1 was observed for Ptr
rd56. The shift of the CO absorption bands suggest that the electron density of Pt is decreased by Pt–Cl interactions. The Cl atom abstracts electrons from Pt, which should thus decrease the number of Pt electrons available for π-bonding to the CO adsorbed on Pt clusters. The decrease in this backbonding from Pt to CO orbital results in a shift of CO to a high frequency [
51]. The decrease of electronic density of the Pt clusters on Ptr
rd56 due to the Pt–Cl interactions, suggested by FT-IR, is consistent with the XANES results.
A change in Pt electronic density due to Pt–Cl interactions may change catalytic properties. For example: (1) the decrease of electron density on Pt induced by metal-support or bimetallic interactions may decrease the affinity of H
2S for Pt clusters, thereby increasing the stability maintenance in sulfur-poisoning catalyst deactivation [
50,
52]; and (2) decreasing Pt electron density may demote the activation of both C=C and C=O functional groups in a hydrogenation reaction [
53]. Hence, an investigation of reduction parameters for different redispersed complexes and their effects on catalytic reactivity should be studied further.
3. Materials and Methods
3.1. Materials and Sample Preparation
The catalyst samples were prepared by using an impregnation technique. The γ-Al2O3 samples (A2U, γ-Al2O3, with a surface area of about 170 m2/g and particle size of about 1 mm, purchased from Osaka Yogyo, Japan) were brought in contact with Pt(NH3)4(NO3)2 (Strem, Newburyport, MA, USA, 99.9%) aqueous solution of 0.0061 mol/L, followed by evacuation (10−1–10−2 torr) at room temperature, and then calcined at 450 °C for 4 h. The resulting material was noted as PtO2/γ-Al2O3 and the samples were stored in vials under N2 environment before reduction. Prior to the study on the effects of reduction temperature, the sintered sample (Ptsin) was prepared by flowing water-containing (4000 ppm) H2 at 50 mL/min with temperature ramping from room temperature to 600 °C and held for 2 h.
The oxychlorination redispersion process for Pt/γ-Al2O3 was conducted in a continuous downflow fixed-bed reactor. The reactor was a stainless-steel tube with an inside diameter of 2.1 cm and volume of 94.0 mL. It was heated electrically and controlled by a PID (proportional, integral, and derivative) temperature controller with a sensor in the center of the catalyst bed. Acid gas in the effluent gas was neutralized by 1.0 N NaOH aqueous solution before purging.
Five grams of the Ptsin sample was mixed with an inert ceramic which was 0.2 cm in diameter at a ratio of 1:10, and the top of the reactor bed was filled with glass balls which were 1.6 mm in diameter to preheat and prevent channeling of the feed. The samples were pre-oxidized in flowing air at 500 °C prior to oxychlorination reaction. The redispersion process was then carried out at an Air/EDC mole ratio of 30, and WHSV of 0.07 h−1 for 16 h at three different temperatures of 460, 520, and 560 ± 15 °C, and the samples were noted as Ptrd46, Ptrd52, and Ptrd56.
In order to study the effects of the Air/EDC ratio and the residence time of the reaction on the structure of the redispersed complex, the redispersion process was also performed at 520 °C for 16 h with WHSV of 0.07 h−1, Air/EDC of 20, and with WHSV of 0.187 h−1, Air/EDC of 30, respectively. These two samples were noted as Ptrd52, Air/EDC=20 and Ptrd52, WHSV=0.187.
3.2. X-ray Absorption Spectroscopy
The X-ray absorption measurements were performed on the wiggler beamline BL17C at the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan). The electron storage ring was operated at an energy of 1.5 GeV, and a beam current between 120 and 200 mA. A Si(111) double-crystal monochromator was employed for energy selection, and higher harmonic radiation was rejected by mirrors. The transmission geometry was arranged using gas-filled ionization chambers to monitor the intensities of the incident, and transmitted x-ray beams. For the collection of a full spectrum, the energy was scanned from 200 eV below the Pt LIII absorption edge (11,564 eV) to 1200 eV above the edge.
The Ptrd46, Ptrd52, and Ptrd56 catalyst samples were pulverized into powder, pressed into wafers, and loaded into an EXAFS cell. The cell was connected to a gas-handling manifold for in situ treatment. After purging with N2, EXAFS spectra were taken. Catalyst samples were then reduced by flowing H2 of 20 mL/min with temperature ramping from room temperature to 400 °C at a rate of 5 °C/min. After the temperature reached the setting temperature, it was maintained for 1 h. The cell was allowed to cool to room temperature, and the EXAFS spectrum was measured. The reduced samples for Ptrd46, Ptrd52, and Ptrd56 were noted as Ptrrd46, Ptrrd52, and Ptrrd56, respectively.
Data reduction and data analysis were performed with the XDAP code developed by Vaarkamp et al. [
37]. Standard procedures were followed to extract the EXAFS data from the measured absorption spectra. The pre-edge was approximated by a modified Victoreen curve, and the post-edge background was subtracted using cubic spline routines. Normalization was performed by dividing the background-subtracted spectrum by the height at 50 eV above the edge [
22,
31,
37]. Phase shifts and backscattering amplitude functions of Pt–Pt, Pt–O and Pt–Cl were generated by the FEFF8 code [
34], and used as reference files for EXAFS data analysis.
3.3. Characterization of Catalyst Samples by FT-IR
Infrared Fourier transform spectra of surface species were recorded with a Shimadzu IR Prestige-21 instrument (Kyoto, Japan), having a spectral resolution of 2 cm−1. Catalyst samples were pulverized into powder and loaded into a diffuse-reflectance infrared Fourier Transform spectroscopy (DRIFT) IR cell, and treatments were done in situ. To characterize the CO-adsorbed catalyst sample, before FT-IR measurement, samples were reduced in H2 flowing at about 20 mL/min at 400 °C for 1 h. After the reduction, the sample was cooled down to room temperature, and CO (flowing at 20 mL/min at 1 atm) was introduced into the cell and maintained for about 20 min. After the CO treatment, the cell was evacuated to a pressure of approximately 10−2–10−3 Torr, and IR spectra were recorded.
4. Conclusions
The fundamental understanding of the detailed oxychlorination redispersion chemistry could help us to develop and/or improve the processes for regeneration of deactivated supported-metal catalysts. In this study, a nearly uniform redispersion (fully redispersed) complex was prepared at an Air/EDC mole ratio of 30, WHSV of 0.07 h
−1 for 16 h at 520 °C. By fully characterizing the structure of this complex, a possible mechanism for oxychlorination redispersion was proposed. The formation of a O–Cl ligand concomitant with the disruption of Pt clusters in the redispersion process, as was evidenced by EXAFS, suggested that chlorine formed from the reaction of HCl with oxygen plays the main role in the redispersion process. Increasing the operation temperature prompts Cl
2 dissociation, thereby enhancing redispersion of the sintered Pt clusters, whereas this positive factor may be counteracted by the reduction in equilibrium conversion of HCl to Cl
2, the decrease in complex-support interactions, and the loss in support surface area. Our experimental results showed that the optimal redispersion temperature is at about 520 °C. By decreasing the redispersion temperature from 520 °C to 460 °C, the coordination number of Pt–Cl* (Pt–O–Cl), CN
Pt–Cl* decreased from 2.6 to 1.4. On the other hand, by increasing the redispersion temperature from 520 °C to 560 °C, Pt–Cl was formed with the decrease of CN
Pt–Cl* to 1.5. Inferred from our experimental results, a lower optimal redispersion temperature for Pt/zeolite and Pt/SiO
2, as opposed to that for Pt/γ-Al
2O [
4,
54,
55], were suggested due to the lower affinity of the redispersed complexes to these supports. Moreover, considering the structure of redispersed complexes and their interactions with supports, the residence time of oxychlorination reaction is also an important parameter that affects redispersion efficacy. High WHSV decreases Cl
2 conversion, which decreases Pt–O–Cl while increasing Pt–Cl formation. After reduction in H
2 at 400 °C, Pt–Cl remains intact. The Cl atom bonded to Pt may abstract electrons from Pt, resulting in a decrease in electronic density of the Pt clusters that could lead to a change in catalyst reactivity. To develop an efficient and economical regeneration process, optimal reduction conditions for the redispersed complexes of different structure should be investigated.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}